

Abstract
Background
National Cancer Institute-Children’s Oncology Group Pediatric Molecular Analysis for Therapy Choice assigns patients aged 1-21 years with refractory solid tumors, brain tumors, lymphomas, and histiocytic disorders to phase II trials of molecularly targeted therapies based on detection of predefined genetic alterations. Patients whose tumors harbored EZH2 mutations or loss of SMARCB1 or SMARCA4 by immunohistochemistry were treated with EZH2 inhibitor tazemetostat.
Methods
Patients received tazemetostat for 28-day cycles until disease progression or intolerable toxicity (max 26 cycles). The primary endpoint was objective response rate; secondary endpoints included progression-free survival and tolerability of tazemetostat.
Results
Twenty patients (median age = 5 years) enrolled, all evaluable for response and toxicities. The most frequent diagnoses were atypical teratoid rhabdoid tumor (n = 8) and malignant rhabdoid tumor (n = 4). Actionable alterations consisted of SMARCB1 loss (n = 16), EZH2 mutation (n = 3), and SMARCA4 loss (n = 1). One objective response was observed in a patient with non-Langerhans cell histiocytosis with SMARCA4 loss (26 cycles, 1200 mg/m2/dose twice daily). Four patients with SMARCB1 loss had a best response of stable disease: epithelioid sarcoma (n = 2), atypical teratoid rhabdoid tumor (n = 1), and renal medullary carcinoma (n = 1). Six-month progression-free survival was 35% (95% confidence interval [CI] = 15.7% to 55.2%) and 6-month overall survival was 45% (95% CI = 23.1% to 64.7%). Treatment-related adverse events were consistent with prior tazemetostat reports.
Conclusions
Although tazemetostat did not meet its primary efficacy endpoint in this population of refractory pediatric tumors (objective response rate = 5%, 90% CI = 1% to 20%), 25% of patients with multiple histologic diagnoses experienced prolonged stable disease of 6 months and over (range = 9-26 cycles), suggesting a potential effect of tazemetostat on disease stabilization.
EZH2 (enhancer of zeste homologue-2) is the catalytic subunit of the polycomb repressive complex 2 (PRC2), a chromatin-based gene regulatory complex responsible for the mono-, di-, and trimethylation of histone 3 lysine 27 (H3K27) (1). Abnormal H3K27 trimethylation is postulated to be oncogenic in multiple human cancers, including non-Hodgkin lymphoma, SMARCB1-deficient tumors, carcinomas, cutaneous melanoma, gliomas, medulloblastoma, and ependymoma (2,3). EZH2 gain-of-function mutations were initially identified in non-Hodgkin lymphoma, the most frequent mutated residues being tyrosine 646 (Y646), alanine 682, and alanine 692 (A682 and A692, respectively) (4,5).
First noted in 1988, a natural antagonistic relationship between the PRC2 and the SWItch/Sucrose Non-Fermentable (SWI/SNF) complexes was confirmed in multiple tumor models (6,7). Loss of either of the SWI/SNF complex subunits, SMARCB1 (also known as INI1) and SMARCA4, has been shown to result in enforced PRC2 signaling and persistent expression of stem cell–associated programs. SMARCB1- or SMARCA4-inactivating mutations underlie the biology of rhabdoid tumors, including atypical teratoid rhabdoid tumors (ATRT) and malignant rhabdoid tumors (MRT), in addition to epithelioid sarcomas (ES) and renal medullary carcinomas (RMCs) (8). Furthermore, genetic suppression of EZH2 has been shown to block tumor formation driven by SMARCB1 loss (7).
Tazemetostat is a selective EZH2 inhibitor that inhibits both wild-type and mutant EZH2 (half-maximal inhibitory concentrations [IC50s] ranging in the 2-38 nM). Preclinical studies have demonstrated cell growth inhibition, induction of apoptosis and differentiation, and dose-dependent regression in a variety of human cancer cell lines and tumor models, including lymphoma, MRT, and SMARCA2/SMARCA4-negative small cell carcinoma of the ovary with hypercalcemia (9-13). In the phase I/II, first-in-human trial (NCT01897571) in adult patients with advanced solid tumors, the recommended pediatric phase II dose was determined to be 800 mg twice daily (14), with tolerable adverse events (AEs) most frequently (>15%) being asthenia, anemia, anorexia, muscle spasms, nausea, and vomiting. Dose exposure was correlated with target inhibition (decreased H3K27Me3 inhibition by immunohistochemistry of skin). Interestingly, 5 of 13 patients (38%) with SMARCB1 or SMARCA4 loss showed clinical benefit, including a complete response in a patient with MRT, a partial response (PR) in a SMARCA4- negative MRT of the ovary, and prolonged stable disease in 2 patients with ES and 1 patient with SMARCA4-negative MRT of the ovary.
The pediatric phase I study of tazemetostat (EZH-102, NCT02601937) was conducted in children with relapsed or refractory MRTs, including ATRT, other SMARCB1-deficient tumors, and synovial sarcoma. Objective responses were seen in 14% of the dose expansion cohort population, with a 24% response in ATRT patients and comparable response rates in patients with chordoma and ES (15). In January 2020, tazemetostat was granted FDA approval for the indication of patients aged 16 years and older with ES in whom complete resection is not feasible. In June 2020, approval was granted for adult patients with relapsed or refractory follicular lymphomas that harbor an EZH2 mutation and who have received at least 2 prior systemic therapies.
The National Cancer Institute-Children’s Oncology Group Pediatric Molecular Analysis for Therapy Choice (NCI-COG Pediatric MATCH) trial was activated in July 2017 to provide a national framework for histology-agnostic trials of molecularly targeted therapies in biomarker-selected populations (16,17). Centralized tumor sequencing is performed through the Pediatric MATCH screening protocol, allowing enrollment of patients from COG institutions in the United States. Patients whose tumors harbor a predetermined actionable genetic alteration are eligible for treatment in a phase II subprotocol trial. Herein, we report the results of subprotocol arm C of the NCI-COG Pediatric MATCH: EZH2 inhibitor tazemetostat for the treatment of patients with tumors harboring alterations in EZH2 or members of the SWI/SNF complex (NCT03213665).
Methods
Study design and eligibility
The NCI-COG Pediatric MATCH screening protocol and all treatment subprotocols were reviewed by the NCI Central Institutional Review Board. The study was conducted in accordance with the Declaration of Helsinki. Written informed consent was obtained from parents or guardians, and assent was also obtained from the patient where appropriate. Details of the screening protocol (including details of tumor molecular testing and subprotocol assignment) and results of the first completed treatment subprotocol (Arm E, selumetinib) were previously published (18,19). Any tumor or histiocytosis histology was eligible for enrollment. The actionable alterations for Arm C (tazemetostat; NCT03213665) were defined as 1) 2 deleterious mutations in either SMARCB1 or SMARCA4, 2) immunohistochemical loss of SMARCB1 or SMARCA4, or 3) hotspot EZH2 mutations (Y646C, Y646F, Y646H, Y646N, Y646S, A682G, A682V, or A692V) (Supplementary Table 1, available online).
In addition to the requirement for detection of an actionable mutation by MATCH screening, criteria for tazemetostat treatment included age 12 months and older, and 21 years and younger, Karnofsky or Lansky performance score at least 50%, radiographically measurable disease, and adequate organ function. Patients were excluded from treatment if they had an uncontrolled infection, abnormal morphologic abnormalities or cytopenias on complete blood count, prior history of myeloid malignancy or T-lymphoblastic lymphoma (T-LBL) or acute lymphoblastic leukemia, a history of solid organ transplantation, or concomitant use of strong CYP3A4-inducing or inhibiting agents.
Tazemetostat was initially orally administered at the recommended pediatric phase II dose of 1200 mg/m2 per dose twice daily (20). Dosing for patients with extracranial tumors was reduced to 520 mg/m2 per dose twice daily in response to the report of secondary T-LBL in a child being treated on a non-MATCH trial; objective response was previously reported at this dose level in the non-MATCH trial. Patients received drug continuously with each cycle lasting 28 days. Patients were eligible to receive therapy for up to 2 years (26 cycles) if there was no evidence of progressive disease or toxicity that met protocol defined criteria for discontinuation of protocol therapy.
AEs and dose modifications
AEs were reported according to the NCI Common Terminology Criteria for Adverse Events (CTCAE) version 5.0. All patients who received at least 1 dose of protocol therapy were considered evaluable for toxicity. Dose limiting toxicity (DLT) was defined differently for hematological and nonhematological toxicity. Treatment could be withheld for DLTs for up to 14 days. If the DLT resolved to baseline or eligibility parameters within 14 days of discontinuing therapy, the patient could resume tazemetostat at a 25% reduced dose; if not, the patient was removed from protocol therapy. If a recurrent or second DLT occurred at a reduced dose of tazemetostat, the patient was removed from protocol therapy. Bromide levels were monitored as tazemetostat is formulated as a bromide salt.
Measurement of response
Any eligible patient who received at least 1 dose of protocol therapy was evaluable for response. Tumor disease evaluations were obtained every other cycle for 3 occurrences, then every 3 cycles. For documentation of objective response (complete response or PR), confirmatory scans were required to be obtained after the next consecutive cycle. The revised Response Evaluation Criteria in Solid Tumors [RECIST, version 1.1 (21)] was used to determine response and progression for solid tumors and 2-dimensional analysis for central nervous system (CNS) tumors. Central review was required for any patient who was deemed by site to have a complete response, PR, or prolonged stable disease, defined as 6 months and longer, and these results were used to determine final assessment of response.
Statistical considerations
The study’s primary endpoint was overall response rate (ORR). The primary study cohort used a single stage design with a minimum of 20 patients. The ORR was compared against a null benchmark value of 5%. With this design (α = 10%), the power was 90% to detect an improvement in response rate from 5%, if the treatment was ineffective, to 25% if the agent was sufficiently effective to warrant further study.
The study’s secondary endpoints included progression-free survival (PFS), defined as time from initiation of protocol therapy until the occurrence of disease progression, disease recurrence, or death from any cause. PFS along with the 95% confidence intervals (CIs) was estimated using the Kaplan-Meier method. Fisher exact tests were used to evaluate the association between patient’s baseline categorical characteristics and treated or untreated cohorts. Continuous data were assessed by Mann-Whitney U-test. All statistical analyses were conducted in SAS (version 9.4) or R (version 4.1).
Results
Patient assignment and characteristics
Between October 2017 and July 2020, 38 patients from 27 study sites had an actionable mutation detected by MATCH screening, and 32 were assigned to Arm C tazemetostat (Figure 1); 6 patients were not assigned during the period when the trial was on hold after report of a patient developing T-LBL on the pediatric phase I clinical trial. Of the 32 patients assigned to arm C, 20 (from 17 different sites) enrolled in the trial and all received protocol therapy. The most common reasons provided for assigned patients not enrolling in the study were worsening clinical status (n = 6) and trial ineligibility (n = 4); the medical team and family of 2 patients chose a different treatment option outside of Pediatric MATCH. All 20 patients treated on arm C were evaluable for response and toxicities. Data as of December 31, 2021, were used for these analyses.
Figure 1.
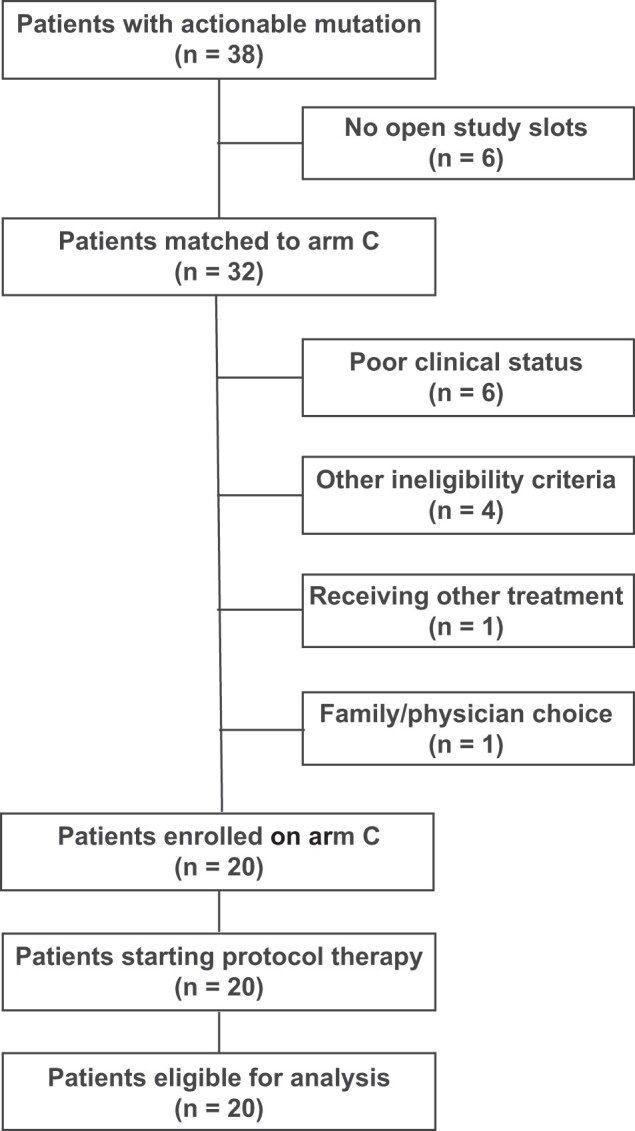
Patient flow diagram of National Cancer Institute-Children’s Oncology Group Pediatric Molecular Analysis for Therapy Choice (NCI-COG Pediatric MATCH) arm C. Patient flow diagram of NCI-COG Pediatric MATCH subprotocol C. Other reasons for ineligibility (n = 1 each) were lack of measurable disease, history of acute myelogenous leukemia, diagnosis of T-cell lymphoma, and prior treatment with tazemetostat.
Demographics and characteristics of the 32 patients assigned to arm C are shown in Table 1. No statistically significant demographic differences were noted between patients who were treated on arm C (n = 20) and those who did not enroll (n = 12). Treated patients ranged in age from 1 to 20 years (median, 5 years) with a slight predominance of male patients. ATRT and MRT were the most common diagnoses for assigned patients (15/32, 46.9% and 6/32, 18.8%, respectively) as well as those who were treated (8/20, 40.0% and 4/20, 20.0%) (Table 1; Figure 2; Supplementary Figure 1, available online). Other diagnoses in treated patients were Ewing sarcoma (n = 2), ES (n = 2), and 1 patient each with RMC, hepatocellular carcinoma, ependymoma, and non-Langerhans cell histiocytosis. Prior therapy of treated patients is listed in Supplementary Table 2 (available online).
Table 1.
Demographics and characteristics of 32 patients assigned to NCI-COG Pediatric MATCH Trial arm Ca
| Treated |
Overall |
P | ||
|---|---|---|---|---|
| Characteristics | Yes (n = 20) | No (n = 12) | Assigned patients (n = 32) | |
| Age at screening protocol enrollment, y | .98 | |||
| Mean (SD) | 8.7 (6.94) | 8.42 (7.25) | 8.59 (6.95) | |
| Median [Min, Max] | 5.00 [1.0, 20.0] | 5.5 [1.0, 21.0] | 5.0 [1.0-21.0] | |
| Sex | .72 | |||
| Female | 7 (35.0%) | 5 (41.7%) | 12 (37.5%) | |
| Male | 13 (65.0%) | 7 (58.3%) | 20 (62.5%) | |
| Race | .51 | |||
| Asian | 0 (0%) | 1 (8.3%) | 1 (3.1%) | |
| Black or African American | 3 (15.0%) | 3 (25.0%) | 6 (18.8%) | |
| Native Hawaiian or Pacific Islander | 1 (5.0%) | 0 (0%) | 1 (3.1%) | |
| White | 16 (80.0%) | 8 (66.7%) | 24 (75.0%) | |
| Ethnicity | .20 | |||
| Hispanic or Latino | 3 (15.0%) | 5 (41.7%) | 8 (25.0%) | |
| Not Hispanic or Latino | 16 (80.0%) | 7 (58.3%) | 23 (71.8%) | |
| Not reported/unknown | 1 (5.0%) | 0 (0%) | 1 (3.1%) | |
| Diagnosis | .83 | |||
| CNS ATRT | 8 (40.0%) | 7 (58.3%) | 15 (46.9%) | |
| Malignant rhabdoid tumor | 4 (20.0%) | 2 (16.7%) | 6 (18.8%) | |
| Ewing sarcoma | 2 (10.0%) | 1 (8.3%) | 3 (9.3%) | |
| Epithelioid sarcoma | 2 (10.0%) | 0 (0%) | 2 (6.3%) | |
| Non-Langerhans cell histiocytosis | 1 (5.0%) | 0 (0%) | 1 (3.1%) | |
| Renal medullary carcinoma | 1 (5.0%) | 0 (0%) | 1 (3.1%) | |
| Hepatocellular carcinoma | 1 (5.0%) | 0 (0%) | 1 (3.1%) | |
| Ependymoma | 1 (5.0%) | 0 (0%) | 1 (3.1%) | |
| Chordoma | 0 (0%) | 1 (8.3%) | 1 (3.1%) | |
| Peripheral T-cell lymphoma | 0 (0%) | 1 (8.3%) | 1 (3.1%) | |
The table reflects data collected at screening enrollment. Data are reported as No. (%) unless otherwise indicated. CNS ATRT = central nervous system atypical teratoid rhabdoid tumor; NCI-COG Pediatric MATCH = National Cancer Institute-Children’s Oncology Group Pediatric Molecular Analysis for Therapy Choice.
Figure 2.

Diagnoses and genetic alterations of arm C. Tumor variants detected by gene and patient. The genes with the variants are indicated for each patient (each column represents a patient). Cooccurring mutations, histology, age, and treated patients as indicated. “Other tumor histologies”: 1 patient each: hepatocellular carcinoma, renal medullary carcinoma, ependymoma, and non-Langerhans cell histiocytosis (n = 1). PD = progressive disease; PR = partial response; SD = stable disease.
Landscape of actionable alterations
The actionable tumor alterations detected in assigned and treated patients are shown in Figure 2 and Supplementary Figure 1 (available online). SMARCB1 loss was most common (27/32 [84%] assigned, 16/20 [80%] treated) followed by EZH2 mutation (4/32 [12%] assigned, 3/20 [15%] treated) and a single treated patient with SMARCA4 loss. Few co-occurring alterations were detected, with TP53 as the only recurrently mutated gene (3/32 assigned, 2/20 treated) (Figure 2). Germline cancer susceptibility gene variants were detected in 2 treated patients: a CHEK2 frameshift variant in a 10 -year-old patient with hepatocellular carcinoma with SMARCB1 loss and a SMARCB1 stop variant in a 3-year-old patient with ATRT (Supplementary Table 3, available online).
Adverse events
Of the 20 enrolled and treated patients, all of whom were evaluable, 6 (30%) experienced a grade 3 or higher AE with possible or probable attribution to tazemetostat (Table 2, Supplementary Table 4, available online). Two patients experienced grade 3 anemia (1 treated with 1200 mg/m2 per dose, the other with 520 mg/m2 per dose). One patient experienced a grade 3 intracranial hemorrhage and another a grade 3 lung infection (treated at 520 mg/m2 per dose) in their first cycle of treatment. One patient developed a grade 3 alkaline phosphatase increase in the follow-up period as their only grade 3 AE. One patient developed multiple grade 3 AEs during cycle 1, which were dyspnea, abdominal pain, increased alanine transaminase (ALT), and decreased platelet count. This same patient had a grade 3 increased gamma-glutamyl transferase (GGT) in the follow-up period. Of the 14 patients who had radiographs showing open tibial growth plates before the start of therapy, 3 remained on therapy long enough to undergo repeat tibial growth plate monitoring, and none of these patients had evidence of growth plate thickening. Three patients experienced serum bromide elevations. No patients on this trial developed a secondary malignancy during this trial or in the follow-up period.
Table 2.
Grade 3 or above adverse events (AEs) potentially associated with the protocol treatment (attribution: possible, probable, or definite) according to Common Terminology Criteria for Adverse Events (CTCAE) v5.0a
| Type of AE | Grade 3 | Grade 4 or 5 |
|---|---|---|
| Dose level 1200 mg/m2, (N = 13) | ||
| Abdominal pain | 1 (7.7%) | 0 |
| Alanine aminotransferase increased | 1 (7.7%) | 0 |
| Alkaline phosphatase increased | 1 (7.7%) | 0 |
| Anemia | 1 (7.7%) | 0 |
| Dyspnea | 1 (7.7%) | 0 |
| GGT increased | 1 (7.7%) | 0 |
| Intracranial hemorrhage | 1 (7.7%) | 0 |
| Platelet count decreased | 1 (7.7%) | 0 |
| Dose level 520 mg/m2, (N = 7) | ||
| Anemia | 1 (14.3%) | 0 |
| Lung infection | 1 (14.3%) | 0 |
Data are reported as number of patients (percentage of the treated patients) separately by dose level. GGT = gamma-glutamyl transferase.
Evaluation of activity and efficacy
Objective response was confirmed in 1 patient (ORR = 5%, 90% CI = 1% to 20%): a PR in a patient with SMARCA4-deficient non-Langerhans cell histiocytosis (Rosai Dorfman disease) who received 1200 mg/m2 per dose twice daily for 26 cycles (maximal decrease: 91% in 2-dimensional size; Table 3, Figures 3 and 4). Prolonged stable disease defined as 6 months and longer was observed in an additional 4 patients, all of whom had tumors with SMARCB1 loss (Table 3). Of these, 2 patients received 520 mg/m2 dosing. One patient with ES completed 26 cycles. Almost all other patients came off tazemetostat within 2 cycles. The median number of cycles completed for the treated cohort was 2 (range = 1-26). Six-month PFS was 35% (95% CI = 15.7% to 55.2%), and 6-month overall survival (OS) was 45% (95% CI = 23.1% to 64.7%) (Figure 3, B and C).
Table 3.
Clinical and genetic details of patients with objective response or prolonged stable diseasea
| Patient ID |
Age (y) | Histology | Genetic alteration | Other mutated genes | Tazemetostat dose | Best response | No. cycles | PFS (mo) | Reason for off- study treatment |
|---|---|---|---|---|---|---|---|---|---|
| 11 | 3 | ATRT | SMARCB1 loss | TSC1 | 1200 mg/m2 | SD | 13 | 13.7 | Physician discretion |
| 4 | 18 | ES | SMARCB1 loss | None | 1200 mg/m2 | SD | 9 | 7.8 | PD |
| 7 | 15 | ES | SMARCB1 loss | None | 520mg/m2 | SD | 26 | 28.3 | Study completion |
| 6 | 19 | RMC | SMARCB1 loss | None | 520mg/m2 | SD | 12 | 10.2 | PD |
| 3 | 10 | Histiocytosis (Non-LCH) | SMARCA4 loss | None | 1200 mg/m2 | PR | 25+ | 42.1 | Physician discretion |
aATRT = atypical teratoid rhabdoid tumor; ES = epithelioid sarcoma; LCH = Langerhans cell histiocytosis; PD = progressive disease; PFS = progression-free survival; RMC = renal medullary carcinoma.
Figure 3.

Outcomes of patients treated on arm C. A) Swimmer’s plot for enrolled patients. B) Kaplan-Meier curve for progression-free survival. C) Kaplan-Meier curve for overall survival.
Figure 4.

Response of patient with non-Langerhans cell histiocytosis over 26 cycles of tazemetostat treatment. Black-outlined arrow denotes the lesion. Percent decrease was calculated by 2-dimensional measurements.
Discussion
In this study we report the results of the NCI-COG Pediatric MATCH phase II subprotocol C with tazemetostat for the treatment of patients with tumors harboring gain-of-function mutations in EZH2 or loss-of-function mutations in SWI/SNF complex subunit members SMARCB1 and SMARCA4. The nationwide molecular screening strategy used in Pediatric MATCH was effective at identifying children and young adults eligible for treatment with tazemetostat, with 32 patients from 18 different COG study sites assigned to arm C. Despite rhabdoid tumors being most common in young children (median age of 3 years for the 12 ATRT-treated and MRT-treated patients), adolescent and young adult patients (aged 15-21 years) were also well represented, comprising 35% of the cohort. Although the expected histologies of ATRT, MRT, and ES were most frequent, patients with diverse tumor types were assigned (10 different diagnoses in 32 patients) and treated (8 diagnoses in 20 patients) in Pediatric MATCH, demonstrating the strength of a national, disease-agnostic screening protocol in matching patients to a precision medicine trial.
As observed in other pediatric and adult trials of tazemetostat, the agent was relatively well tolerated (14,20,22,23). Toxicities seen in Pediatric MATCH were consistent with previous studies, including thrombocytopenia, anemia, and liver laboratory abnormalities (Table 2). In this cohort of children with relapsed tumors harboring EZH2 mutations or loss of SMARCB1 or SMARCA4, the primary efficacy endpoint was not reached with single agent tazemetostat, which produced only 1 objective response (ORR = 5%). This was a PR in a single patient with non-Langerhans cell histiocytosis and evidence of tumor SMARCA4 loss by IHC who received tazemetostat for 2 years with minimal side effects. Although infrequent, variants in SMARCA4 have previously been reported in Rosai Dorfman disease (24,25), highlighting the value of molecular profiling such as that used in Pediatric MATCH to identify rare variants occurring in uncommon cancer types. In addition to this PR, prolonged stable disease of 6 months or longer (range = 9-26 cycles) was observed in 4 of 20 (20%) treated patients, including 2 patients at the lower dose of 520 mg/m2, suggesting a potential role of tazemetostat in disease stabilization, specifically as a maintenance therapy or in combination with other agents.
The diversity of tumor diagnoses and molecular alterations in this 20-patient Pediatric MATCH cohort makes conclusions regarding the efficacy of tazemetostat for specific tumor types and/or alterations challenging. Prolonged disease stabilization was seen in 4 of 16 (25%) patients with SMARCB1-deficient tumors (including 1 ATRT patient, both patients with ES, and the only patient with RMC), despite the lack of objective responses in this group. The initial FDA approval of tazemetostat in January 2020 for treatment of ES with SMARCB1 loss was based on an ORR of 15% (95% CI = 7% to 26%) and the observation that 67% of responses lasted 6 months or longer (22). Preliminary results from the expansion phase of a phase I multicenter pediatric study of tazemetostat (NCT2601937) have included an ORR of 14% (9/63) including 5 of 21 ATRT, 2 of 6 chordomas, 2 of 9 ES, and 0 of 21 non-CNS rhabdoid tumors (15). In combination, these data suggest that among pediatric cancer types with SMARCB1 loss, responses to tazemetostat are more frequently observed in ATRT and ES compared with non-CNS MRT (responses in 0/4 patients in Pediatric MATCH and 0/21 patients in the non-MATCH pediatric trial). One consideration, however, is the potential impact of the tazemetostat dose reduction from 1200 mg/m2 per dose to 520 mg/m2 per dose twice daily for patients with non-CNS disease on response outcomes and efficacy. On our study, 3 of 5 of the patients who had PR or stable disease were treated at the 1200-mg/m2 per dose regimen.
All 3 patients with activating EZH2 mutations treated in Pediatric MATCH (Ewing sarcoma × 2, ependymoma) came off study at 2 cycles for tumor progression, in marked contrast to the ORR of approximately 70% observed for adult patients with refractory EZH2-mutant follicular lymphomas (that led to FDA approval for that cancer type). Although EZH2 (and the PRC2 complex, more broadly) has been proposed as a potential target in diverse pediatric cancer types (12,26), its role as a primary biological driver of tumorigenesis in Ewing sarcoma and ependymoma is unknown, and our small cohort size limits ability to generalize these findings.
Other studies of tazemetostat have revealed that tumor regression in response to alteration of the chromatin landscape can be a relatively slow process: 3.7 months to first response in patients with follicular lymphoma (23) and 3.9 months in patients with ES (22). In this refractory cancer cohort where most patients did not receive more than 2 cycles of study drug due to tumor progression, the potential impact of tazemetostat monotherapy appears limited, though encouraging activity was noted on the pediatric phase 1 EZH-102 trial (15). A combination therapy strategy has proven beneficial for another class of epigenetic modifiers: DNMT3A inhibitors with venetoclax (27). Given the signals of activity observed for this generally well-tolerated oral targeted agent, future trials that incorporate tazemetostat in a combinatorial approach with conventional chemotherapy, radiation therapy, or other biologic or immunologic agents may offer meaningful clinical benefit to patients with these rapidly aggressive tumors and few curative treatment options. The critical role that EZH2 plays in the immune system (28), particularly in rhabdoid tumors, suggests that the combination of an EZH2 inhibitor with immunotherapy may be a particularly attractive approach.
Overall, the 20 patients enrolled on this trial represented diverse and ultra-rare pediatric cancer diagnoses with small numbers within each disease cohort. Integration of Pediatric MATCH data with other trials reporting outcomes in molecularly characterized pediatric and adult patients and laboratory studies to identify biological and genetic explanations for this diversity of responses will be critical to better understand the potential role of tazemetostat for treatment of pediatric cancers. In addition, as tazemetostat continues to be further evaluated in other pediatric trials, particularly in SMARCB1-deficient tumors, our findings bolster its potential in future combinatorial approaches for these difficult-to-treat diseases.
Supplementary Material
Acknowledgements
Much thanks and appreciation to all patients and families for their participation in this clinical trial and for their courage in battling some of the most aggressive diseases of childhood. The funders did not play a role in the design of the study; the collection, analysis, and interpretation of the data; the writing of the manuscript; and the decision to submit the manuscript for publication. Prior presentation: ASCO Annual Meeting, June 2022 in Chicago, IL. Abstract #10009.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Susan N Chi, Department of Pediatrics, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Harvard Medical School, Boston, MA, USA.
Joanna S Yi, Department of Pediatrics, Texas Children’s Cancer and Hematology Center, Baylor College of Medicine, Houston, TX, USA.
P Mickey Williams, Molecular Characterization Laboratory, Frederick National Laboratory for Cancer Research, Frederick, MD, USA.
Sinchita Roy-Chowdhuri, Department of Pathology and Laboratory Medicine, University of Texas MD Anderson Cancer Center, Houston, TX, USA.
David R Patton, Center for Biomedical Informatics and Information Technology, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Brent D Coffey, Center for Biomedical Informatics and Information Technology, National Cancer Institute, National Institutes of Health, Bethesda, MD, USA.
Joel M Reid, Department of Molecular Pharmacology and Experimental Therapeutics, Mayo Clinic, Rochester, MN, USA.
Jin Piao, Department of Biostatistics, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA.
Lauren Saguilig, Children’s Oncology Group Statistical Center, Monrovia, CA, USA.
Todd A Alonzo, Department of Biostatistics, Keck School of Medicine, University of Southern California, Los Angeles, CA, USA.
Stacey L Berg, Department of Pediatrics, Texas Children’s Cancer and Hematology Center, Baylor College of Medicine, Houston, TX, USA.
Nilsa C Ramirez, Biopathology Center, Research Institute at Nationwide Children’s Hospital, Columbus, OH, USA.
Alok Jaju, Department of Radiology, Ann and Robert H. Lurie Children's Hospital, Chicago, IL, USA.
Joyce C Mhlanga, Department of Radiology, Washington University School of Medicine, St Louis, MO, USA.
Elizabeth Fox, Department of Oncology, St Jude Children’s Research Hospital, Memphis, TN, USA.
Douglas S Hawkins, Department of Hematology-Oncology, Seattle Children’s Hospital, University of Washington, Seattle, WA, USA.
Margaret M Mooney, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Cancer Therapy Evaluation Program, Bethesda, MD, USA.
Naoko Takebe, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Cancer Therapy Evaluation Program, Bethesda, MD, USA.
James V Tricoli, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Bethesda, MD, USA.
Katherine A Janeway, Department of Pediatrics, Dana-Farber/Boston Children’s Cancer and Blood Disorders Center, Harvard Medical School, Boston, MA, USA.
Nita L Seibel, Division of Cancer Treatment and Diagnosis, National Cancer Institute, Cancer Therapy Evaluation Program, Bethesda, MD, USA.
D Williams Parsons, Department of Pediatrics, Texas Children’s Cancer and Hematology Center, Baylor College of Medicine, Houston, TX, USA.
Data availability
The Children’s Oncology Group Data Sharing policy describes the release and use of COG individual patient data for use in research projects in accordance with National Clinical Trials Network (NCTN) Program and NCI Community Oncology Research Program (NCORP) Guidelines. Only data expressly released from the oversight of the relevant COG Data and Safety Monitoring Committee (DSMC) are available to be shared. Data sharing will ordinarily be considered only after the primary study manuscript is accepted for publication. For phase III studies, individual-level deidentified data sets that would be sufficient to reproduce results provided in a publication containing the primary study analysis can be requested from the NCTN/NCORP Data Archive at https://nctn-data-archive.nci.nih.gov/. Data are available to researchers who wish to analyze the data in secondary studies to enhance the public health benefit of the original work and agree to the terms and conditions of use. For non–phase III studies, data are available following the primary publication. An individual-level deidentified data set containing the variables analyzed in the primary results article can be expected to be available upon request. Requests for access to COG protocol research data should be sent to datarequest@childrensoncologygroup.org. Data are available to researchers whose proposed analysis is found by COG to be feasible and of scientific merit and who agree to the terms and conditions of use. For all requests, no other study documents, including the protocol, will be made available and no end date exists for requests. In addition to above, release of data collected in a clinical trial conducted under a binding collaborative agreement between COG or the NCI Cancer Therapy Evaluation Program (CTEP) and a pharmaceutical or biotechnology company must comply with the data sharing terms of the binding collaborative and contractual agreement and must receive the proper approvals.
Author contributions
Susan N. Chi, MD (Conceptualization; Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Validation; Visualization; Writing—original draft; Writing—review & editing), Katherine A. Janeway, MD (Formal analysis; Investigation; Methodology; Supervision; Writing—review & editing), James V. Tricoli, PhD (Visualization; Writing—review & editing), Naoko Takebe, MD, PhD (Visualization; Writing—review & editing), Margaret M. Mooney, MD (Visualization; Writing—review & editing), Douglas S. Hawkins, MD (Funding acquisition; Investigation; Project administration; Supervision; Writing—review & editing), Elizabeth Fox, MD (Data curation; Formal analysis; Investigation; Project administration; Supervision; Writing—review & editing), Joyce C. Mhlanga, MBChB (Visualization; Writing—review & editing), Alok Jaju, MD (Writing—review & editing), Nilsa C. Ramirez, MD (Data curation; Visualization; Writing—review & editing), Stacey L. Berg, MD (Conceptualization; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Writing—review & editing), Todd A. Alonzo, PhD (Data curation; Formal analysis; Writing—review & editing), Lauren Saguilig, MS (Project administration; Writing—review & editing), Jin Piao, PhD (Formal analysis; Methodology; Visualization; Writing—review & editing), Joel M. Reid, PhD (Formal analysis; Methodology; Writing—review & editing), Brent D. Coffey, MS, MBA (Data curation; Formal analysis; Writing—review & editing), David R. Patton, BS (Formal analysis; Writing—review & editing), Sinchita Roy-Chowdhuri, MD, PhD (Data curation; Formal analysis; Writing—review & editing), P. Mickey Williams, PhD (Data curation; Writing—review & editing), Joanna Yi, MD (Data curation; Formal analysis; Investigation; Methodology; Project administration; Supervision; Visualization; Writing—original draft; Writing—review & editing), Nita L. Seibel, MD (Conceptualization; Project administration; Supervision; Writing—review & editing), D. Williams Parsons, MD, PhD (Conceptualization; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Project administration; Software; Supervision; Visualization; Writing—original draft; Writing—review & editing)
Funding
This work was supported by the NCTN Operations Center Grant U10CA180886, the COG Biospecimen Bank Grant U24CA196173, the NCTN Statistics and Data Center Grant U10CA180899, and the St. Baldrick’s Foundation.
Conflicts of interest
Susan Chi: Consulting or advisory role—Epizyme; Blueprint Medicines. Stacey L. Berg: travel, accommodations, expenses—nonprofit. Other relationship—member of the COG Developmental Therapeutics Steering Committee, through which some clinical trials may be partially industry funded, institution may receive some funding, Pediatric Early Phase Clinical Trials Network. Elizabeth Fox: other relationship—Helsinn Therapeutics. Douglas S. Hawkins: research funding—Amgen; Bayer (Inst); Bristol-Myers Squibb (Inst); Eisai (Inst); Incyte; Jazz Pharmaceuticals; Lilly (Inst); Loxo (Inst); Merck Sharp & Dohme (Inst); Seattle Genetics. Katherine A. Janeway: honoraria—Foundation Medicine and Takeda. Consulting or advisory role—Bayer; Ipsen. Travel, accommodations, expenses—Bayer. D. Williams Parsons: Patents, royalties, other intellectual property—Co-inventor on current and pending patents related to cancer genes discovered through sequencing of several adult cancer types. Participates in royalty sharing related to those patents.
Elizabeth Fox, a JNCI Associate Editor an coauthor on this article, was not involved in the editorial review or decision to accept the manuscript for publication.
References
- 1. Sneeringer CJ, Scott MP, Kuntz KW, et al. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci USA. 2010;107(49):20980-20985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Volkel P, Dupret B, Le Bourhis X, Angrand PO. Diverse involvement of EZH2 in cancer epigenetics. Am J Transl Res. 2015;7(2):175-193. [PMC free article] [PubMed] [Google Scholar]
- 3. Gajjar A, Pfister SM, Taylor MD, Gilbertson RJ. Molecular insights into pediatric brain tumors have the potential to transform therapy. Clin Cancer Res. 2014;20(22):5630-5640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Morin RD, Johnson NA, Severson TM, et al. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 2010;42(2):181-185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Morin RD, Mendez-Lago M, Mungall AJ, et al. Frequent mutation of histone-modifying genes in non-Hodgkin lymphoma. Nature. 2011;476(7360):298-303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kennison JA, Tamkun JW. Dosage-dependent modifiers of polycomb and antennapedia mutations in Drosophila. Proc Natl Acad Sci USA. 1988;85(21):8136-8140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wilson BG, Wang X, Shen X, et al. Epigenetic antagonism between polycomb and SWI/SNF complexes during oncogenic transformation. Cancer Cell. 2010;18(4):316-328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cheng JX, Tretiakova M, Gong C, Mandal S, Krausz T, Taxy JB. Renal medullary carcinoma: rhabdoid features and the absence of INI1 expression as markers of aggressive behavior. Mod Pathol. 2008;21(6):647-652. [DOI] [PubMed] [Google Scholar]
- 9. Knutson SK, Wigle TJ, Warholic NM, et al. A selective inhibitor of EZH2 blocks H3K27 methylation and kills mutant lymphoma cells. Nat Chem Biol. 2012;8(11):890-896. [DOI] [PubMed] [Google Scholar]
- 10. Knutson SK, Warholic NM, Wigle TJ, et al. Durable tumor regression in genetically altered malignant rhabdoid tumors by inhibition of methyltransferase EZH2. Proc Natl Acad Sci USA. 2013;110(19):7922-7927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chan-Penebre E, Armstrong K, Drew A, et al. Selective killing of SMARCA2- and SMARCA4-deficient small cell carcinoma of the ovary, hypercalcemic type cells by inhibition of EZH2: in vitro and in vivo preclinical models. Mol Cancer Ther. 2017;16(5):850-860. [DOI] [PubMed] [Google Scholar]
- 12. Kurmasheva RT, Sammons M, Favours E, et al. Initial testing (stage 1) of tazemetostat (EPZ-6438), a novel EZH2 inhibitor, by the Pediatric Preclinical Testing Program. Pediatr Blood Cancer. 2017;64(3):e26218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Knutson SK, Kawano S, Minoshima Y, et al. Selective inhibition of EZH2 by EPZ-6438 leads to potent antitumor activity in EZH2-mutant non-Hodgkin lymphoma. Mol Cancer Ther. 2014;13(4):842-854. [DOI] [PubMed] [Google Scholar]
- 14. Italiano A, Soria JC, Toulmonde M, et al. Tazemetostat, an EZH2 inhibitor, in relapsed or refractory B-cell non-Hodgkin lymphoma and advanced solid tumours: a first-in-human, open-label, phase 1 study. Lancet Oncol. 2018;19(5):649-659. [DOI] [PubMed] [Google Scholar]
- 15. Chi SN, Bourdeaut F, Casanova M, et al. Update on phase 1 study of tazemetostat, an enhancer of zeste homolog 2 inhibitor, in pediatric patients with relapsed or refractory integrase interactor 1–negative tumors. J Clin Oncol 2022;40(suppl 16):10040. [Google Scholar]
- 16. Allen CE, Laetsch TW, Mody R, et al. Target and agent prioritization for the Children's Oncology Group-National Cancer Institute pediatric MATCH trial. J Natl Cancer Inst. 2017;109(5):djw274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Seibel NL, Janeway K, Allen CE, et al. Pediatric oncology enters an era of precision medicine. Curr Probl Cancer. 2017;41(3):194-200. [DOI] [PubMed] [Google Scholar]
- 18. Parsons DW, Janeway KA, Patton DR, et al. ; the NCI-COG Pediatric MATCH Team. Actionable tumor alterations and treatment protocol enrollment of pediatric and young adult patients with refractory cancers in the National Cancer Institute-Children's Oncology Group Pediatric MATCH Trial. J Clin Oncol. 2022;40(20):2224-2234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eckstein OS, Allen CE, Williams PM, et al. Phase II study of selumetinib in children and young adults with tumors harboring activating mitogen-activated protein kinase pathway genetic alterations: arm E of the NCI-COG pediatric MATCH trial. J Clin Oncol 2022;40(20):2235-2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chi SB, Laetsch TW, Fouladi M, et al. Phase I study of tazemetostat, an enhancer of zeste homolog-2 inhibitor, in pediatric pts with relapsed/refractory integrase interactor 1-negative tumors. In: American Society of Clinical Oncology: Poster # 10205 2020; Virtual; 2020. https://ascopubs.org/doi/10.1200/JCO.2020.38.15_suppl.10525.
- 21. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45(2):228-247. [DOI] [PubMed] [Google Scholar]
- 22. Gounder M, Schoffski P, Jones RL, et al. Tazemetostat in advanced epithelioid sarcoma with loss of INI1/SMARCB1: an international, open-label, phase 2 basket study. Lancet Oncol. 2020;21(11):1423-1432. [DOI] [PubMed] [Google Scholar]
- 23. Morschhauser F, Tilly H, Chaidos A, et al. Tazemetostat for patients with relapsed or refractory follicular lymphoma: an open-label, single-arm, multicentre, phase 2 trial. The Lancet Oncology. 2020;21(11):1433-1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Abla O, Jacobsen E, Picarsic J, et al. Consensus recommendations for the diagnosis and clinical management of Rosai-Dorfman-Destombes disease. Blood. 2018;131(26):2877-2890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Goyal G, Ravindran A, Young JR, et al. ; Mayo Clinic Histiocytosis Working Group Clinicopathological features, treatment approaches, and outcomes in Rosai-Dorfman disease. Haematologica. 2020;105(2):348-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Paskeh MDA, Mehrabi A, Gholami MH, et al. EZH2 as a new therapeutic target in brain tumors: molecular landscape, therapeutic targeting and future prospects. Biomed Pharmacother. 2022;146:112532. [DOI] [PubMed] [Google Scholar]
- 27. Samra B, Konopleva M, Isidori A, Daver N, DiNardo C. Venetoclax-based combinations in acute myeloid leukemia: current evidence and future directions. Front Oncol. 2020;10:562558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Leruste A, Tosello J, Ramos RN, et al. Clonally expanded T cells reveal immunogenicity of rhabdoid tumors. Cancer Cell. 2019;36(6):597-612 e598. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Children’s Oncology Group Data Sharing policy describes the release and use of COG individual patient data for use in research projects in accordance with National Clinical Trials Network (NCTN) Program and NCI Community Oncology Research Program (NCORP) Guidelines. Only data expressly released from the oversight of the relevant COG Data and Safety Monitoring Committee (DSMC) are available to be shared. Data sharing will ordinarily be considered only after the primary study manuscript is accepted for publication. For phase III studies, individual-level deidentified data sets that would be sufficient to reproduce results provided in a publication containing the primary study analysis can be requested from the NCTN/NCORP Data Archive at https://nctn-data-archive.nci.nih.gov/. Data are available to researchers who wish to analyze the data in secondary studies to enhance the public health benefit of the original work and agree to the terms and conditions of use. For non–phase III studies, data are available following the primary publication. An individual-level deidentified data set containing the variables analyzed in the primary results article can be expected to be available upon request. Requests for access to COG protocol research data should be sent to datarequest@childrensoncologygroup.org. Data are available to researchers whose proposed analysis is found by COG to be feasible and of scientific merit and who agree to the terms and conditions of use. For all requests, no other study documents, including the protocol, will be made available and no end date exists for requests. In addition to above, release of data collected in a clinical trial conducted under a binding collaborative agreement between COG or the NCI Cancer Therapy Evaluation Program (CTEP) and a pharmaceutical or biotechnology company must comply with the data sharing terms of the binding collaborative and contractual agreement and must receive the proper approvals.