

The first ligand-exchange rate measurements of lanthanide ions in an ionic liquid are reported here. The trend of water-exchange rates in the ionic liquid is the opposite of the trend in water.
Room temperature ionic liquids have the potential to impact catalysis,1 energy storage,2 organic synthesis,3 separation science,4 and chemical sensing.5 These liquids influence the properties of solutes, and the lanthanides are an important set of solutes because of their luminescence,6 catalytic,7 and magnetic properties.8 These properties are influenced by coordination chemistry that is often unique in ionic liquids relative to molecular solvents.6a,b,7h,8a Consequently, it is important to study the coordination chemistry of lanthanide ions in ionic liquids, including ligand-exchange rates that are crucial to catalysis.9 The coordination environments of lanthanide ions in ionic liquids have been reported;10 however, the ligand-exchange rates of lanthanide ions in ionic liquids, to the best of our knowledge, have not been reported. We expected that 17O-NMR spectroscopy could be used to study the ligand- exchange rates of lanthanide ions in ionic liquids, because this technique has been used to determine water-exchange rates of metal ions in aqueous and organic solvents.11
To test our hypothesis, we chose 1-ethyl-3-methylimidazolium ethyl sulfate (EMIES) as the room temperature ionic liquid because of its well-studied properties and the high solubility of lanthanide ions in this solvent.12 Water is known to bind to lanthanide ions and is a common impurity in room temperature ionic liquids;10b,13 therefore, water was selected as a candidate ligand for this study.
Before determining the water-exchange rates of lanthanide ions, it is necessary to know the number of water molecules coordinated to the lanthanide ions in EMIES. Europium triflate [Eu(OTf)3] was used for this purpose because the binding of water to Eu3+ causes changes in the luminescence emission of the ion: the electric dipole-governed emission (5D0→7F2, λ = 615 nm) is sensitive to changes to the inner-sphere of Eu3+, but the magnetic dipole-dominated emission (5D0→7F1, λ = 591 nm) is relatively insensitive to its surroundings.14 Consequently, the ratio of these emission intensities (I615/I591) has been used to study the inner-sphere change of Eu3+.15 We studied the luminescence of Eu3+ in EMIES to investigate the inner-sphere change of Eu3+ in the absence and presence of water (Fig. 1). The I615/I591 ratio decreased from 1.69 to 1.27 when water was added, suggesting that the coordination environment of Eu3+ changed upon the addition of water. This change was attributed to the replacement of inner-sphere ligand, either triflate or ethyl sulfate, by water. Further, luminescence-decay measurements of Eu(OTf)3 and Tb(OTf)3 in water–EMIES (1 : 19, v/v) indicated that the average water-coordination number increased from 0 to 1.6 upon the addition of water. Because all lanthanide ions in the same solvent tend to differ in coordination number by at most one, with smaller differences for lanthanides that are adjacent to each other in atomic number, we expect the trivalent ions in this study to have the same water-coordination numbers in water–EMIES (1 : 19, v/v) as Eu3+ and Tb3+.
Fig. 1.
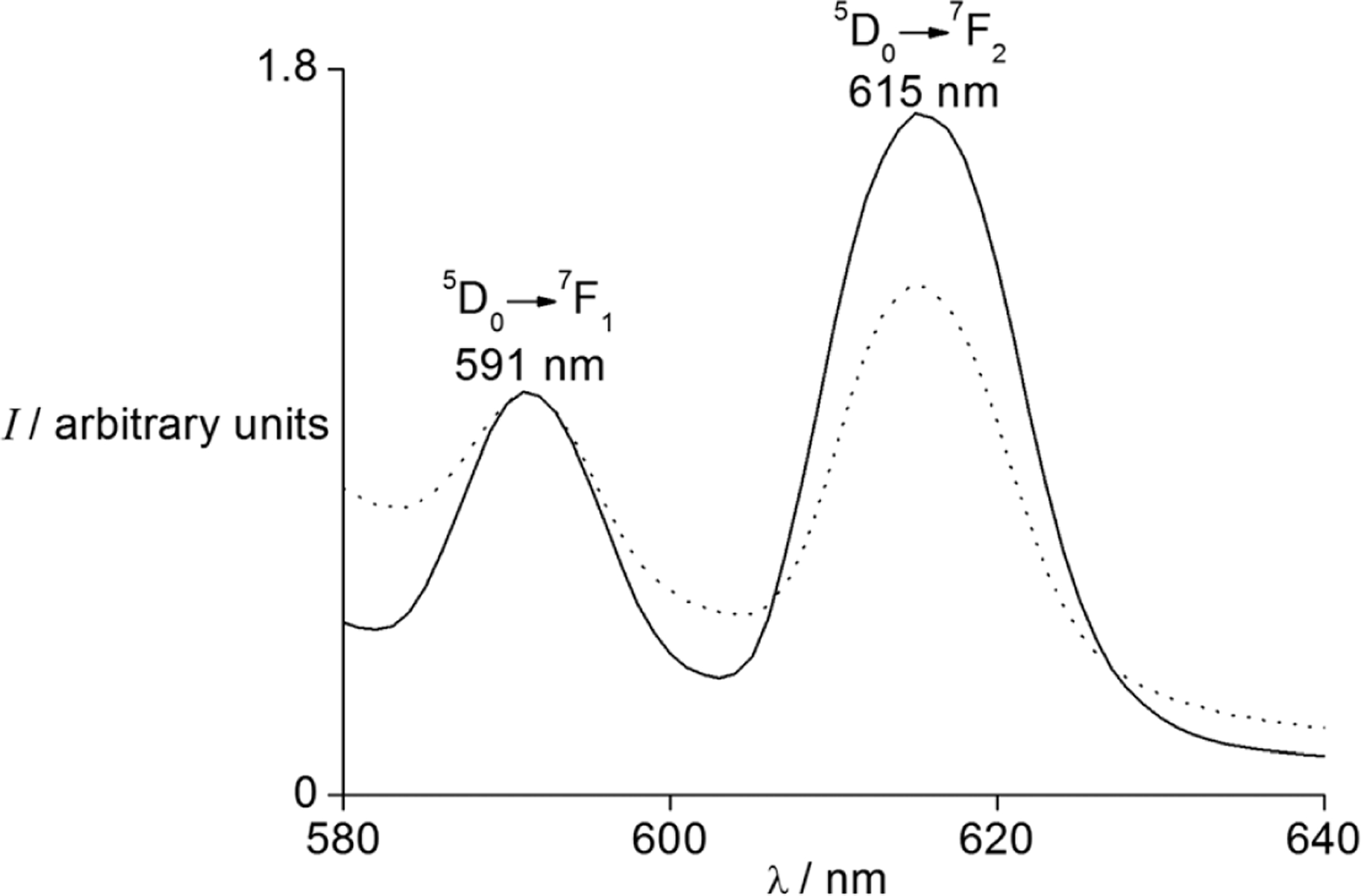
Normalized emission spectra (λex = 395 nm) of Eu(OTf)3 (5 mM) in EMIES (–) and H2O–EMIES (1 : 19, v/v) (⋯). The emission intensities of Eu3+ at 591 nm are normalized to 1.
Because water coordinates to lanthanide ions in EMIES, we used variable-temperature 17O-NMR spectroscopy to determine the water-exchange rates in EMIES. The water-exchange rate (kex) between bound and bulk water in EMIES was determined from the residency lifetime of the bound water (kex = life-time−1). The residency lifetime of water was probed by measuring the transverse relaxation rates of water in the presence of lanthanide ions as a function of temperature (see ESI† for Experimental details). We measured the water-exchange rates of Gd(OTf)3, Tb(OTf)3, Dy(OTf)3, Ho(OTf)3, and Er(OTf)3 in water–EMIES (1 : 19, v/v). Attempts to measure the water-exchange rates for other lanthanide ions were not successful because of small and irregular differences between the line widths of the bulk water peaks at half height for the other lanthanide ions and the diamagnetic reference. The inability to measure the other ions is consistent with measurements in water.16a The mean water-exchange rates of the studied lanthanide ions in water–EMIES (1 : 19, v/v) at 298.15 K were plotted versus the charge density of lanthanide ions (Fig. 2a). The water-exchange rates of the ions increased as a function of charge density with exception of the terbium ion. Interestingly, the trend of water-exchange rates in EMIES is the opposite of lanthanide ions in water (Fig. 2b).16
Fig. 2.
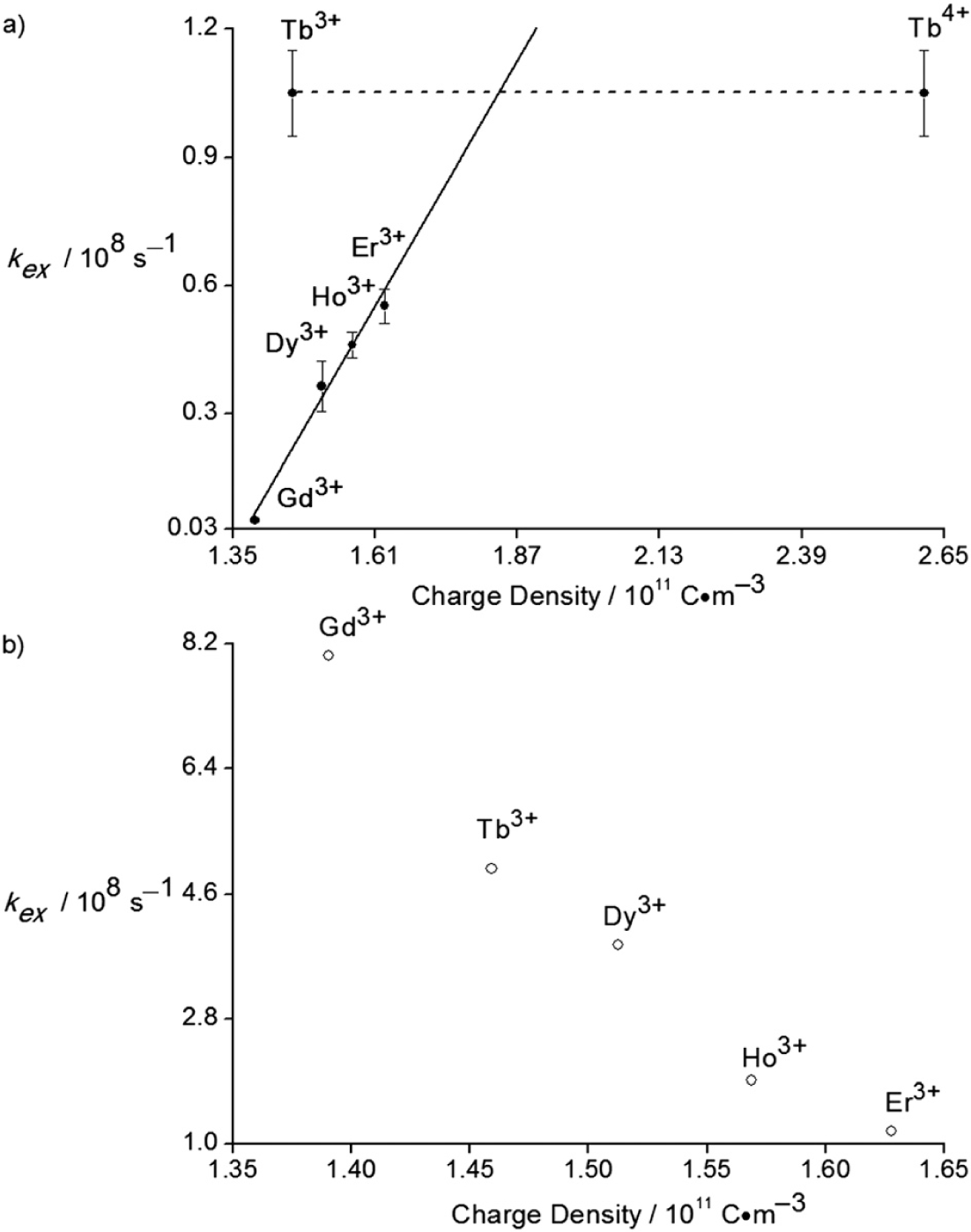
(a) Water-exchange rates of lanthanide ions in H2O–EMIES (1 : 19, v/v) at 298.15 K as a function of charge density. Solid dots represent the means of three or four independently prepared measurements. Error bars represent standard errors of the means (error bars for Gd3+ are smaller than the dot). The solid line is the linear fitting of the water-exchange rates of Gd3+, Dy3+, Ho3+, and Er3+. The dashed line is a guide for the eyes to connect the points for Tb3+ and Tb4+. The intersection of the solid and dashed line is the estimated average charge density of the Tb species in EMIES based on fitting. (b) Water-exchange rate of lanthanide ions in water at 298.15 K as a function of charge density.16
For lanthanide aquo ions, the decreasing water-exchange rates with respect to increasing charge density can be accounted for by two possible explanations (electrostatic or steric) or a combination of these explanations:17 first, because the water exchange of lanthanide aquo ions is associative, association of water to the lanthanide ions is the rate limiting step. As the charge densities increase from Gd3+ to Er3+, the attractions between water molecules and lanthanide ions increase, resulting in fast water-exchange. Second, the steric crowding of the smaller lanthanide aquo ions tends to hinder the water molecules from coordinating to the lanthanide ions, resulting in slower exchange rates. The steric effects outweigh the electrostatic effects in water.
In contrast to aqueous solution, the water-exchange rates of lanthanide ions in EMIES increase as a function of increasing charge density. This trend is likely caused by increased steric crowding in EMIES of lanthanide complexes that have higher charge densities compared to lanthanide complexes with lower charge densities. The increasing steric crowding from Gd3+ to Er3+ in EMIES is illustrated in Fig. 3. Based on luminescence-decay measurements, the average water-coordination number of lanthanide ions in water–EMIES (1 : 19, v/v) is 1.6; therefore, the remaining coordination sites were assigned to ethyl sulfate or triflate. Because the charge density of Gd3+ is smaller than that of Er3+, ethyl sulfate tends to bind less tightly to Gd3+ than Er3+. As a consequence, ethyl sulfate likely forms less compact structures with Gd3+ than Er3+, with the other lanthanide ions falling between these two extremes. This hypothesis is consistent with the report of lanthanide ions with high charge densities forming compact structures in imidazolium-based ionic liquids.18 For the relatively compact Er3+ complex, the steric crowding of the complex could hinder water binding and cause a fast water-exchange rate. These observations suggest a dissociative exchange mechanism, but variable pressure experiments would be needed in the future to confirm an exact mechanism of exchange. An alternative explanation for our observations is that the smaller cations have more well-ordered water in their second sphere than the larger cations, leading to faster exchange as observed in aqueous solution with variations in anions.19
Fig. 3.
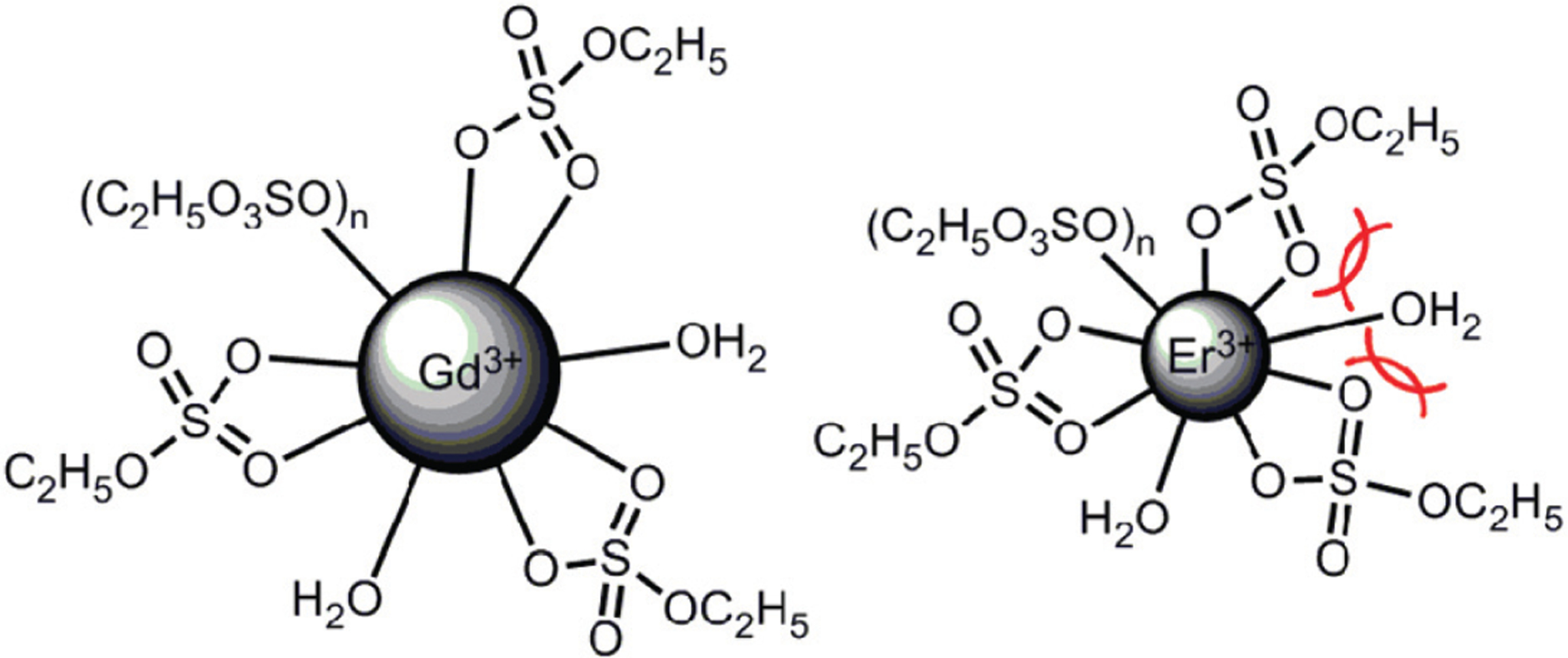
Cartoon representation of the proposed inner-sphere environments of Gd3+ and Er3+ in H2O–EMIES, where n represents for the number of anions (ethyl sulfate or triflate) minus three in the inner-sphere of LnIII. These are simplified depictions for illustration: for example, ethyl sulfate can also bind to lanthanide ions in a monodentate fashion,20 and other species (such as triflate) might be bound.
Notably, the water-exchange rate of Tb(OTf)3 in water–EMIES (1 : 19, v/v) was unexpectedly higher than the other lanthanide triflates and did not follow the trend based on the charge density of Tb3+. This observation motivated us to consider the presence of Tb4+, which is smaller than any Ln3+ ion and relatively stable.21 We measured the L3-edge using X-ray absorption near-edge structure (XANES) spectroscopy of Tb(OTf)3 in water–EMIES (1 : 19, v/v) (Fig. 4). In the normalized L3-edge XANES spectra the small peak around 7533 eV, which is associated with the Tb4+ oxidation state, indicated the presence of Tb4+ in all three samples. The Tb4+ in water–EMIES was likely from the original Tb(OTf)3 starting material, for which the presence of this XANES feature also indicates the presence of Tb4+. The percentage of Tb4+ in Tb(OTf)3 in the water–EMIES (1 : 19, v/v) is estimated to be less than 33% based on the linear fitting of water-exchange rate versus charge density (the average charge density of Tb was estimated to be the intersection of the solid and dashed line in Fig. 2). The stability of a small amount of Tb4+ in ionic liquids relative to aqueous solution, where Tb4+ is relatively unstable, can be rationalized by the presence of a large excess of imidazolium cations in the ionic liquid that have a strong electron-withdrawing ability to stabilize Tb4+. However, the amount of Tb4+ is likely overestimated in Tb(OTf)3 because the water-exchange rate of Tb4+ is out of the linear range of the other measured tri- valent lanthanide ions. Furthermore, because Tb4+ might follow a non-linear trend with the trivalent ions, a small amount of this ion (suggested by XANES) could be responsible for a large increase in average exchange rate.
Fig. 4.
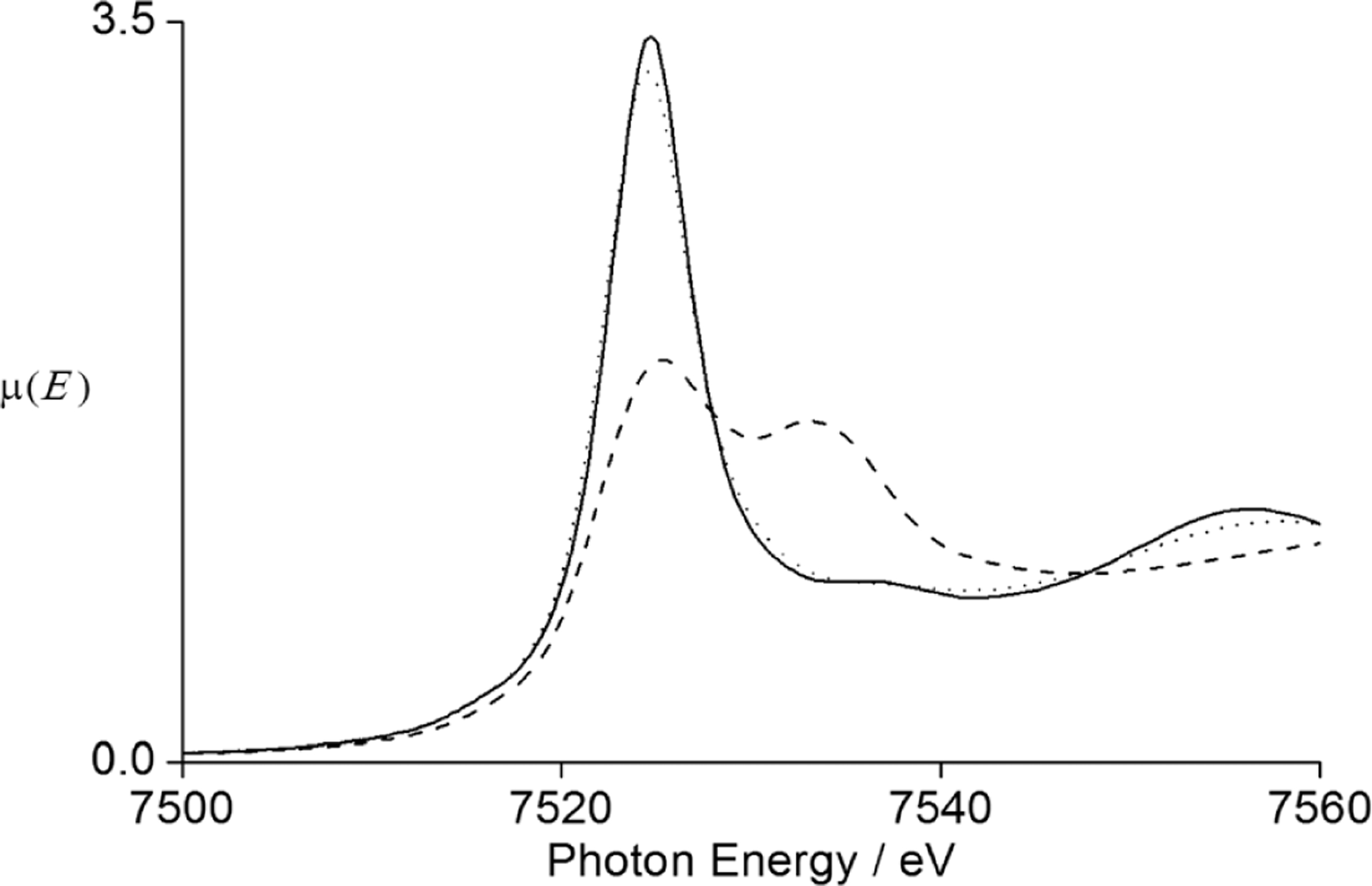
Normalized (edge step set to 1) L3-edge XANES spectra of solid Tb(OTf)3 (—), solid Tb4O7 (– –), and Tb(OTf)3 in water–EMIES (1 : 19, v/v) (⋯). The features at approximately 7533 and 7524 eV are associated with Tb4+ and Tb3+, respectively.
We have measured the water-exchange rates of lanthanide ions in water–EMIES (1 : 19, v/v) using 17O-NMR spectroscopy. The water-exchange rates of the studied lanthanide ions show an inverse trend relative to lanthanide aquo ions and the unexpectedly high water-exchange rate of Tb(OTf)3 in water–EMIES was likely caused by the presence of Tb4+, which was confirmed by L3-edge XANES spectra. Our study suggests that water-exchange rates of lanthanide ions can be manipulated by the solvent to the point of reversing trends in exchange rate. We suspect that this tunability of ligand-exchange rates with respect to solvent will be useful in the selection of ionic liquids for catalysis, separation of lanthanide ions, and other applications.
Supplementary Material
Acknowledgments
This research was supported by a CAREER Award from the National Science Foundation (CHE-0955000) and Cambridge Isotope Laboratories, Inc. The authors gratefully acknowledge the Lumingen Instrument Center at Wayne State University. We thank the support from the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. Use of the Advanced Photon Source at Argonne National Laboratory was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. The authors would like to thank Drs S. Lee and B. Reinhart for their assistance at Beamline 12BM.
Footnotes
Electronic supplementary information (ESI) available: Experimental procedures, luminescence-decay rate data, and 17O-NMR data. See DOI: 10.1039/c4dt02492c
Notes and references
- 1.(a) Hu Z, Zhang J, Song Y, Zhou Y. and Han B, Green Chem., 2009, 11, 1746; [Google Scholar]; (b) Dzudza A. and Marks TJ, Org. Lett, 2009, 11, 1523; [DOI] [PubMed] [Google Scholar]; (c) Kang X, Zhang J, Shang W, Wu T, Zhang P, Han B, Wu Z, Mo G. and Xing X, J. Am. Chem. Soc, 2014, 136, 3768; [DOI] [PubMed] [Google Scholar]; (d) Schneider MJ, Lijewski M, Woelfel R, Haumann M. and Wasserscheid P, Angew. Chem., Int. Ed, 2013, 52, 6996; [DOI] [PubMed] [Google Scholar]; (e) Rosen BA, Salehi-Khojin A, Thorson MR, Zhu W, Whipple DT, Kenis PJ and Masel RI, Science, 2011, 334, 643. [DOI] [PubMed] [Google Scholar]
- 2.(a) Lin R, Taberna P-L, Fantini S, Presser V, Pérez CR, Malbosc F, Rupesinghe NL, Teo KB, Gogotsi Y. and Simon P, J. Phys. Chem. Lett., 2011, 2, 2396; [Google Scholar]; (b) Armand M, Endres F, MacFarlane DR, Ohno H. and Scrosati B, Nat. Mater, 2009, 8, 621. [DOI] [PubMed] [Google Scholar]
- 3.Hallett JP and Welton T, Chem. Rev, 2011, 111, 3508. [DOI] [PubMed] [Google Scholar]
- 4.(a) Poole CF, Chromatogr J., A, 2004, 1037, 49; [DOI] [PubMed] [Google Scholar]; (b) Hoogestraete TV, Wellens S, Verachtert K. and Binnemans K, Green Chem., 2013, 15, 919. [Google Scholar]
- 5.(a) Rehman A, Hamilton A, Chung A, Baker GA, Wang Z. and Zeng X, Anal. Chem, 2011, 83, 7823; [DOI] [PubMed] [Google Scholar]; (b) Chen L, Huang D, Ren S, Chi Y. and Chen G, Anal. Chem, 2011, 83, 6862; [DOI] [PubMed] [Google Scholar]; (c) Chen L, Zhang Y, Ren S, Huang D, Zhou C, Chi Y. and Chen G, Analyst, 2013, 138, 7006; [DOI] [PubMed] [Google Scholar]; (d) Zhao Q, Yin M, Zhang AP, Prescher S, Antonietti M. and Yuan J, J. Am. Chem. Soc, 2013, 135, 5549. [DOI] [PubMed] [Google Scholar]
- 6.(a) Tang S, Babai A. and Mudring AV, Angew. Chem., Int. Ed, 2008, 47, 7631; [DOI] [PubMed] [Google Scholar]; (b) Lunstroot K, Nockemann P, Hecke KV, Meervelt LV, Görller-Walrand C, Binnemans K. and Driesen K, Inorg. Chem, 2009, 48, 3018; [DOI] [PubMed] [Google Scholar]; (c) Foucault-Collet A, Gogick KA, White KA, Villette S, Pallier A, Collet G, Kieda C, Li T, Geib SJ and Rosi NL, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 17199; [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) de Bettencourt-Dias A., Barber PS and Bauer S, J. Am. Chem. Soc., 2012, 134, 6987. [DOI] [PubMed] [Google Scholar]
- 7.(a) Robinson JR, Fan X, Yadav J, Carroll PJ, Wooten AJ, Pericàs MA, Schelter EJ and Walsh PJ, J. Am. Chem. Soc, 2014, 136, 8034; [DOI] [PubMed] [Google Scholar]; (b) Mei Y, Dissanayake P. and Allen MJ, J. Am. Chem. Soc, 2010, 132, 12871; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Mei Y, Averill DJ and Allen MJ, J. Org. Chem, 2012, 77, 5624; [DOI] [PubMed] [Google Scholar]; (d) Mi X, Luo S, He J. and Cheng J-P, Tetrahedron Lett., 2004, 45, 4567; [Google Scholar]; (e) Atesin AC, Ray NA, Stair PC and Marks TJ, J. Am. Chem. Soc, 2012, 134, 14682; [DOI] [PubMed] [Google Scholar]; (f) Li Z, Assary RS, Atesin AC, Curtiss LA and Marks TJ, J. Am. Chem. Soc, 2014, 136, 104; [DOI] [PubMed] [Google Scholar]; (g) Kobayashi S. and Hachiya I, J. Org. Chem, 1994, 59, 3590; [Google Scholar]; (h) Song CE, Shim WH, Roh EJ and Choi JH, Chem. Commun, 2000, 1695. [Google Scholar]
- 8.(a) Mallick B, Balke B, Felser C. and Mudring AV, Angew. Chem., Int. Ed, 2008, 47, 7635; [DOI] [PubMed] [Google Scholar]; (b) Blagg RJ, Ungur L, Tuna F, Speak J, Comar P, Collison D, Wernsdorfer W, McInnes EJ, Chibotaru LF and Winpenny RE, Nat. Chem, 2013, 5, 673; [DOI] [PubMed] [Google Scholar]; (c) Kielar F, Tei L, Terreno E. and Botta M, J. Am. Chem. Soc, 2010, 132, 7836; [DOI] [PubMed] [Google Scholar]; (d) Le Roy JJ, Ungur L, Korobkov I, Chibotaru LF and Murugesu M, J. Am. Chem. Soc, 2014, 136, 8003; [DOI] [PubMed] [Google Scholar]; (e) Ungur L, Le Roy JJ, Korobkov I, Murugesu M. and Chibotaru LF, Angew. Chem., Int. Ed, 2014, 53, 4413; [DOI] [PubMed] [Google Scholar]; (f) Rinehart JD, Fang M, Evans WJ and Long JR, J. Am. Chem. Soc, 2011, 133, 14236; [DOI] [PubMed] [Google Scholar]; (g) Rinehart JD, Fang M, Evans WJ and Long JR, Nat. Chem, 2011, 3, 538. [DOI] [PubMed] [Google Scholar]
- 9.Kobayashi S, Nagayama S. and Busujima T, J. Am. Chem. Soc, 1998, 120, 8287. [Google Scholar]
- 10.(a) Gaillard C, Billard I, Chaumont A, Mekki S, Ouadi A, Denecke MA, Moutiers G. and Wipff G, Inorg. Chem, 2005, 44, 8355; [DOI] [PubMed] [Google Scholar]; (b) Billard I, Mekki S, Gaillard C, Hesemann P, Moutiers G, Mariet C, Labet A. and Bünzli JCG, Eur. J. Inorg. Chem, 2004, 2004, 1190. [Google Scholar]
- 11.(a) Zhang S, Wu K. and Sherry AD, J. Am. Chem. Soc, 2002, 124, 4226; [DOI] [PubMed] [Google Scholar]; (b) Urbanczyk-Pearson LM, Femia FJ, Smith J, Parigi G, Duimstra JA, Eckermann AL, Luchinat C. and Meade TJ, Inorg. Chem, 2008, 47, 56; [DOI] [PubMed] [Google Scholar]; (c) Caravan P, Parigi G, Chasse JM, Cloutier NJ, Ellison JJ, Lauffer RB, Luchinat C, McDermid SA, Spiller M. and McMurry TJ, Inorg. Chem, 2007, 46, 6632; [DOI] [PubMed] [Google Scholar]; (d) Gale EM, Zhu J. and Caravan P, J. Am. Chem. Soc, 2013, 135, 18600; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Garcia J, Neelavalli J, Haacke EM and Allen MJ, Chem. Commun, 2011, 47, 12858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.(a) González EJ, González B, Calvar N. and Domínguez Á, J. Chem. Eng. Data, 2007, 52, 1641–1648; [Google Scholar]; (b) Gómez E, González B, Calvar N, Tojo E. and Domínguez Á, J. Chem. Eng. Data, 2006, 51, 2096; [Google Scholar]; (c) Zhang Q-G, Wang N-N and Yu Z-W, J. Phys. Chem. B, 2010, 114, 4747. [DOI] [PubMed] [Google Scholar]
- 13.(a) Chaumont A. and Wipff G, Inorg. Chem, 2004, 43, 5891; [DOI] [PubMed] [Google Scholar]; (b) Zhao C, Bond AM and Lu X, Anal. Chem, 2012, 84, 2784. [DOI] [PubMed] [Google Scholar]
- 14.Lin Z. and Allen MJ, Dyes Pigm., 2014, 110, 261. [Google Scholar]
- 15.(a) Werts MH, Jukes RT and Verhoeven JW, Phys. Chem. Chem. Phys, 2002, 4, 1542; [Google Scholar]; (b) Averill DJ and Allen MJ, Inorg. Chem, 2014, 53, 6257; [DOI] [PubMed] [Google Scholar]; (c) Parker D. and Yu J, Chem. Commun, 2005, 3141; [DOI] [PubMed] [Google Scholar]; (d) Bretonniere Y, Cann MJ, Parker D. and Slater R, Org. Biomol. Chem, 2004, 2, 1624. [DOI] [PubMed] [Google Scholar]
- 16.(a) Cossy C, Helm L. and Merbach AE, Inorg. Chem, 1988, 27, 1973; [Google Scholar]; (b) Powell DH, Ni Dhubhghaill OM, Pubanz D, Helm L, Lebedev YS, Schlaepfer W. and Merbach AE, J. Am. Chem. Soc, 1996, 118, 9333. [Google Scholar]
- 17.Cossy C, Helm L. and Merbach AE, Inorg. Chem, 1989, 28, 2699. [Google Scholar]
- 18.Chaumont A. and Wipff G, Phys. Chem. Chem. Phys, 2003, 5, 3481. [Google Scholar]
- 19.Thompson AL, Parker D, Fulton DA, Howard JAK, Pandya SU, Puschmann H, Senanayake K, Stenson PA, Badari A, Botta M, Avedano S. and Aime S, Dalton Trans., 2006, 5605. [DOI] [PubMed] [Google Scholar]
- 20.He Z, Gao E, Wang Z, Yan C. and Kurmoo M, Inorg. Chem, 2005, 44, 862. [DOI] [PubMed] [Google Scholar]
- 21.Hobart D, Samhoun K, Young J, Norvell V, Mamantov G. and Peterson J, Inorg. Nucl. Chem. Lett, 1980, 16, 321. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.