

Abstract
Several studies have shown that ethanol (EtOH) can enhance the activity of GABAergic synapses via presynaptic mechanisms, including in hippocampal CA1 neurons. The serotonin type 3 receptor (5-HT3-R) has been implicated in the neural actions of ethanol (EtOH) and in modulation of GABA release from presynaptic terminals. In the present study, we investigated EtOH modulation of GABA release induced by 5-HT3-R activation using the mechanically isolated neuron/bouton preparation from the rat CA1 hippocampal subregion. EtOH application before and during exposure to the selective 5-HT3 receptor agonist, m-chlorophenylbiguanide (mCPBG) potentiated the mCPBG-induced increases in the peak frequency and charge transfer of spontaneous GABAergic inhibitory postsynaptic currents. Interestingly, the potentiation was maintained even after EtOH was removed from the preparation. A protein kinase A inhibitor reduced the magnitude of EtOH potentiation. Fluorescent Ca2+ imaging showed that Ca2+ transients in the presynaptic terminals increased during EtOH exposure. These findings indicate that EtOH produces long-lasting potentiation of 5-HT3-induced GABA release by modulating calcium levels, via a process involving cAMP-mediated signaling in presynaptic terminals.
Keywords: Alcohol, GABAergic synapse, Serotonin, Neuron/bouton, Hippocampus
1. Introduction
Serotonin (5-hydroxytryptamine, 5-HT) is a prominent neurotransmitter that regulates a variety of central and peripheral functions (Hoyer et al., 2002). The 5-HT3 receptor is the only serotonin-activated ligand-gated ion channel (LGIC) (Barnes et al., 2009), and it is a member of the Cys-loop LGIC superfamily that forms a cation-selective ion channel (Maricq et al., 1991). To date, five 5-HT3 receptor subunits (A, B, C, D, and E) have been cloned (Davies et al., 1999; Karnovsky et al., 2003; Maricq et al., 1991; Niesler et al., 2003). However, only homomeric 5-HT3A receptors and heteromeric 5-HT3A/3B receptors are known to form functional LGICs (Davies et al., 1999; Dubin et al., 1999).
The 5-HT3 receptor is found in many brain regions (Maricq et al., 1991; Morales et al., 1996; Morales and Bloom, 1997), and is implicated in functions and disorders including cognition, appetite, anxiety, emesis, pain, schizophrenia, and drug abuse (Barnes et al., 1992; Faerber et al., 2007; Grant, 1995; Jackson and Yakel, 1995). 5-HT3Rs are mainly expressed by GABAergic interneurons in the hippocampus and other forebrain nuclei (Vucurovic et al., 2010) at both pre- and postsynaptic sites. The presynaptic 5-HT3 receptors are believed to regulate the release of several neurotransmitters including GABA, dopamine, cholecystokinin, glutamate, and acetylcholine (Blandina et al., 1989; Giovannini et al., 1998; Morales and Bloom, 1997; Paudice and Raiteri, 1991; Ropert and Guy, 1991; Wan and Browning, 2008). In the hippocampus, 5-HT3 receptors stimulate GABA release from select interneuron subtypes, as shown using vibrodissociated neurons (Choi et al., 2007; Koyama et al., 2000).
The 5-HT3 receptor has been implicated in neuronal responses to alcohol. Acute EtOH exposure potentiates 5-HT3 receptor function in a variety of preparations (Lovinger et al., 2000; Lovinger and Zhou, 1993, 1994). However, to date there is no direct evidence of EtOH potentiation of native CNS 5-HT3Rs. Furthermore, all previous studies have focused on EtOH actions directly on 5-HT3Rs in neuronal or cultured cell somata, and thus it is not yet known how EtOH affects presynaptic 5-HT3 Antagonism of 5-HT3 receptors reduces alcohol craving and alters human self-reports of affective states that are relevant to alcohol disorder (Johnson et al., 1993; LeMarquand et al., 1994a; Sellers et 1994). These receptors have also been shown to reduce alcohol intake and alcohol-related aggression in rodents (Fadda et al., 1991; LeMarquand et al., 1994b). Interoceptive cues that signal intoxication during acute EtOH exposure involve 5-HT3 receptors (Grant and Barrett, 1991; Shelton et al., 2004).
In the present study, we used a vibrodissociated neuron/bouton preparation from the rat hippocampus to directly examine 5-HT3− mediated GABA release from presynaptic terminals and EtOH effects this receptor-induced response. We then examined EtOH effects calcium transients in presynaptic boutons in the neuron/bouton preparation. Ethanol potentiated 5-HT3 receptor-mediated GABAergic transmission in a concentration-dependent manner. Surprisingly, potentiation persisted even after EtOH was removed from the preparation, indicating ethanol-induced long-term synaptic plasticity. We examined the role of presynaptic calcium and PKA signaling in EtOH/5-HT3-dependent long-lasting potentiation.
2. Material and methods
2.1. Postsynaptic neuron/presynaptic bouton preparation
Coronal brain slices containing hippocampus were prepared from individual P13–P17 Sprague-Dawley rats. Rat pups were selected from litters without sex identification, and thus we assume that approximately equal numbers of female and male rats were used but we do not know the actual proportion of each sex. It should be noted that rats are not fully sexually mature at this pre-weaning age, and thus it is unclear if they would express sex differences that might be seen in adult animals. Animals were decapitated while anesthetized with halothane. All procedures were performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals, and were approved under protocol LIN-DL-1. Extracted brains were sectioned (400 μm thickness) using a VT1200S vibratome (Leica Microsystems Inc., Bannockburn, IL) in buffer containing (in mM): 194 sucrose, 30 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, and 10 D-Glucose, oxygenated with 95% O2/5% CO2. Slices were maintained for at least 1 h in oxygenated artificial cerebrospinal fluid (ACSF) containing (in mM): 124 NaCl, 4.5 KCl, 1 MgCl2, 26 NaHCO3, 1.2 NaH2PO4, 10 Glucose, 2 CaCl2, osmolarity adjusted to 320 mOsm with sucrose at room temperature.
2.2. Mechanical neuronal dissociation
Individual hippocampal neurons were isolated from the CA1 region using an enzyme-free mechanical dissociation procedure, as described previously (Akaike and Moorhouse, 2003; Jun et al., 2011; Vorobjev, 1991). Briefly, slices including hippocampus were transferred into a 35 mm culture dish containing an external recording buffer with the following composition (in mM): 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 D-Glucose, pH adjusted to 7.4 with NaOH, osmolarity adjusted to 320 mOsm with sucrose. A fire-polished glass micropipette was placed on the surface of the CA1 region. The pipette tip was vibrated horizontally within the CA1 region at 30Hz over a distance of 100–200 μm for ~2 min using a piezoelectric manipulator (LSS-3000, Burleigh/Exfo, Quebec, Canada) triggered by a Grass SD9K stimulator (Grass Technologies, West Warwick, RI). After the slice was removed, the isolated neurons were allowed to settle on the bottom of the dish for 10–15 min. For purely electrophysiological experiments, cells were isolated/examined in standard 35 mm cell culture dishes, (BD Biosciences, San Jose, CA). For experiments involving imaging, cells were isolated/examined in 35 mm cell culture dishes containing a glass coverslip insert (WillCo Wells B.V., Amsterdam, Netherlands). Cover-slips were coated with poly-l-lysine prior to use.
2.3. Electrophysiology
Synaptic currents were measured in the conventional whole-cell voltage-clamp mode from the isolated neurons with a glass pipette electrode (2–5 MΩ resistance) at a holding potential of −60 mV. Only neurons that exhibited somata and proximal dendrites characteristic of CA1 pyramidal neurons were selected for recording (Sheinin et al., 2008). All recordings were performed at room temperature. The intracellular whole-cell patch pipette solution contained the following (in mM): 150 CsCl, 2 MgCl2, 0.3 Na-GTP, 3 Mg-ATP, 0.2 BAPTA, and 10 HEPES, pH adjusted to 7.23 with CsOH, and osmolarity adjusted to 300 mOsm with sucrose. The composition of the extracellular solution was (in mM): 150 NaCl, 5 KCl, 2.5 CaCl2, 1 MgCl2, 10 HEPES, and 10 d-Glucose, pH adjusted to 7.4 with NaOH, and osmolarity adjusted to 320 mOsm with sucrose. To isolate spontaneous inhibitory postsynaptic currents (sIPSCs), 2,3-dioxo-6-nitro-1,2,3,4-tetrahydrobenzo[f]quinoxa-line-7-sulfonamide disodium (NBQX, 5 μM) and D-2-amino-5--phosphonopentanoic acid (AP5, 50 μM) were locally applied to the target neurons using a multi-barreled array of square glass applicators mounted on a Perfusion Fast-Step system (Warner Instruments, Hamden, CT) with different solutions gravity-driven through the applicator from wells placed above the preparation.
Current recorded via the patch pipette was amplified and low-pass filtered with a cutoff frequency of 2 kHz using an Axopatch 200B amplifier (Molecular Devices, Sunnyvale, CA), and the data were stored using pClamp8 software after digitization at 5 kHz using a Digidata 1322 interface (Molecular Devices, Sunnyvale, CA). The spontaneous synaptic currents were detected using MiniAnalysis Software (Synaptosoft, Decatur, GA) with a threshold detection of 20 pA and manual detection for smaller events. Measures of sIPSC amplitude, rise time and decay time were also calculated using MiniAnalysis. Agonists, antagonists and EtOH were delivered via the Perfusion Fast-Step system, enabling solution exchanges within 100 ms. After normalization to the baseline frequency, sIPSC frequency, amplitude, rise and decay time histograms were averaged and plotted with GraphPad Prism software (GraphPad Software, Inc., La Jolla, CA). The peak sIPSC frequency and the charge transfer during agonist application were used as measures to compare the IPSCs induced by 5-HT3R activation under different conditions. The charge transfer was calculated by measuring the total area of all sISPCs relative to the holding current during 5-HT applications using Clampfit 9 software (Molecular Devices, Sunnyvale, CA). The charge transfer value was normalized to the charge transfer during initial 5-HT3R activation for each cell before exposure to EtOH or other drugs. Average sIPSC measure values are expressed as mean+/−SEM unless otherwise stated.
2.4. Calcium imaging
To directly assess presynaptic terminal function, we measured calcium transients in terminals in the neuron/bouton preparation after refining the previously reported techniques for loading an AM-esterified calcium indicator dye into presynaptic elements (Ye et al., 2004). Ten min after isolation, vibrodissociated neurons were incubated in HEPES-buffered saline solution containing acetoxymethyl (AM) esterified calcium indicator (Fluo-4AM, 2 μM, Molecular Probes™, Eugene, OR) at 37 °C for 15 min. Prior to fluorescence measurement, cells were washed in indicator-free medium to remove dye nonspecifically associated with the cell surface. Dishes containing isolated neurons were placed on the stage of an inverted microscope (Olympus) with a 60x, 1.4NA oil-immersion objective. An EMCCD camera (Andor iXon+, model DU-888) was used for image collection. Whole-cell recording in the postsynaptic cell was performed to measure sIPSCs as well as to wash the dye out of the postsynaptic cytosol, thus enabling the selective visualization of dye-loaded presynaptic puncta. Fluorescence was detected with 1.2% of maximum illumination intensity using an X-Cite® illuminator with a GFP filter set (Chroma, Bellows Falls, VT). Images were captured at a sampling rate of 15Hz. External buffer or drug solutions were locally superfused with the multi-barrel fast applicator system described previously. Stored images were analyzed offline with ImageJ. The images were collected over 60 s epochs before, during and after agonist/drug treatment. After collecting all images from the same preparations, circular regions of interest (ROI, 1–2 μm diameter) were defined that encompassed fluorescent puncta showing transient changes in brightness during the pre-drug baseline period or during EtOH application. The mean fluorescence intensity of pixels inside the ROI was calculated and plotted over time. Data were not included if the punctum moved significantly during imaging or escaped from the ROI. Some puncta showed detectable fluorescence prior to, and throughout EtOH application with no apparent transients during the entire experiment. These puncta were excluded from further analysis.
2.5. Statistics
The quantified data are presented as mean ± standard error of the mean (SEM). Student’s t-test was used for between-group comparisons with GraphPad Prism (GraphPad Software, Inc.). P value < 0.05 was considered statistically significant when paired t-tests were performed, for example when comparing baseline to drug effects within the same neuron in electrophysiological and imaging experiments. To analyze the effect of the EtOH concentration on mCPBG-induced sIPSCs, analysis of variance (ANOVA), Friedman test and multiple comparisons were performed using the Wilcoxon Signed-Rank Test for Matched Pairs with the level of significance set at P < 0.05. The Kolmogorov-Smirnov test was used to compare cumulative probability histograms for the sISPC measurements, with the level of significance set at P < 0.05.
3. Results
3.1. 5-HT facilitates spontaneous IPSCs through the activation of 5-HT3 receptors in the neuron/bouton preparation
During whole-cell recordings from vibrodissociated CA1 pyramidal neurons, spontaneous synaptic currents were recorded in the presence of ionotropic glutamate receptor antagonists. In previous studies, we and others have shown that these are GABAAR-mediated sIPSCs (Akaike et al., 2002; Sheinin et al., 2008). Mean baseline sIPSC frequency was 3.6 ± 0.6 Hz, and mean baseline amplitude was 61.3 ± 13.9 pA. Application of 5-HT (1, 3, and 30 μM) using fast perfusion resulted in a burst-like increase of the frequency and amplitude of sIPSCs as shown in Fig. 1A. The peak sIPSC amplitude during 5-HT applications averaged 109 ± 17, 305 ± 40, and 516 ± 11 pA for 1, 3, and 30 μM 5-HT, respectively (Fig. 1B, n = 3 for each condition). The peak frequencies of sIPSCs during 5-HT application averaged 9.6 ± 0.4, 12.6 ± 1.9, and 28.7 ± 3.3 Hz for 1, 3 and 30 μM 5-HT, respectively (Fig. 1C). The sIPSC frequency returned toward baseline levels during continuous agonist applications lasting 30 s. The total area of all sISPCs relative to the holding current during 5-HT applications was calculated and used as the charge transfer measure. 1, 3 and 30 μM 5-HT induced windows of current with charge transfer values of 2.4 ± 0.9, 7.4 ± 2.7, and 11.8 ± 4.8 μC respectively (Fig. 1D). Fig. 1E–G shows cumulative probability histograms demonstrating that 5-HT induces a concentration-dependent increase in IPSC amplitude with no change in rise time but a detectable change in decay time. Kolmogorov-Smirnov (K–S) tests indicated significant increases in amplitude at all three 5-HT concentrations (K–S p values: 1 μM < 0.0001, 3 μM < 0.0001, 30 μM < 0.0001), no differences in rise time (1 μM > 0.9999, 3 μM > 0.09996, 30 μM > 0.9986), and increases in decay time (K–S p values: 1 μM < 0.0001, 3 μM < 0.0001, 30 μM = 0.0002).
Fig. 1. 5-HT increases the frequency and amplitude of spontaneous IPSCs.
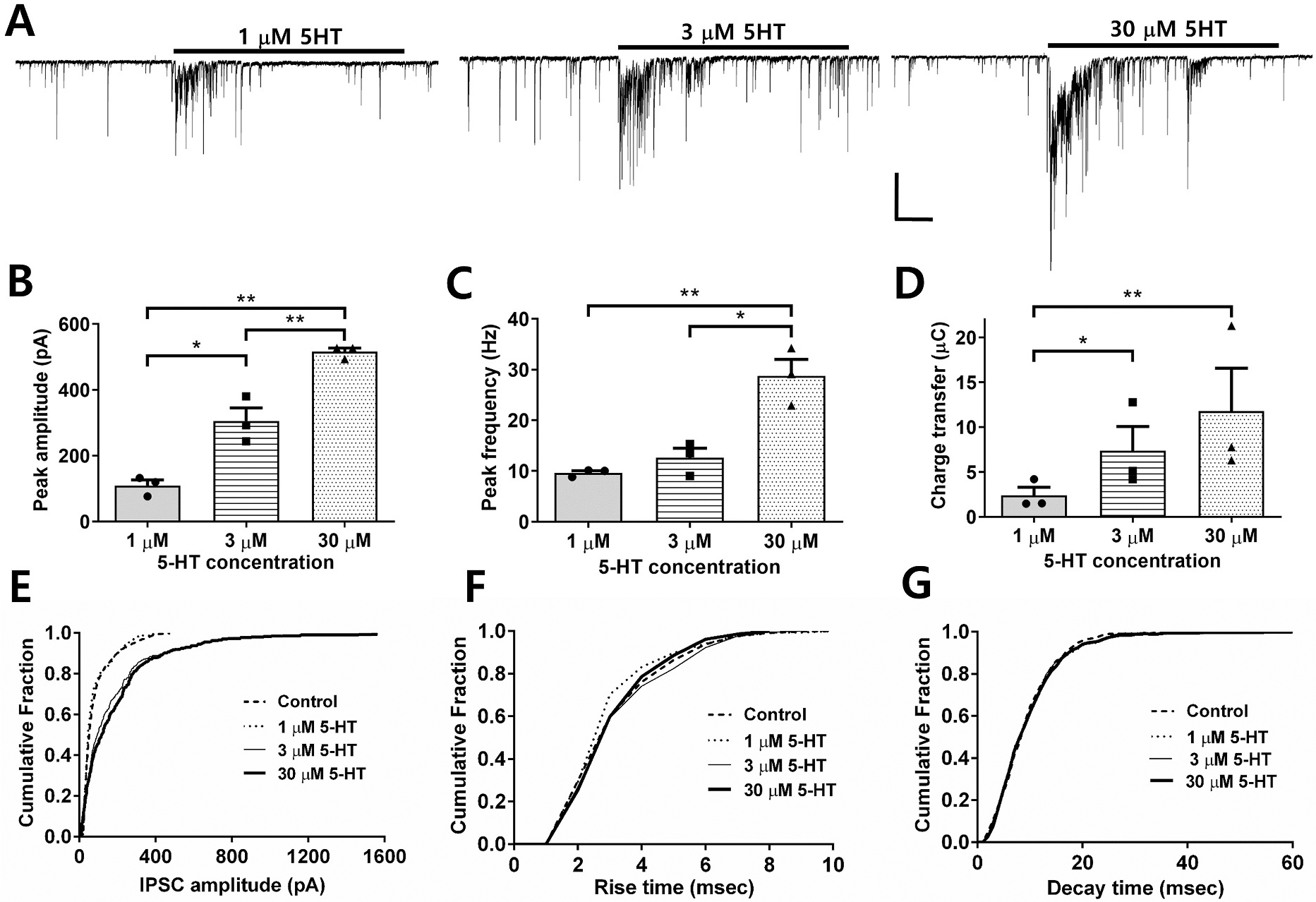
A. Representative sIPSC traces before, during and after exposure to different concentrations of 5-HT. Scale bar = 100 pA, 5 s. B. Bar graph showing peak amplitude of IPSCs evoked by the different 5-HT concentrations (n = 3) C. Bar graph showing peak frequency of IPSCs evoked by the different 5-HT concentrations (n = 3) D. Bar graph showing charge transfer of IPSCs evoked by the different 5-HT concentrations (n = 3). (unpaired t-tests *P < 0.05, **P < 0.001). Cumulative probability distributions for E. IPSC amplitude, F. Rise time, and G. Decay time.
The GABAA receptor antagonist gabazine (10 μM) suppressed all sIPSCs both before and during 5-HT application (Fig. 2A), consistent with previous reports that 5-HT acts to increase GABA release leading to postsynaptic GABA-R activation in this preparation. This finding also indicates that 5-HT does not induce a direct postsynaptic current in isolated CA1 pyramidal neurons (Turner et al., 2004). In the presence of the 5-HT3 antagonist, MDL 72,222 (0.5 μM), the facilitation by 5-HT was not observed while baseline sIPSC frequency was unaffected (93 ± 35% of control, Fig. 2B and C).
Fig. 2. 5-HT transiently increases the frequency of GABAergic sIPSCs through the activation of 5-HT3 receptors and ethanol potentiates the increase induced by a selective 5-HT3 agonist.
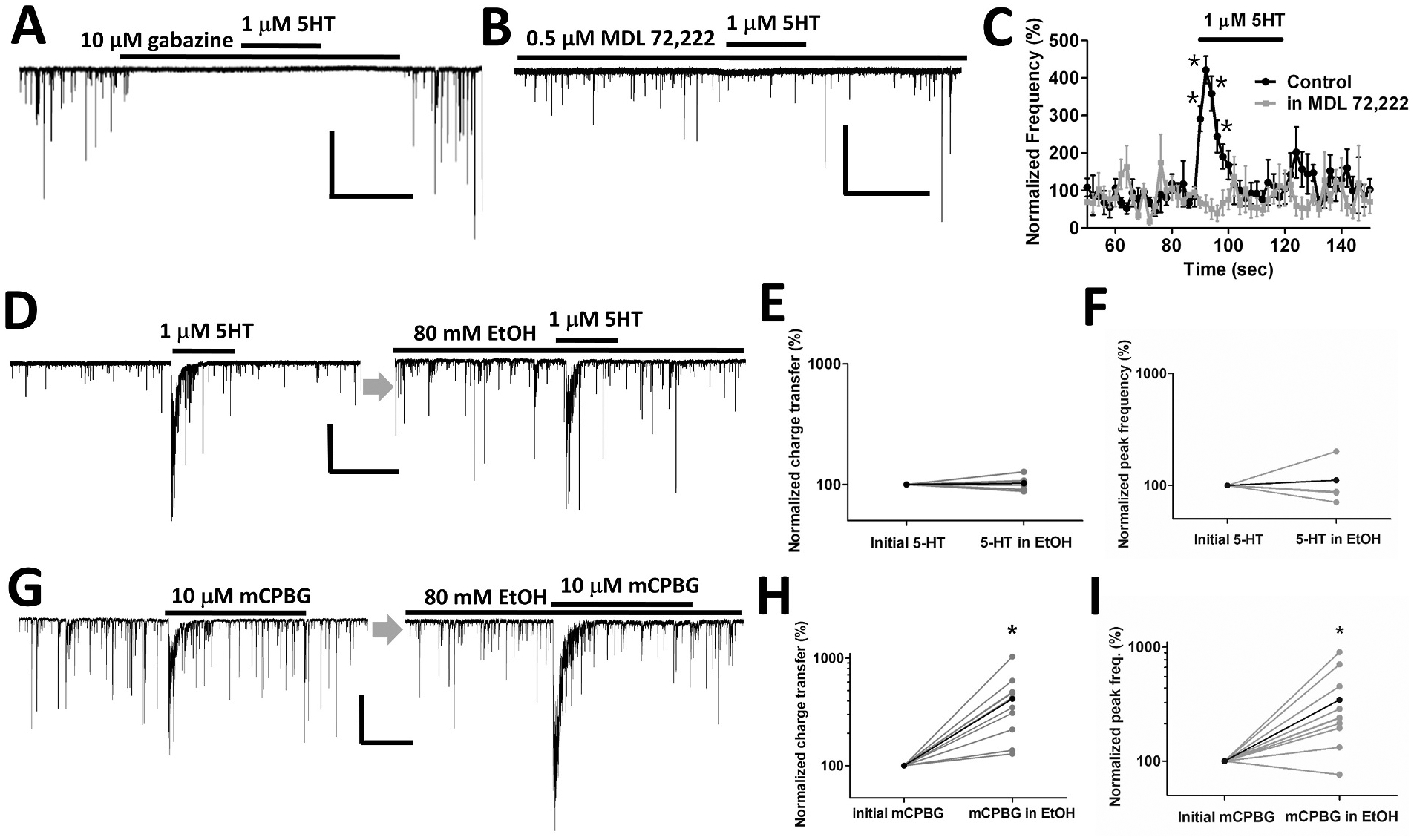
A. The GABAA receptor antagonist gabazine suppressed all sIPSCs as well as the response to 5-HT. Scale bar = 100 pA, 30 s. B, C. The response to 5-HT was blocked in the presence of the selective 5-HT3R antagonist MDL 72222 while baseline sIPSCs remained (n = 5). Scale bar = 200 pA, 30 s (unpaired t-tests *P < 0.05). D. Representative sIPSC waveforms in response to 5-HT in the absence and presence of 80 mM EtOH. sIPSCs induced by 5-HT. Scale bar = 200 pA, 30 s. E, F. No difference was observed in the charge transfer and peak frequency of sIPSC responses to 5-HT before and during EtOH incubation (n = 5). G. Representative sIPSC waveforms in response to the selective 5-HT3 receptor agonist, mCPBG in the absence and presence of 80 mM EtOH. Scale bar = 200 pA, 30 s. H, I. EtOH potentiates the charge transfer and peak frequency of the 5-HT3 agonist-induced increase in sIPSCs (paired t-tests *P < 0.05, n = 10).
Application of the selective 5-HT3 agonist mCPBG (10 μM) potentiated the peak frequency of GABAergic sIPSCs to 362 ± 65% of pre-agonist baseline values, which was similar to the sIPSC frequency increase elicited by 1 μM 5-HT (313 ± 32%) as shown in Fig. 2D, G (left). This agonist also produced a window of increased total charge transfer for GABAergic sIPSCs of 4.6 ± 1.6 μC (n = 10) comparable to that induced by 1 μM 5-HT (2.4 ± 0.9 μC, n = 3). To allow for sufficient recovery from desensitization between responses, successive applications of 5-HT or mCPBG were given more than 6 min apart.
3.2. Ethanol potentiates 5-HT3 agonist-induced increases in sIPSCs
The effect of EtOH on 5-HT enhancement of sIPSCs was examined by first recording at least 2 responses to agonist before EtOH application, then applying 80 mM EtOH for 3.5 min from one of the barrels of the rapid perfusion system, then applying 5-HT at two time points during continued EtOH exposure, followed by removal of EtOH and subsequent measurement of at least two more responses to agonist. In these experiments, 1 μM 5-HT was used to avoid full occupancy of the agonist site. Past studies showed that EtOH potentiation was most robust at this 5-HT concentration (Lovinger and White, 1991). The frequency of sIPSCs in the absence of agonist was slightly increased to 134 ± 15% of baseline during exposure to 80 mM EtOH alone. When examining individual neurons, clear increases to 156 ± 15% were observed in 14 of 20 cells examined. The sIPSC frequency returned to baseline levels following washout of EtOH. When 5-HT was applied to the neurons, no distinct EtOH effect on the agonist-induced change in sIPSCs was observed using either the peak frequency or the charge transfer measure (Fig. 2D, E, F). The normalized charge transfer and peak frequency values of 5-HT responses in 80 mM EtOH were 103 ± 2% and 111 ± 23% of the values in the absence of EtOH.
We then examined EtOH effects on responses to the selective 5-HT3 agonist mCPBG, to assess effects on receptor function while avoiding possible interference from activation of other 5-HT receptors. Exposure to 80 mM ethanol produced a more than 3-fold increase in the mCPBG-induced sIPSC charge transfer and peak frequency compared with the initial response to mCPBG alone (418 ± 85% for charge transfer and 343 ± 84% for peak frequency of the control mCPBG response, paired t-test P < 0.05, n = 10) (Fig. 2G, H, I). To ensure the stability of responses to mCPBG alone, the application of mCPBG in ethanol was performed after two consecutive applications of mCPBG with the same fixed interval between applications (6.5 min). The sIPSC charge transfer and peak frequency were stable prior to EtOH application (Fig. 3A and B).
Fig. 3. The sIPSC response to mCPBG depends on EtOH exposure and the exposure time.
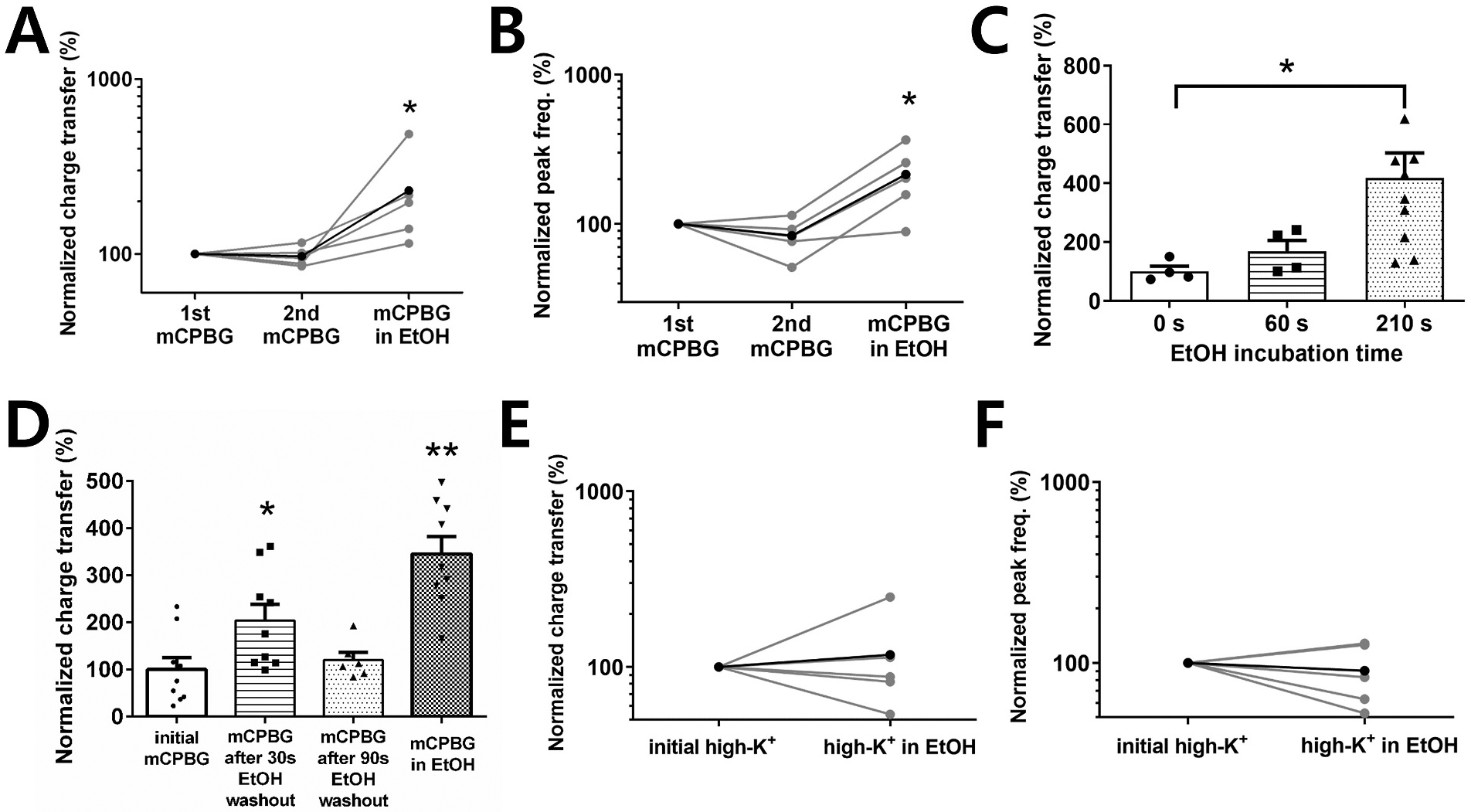
A, B. The charge transfer and peak frequency of sIPSCs were not potentiated until EtOH was applied (paired t-tests *P <0.05, n = 5). C. EtOH potentiation of sIPSC charge transfer with different EtOH incubation times (unpaired t-tests *P <0.05, n = 4 for 0, 60 s conditions, n = 10 for 210 s condition). D. When a second mCPBG application was given at different time points after EtOH exposure, the potentiation of the mCPBG response was smaller at longer EtOH wash-out times (unpaired t-tests *P <0.05, **P <0.001, n = 6 for 90s condition, n = 9 for other conditions). E, F. The charge transfer and peak frequency of sIPSC responses to high K+ were not potentiated by EtOH exposure (n = 5).
The duration and relative timing of EtOH exposure required to produce potentiation was next examined. Initiating EtOH (80 mM) exposure at the same time as the onset of mCPBG application did not result in detectable potentiation of the response to mCPBG (Fig. 3C). Exposure to EtOH for 1 min or 3.5 min prior to mCBPG application yielded increases in charge transfer of 169 ± 36% of agonist alone and 418 ± 85% of agonist alone, respectively (Fig. 3C). To determine if potentiation requires that EtOH be present during agonist application we first recorded the baseline response to mCPBG as usual and applied EtOH for 3.5 min with no agonist application, then washed the preparation for 30 or 90 s before re-applying mCPBG. The mCPBG-induced charge transfer values were 204 ± 34% of agonist-alone and 121 ± 16% of baseline agonist responses for the 30 s and 90 s washout conditions, respectively (Fig. 3D). These data indicate that EtOH does not have to be present at the time of mCPBG application for potentiation to occur. However, the process initiated by EtOH exposure that leads to potentiation diminishes within 90 s if agonist is not applied.
To determine if EtOH potentiation requires 5-HT3R activation, in preference to other mechanisms of presynaptic depolarization, high-K+ stimulation was applied to vibrodissociated neurons instead of mCPBG. The application of high-K+ (10 mM) solution increased the charge transfer of sIPSCs. However, the KCl stimulation of sIPSCs was not potentiated in the presence of 80 mM EtOH. The charge transfer and peak frequency during high-K+ exposure in the presence of EtOH averaged 117 ± 35% and 90 ± 14% of the control high-K+ response, respectively (Fig. 3E and F).
3.3. Long-lasting EtOH potentiation of 5-HT3 receptor-mediated responses
Potentiation of 5-HT3 receptor-mediated increases in GABA release was maintained even after washout of EtOH as shown in Fig. 4A. Following the second application of mCPBG in the presence of 80 mM EtOH, the cells were superfused with external solution without EtOH for 3.5 min, a time period more than sufficient to allow for reversal of EtOH effects in previous studies on isolated neurons (Criswell et al., 2008; Lovinger and White, 1991; Zhu and Lovinger, 2006). In this experiment, exposure to EtOH (80 mM) alone had no effect on baseline sIPSC frequency to 100 ± 2.6% of baseline response (n = 6). However, the response to mCPBG remained potentiated even after EtOH washout. The normalized peak sIPSC frequency during mCPBG exposure in the presence of EtOH and after EtOH washout were 319 ± 117% and 481 ± 274% of baseline, respectively (Fig. 4B). The charge transfer values for mCPBG stimulation of sIPSCs in the presence of EtOH and after EtOH washout were 479 ± 127% and 494 ± 182% of the value of initial charge transfer without EtOH exposure, respectively (Fig. 4C). The increases in sIPSC charge transfer and peak frequency during EtOH exposure and after washout were statistically significant relative to the pre-EtOH values (paired t-test P < 0.05, n = 6). The effect of EtOH potentiation persisted for as long as recorded neurons remained viable (up to 30 min after EtOH washout was the longest time point examined).
Fig. 4. Potentiation of the response to mCPBG was maintained even after EtOH washout.
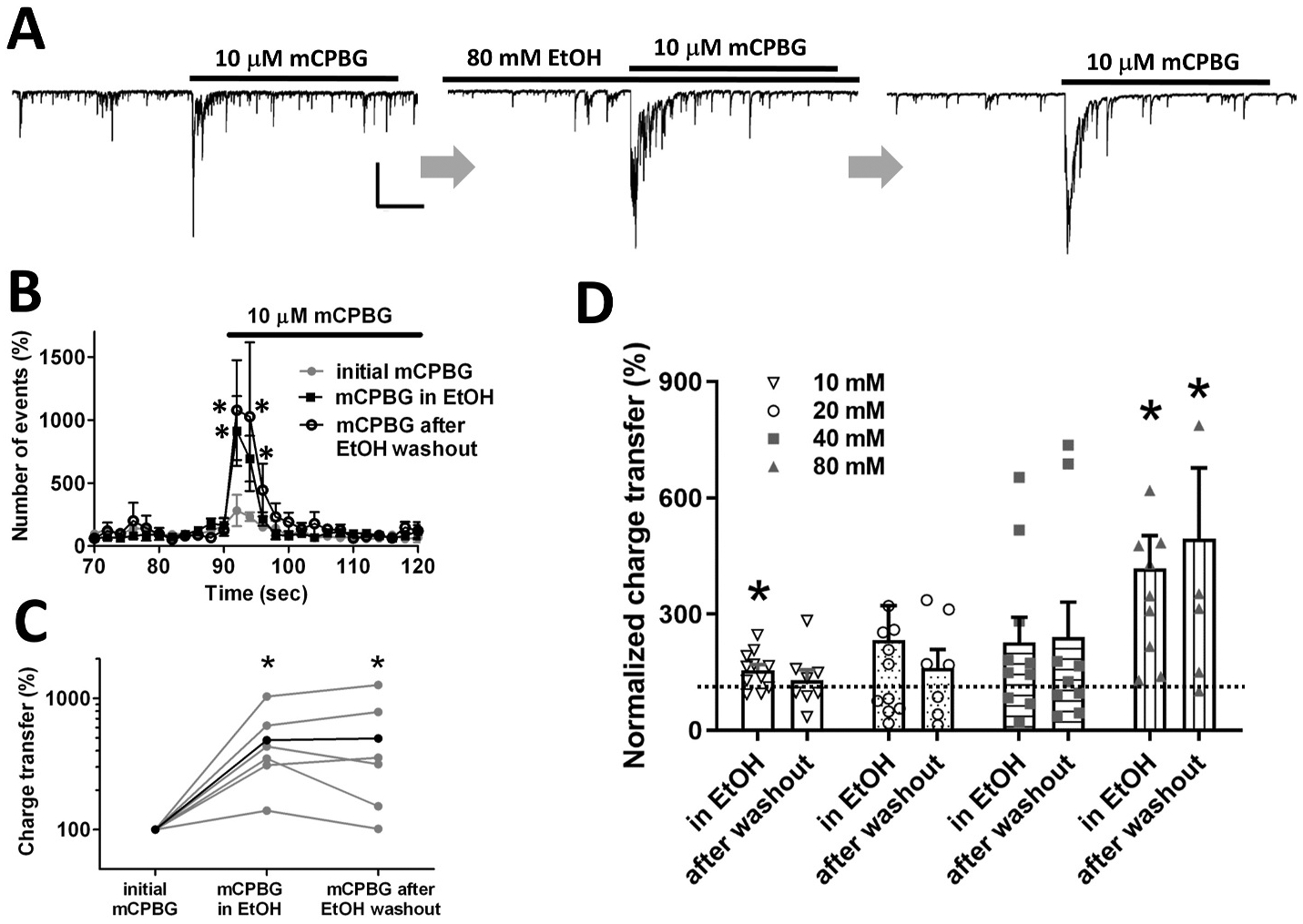
A. Representative waveforms of sIPSCs in response to 10 μM mCPBG before, during and after washout of 80 mM EtOH. Scale bar = 400 pA, 10 s. B. Plots of sIPSC frequency (paired t-tests *P < 0.05, n = 6). C. EtOH potentiation of sIPSC charge transfer and peak frequency was maintained even after EtOH washout (paired t-tests *P < 0.05, n = 6). D. EtOH increases the charge transfer of sIPSC responses to mCPBG in a concentration-dependent manner. The post-EtOH-washout potentiation was also more distinct with higher EtOH concentrations. (paired t-tests *P < 0.05, n = 9, 9, 8 and 6 for 10, 20, 40 and 80 mM EtOH, respectively).
To examine the concentration dependence of EtOH actions, the effects of 10, 20, and 40 mM EtOH were also determined using the EtOH exposure, agonist exposure and washout paradigms described above. The EtOH potentiation of the charge transfer during mCPBG exposure increased in a concentration-dependent manner as shown in Fig. 4D. The changes in charge transfer averaged 155 ± 14%, 233 ± 89%, 227 ± 64%, and 418 ± 84% for 10, 20, 40 and 80 mM EtOH, respectively. The potentiation persisted after EtOH washout after exposure to 40 and 80 mM EtOH, while responses after washout were generally similar to baseline values after the end of exposure to the 10 and 20 mM EtOH concentrations. The normalized charge transfer during mCPBG exposure was compared with the responses before and during EtOH exposure and after EtOH washout for the different EtOH concentrations.
The concentration dependence of the EtOH effect on mCPBG-induced sIPSCs was analyzed using a non-parametric analysis considering small and unequal sample sizes for each condition. Friedman tests were conducted to test whether there were significant differences among the three stages (initial, exposure to EtOH, wash-out) for each intensity (10, 20, 40, and 80 mM EtOH). Friedman tests revealed that there were significant differences only at 80 mM EtOH among the three stages (Chi-square = 9.0, df = 2, p = 0.011). As a post-hoc pairwise comparison, Wilcoxon signed rank tests were performed to identify the conditions which led to the significant results from the Friedman test. Results revealed that there were significant differences between initial response and exposure to EtOH (Z = −2.803, p=.005) and between initial response and washout conditions (Z = −2.201, p=.028), whereas differences between wash-out and exposure to EtOH were not significant (Z = −.734, p=.463). From the Friedman tests, none of the comparisons were significant at 10 mM (Chi-square = 1.75, df = 2, p=.417), 20 mM (Chi-square = 0.286, df = 2, p=.867), and 40 mM EtOH (Chi-square = 0.667, df = 2, p=.717).
3.4. Voltage-gated sodium channels and voltage-gated calcium channels are involved in 5-HT3 receptor-mediated synaptic potentiation
Previously, it was reported that activation of 5-HT3 receptors also enhances the frequency of GABAergic miniature inhibitory postsynaptic currents (mIPSCs) measured in the presence of tetrodotoxin (TTX) in mechanically dissociated rat amygdala neurons (Koyama et al., 2000). Furthermore, application of the nonspecific channel blocking ion cadmium did not prevent 5-HT3 facilitation of release. These findings were interpreted as indicating that GABA release could be stimulated by direct calcium influx through the 5-HT3 receptor activation. To determine if voltage-gated calcium channels are involved in 5-HT3-stimulated GABA release, we examined effects of the CaV2.2 N-type voltage-gated Ca2+ channel blocker, ω-conotoxin-GVIA (ω-Ctx) and the CaV2.1 P/Q-type voltage-gated Ca2+ channel blocker, ω-agatoxin-IVA (ω-AgaTx) on responses to mCPBG. These experiments were carried out in the absence of TTX. In the presence of 500 nM ω-Ctx, mCPBG produced little or no increase in sIPSC frequency or charge transfer (Fig. 5A). The charge transfer and peak frequency values of mCPBG responses during ω-Ctx exposure were 16 ± 4% and 38 ± 10% of the initial mCPBG responses, respectively (Fig. 5B and C). The baseline frequency of sIPSCs, prior to the second agonist application, was decreased in the presence of ω-Ctx to 75 ± 9% of pre-drug value (n = 5). These findings indicate the involvement of CaV2.2 calcium channels in spontaneous and 5-HT3R-mediated GABA release in the hippocampal neuron-bouton preparation. Application of EtOH (80 mM) in the presence of ω-Ctx did not increase sIPSC frequency or restore responses to mCPBG. Thus, the activity of CaV2.2 calcium channels appears to be necessary for stimulated release even under the EtOH treatment condition. Application of ω-AgaTx (300 nM) did not significantly alter the baseline frequency of sIPSCs (90 ± 9% of pre-drug value, n = 7) or the charge transfer and peak frequency during mCPBG application (98 ± 27% (charge transfer) and 87 ± 7% (peak frequency) of pre-drug value, (n = 7), suggesting that CaV2.1 channels are not mediating GABA release under these circumstances in this preparation (Fig. 5D, E, F). These findings suggest that 5-HT3R-mediated synaptic transmission involves activation of voltage-gated calcium channels, perhaps secondary to depolarization produced directly by activation of the ligand-gated channel itself.
Fig. 5. Voltage-gated calcium channels and voltage-gated sodium channels are involved in 5-HT3 receptor-mediated IPSC increases.
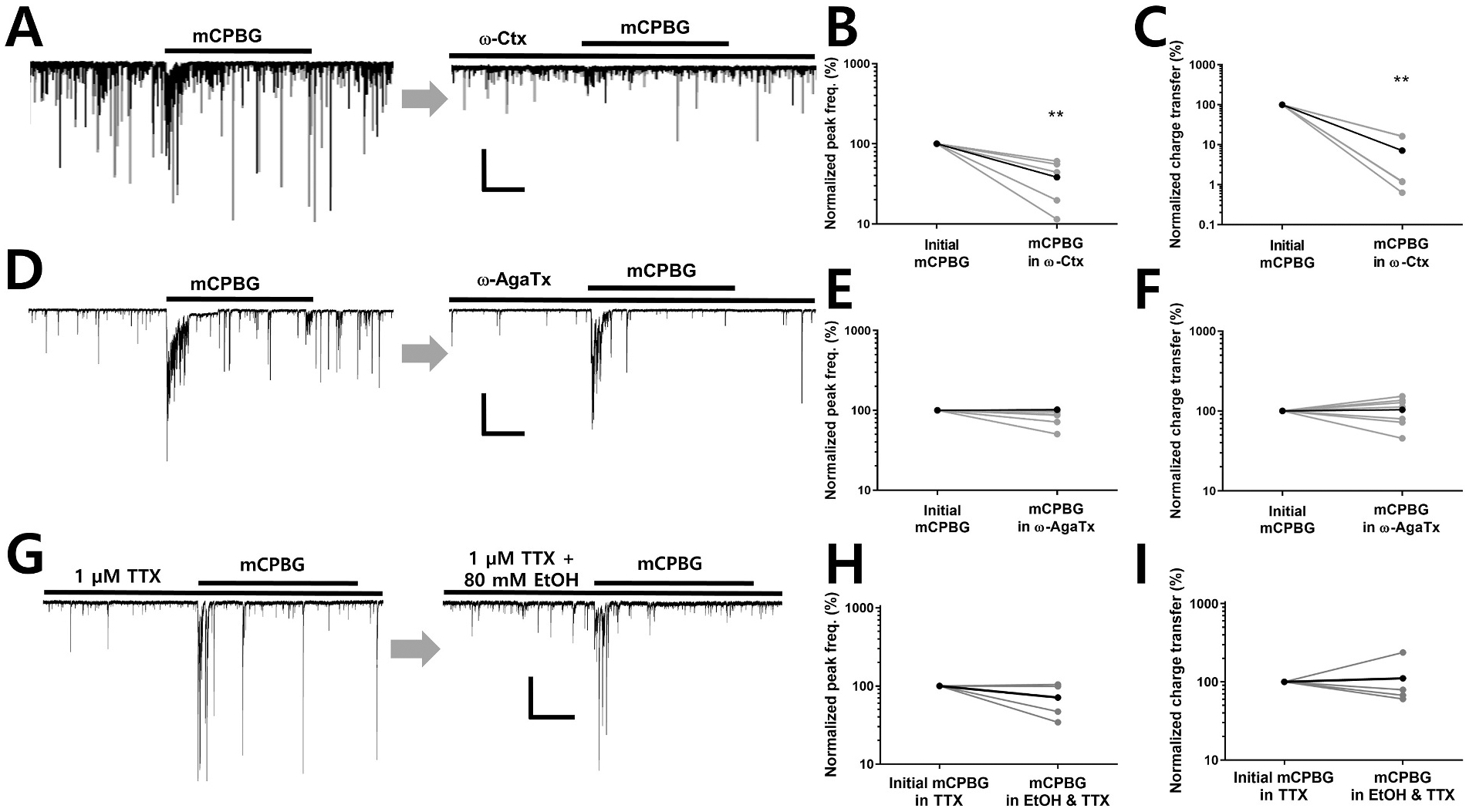
A. CaV2.2 N-type voltage-gated Ca2+ channel blocker ω-conotoxin-GVIA (ω-Ctx) suppressed baseline sIPSCs as well as the mCPBG response. Scale bar = 400 pA, 10 s. B, C. ω-Ctx significantly decreased the charge transfer and peak frequency of the mCPBG-induced increase in sIPSCs (paired t-tests **P < 0.01, n = 5). D. CaV2.1 P/Q-type voltage-gated Ca2+ channel blocker, ω-agatoxin-IVA (ω-AgaTx) had little effect on sIPSC responses to mCPBG. Scale bar = 400 pA, 10 s. E, F. No difference was observed in the charge transfer and peak frequency of sIPSC responses to mCPBG in the presence of ω-AgaTx (n = 7). G. Representative waveforms of mIPSC responses to mCPBG before and during ethanol exposure in the presence of TTX. Scale bar = 200 pA, 10 s. H, I. In the presence of TTX, EtOH did not potentiate mIPSC charge transfer during mCPBG application (n = 4).
The dependence of EtOH effects on voltage-gated sodium channel activation was examined by applying mCPBG and EtOH in the continuous presence of TTX. In the presence of TTX (1 μM), the baseline frequency of sIPSCs decreased to 29 ± 13% (n = 4) of the values recorded in the absence of the toxin. This is consistent with previous studies showing that TTX strongly decreases sIPSC frequency in the neuron/bouton preparation (Zhu and Lovinger, 2006). As shown in Fig. 5G, the application of mCPBG increased mIPSC frequency and charge transfer in the presence of 1 μM TTX. However, the increases induced by the 5-HT3 receptor agonist were not affected by acute exposure to 80 mM EtOH under this condition as shown in Fig. 5H and I. This finding indicates that the depolarization of presynaptic boutons through the opening of voltage-gated sodium channel is not necessary for mCPBG actions, but is needed for potentiation by EtOH.
3.5. Ethanol potentiation of 5-HT3 receptor-mediated GABA release is dependent on cAMP actions
Direct effects of EtOH on ion channels such as the 5-HT3 receptor are rapid in onset (initiated within msec) and rapidly reverse (Lovinger, 1991; Zhou et al., 1998). The finding that EtOH potentiation of 5-HT3-mediated GABA release required minutes of ethanol exposure, and was long-lasting, suggested a possible role for intracellular signaling mechanisms in the persistent release increase. Previous studies indicated that EtOH can stimulate the activity of adenylyl cyclase (Hasanuzzaman and Yoshimura, 2010; Rabin and Molinoff, 1983), and that stimulation of this enzyme to produce cyclic AMP (cAMP) may contribute to EtOH-induced increases in GABA release at CNS synapses (Tabakoff et al., 2001; Yoshimura and Tabakoff, 1999). If cAMP signaling contributes to EtOH potentiation of the 5-HT3-mediated response, then this effect might be prevented by a blocker of the cAMP-activated protein kinase A (PKA). In the presence of Rp-cAMPs (15 μM), a selective cell permeant PKA inhibitor, EtOH potentiation of mCPBG responses was abolished when examined with both the frequency and charge transfer measures (Fig. 6A). The normalized charge transfer value and the peak frequency of sIPSCs during mCPBG stimulation in the presence of EtOH and Rp-cAMPs were 102 ± 24% and 100 ± 19% of initial values before EtOH exposure, respectively (Fig. 6B and C). Application of this inhibitor did not alter baseline sIPSC frequency prior to mCPBG or EtOH application (92 ± 8% of control, n = 3). The inhibitor also did not alter the response to mCPBG in the absence of EtOH. This result suggests that PKA activation plays an important role in EtOH potentiation of 5-HT3–stimulated GABA release.
Fig. 6. Ethanol potentiation involves the cAMP-dependent protein kinase.
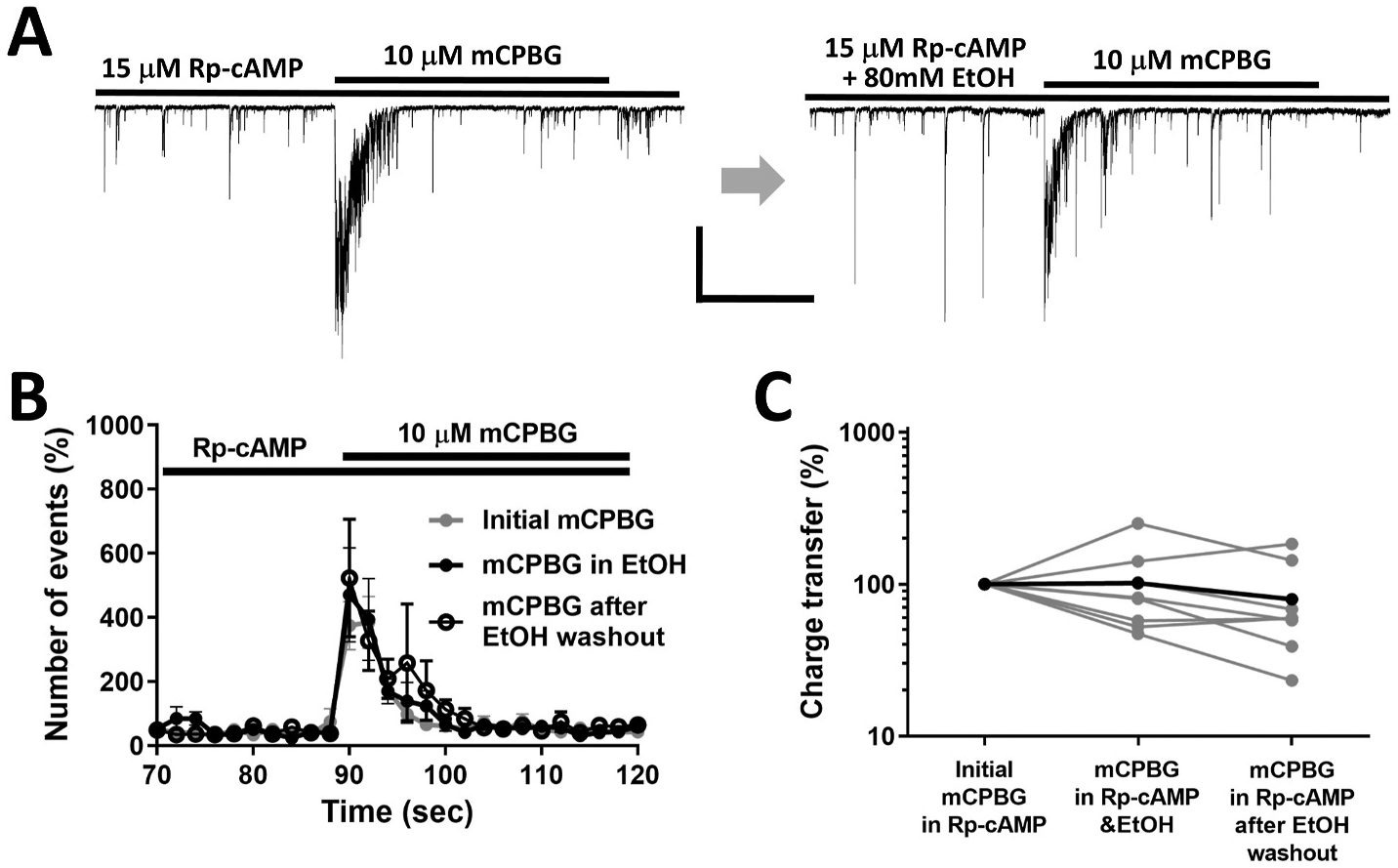
A. Representative waveforms of sIPSC responses to mCPBG and mCPBG + EtOH in the presence of the competitive PKA inhibitor, Rp-cAMP. Scale bar = 400 pA, 10s. B. Plots of sIPSC frequency in the presence of Rp-cAMP (n = 6) before, during and after EtOH exposure. C. EtOH potentiation of the 5-HT3 receptor-induced increase in charge trransfer was inhibited in the presence of Rp-cAMP (n = 6).
3.6. Spontaneous Ca2+ transients in presynaptic boutons are enhanced by acute EtOH exposure
After loading vibrodissociated neurons with AM-esterified Fluo-4 (Fig. 7A) and diluting postsynaptic dye via the intracellular patch-pipette solution (Fig. 7B), several bright dye-filled boutons were observed on the soma and dendrites of isolated neurons (Fig. 7C). In a subpopulation of the boutons on each neuron, spontaneous transient increases in fluorescence were observed (Fig. 7D). Tetrodotoxin (TTX, 1 μM) eliminated these spontaneous transients, showing that they are mediated by depolarization of the presynaptic terminals initiated by the activation of voltage-gated sodium currents (Fig. 7E). The Fluo-4 loaded puncta (presumed boutons) were categorized into three groups: i) puncta that exhibited fluorescence transients before and during EtOH exposure (e.g. Fig. 7F); ii) puncta that could not be detected prior to EtOH application, but exhibited detectable fluorescent transients during EtOH application, and iii) puncta that showed invariant fluorescence throughout the experiment. In 32 fluorescent puncta selected as ROI from 6 neurons with Fluo-4 loaded presynaptic terminals, 12 puncta showed fluorescence transients (i.e. fit into categories i and ii), while no transient was observed in 20 puncta (and these puncta were not included in the analysis of EtOH effects). To examine the effect of EtOH exposure on the calcium transients, the boutons with fluorescence transients were monitored before and during EtOH application. The frequency of calcium transients in the presynaptic terminals significantly increased in the presence of 80 mM ethanol (Fig. 7F). The integrated normalized fluorescence intensity (total area under the curves for all the transient waveforms observed during a 2 min recording epoch) was calculated and plotted for control conditions and EtOH exposure. After 3 min exposure to 80 mM ethanol, the total area of the calcium transients increased from 6.4 ± 2.3 a.u. to 49.2 ± 22.2 a.u. (paired t-test P < 0.05, n = 12, Fig. 7G). No change in the amplitude or area of each individual transient was observed. In contrast, in a control condition without EtOH exposure, the total area of fluorescence transients decreased over time from 112 ± 15 a.u. to 66 ± 9 a.u. presumably due to photobleaching (Fig. 7H). It should be noted that the data in Fig. 7G include puncta in which fluorescence was extremely low before EtOH application, but became readily detectable after drug administration. In the control data only include puncta where fluorescence was easily observed from the beginning of the recording are included. Thus, the range of y-axis values is different in the two panels.
Fig. 7. Calcium imaging shows that calcium entry is increased in presynaptic boutons after EtOH incubation.
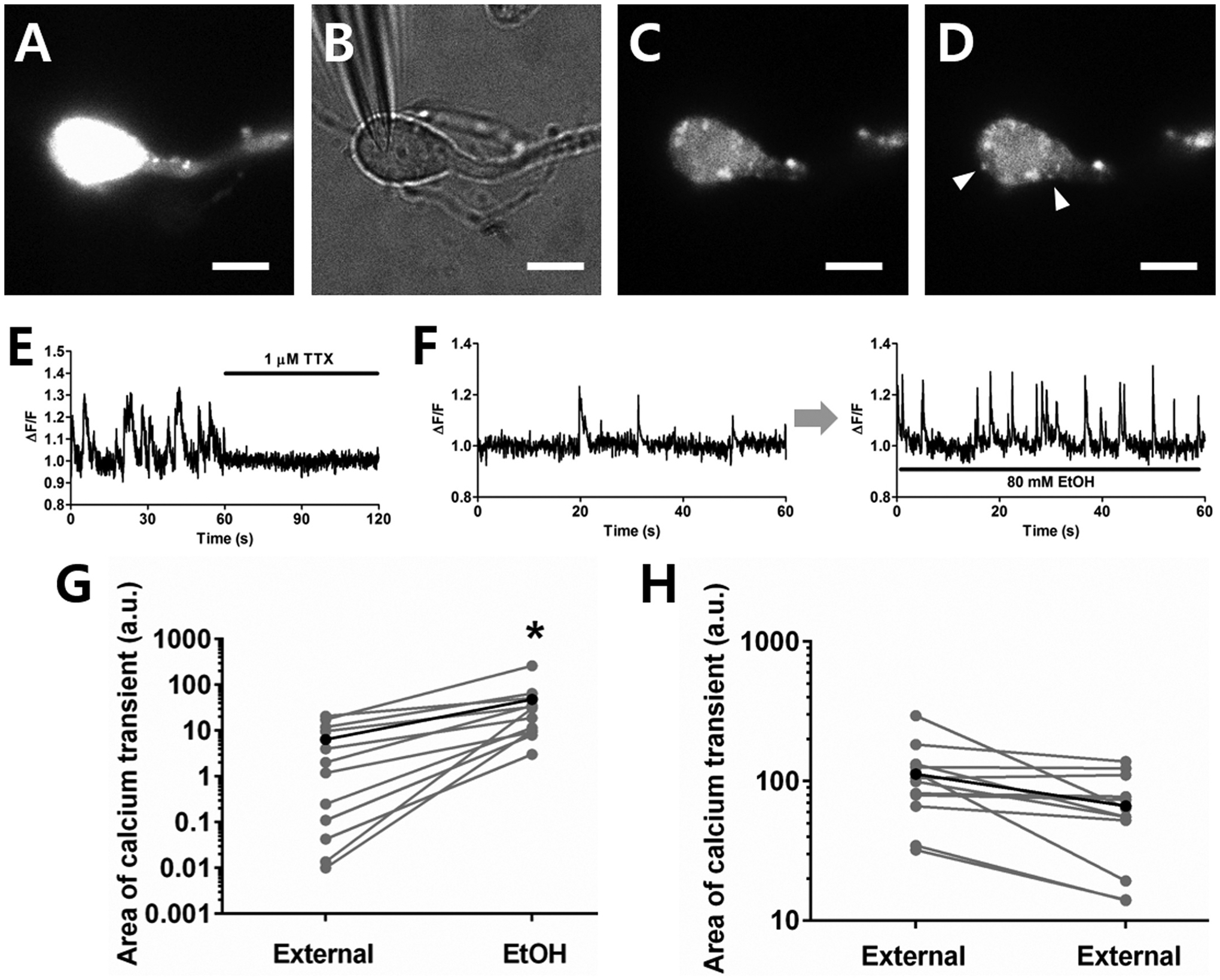
A. Fluorescence image after calcium dye loading in a mechanically-dissociated neuron. B. Wide-field image of patch pipette with giga-ohm seal on the neuron. C. Fluorescence image 2 min after opening the membrane for whole-cell recording. D. Calcium transients were observed from a subpopulation of dye-loaded puncta (white arrowheads) after diluting the dye in the postsynaptic neuron. Scale bar = 10 μm. E. The spontaneous calcium transients were suppressed by TTX. F. Representative spontaneous normalized Ca2+ transients before and during EtOH incubation. G. The spontaneous calcium entry increased in the presence of 80 mM EtOH (paired t-tests *P < 0.05, n = 12). H. In control experiments without EtOH exposure, no increase of calcium entry was observed. The small decrease in transients in the control condition is likely due to photo bleaching (n = 12).
4. Discussion
The present study shows EtOH potentiation of presynaptic 5-HT3 receptor-mediated synaptic GABA release in a CA1 hippocampal pyramidal neuron/bouton preparation. Potentiation was observed at concentrations associated with acute intoxication (10–80 mM), and the potentiation persisted even after EtOH was removed from the preparation following exposure to the highest EtOH concentrations. As predicted from previous studies, activation of presynaptic 5-HT3 receptors increased the frequency of both sIPSCs and mIPSCs (Choi et al., 2007; Koyama et al., 2000), indicating that receptors increase GABA release independent of voltage-gated sodium channel function. EtOH did not potentiate 5-HT3 effects on mIPSCs measured in the presence of TTX. Blockade of N-type calcium channels strongly suppressed 5-HT3R-mediated GABA release. Previous reports suggested that 5-HT3 receptors on presynaptic nerve terminals have high calcium permeability because the blockage of calcium channels did not affect GABA release induced by 5-HT3R activation (Koyama et al., 2000; Ronde and Nichols, 1998; Turner et al., 2004). In this regard, our findings may appear surprising. However, the evidence supporting a role for calcium influx directly through 5-HT3R in potentiated release comes mainly from experiments using the nonspecific channel blocker cadmium. In addition, the Ronde and Nichols (1998) and the Turner et al. (2004) studies examined the effects of conotoxin in synaptosomal preparations, where effects on release may differ relative to intact synapses. The calcium permeability of the 5HT3R may also vary with subunit composition of the 5-HT3R and the brain region examined (Nayak et al., 1999; Stewart et al., 2003). These factors might contribute to the apparent discrepancies between our findings and the previous studies by Koyama et al. (2000) in amygdala and Ronde and Nichols (1998) in striatum. Overall, our observations indicate that the increase in GABA release is unlikely to be a direct result of calcium entering through the 5-HT3R pore. Instead, it appears that 5-HT3R activation triggers activation of voltage-gated N-type calcium channels, probably via depolarization of the presynaptic terminal.
Previous findings indicate that EtOH directly potentiates postsynaptic 5-HT3 function (Lovinger and White, 1991). However, the present findings with TTX and N-type Ca2+ channel blockade suggest that this simple interpretation cannot account for the presynaptic potentiation by EtOH. The blockade of EtOH actions by TTX, despite the fact that 5-HT3R-driven GABA release still occurs under this condition, indicates that voltage-gated sodium channels downstream of the receptor likely participate in the potentiation. The loss of stimulated GABA release during N-type Ca2+ channel blockade, regardless of the presence or absence of EtOH, indicates that potentiation of 5-HT3R function is not sufficient to produce enhanced release. Other characteristics of the EtOH potentiation are also inconsistent with the receptor being the predominant mediator of this effect. Direct EtOH potentiation of 5-HT3R function is readily reversible when examined with whole-cell recording in cell lines or neurons (Lovinger and White, 1991; Rusch et al., 2007; Sung et al., 2000), but persisted well beyond the period of simultaneous application of EtOH and agonist in the neuron/bouton preparation. Thus presynaptic potentiation is not simply due to the reversible enhancement of receptor function. In previous studies showing direct EtOH potentiation of 5-HT3Rs, this effect was only observed at relatively low agonist concentrations. However, the concentration of mCPBG used in the present study was close to saturating (Koyama et al., 2000), indicating that the presynaptic EtOH potentiation may not involve only direct actions on 5-HT3 receptors. It seems very likely that factors downstream of the receptor contribute to this potentiation.
Several studies have reported that EtOH activates cAMP-PKA signaling in the CNS (Asher et al., 2002; Asyyed et al., 2006; Lin et al., 2006). The observation that a cell permeable PKA blocker prevents EtOH potentiation of 5-HT3-mediated GABA release indicates that cAMP/PKA signaling plays a role in this plasticity. We did not attempt to block only postsynaptic PKA activation, and thus we cannot say at the present time if the PKA activation that is relevant to potentiation takes place in a pre- or postsynaptic neuronal compartment. It is interesting to note that Koyama et al. (2000) observed faster recovery from desensitization of presynaptic 5-HT3R-mediated responses and increased mIPSC frequency in the presence of a cell-permeable cAMP mimic. Thus, there is precedent for modulation of 5-HT3R actions by the cAMP/PKA pathway. The observed effect of N-type Ca2+ channel blockers on EtOH potentiation of GABA release, as well as the EtOH-induced increases in presynaptic calcium transients suggests that calcium signaling also contributes to potentiation.
It is notable that EtOH did not potentiate 5-HT-induced GABA release while the GABA release induced by the selective agonist mCPBG was clearly potentiated by EtOH. To determine if 5-HT3R activation is a crucial factor in the long-lasting EtOH potentiation of GABAergic transmission, high K+ (20 mM) stimulation was employed in the absence or presence of EtOH to depolarize the presynaptic terminals instead of the activation of 5-HT3 receptors. High-K+-induced GABA release was not increased after EtOH exposure, indicating that the EtOH effect is not observed under every condition where presynaptic function is enhanced, and thus has some specificity for 5-HT3-induced responses. The reason for the lack of EtOH potentiation when receptors are activated by 5-HT, as opposed to mCPBG, is unclear. However, Gi/o-coupled 5-HT1A receptors reduce GABA release in this preparation (Katsurabayashi et al., 2003). Activation of this receptor subtype may limit ethanol effects when 5-HT is applied. This scenario would also be consistent with findings from several laboratories showing that activation of Gi/o-coupled metabotropic receptors reduces ethanol effects at GABAergic synapses in several brain regions (Ariwodola and Weiner, 2004; Kelm et al., 2008; Roberto et al., 2010; Zhu and Lovinger, 2006; Wan et al., 1996).
Ethanol has been shown to directly potentiate GABAA receptors on postsynaptic neurons, at least for some receptor subtypes in particular brain regions (Lobo and Harris, 2008). However, ethanol enhancement of GABAA receptors is not uniformly observed throughout the nervous system, and EtOH potentiation of GABAergic transmission in the hippocampal CA1 region involves presynaptic as well as postsynaptic effects (Fleming et al., 2009; Glykys and Mody, 2007; Silberman et al., 2008; Wafford and Whiting, 1992). The potentiation we observed clearly involved increased sIPSC frequency and increased presynaptic calcium transients, and occurred in the absence of any postsynaptic 5-HT3R-mediated current or measureable EtOH potentiation of postsynaptic GABAAR function. Thus, the long-lasting potentiation of 5-HT3R-stimulated GABA release appears to be mainly due to presynaptic mechanisms.
Imaging presynaptic terminals showed that the frequency of calcium transients increased during EtOH (80 mM) exposure without explicit 5-HT3R activation. This EtOH effect likely contributes to the enhancement of 5-HT3-induced GABA release (possibly even if the agonist exposure is delayed until after the end of the EtOH application). Unfortunately, calcium increases produced by 5-HT3 agonist alone could not be detected in the putative presynaptic terminals we examined despite the electrophysiological data indicating that 5-HT3 activation increases calcium-dependent GABA release. The relatively low sensitivity of the calcium measurements, perhaps due to Ca2+-buffering by Fluo-4, is one likely factor accounting for the failure to observe these responses. Dye loading with low concentrations or for short times did not impair sIPSCs, but resulted in low fluorescence intensity and poor detection of loading in terminals. Although spontaneous calcium transients were observed from several puncta after dye loading, most of the puncta on a given neuron showed bright fluorescence immediately after the loading procedure and did not change intensity with any treatment, probably due to saturation of the dye binding to Ca2+ ions. Thus, it is not yet clear if presynaptic calcium increases stimulated by the receptor are increased during EtOH exposure.
The experiments described at present were performed using isolated neurons from pre-weanling rats as the mechanical dissociation procedure produces few, if any, viable neurons from adults. Thus, it is not yet clear if a similar ethanol potentiation of 5-HT3 actions would be observed in adult rats. Future experiments could examine this issue using brain slices.
Acute actions of alcohol on the central nervous system (CNS) produce intoxication that directly contributes to alcohol use disorder and plays a role in problems involving chronic alcohol abuse. Acute exposure to ethanol (EtOH) affects many neuronal molecules (Abrahao et al., 2017). Among these effects, increased GABAergic inhibitory synaptic transmission has been implicated in nervous system depression, leading to sedation (Chastain, 2006; Hoffman and Tabakoff, 1996). Potentiation of the function of GABAA receptors has been suggested to be the main contributor to these EtOH actions (Golovko et al., 2002; Hakkinen and Kulonen, 1959). However, studies in the last fifteen years indicate that presynaptic EtOH actions leading to increased GABA release are also important components of the increased CNS inhibition produced by the drug (Criswell et al., 2008; Roberto et al., 2003; Silberman et al., 2008; Zhu and Lovinger, 2006). Activation of the cAMP/PKA signaling pathway is a strong candidate to mediate these effects (Kelm et al., 2008). As previously mentioned, 5-HT3 receptors have been implicated in the acute neural actions of EtOH as well as in EtOH drinking and preference (Fadda et al., 1991; Grant and Barrett, 1991; Johnson et al., 1993; LeMarquand et al., 1994a, b; McKenzie-Quirk et al., 2005; Sellers et al., 1994; Shelton et al., 2004). Thus, the EtOH effects on presynaptic 5-HT3 receptors described in the present study might play an important role in EtOH intoxication by enhancing GABAergic transmission as well as 5-HT3-modulation of release of other neurotransmitters.
Acknowledgements
This work was supported by the Division of Intramural Clinical and Biological Research of the NIAAA (ZIA AA000407), the National Research Foundation of Korea grant (NRF-2019R1A2C1088909, NRF-2019M3C1B8090805) to S.B.J.
Footnotes
Declaration of competing interestCOI
The authors declare no competing financial interests.
CRediT authorship contribution statement
Sang Beom Jun: designed and performed experiments, analyzed data and wrote manuscript. Stephen R. Ikeda: designed and performed experiments and assisted in manuscript preparation. Jee Eun Sung: assisted with data analysis and statistics and assisted in manuscript preparation.
References
- Abrahao KP, Salinas AG, Lovinger DM, 2017. Alcohol and the brain: neuronal molecular targets, synapses, and circuits. Neuron 96, 1223–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akaike N, Moorhouse AJ, 2003. Techniques: applications of the nerve-bouton preparation in neuropharmacology. Trends Pharmacol. Sci 24, 44–47. [DOI] [PubMed] [Google Scholar]
- Akaike N, Murakami N, Katsurabayashi S, Jin YH, Imazawa T, 2002. Focal stimulation of single GABAergic presynaptic boutons on the rat hippocampal neuron. Neurosci. Res 42, 187–195. [DOI] [PubMed] [Google Scholar]
- Ariwodola OJ, Weiner JL, 2004. Ethanol potentiation of GABAergic synaptic transmission may be self-limiting: role of presynaptic GABA(B) receptors. J. Neurosci 24, 10679–10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asher O, Cunningham TD, Yao L, Gordon AS, Diamond I, 2002. Ethanol stimulates cAMP-responsive element (CRE)-mediated transcription via CRE-binding protein and cAMP-dependent protein kinase. J. Pharmacol. Exp. Therapeut 301, 66–70. [DOI] [PubMed] [Google Scholar]
- Asyyed A, Storm D, Diamond I, 2006. Ethanol activates cAMP response element-mediated gene expression in select regions of the mouse brain. Brain Res 1106, 63–71. [DOI] [PubMed] [Google Scholar]
- Barnes JM, Barnes NM, Cooper SJ, 1992. Behavioural pharmacology of 5-HT3 receptor ligands. Neurosci. Biobehav. Rev 16, 107–113. [DOI] [PubMed] [Google Scholar]
- Barnes NM, Hales TG, Lummis SC, Peters JA, 2009. The 5-HT3 receptor–the relationship between structure and function. Neuropharmacology 56, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandina P, Goldfarb J, Craddock-Royal B, Green JP, 1989. Release of endogenous dopamine by stimulation of 5-hydroxytryptamine3 receptors in rat striatum. J. Pharmacol. Exp. Therapeut 251, 803–809. [PubMed] [Google Scholar]
- Chastain G, 2006. Alcohol, neurotransmitter systems, and behavior. J. Gen. Psychol 133, 329–335. [DOI] [PubMed] [Google Scholar]
- Choi IS, Cho JH, Kim JT, Park EJ, Lee MG, Shin HI, Choi BJ, Jang IS, 2007. Serotoninergic modulation of GABAergic synaptic transmission in developing rat CA3 pyramidal neurons. J. Neurochem 103, 2342–2353. [DOI] [PubMed] [Google Scholar]
- Criswell HE, Ming Z, Kelm MK, Breese GR, 2008. Brain regional differences in the effect of ethanol on GABA release from presynaptic terminals. J. Pharmacol. Exp. Therapeut 326, 596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies PA, Pistis M, Hanna MC, Peters JA, Lambert JJ, Hales TG, Kirkness EF, 1999. The 5-HT3B subunit is a major determinant of serotonin-receptor function. Nature 397, 359–363. [DOI] [PubMed] [Google Scholar]
- Dubin AE, Huvar R, D’Andrea MR, Pyati J, Zhu JY, Joy KC, Wilson SJ, Galindo JE, Glass CA, Luo L, Jackson MR, Lovenberg TW, Erlander MG, 1999. The pharmacological and functional characteristics of the serotonin 5-HT3A receptor are specifically modified by a 5-HT3B receptor subunit. J. Biol. Chem 274, 30799–30810. [DOI] [PubMed] [Google Scholar]
- Fadda F, Garau B, Marchei F, Colombo G, Gessa GL, 1991. MDL 72222, a selective 5-HT3 receptor antagonist, suppresses voluntary ethanol consumption in alcohol-preferring rats. Alcohol Alcohol 26, 107–110. [DOI] [PubMed] [Google Scholar]
- Faerber L, Drechsler S, Ladenburger S, Gschaidmeier H, Fischer W, 2007. The neuronal 5-HT3 receptor network after 20 years of research – Evolving concepts in management of pain and inflammation. Eur. J. Pharmacol 560, 1–8. [DOI] [PubMed] [Google Scholar]
- Fleming RL, Manis PB, Morrow AL, 2009. The effects of acute and chronic ethanol exposure on presynaptic and postsynaptic gamma-aminobutyric acid (GABA) neurotransmission in cultured cortical and hippocampal neurons. Alcohol 43, 603–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giovannini MG, Ceccarelli I, Molinari B, Cecchi M, Goldfarb J, Blandina P, 1998. Serotonergic modulation of acetylcholine release from cortex of freely moving rats. J. Pharmacol. Exp. Therapeut 285, 1219–1225. [PubMed] [Google Scholar]
- Glykys J, Mody I, 2007. The main source of ambient GABA responsible for tonic inhibition in the mouse hippocampus. J. Physiol 582, 1163–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovko AI, Golovko SI, Leontieva LV, Zefirov SY, 2002. The influence of ethanol on the functional status of GABA(A) receptors. Biochemistry (Mosc.) 67, 719–729. [DOI] [PubMed] [Google Scholar]
- Grant KA, 1995. The role of 5-HT3 receptors in drug dependence. Drug Alcohol Depend 38, 155–171. [DOI] [PubMed] [Google Scholar]
- Grant KA, Barrett JE, 1991. Blockade of the discriminative stimulus effects of ethanol with 5-HT3 receptor antagonists. Psychopharmacology (Berl) 104, 451–456. [DOI] [PubMed] [Google Scholar]
- Hakkinen HM, Kulonen E, 1959. Increase in the gamma-aminobutyric acid content of rat brain after ingestion of ethanol. Nature 184 (Suppl. 10), 726. [DOI] [PubMed] [Google Scholar]
- Hasanuzzaman M, Yoshimura M, 2010. Effects of straight chain alcohols on specific isoforms of adenylyl cyclase. Alcohol Clin. Exp. Res 34, 743–749. [DOI] [PubMed] [Google Scholar]
- Hoffman PL, Tabakoff B, 1996. To be or not to be: how ethanol can affect neuronal death during development. Alcohol Clin. Exp. Res 20, 193–195. [DOI] [PubMed] [Google Scholar]
- Hoyer D, Hannon JP, Martin GR, 2002. Molecular, pharmacological and functional diversity of 5-HT receptors. Pharmacol. Biochem. Behav 71, 533–554. [DOI] [PubMed] [Google Scholar]
- Jackson MB, Yakel JL, 1995. The 5-HT3 receptor channel. Annu. Rev. Physiol 57, 447–468. [DOI] [PubMed] [Google Scholar]
- Johnson BA, Campling GM, Griffiths P, Cowen PJ, 1993. Attenuation of some alcohol-induced mood changes and the desire to drink by 5-HT3 receptor blockade: a preliminary study in healthy male volunteers. Psychopharmacology 112, 142–144. [DOI] [PubMed] [Google Scholar]
- Jun SB, Cuzon Carlson V, Ikeda S, Lovinger D, 2011. Vibrodissociation of neurons from rodent brain slices to study synaptic transmission and image presynaptic terminals. J Vis Exp 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karnovsky AM, Gotow LF, McKinley DD, Piechan JL, Ruble CL, Mills CJ, Schellin KA, Slightom JL, Fitzgerald LR, Benjamin CW, Roberds SL, 2003. A cluster of novel serotonin receptor 3-like genes on human chromosome 3. Gene 319, 137–148. [DOI] [PubMed] [Google Scholar]
- Katsurabayashi S, Kubota H, Tokutomi N, Akaike N, 2003. A distinct distribution of functional presynaptic 5-HT receptor subtypes on GABAergic nerve terminals projecting to single hippocampal CA1 pyramidal neurons. Neuropharmacology 44, 1022–1030. [DOI] [PubMed] [Google Scholar]
- Kelm MK, Criswell HE, Breese GR, 2008. The role of protein kinase A in the ethanol-induced increase in spontaneous GABA release onto cerebellar Purkinje neurons. J. Neurophysiol 100, 3417–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama S, Matsumoto N, Kubo C, Akaike N, 2000. Presynaptic 5-HT3 receptor-mediated modulation of synaptic GABA release in the mechanically dissociated rat amygdala neurons. J. Physiol 529 Pt 2, 373–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeMarquand D, Pihl RO, Benkelfat C, 1994a. Serotonin and alcohol intake, abuse, and dependence: Clinical evidence. Biol. Psychiatr 36, 326–337. [DOI] [PubMed] [Google Scholar]
- LeMarquand D, Pihl RO, Benkelfat C, 1994b. Serotonin and alcohol intake, abuse, and dependence: findings of animal studies. Biol. Psychiatr 36, 395–421. [DOI] [PubMed] [Google Scholar]
- Lin HH, Chang SJ, Shie HJ, Lai CC, 2006. Ethanol inhibition of NMDA-induced responses and acute tolerance to the inhibition in rat rostral ventrolateral medulla in vivo: involvement of cAMP-dependent protein kinases. Neuropharmacology 51, 747–755. [DOI] [PubMed] [Google Scholar]
- Lobo IA, Harris RA, 2008. GABA(A) receptors and alcohol. Pharmacol. Biochem. Behav 90, 90–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovinger DM, 1991. Ethanol potentiates ion current mediated by 5-HT3 receptors on neuroblastoma cells and isolated neurons. Alcohol Alcohol Suppl 1, 181–185. [PubMed] [Google Scholar]
- Lovinger DM, Sung KW, Zhou Q, 2000. Ethanol and trichloroethanol alter gating of 5-HT3 receptor-channels in NCB-20 neuroblastoma cells. Neuropharmacology 39, 561–570. [DOI] [PubMed] [Google Scholar]
- Lovinger DM, White G, 1991. Ethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in neuroblastoma cells and isolated adult mammalian neurons. Mol. Pharmacol 40, 263–270. [PubMed] [Google Scholar]
- Lovinger DM, Zhou Q, 1993. Trichloroethanol potentiation of 5-hydroxytryptamine3 receptor-mediated ion current in nodose ganglion neurons from the adult rat. J. Pharmacol. Exp. Therapeut 265, 771–776. [PubMed] [Google Scholar]
- Lovinger DM, Zhou Q, 1994. Alcohols potentiate ion current mediated by recombinant 5-HT3RA receptors expressed in a mammalian cell line. Neuropharmacology 33, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Maricq AV, Peterson AS, Brake AJ, Myers RM, Julius D, 1991. Primary structure and functional expression of the 5HT3 receptor, a serotonin-gated ion channel. Science 254, 432–437. [DOI] [PubMed] [Google Scholar]
- McKenzie-Quirk SD, Girasa KA, Allan AM, Miczek KA, 2005. 5-HT(3) receptors, alcohol and aggressive behavior in mice. Behav. Pharmacol 16, 163–169. [DOI] [PubMed] [Google Scholar]
- Morales M, Battenberg E, De Lecea L, Bloom FE, 1996. The type 3 serotonin receptor is expressed in a subpopulation of GABAergic neurons in the rat neocortex and hippocampus. Brain Res 731, 199–202. [DOI] [PubMed] [Google Scholar]
- Morales M, Bloom FE, 1997. The 5-HT3 receptor is present in different subpopulations of GABAergic neurons in the rat telencephalon. J. Neurosci 17, 3157–3167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayak SV, Ronde P, Spier AD, Lummis SC, Nichols RA, 1999. Calcium changes induced by presynaptic 5-hydroxytryptamine-3 serotonin receptors on isolated terminals from various regions of the rat brain. Neuroscience 91, 107–117. [DOI] [PubMed] [Google Scholar]
- Niesler B, Frank B, Kapeller J, Rappold GA, 2003. Cloning, physical mapping and expression analysis of the human 5-HT3 serotonin receptor-like genes HTR3C, HTR3D and HTR3E. Gene 310, 101–111. [DOI] [PubMed] [Google Scholar]
- Paudice P, Raiteri M, 1991. Cholecystokinin release mediated by 5-HT3 receptors in rat cerebral cortex and nucleus accumbens. Br. J. Pharmacol 103, 1790–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabin RA, Molinoff PB, 1983. Multiple sites of action of ethanol on adenylate cyclase. J. Pharmacol. Exp. Therapeut 227, 551–556. [PubMed] [Google Scholar]
- Roberto M, Cruz M, Bajo M, Siggins GR, Parsons LH, Schweitzer P, 2010. The endocannabinoid system tonically regulates inhibitory transmission and depresses the effect of ethanol in central amygdala. Neuropsychopharmacology 35, 1962–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberto M, Madamba SG, Moore SD, Tallent MK, Siggins GR, 2003. Ethanol increases GABAergic transmission at both pre- and postsynaptic sites in rat central amygdala neurons. Proc. Natl. Acad. Sci. U. S. A 100, 2053–2058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronde P, Nichols RA, 1998. High calcium permeability of serotonin 5-HT3 receptors on presynaptic nerve terminals from rat striatum. J. Neurochem 70, 1094–1103. [DOI] [PubMed] [Google Scholar]
- Ropert N, Guy N, 1991. Serotonin facilitates GABAergic transmission in the CA1 region of rat hippocampus in vitro. J. Physiol 441, 121–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rusch D, Musset B, Wulf H, Schuster A, Raines DE, 2007. Subunit-dependent modulation of the 5-hydroxytryptamine type 3 receptor open-close equilibrium by n-alcohols. J. Pharmacol. Exp. Therapeut 321, 1069–1074. [DOI] [PubMed] [Google Scholar]
- Sellers EM, Toneatto T, Romach MK, Somer GR, Sobell LC, Sobell MB, 1994. Clinical efficacy of the 5-HT3 antagonist ondansetron in alcohol abuse and dependence. Alcohol Clin. Exp. Res 18, 879–885. [DOI] [PubMed] [Google Scholar]
- Sheinin A, Talani G, Davis MI, Lovinger DM, 2008. Endocannabinoid- and mGluR5-dependent short-term synaptic depression in an isolated neuron/bouton preparation from the hippocampal CA1 region. J. Neurophysiol 100, 1041–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton KL, Dukat M, Allan AM, 2004. Effect of 5-HT3 receptor over-expression on the discriminative stimulus effects of ethanol. Alcohol Clin. Exp. Res 28, 1161–1171. [DOI] [PubMed] [Google Scholar]
- Silberman Y, Shi L, Brunso-Bechtold JK, Weiner JL, 2008. Distinct mechanisms of ethanol potentiation of local and paracapsular GABAergic synapses in the rat basolateral amygdala. J. Pharmacol. Exp. Therapeut 324, 251–260. [DOI] [PubMed] [Google Scholar]
- Stewart A, Davies PA, Kirkness EF, Safa P, Hales TG, 2003. Introduction of the 5-HT3B subunit alters the functional properties of 5-HT3 receptors native to neuroblastoma cells. Neuropharmacology 44, 214–223. [DOI] [PubMed] [Google Scholar]
- Sung KW, Engel SR, Allan AM, Lovinger DM, 2000. 5-HT(3) receptor function and potentiation by alcohols in frontal cortex neurons from transgenic mice overexpressing the receptor. Neuropharmacology 39, 2346–2351. [DOI] [PubMed] [Google Scholar]
- Tabakoff B, Nelson E, Yoshimura M, Hellevuo K, Hoffman PL, 2001. Phosphorylation cascades control the actions of ethanol on cell cAMP signalling. J. Biomed. Sci 8, 44–51. [DOI] [PubMed] [Google Scholar]
- Turner TJ, Mokler DJ, Luebke JI, 2004. Calcium influx through presynaptic 5-HT3 receptors facilitates GABA release in the hippocampus: in vitro slice and synaptosome studies. Neuroscience 129, 703–718. [DOI] [PubMed] [Google Scholar]
- Vorobjev VS, 1991. Vibrodissociation of sliced mammalian nervous tissue. J. Neurosci. Methods 38, 145–150. [DOI] [PubMed] [Google Scholar]
- Vucurovic K, Gallopin T, Ferezou I, Rancillac A, Chameau P, van Hooft JA, Geoffroy H, Monyer H, Rossier J, Vitalis T, 2010. Serotonin 3A receptor subtype as an early and protracted marker of cortical interneuron subpopulations. Cerebr. Cortex 20, 2333–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wafford KA, Whiting PJ, 1992. Ethanol potentiation of GABAA receptors requires phosphorylation of the alternatively spliced variant of the gamma 2 subunit. FEBS Lett 313, 113–117. [DOI] [PubMed] [Google Scholar]
- Wan FJ, Berton F, Madamba SG, Francesconi W, Siggins GR, 1996. Low ethanol concentrations enhance GABAergic inhibitory postsynaptic potentials in hippocampal pyramidal neurons only after block of GABAB receptors. Proc. Natl. Acad. Sci. U. S. A 93, 5049–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan S, Browning KN, 2008. Glucose increases synaptic transmission from vagal afferent central nerve terminals via modulation of 5-HT3 receptors. Am. J. Physiol. Gastrointest. Liver Physiol 295, G1050–G1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye JH, Wang F, Krnjevic K, Wang W, Xiong ZG, Zhang J, 2004. Presynaptic glycine receptors on GABAergic terminals facilitate discharge of dopaminergic neurons in ventral tegmental area. J. Neurosci 24, 8961–8974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshimura M, Tabakoff B, 1999. Ethanol’s actions on cAMP-mediated signaling in cells transfected with type VII adenylyl cyclase. Alcohol Clin. Exp. Res 23, 1457–1461. [PubMed] [Google Scholar]
- Zhou Q, Verdoorn TA, Lovinger DM, 1998. Alcohols potentiate the function of 5-HT3 receptor-channels on NCB-20 neuroblastoma cells by favouring and stabilizing the open channel state. J. Physiol 507 (Pt 2), 335–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu PJ, Lovinger DM, 2006. Ethanol potentiates GABAergic synaptic transmission in a postsynaptic neuron/synaptic bouton preparation from basolateral amygdala. J. Neurophysiol 96, 433–441. [DOI] [PubMed] [Google Scholar]