

Abstract
Background
Dilated cardiomyopathy (DCM) is the leading cause of heart failure with a poor prognosis. Recent studies suggest that endothelial to mesenchymal transition (EndMT) may be involved in the pathogenesis and cardiac remodeling during DCM development. EDIL3 (epidermal growth factor‐like repeats and discoidin I‐like domains 3) is an extracellular matrix glycoprotein that has been reported to promote EndMT in various diseases. However, the roles of EDIL3 in DCM still remain unclear.
Methods and Results
A mouse model of DCM and human umbilical vein endothelial cells were used to explore the roles and mechanisms of EDIL3 in DCM. The results indicated that EndMT and EDIL3 were activated in DCM mice. EDIL3 deficiency attenuated cardiac dysfunction and remodeling in DCM mice. EDIL3 knockdown alleviated EndMT by inhibiting USP10 (ubiquitin specific peptidase 10) dependent Smad4 deubiquitination in vivo and in vitro. Recombinant human EDIL3 promoted EndMT via reinforcing deubiquitination of Smad4 in human umbilical vein endothelial cells treated with IL‐1β (interleukin 1β) and TGF‐β (transforming growth factor beta). Inhibiting USP10 abolished EndMT exacerbated by EDIL3. In addition, recombinant EDIL3 also aggravates doxorubicin‐induced EndMT by promoting Smad4 deubiquitination in HUVECs.
Conclusions
Taken together, these results indicate that EDIL3 deficiency attenuated EndMT by inhibiting USP10 dependent Smad4 deubiquitination in DCM mice.
Keywords: dilated cardiomyopathy, EDIL3, EndMT, Smad4 deubiquitination, USP10
Subject Categories: Cell Signalling/Signal Transduction
Nonstandard Abbreviations and Acronyms
- αSMA
α‐smooth muscle actin
- BMP
bone morphogenetic protein
- DCM
dilated cardiomyopathy
- DEL‐1
developmental endothelial locus‐1
- EDIL3
epidermal growth factor‐like repeats and discoidin I‐like domains 3
- EndMT
endothelial to mesenchymal transition
- EMT
epithelial‐mesenchymal transition
- HUVEC
human umbilical vein endothelial cell
- HW
heart weight
- ITGB3
integrin β3
- TGF‐β
transforming growth factor beta
- USP10
ubiquitin‐specific peptidase 10
Research Perspective.
What Is New?
EDIL3 (epidermal growth factor‐like repeats and discoidin I‐like domains 3) deficiency attenuated endothelial to mesenchymal transition by inhibiting USP10 (ubiquitin‐specific peptidase 10) dependent Smad4 deubiquitination in mice with dilated cardiomyopathy.
What Question Should Be Addressed Next?
Developing drugs that inhibit EDIL3 expression may help treat dilated cardiomyopathy.
Dilated cardiomyopathy (DCM) is a cardiomyopathy characterized by left ventricular (LV) or biventricular dilation and systolic dysfunction without pressure or volume overload or coronary artery disease sufficient to explain the dysfunction. 1 Most common pathologies underlie reactive changes such as inflammation (viral myocarditis or autoimmune disease), nutritional toxic influences (alcohol, drugs, chemotactic toxins), and metabolic derangements. 2 DCM is one of the most common causes of heart failure with an estimated prevalence of ≈1:250 to 2500 in the general population. 3 , 4 Despite advances in prevention, diagnosis, and pharmacotherapy (including angiotensin‐converting enzyme inhibitors, angiotensin receptor antagonists, beta blockers, etc), 5 emerging evidence suggests that some patients remain vulnerable to sudden cardiac death and refractory heart failure, requiring cardiac transplantation or mechanical circulatory support. 6 Therefore, further exploration of the pathogenesis of DCM is helpful for the development of new therapeutic strategies.
Endothelial cells (ECs) and mesenchymal cells are 2 distinct cell lineages, both derived from the mesoderm. ECs can be distinguished by the expression of cell–cell adhesion molecules, including CD31/PECAM‐1 (platelet/EC adhesion molecule‐1), vascular endothelial (VE)‐cadherin, vWF (von Willebrand factor), TIE1 (tyrosine kinase with immunoglobulin‐like and EGF [epidermal growth factor]‐like domain 1) and TIE2. 7 Mesenchymal cells, commonly referred to as mesenchymal stem cells, have been reported to have the ability to differentiate into cartilage cells, bone cells, and adipocytes, which express mesenchymal‐specific markers such as N‐cadherin, α‐smooth muscle actin (αSMA), vimentin, FSP‐1 (fibroblast‐specific protein‐1), fibronectin, and SM22α (smooth muscle protein 22α). 7 The term “endothelial to mesenchymal transition” (EndMT) is defined as the process by which endothelial cells (ECs) differentiate into mesenchymal cells. 8 Under normal physiological conditions, this cell fate switch is necessary for the correct formation of the heart valves of the developing heart. 9 However, EndMT also plays an important role in various cardiovascular diseases, such as atherosclerosis, valvular disease in adults, myocardial fibrosis, and pulmonary arterial hypertension. 10 Recent studies suggest that EndMT may be involved in the pathogenesis and cardiac remodeling during DCM development, suggesting a potential therapeutic target for DCM treatment. 11 , 12 , 13
EDIL3 (epidermal growth factor‐like repeats and discoidin I‐like domains 3) is a 52 kDa extracellular matrix glycoprotein consisting of 3 N‐terminal EGF‐like repeats and 2 C‐terminal discoidin I‐like domains, also known as DEL‐1 (developmental endothelial locus‐1). 14 EDIL3 is mainly produced by ECs during embryonic vascular development but also by macrophages, neurons, osteoclasts, and some hematopoietic microenvironmental cells. 15 EDIL3 interacts with integrins on the membrane of ECs through the RGD motif in its second EGF repeat, affecting ECs functions, including adhesion, migration, and angiogenesis. 16 , 17 , 18 EDIL3‐mediated angiogenesis plays an important role in early embryogenesi and the initiation and development of tumors and ischemic tissues. 16 , 19 , 20 EDIL3 has also been identified as a novel regulator of epithelial mesenchymal transition (EMT), promoting EMT by mediating TGF‐β (transforming growth factor beta) activation and ERK signaling. 21 However, EDIL3 depletion or deletion inhibited the occurrence of EMT. 22 , 23 EDIL3‐mediated EMT is involved in the occurrence and progression of various diseases. 22 , 23 , 24 , 25 In addition, EDIL3, as an important immunomodulatory molecule, accelerates the resolution of inflammation and thus plays a protective role in various inflammation‐related diseases. 26 , 27 However, the role of EDIL3 in cardiovascular disease remains controversial. EDIL3 attenuates angiotensin II‐induced hypertension and myocardial remodeling by inhibiting inflammation. 28 However, EDIL3 deficiency ameliorates adverse cardiac healing via neutrophil extracellular traps mediated proinflammatory macrophage polarization. 29 These evidences suggest that the function of EDIL3 remains controversial. The role of EDIL3 in DCM is unclear and requires further exploration.
In this study, we explored the role and mechanism of EDIL3‐mediated EndMT in the occurrence and development of DCM through in vivo and in vitro experiments. We found that EDIL3 deficiency suppressed EndMT in DCM mouse heart tissue, thereby attenuating cardiac dysfunction and remodeling. Mechanistically, EDIL3 promotes USP10 (ubiquitin‐specific peptidase 10)‐mediated deubiquitination of Smad4, thereby promoting EndMT. These results suggest that EDIL3 may be a potential target for the treatment of DCM.
Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request. Some data may not be made available because of privacy or ethical restrictions.
Reagents
Doxorubicin (cat no. HY‐15142A) and Spautin 1 (cat no. HY‐12990) were purchased from MedChemExpress (Monmouth Junction, NJ). Recombinant human EDIL3 (cat no. 6046‐ED) was purchased from R&D Systems (Minneapolis, MN). All the information of primary antibodies (name, company, catalog no., molecular weight) is shown in Table S1.
Animals and Animal Models
EDIL3 knockout (EDIL3−/− KO) mice on the C57BL/6 background were purchased from the Gempharmatech Co., Ltd. (Nanjing, China). Wild‐type (WT) mice in the same brood were used as controls. All animals were housed at constant room temperature on a 12:12 hour light–dark cycle at the Animal Center of Wuhan University Renmin Hospital and fed a standard rodent diet and water. All animals received humane care according to National Institutes of Health (USA) guidelines. All animal care and experimental procedures were approved by the Animal Policy and Welfare Committee of Wuhan University Renmin Hospital (WDRM20230202D). To model the DCM, EDIL3−/− and WT mice at 10 weeks of age were treated with intraperitoneal injection of doxorubicin (2.5 mg/kg per week) for 6 weeks (n=10 in each group). 30 , 31 Sham mice received the same volume of normal saline (n=10 in each group) (Figure S1). Euthanasia was performed under pentobarbital sodium anesthesia. Heart tissue was collected and fixed in 4% paraformaldehyde for pathological analysis or snap‐frozen in liquid nitrogen for gene and protein expression analysis.
Bioinformatic Tools
Data from Gene Expression Omnibus series GSE84796 and GSE111544 were downloaded (http://www.ncbi.nlm.nih.gov/geo) and analyzed using gene set enrichment analysis. Gene Ontology analysis was performed with DAVID Bioinformatics Resources 6.7 (http://david.abcc.ncifcrf.gov/) and gene set enrichment analysis was performed with GSEA v2.0.13 software to analyze the differentially expressed genes in myocardial tissues between the patients with DCM and healthy controls.
Echocardiography
Systolic and diastolic cardiac function in anesthetized mice were measured noninvasively by transthoracic echocardiography 1 day before euthanasia as described. 32 LV ejection fraction, fractional shortening, and LV posterior wall thickness were estimated.
Histology and Tissue Staining
Heart tissue was fixed with 4% paraformaldehyde, embedded in paraffin, and sectioned at 5 μm thickness. After dehydration, sections were stained with hematoxylin and eosin and Masson staining as previous described. 33 For immunofluorescence staining, harvested mice hearts were embedded in paraffin and cut into 5‐micron thick sections. Sections were incubated with primary antibodies overnight in a humidified chamber at 4 °C and then incubated with secondary antibody for 1 hour in the dark. Sections were evaluated using a fluorescence microscope (Olympus DX51, Tokyo, Japan) and further analyzed using Image‐Pro Plus software (version 6.0).
Cell Culture and Treatments
Human umbilical vein ECs (HUVECs) were purchased from Procell Life Science & Technology Co., Ltd. (Wuhan, China) and cultured in a fully supplemented endothelial growth medium‐2 purchased from Warner Bio (Wuhan, China) Co., Ltd. For EndMT models, HUVECs were plated at a density of 7×104 cells/well in collagen coated 6‐well culture plates and were treated with TGF‐β2 (10 ng/mL, PeproTech NJ, USA) and IL‐1β (interleukin‐1β; 1 ng/mL, PeproTech NJ, USA) for 7 consecutive days in endothelial growth medium‐2 supplemented with 10% FBS and refreshed every 24 hours. 34 Recombinant human EDIL3 treatment (0.5 μg/mL) were added into the culture medium to figure out the role of EDIL3 on EndMT. 35 For USP10 inhibition, Spautin 1 (10 μmol/L) was added to the (endothelial growth medium) ECM‐2. 36 MG132 (20 μmol/L) was added to the endothelial growth medium‐2 medium to examine the ubiquitination of Smad4. 37 To examine the role of EDIL3 in doxorubicin treated HUVECs, doxorubicin was used at 1 μmol/L for 48 hours. 38
To silence EDIL3 and ITGB3 (integrin β3) in HUVECs, transfection was carried out according to the manufacturer's protocol. Briefly, HiperFect transfection reagent (QIAGEN, Dusseldorf, Germany) mixed with EDIL3 or ITGB3 siRNA (small interfering RNA, GenePharma, Shanghai, China) in endothelial growth medium‐2. A mixture of transfection reagent and siRNA was incubated for 15 minutes at 37 °C, then added to the 6‐well plate. The negative control group was incubated with a mixture of transfection reagent and negative control RNA. The silence EDIL3 and ITGB3 efficacy was evaluated by western blotting. EDIL3 siRNA sequences are as follows: 5′‐CCCAUCUAUGCACGACACAUATT‐3′, ITGB3 siRNA sequences are as follows: 5′‐GAUGCAGUGAAUUGUACCUAUTT‐3′, negative control siRNA: 5′‐UUCUCCGAACGUGUCACGUTT‐3′. To silence ERK and AKT, siERK (sc‐29 307) and siAKT (sc‐29 195) were purchased from Santa Cruz Biotechnology and used to treat HUVECs.
Western Blotting and Coimmunoprecipitation
Western blots were performed to assess protein expression levels. Lysed cells or homogenized cardiac tissue were collected for total protein extraction. Protein concentration was assessed using the BCA protein assay kit (23 227, Thermo Fisher Scientific, Waltham, MA) and normalized before Western blotting. Protein samples (50 μg) were separated on 10% SDS‐PAGE and subsequently transferred to polyvinylidene fluoride membranes (FL00010, Millipore, Billerica, MA). Polyvinylidene fluoride membranes were blocked with 5% nonfat milk for 1 hour at room temperature to block nonspecific binding sites, then incubated with primary antibody overnight at 4 °C. The protein expression level of GAPDH was used as an internal standard. The next day, the Western blot was incubated with the secondary antibody for 1 hour at room temperature. Finally, blots were obtained and scanned by Image Lab software (Bio‐Rad Laboratories, Inc., Hercules, CA) to assess protein expression.
The coimmunoprecipitation test was performed according to manufacturer's protocol (Protein A/G Magnetic Beads, HY‐K0202, MCE). Briefly, the tissues of hearts and HUVECs were lysed in octyl‐d‐glucoside (2%) buffer. The protein concentration was diluted into 1 μg/μL for immunoprecipitation. The lysates were incubated with 2 μL primary antibodies overnight then 30 μL protein A+G magnetic beads added for 4 to 6 hours. A magnetic rack was used to discard the supernatant and the beads were washed 3 times with RIPA buffer. A 30 μL 1×loading buffer was added and boiled for 10 minutes, then the supernatant was collected and analyzed by Western blotting.
Real‐Time Quantitative Polymerase Chain Reaction
Cardiac tissues or HUVECs were homogenized in TRIzol (15596–026, Invitrogen Corporation, Waltham, MA), followed by a complementary DNA synthesis kit (489 703 001, Basel, Switzerland) reverse transcription of mRNA into complementary DNA. The LightCycler 480 system (04896866001, Roche) was used for quantitative polymerase chain reaction analysis. Primers for target genes are listed in Table S2. The amount of each gene was determined and normalized to the amount of GAPDH.
Flow Cytometry
Flow cytometry was used to analyze the ratio of vimentin‐positive HUVECs as described in our previous study. 39 In brief, cells in culture dishes were digested with 0.25% trypsin in culture dishes to prepare HUVEC cell suspensions. Then, CoraLite Plus 488‐conjugated Vimentin antibody was used for flow cytometry. After the primary antibodies were incubated for 30 minutes in the dark, the cells were analyzed with CytExpert (Beckman, USA).
Biochemical Determination
Cardiac tissue was triturated in phosphate buffered saline to obtain tissue homogenate. Cardiac tissue concentrations of creatine kinase‐myocardial band (Jiancheng Bioengineer, China) and lactate dehydrogenase were detected according to the manufacturer's instructions.
Statistical Analysis
All experiments were randomized and blinded. All statistical analyses in this study were performed using SPSS version 25.0 software and expressed as the mean±SD. n numbers are biological replicates. For all in vivo and in vitro analyses, normal distribution (Gaussian distribution) was first determined by the Shapiro–Wilk test. Data were then analyzed using Student t test for comparisons between 2 experimental groups, and 1‐way or 2‐way ANOVA with post hoc Tukey test for comparisons among multiple experimental groups. P<0.05 was considered statistically significant.
Results
EndMT and EDIL3 Are Activated in Patients and Mice With DCM
To check if EndMT signals are involved in DCM progression, we first performed gene set enrichment analysis using data from a data set of patients with DCM (GSE84796 and GSE111544). We found that the Hallmark EMT pathway is enriched in patients with DCM (Figure 1A). We sorted out the differentially expressed genes on this pathway and found that EDIL3 was upregulated in patients with DCM, indicating that EndMT and EDIL3 are associated with DCM progression (Figure 1B). To explore whether EndMT and EDIL3 activations are present in DCM hearts, myocardial specimens were collected from sham and DCM mice. Compared with sham mice, CD31 expression decreased and αSMA and EDIL3 expression increased (≈3‐fold) in DCM mice (Figure 1C). These results strongly indicated that EndMT and EDIL3 are activated in the myocardial tissues of DCM mice, which may contribute to cardiac dysfunction. We also examined the expression of EDIL3 in HUVECs models of EndMT. Consistent with results in vivo, CD31 expression decreased and αSMA and EDIL3 expression increased (≈2‐fold) in EndMT group compared with sham group (Figure 1D).
Figure 1. EndMT and EDIL3 are activated in patients and mice with DCM.
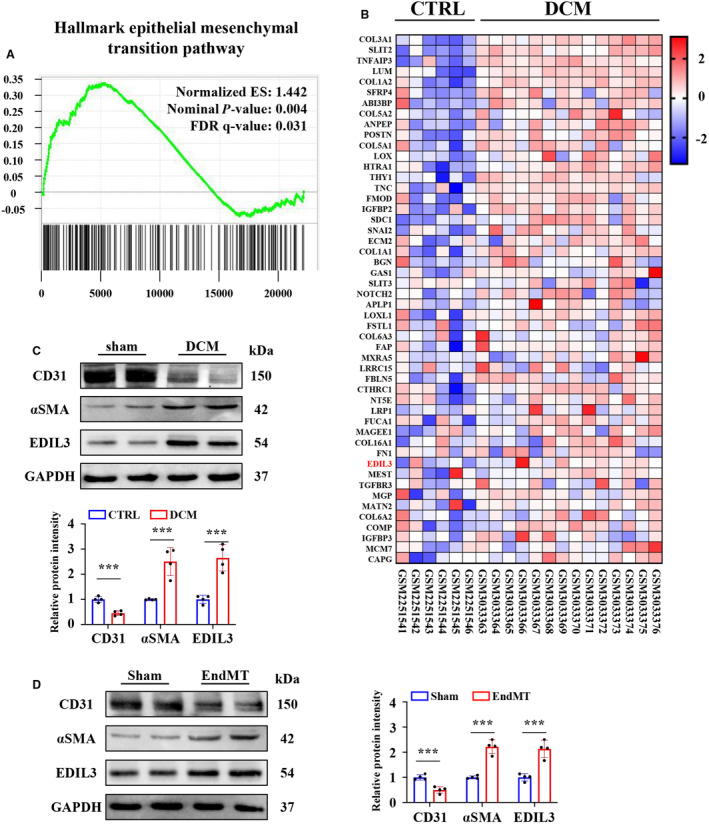
A, Gene set enrichment analysis is performed using data from Gene Expression Omnibus data set GSE84796 and GSE111544 to check if EndMT signals are involved in DCM progression. The Hallmark epithelial mesenchymal transition pathway is significantly enriched in the cardiac tissues of patients with DCM. B, Heatmap showing the expression of genes in Hallmark epithelial mesenchymal transition pathway. C, Representative immunoblots and corresponding quantification showing cardiac tissue levels of CD31, αSMA, and EDIL3 in DCM and sham mice (n=4). D, Representative immunoblots and corresponding quantification showing CD31, αSMA, and EDIL3 in EndMT and sham HUVECs (n=4). Data are presented as mean±SD and were analyzed using unpaired Student t test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001. DCM indicates dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; FDR, false discovery rate; HUVEC, human umbilical vein endothelial cell; and αSMA, α‐smooth muscle actin.
EDIL3 Deficiency Attenuates Cardiac Dysfunction and Remodeling in DCM Mice
EDIL3 gene KO mice were constructed to explore the role of EDIL3 in DCM progression (Figure 2A). Six weekly injections of doxorubicin (2.5 mg/kg body weight, intraperitoneally) were administered to WT and KO mice. Consistent with previous findings, 31 all mice regardless of genotype exhibited a significant decrease in body weight at 3 weeks following completion of doxorubicin challenge (Table S3). The weights of hearts (HW) isolated from WT and KO mice decreased significantly with doxorubicin when compared with saline controls (Table S3). EDIL3 KO mice showed higher HW compared with WT mice (Table S3). However, when comparing the ratio of HW to body weight as a percentage there was no change between WT‐DCM and KO‐DCM groups, suggesting that doxorubicin‐treated hearts decrease proportionally with body size (Table S3). The ratio of HW to tibia length decreased significantly with doxorubicin when compared with saline controls (Table S3). KO‐DCM mice showed higher HW/tibia length ratio compared with WT‐DCM mice (Table S3).
Figure 2. EDIL3 deficiency attenuates cardiac dysfunction and remodeling in DCM mice.
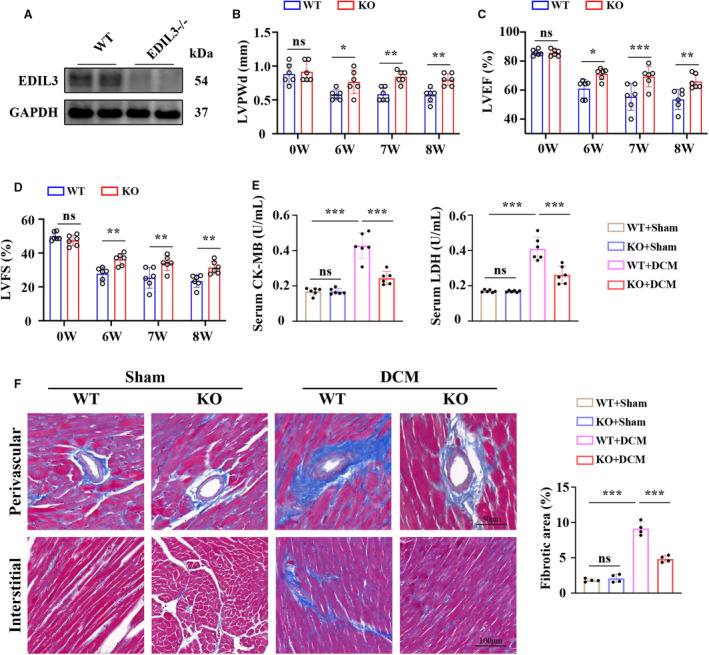
A, Representative immunoblots showing cardiac tissue levels EDIL3 (n=2). B through D, LVPWd, ejection fraction, and FS at 0, 6, 7, and 8 weeks (n=6). E, The cardiac levels of CK‐MB and LDH in each group at 8 weeks (n=6). F, Representative Masson staining for cardiac fibrosis at 8 weeks (n=4). Data are presented as mean±SD. In B through D, data were analyzed by unpaired Student t test. In (E through F), data were analyzed using 1‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. CK‐MB indicates creatine kinase‐myocardial band; DCM, dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; FS, fractional shortening; KO, knockout; LDH, lactate dehydrogenase; LVEF, left ventricular ejection fraction; LVPWd, left ventricular posterior wall thickness during diastole; and WT, wild type.
Transthoracic echocardiography measurements were performed on mice from all 4 experimental groups and revealed severe cardiac remodeling in WT‐DCM mice as indicated by decreased LV posterior wall thickness (Figure 2B, Table S3). EDIL3 deficiency partially attenuated doxorubicin‐induced cardiac remodeling (Figure 2B, Table S3). In WT mice, doxorubicin treatment reduced ejection fraction and fractional shortening compared with saline controls, which were increased by EDIL3 deficiency (Figure 2C and 2D, Table S3). Doxorubicin treatment induced cardiac remodeling as well as cardiac dysfunction in mice that was characteristic of DCM. These findings show that EDIL3 deficiency preserved cardiac function and inhibited cardiac remodeling in the presence of doxorubicin.
Cardiac injury was assessed by the level of serum creatine kinase‐myocardial band and lactate dehydrogenase level. In WT mice, doxorubicin treatment increased the level of serum creatine kinase‐myocardial band and lactate dehydrogenase compared with saline controls, which were attenuated by EDIL3 deficiency (Figure 2E). To investigate whether cardiac dysfunction was a result of doxorubicin‐induced fibrosis, we performed Masson staining of cardiac tissue in the mice (Figure 2F). WT‐DCM mice showed increased perivascular and interstitial fibrosis area compared with sham mice. KO‐DCM hearts exhibited significantly less fibrotic staining when compared with hearts from WT‐DCM mice. These findings indicated that EDIL3 deficiency attenuated cardiac injury and fibrosis in DCM mice.
EDIL3 Deficiency Inhibited EndMT In Vivo and In Vitro
EDIL3 deficiency inhibited the CD31+ ECs differentiate into αSMA+ mesenchymal cells in DCM mice as shown by the immunofluorescence staining (Figure 3A). EDIL3 deletion also increased CD31 and VE‐cadherin expression, and reduced αSMA and vimentin expression in DCM mice (Figure 3B). In addition, EDIL3 deficiency also reduced the expression of EndMT biomarkers, including snail1 and twist1 in DCM mice (Figure 3C). In vitro, EDIL3 knockdown via siEDIL3 (Figure 3D) reduced the ratio of vimentin‐positive HUVECs with EndMT treatment (Figure 3E). EDIL3 deficiency also increased CD31 and VE‐cadherin expression, and reduced αSMA and vimentin expression in EndMT HUVECs (Figure 3F). EDIL3 deficiency also inhibited the expression of EndMT biomarkers in HUVECs (Figure 3F). All these results indicated that EDIL3 deficiency inhibited EndMT in vivo and in vitro.
Figure 3. EDIL3 deficiency inhibited EndMT in vivo and in vitro.
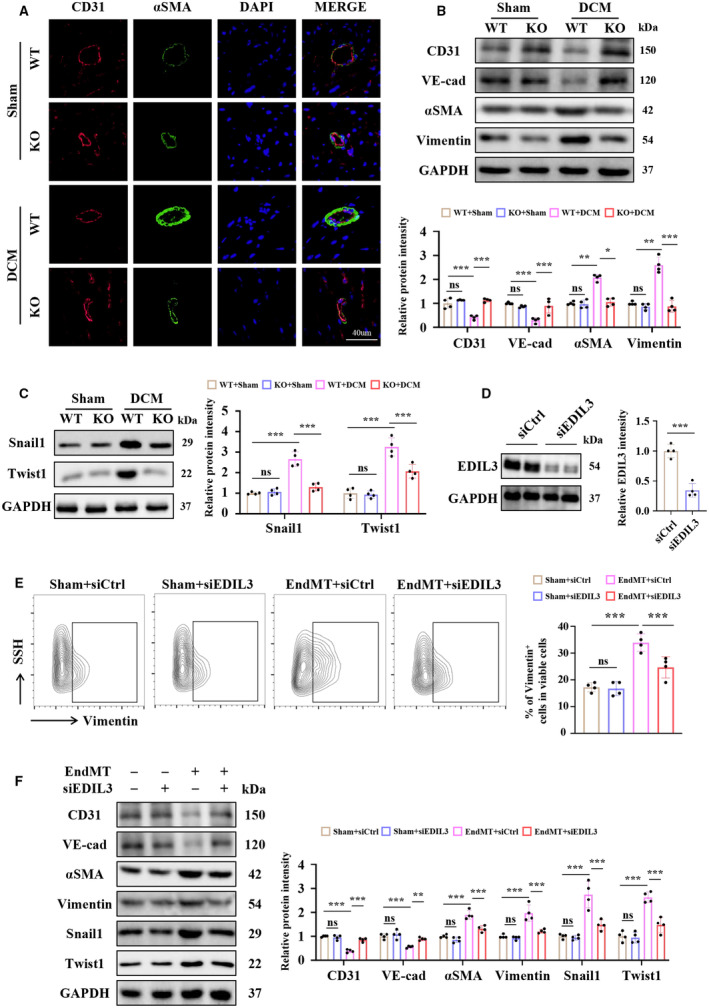
A, Representative immunofluorescence staining images of cardiac tissues for CD31 and αSMA (n=4). B, Representative immunoblots and corresponding quantification showing cardiac CD31, VE‐cadherin, αSMA, and vimentin (n=4). C, Representative immunoblots and corresponding quantification showing cardiac Snail1 and Twist1 (n=4). D, Representative immunoblots and corresponding quantification showing EDIL3 in HUVECs (n=4). E, Representative flow cytometry images and corresponding quantification showing the ratio of vimentin‐positive HUVECs (n=4). F, Representative immunoblots and corresponding quantification showing CD31, VE‐cadherin, αSMA, vimentin, Snail1, and Twist1 in HUVECs (n=4). Data are presented as mean±SD. In (D), data were analyzed by unpaired Student t test. In (E), data were analyzed using 1‐way ANOVA followed by Tukey test. In (B), (C) and (F), data were analyzed using 2‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. DCM indicates dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; HUVEC, human umbilical vein endothelial cell; KO, knockout; LDH, lactate dehydrogenase; LVEF, left ventricular ejection fraction; VE‐cadherin, vascular endothelial‐cadherin; WT, wild type; and αSMA, α‐smooth muscle Actin.
EDIL3 Deficiency Reinforces Ubiquitination of Smad4 by Inhibiting USP10 in HUVECs
Tgf‐β signaling is the most well‐defined pathway known to induce EMT and acts through various intracellular messengers. 40 Signaling is normally activated by the Tgf‐β ligand superfamily, which includes 3 isoforms of Tgf‐β (Tgf‐β1, 2, and 3) and 6 isoforms of Bmp (bone morphogenetic protein; Bmp2 to Bmp7). 40 Therefore, we examined the expression of Tgf‐β1, Bmp2, Bmp4, and Bmp7 in HUVECs (Figure 4A and 4B). EDIL3 deficiency reduced the expression of Tgf‐β1 but had no effect on Bmp2, Bmp4, and Bmp7 expression. We also examined the level P‐smad2/3, Smad4, and Smad7 and found that only the level of Smad4 was reduced significantly in EDIL3 deficiency HUVECs with EndMT (Figure 4A and 4C). Interestingly, EDIL3 depletion did not affect Smad4 transcription, which conflicts with the protein measurements (Figure 4D). Therefore, we speculate that EDIL3 may affect the degradation of Smad4. Given that ubiquitination of Smad4 has been identified as regulating cardiac fibrosis, 41 we next investigated the effect of EDIL3 knockdown on Smad4 ubiquitination in HUVEC with EndMT. EDIL3 knockdown markedly promoted ubiquitination and degradation of Smad4, which led to the reduction of Smad4 (Figure 4E). Previous studies have shown that USPs are an important class of deubiquitinating enzymes that contribute to the deubiquitination of Smad4 and hepatocellular carcinoma metastasis. 42 We examined the mRNA levels of USP4, USP10, and USP14 and found that EDIL3 deficiency significantly reduced the level of USP10 in EndMT HUVECs (Figure 4F). Moreover, EDIL3 knockdown reduced the level of USP10 bound to Smad4 in HUVECs with EndMT (Figure 4G). Taken together, these results indicated that EDIL3 deficiency reinforced ubiquitination of Smad4 by inhibiting USP10 in HUVECs.
Figure 4. EDIL3 deficiency reinforces ubiquitination of Smad4 by inhibiting USP10 in HUVECs.
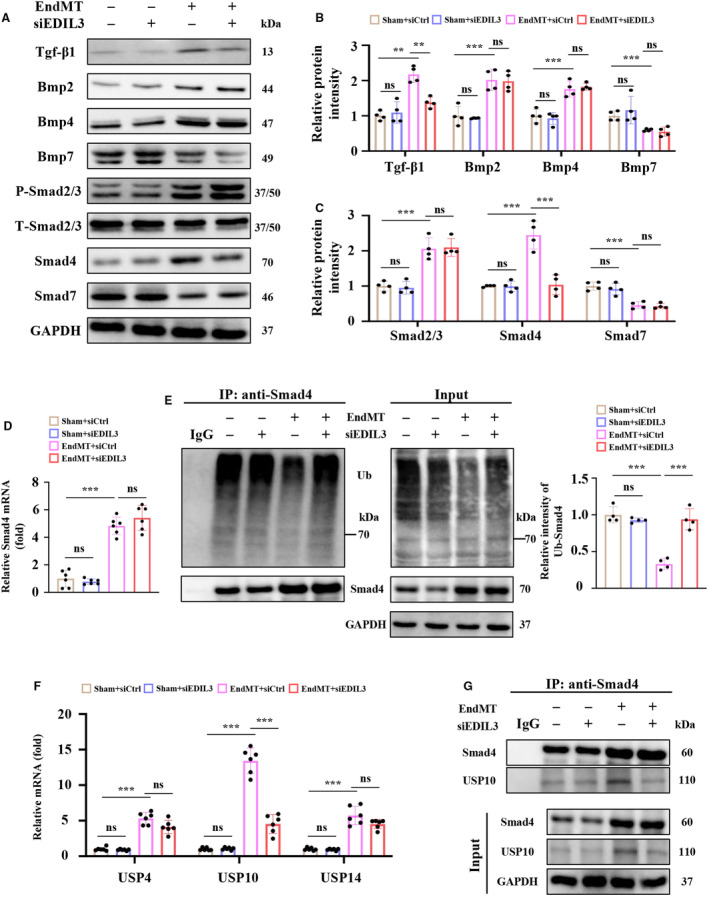
A through C, Representative immunoblots and corresponding quantification showing Tgf‐β1, Bmp2, Bmp4, Bmp7, P‐smad2/3, Smad4, and Smad7 in HUVECs (n=4). D, mRNA levels of Smad4 in HUVECs (n=6). E, Representative immunoblots and corresponding quantification showing ubiquitination of Smad4. F, mRNA levels of cardiac USP4, USP10, and USP14 (n=6). G, Representative immunoblots and corresponding quantification showing USP10 bound to Smad4 in HUVECs (n=4). Data are presented as mean±SD. In (D) and (E), data were analyzed using 1‐way ANOVA followed by Tukey test. In (B), (C) and (F), data were analyzed using 2‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. Bmp indicates bone morphogenetic protein; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; HUVEC, human umbilical vein endothelial cell; Tgf‐β, transforming growth factor beta; Ub, ubiquitin; and USP, ubiquitin specific peptidase.
EDIL3 Deficiency Reinforces Ubiquitination of Smad4 in DCM Mice
EDIL3 depletion significantly reduced the cardiac Smad4 protein expression in DCM mice but not mRNA level (Figure 5A and 5B). EDIL3 knockout markedly promoted ubiquitination of Smad4 in DCM mice heart tissue (Figure 5C). Then, we uncovered that USP10 directly interacted with Smad4 in DCM mouse heart tissues (Figure 5D). In addition, EDIL3 deficiency inhibited the cardiac expression of USP10 in DCM mice (Figure 5E). These results showed that EDIL3 deficiency reinforces ubiquitination of Smad4 in DCM mice.
Figure 5. EDIL3 deficiency reinforces ubiquitination of Smad4 in DCM mice.
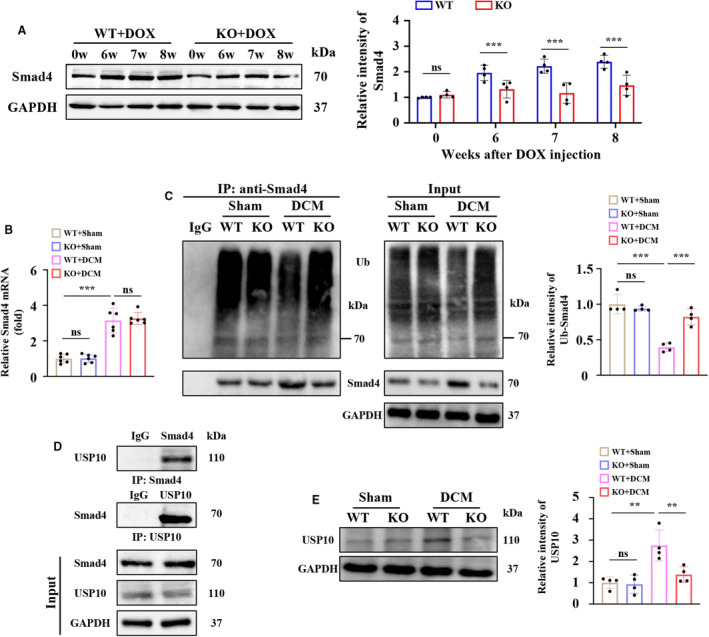
A, Representative immunoblots and corresponding quantification showing cardiac Smad4 at 0, 6, 7, and 8 weeks (n=6). B, mRNA levels of cardiac Smad4 (n=6). C, Representative immunoblots and corresponding quantification showing ubiquitination of Smad4 (n=4). D, Cardiac tissues of DCM mice were immunoprecipitated with anti‐Smad4 or USP10 antibodies and probed with anti‐USP10 or anti‐Smad4 antibodies. E, Representative immunoblots and corresponding quantification showing cardiac USP10 at 8 weeks (n=4). Data are presented as mean±SD. In (A), data were analyzed by unpaired Student t test. In (B), (C) and (E), data were analyzed using one‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. DCM indicates dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; HUVEC, human umbilical vein endothelial cell; KO, knockout; Ub, ubiquitin; USP, ubiquitin specific peptidase; and WT, wild type.
Recombinant Human EDIL3 Promotes EndMT Via Reinforcing Deubiquitination of Smad4 in HUVECs
We next treated HUVECs with recombinant human EDIL3 and found that EDIL3 promoted EndMT as shown by the increased ratio of vimentin‐positive HUVECs with EndMT (Figure 6A). Recombinant EDIL3 reduced CD31 and VE‐cadherin expression, and increased αSMA and vimentin (Figure 6B). The biomarker of EndMT was also upregulated by EDIL3 treatment, indicating that EDIL3 promoted the development of EndMT (Figure 6C). Mechanically, EDIL3 increased the protein levels of Smad4 (Figure 6D). Further studies showed that EDIL3 inhibited the ubiquitination of Smad4 (Figure 6E). In addition, EDIL3 promoted the level of USP10 bound to Smad4 in HUVECs with EndMT (Figure 6F). Taken together, these results indicated that EDIL3 promoted EndMT via reinforcing deubiquitination of Smad4 in HUVECs.
Figure 6. Recombinant human EDIL3 promotes EndMT via reinforcing deubiquitination of Smad4 in HUVECs.
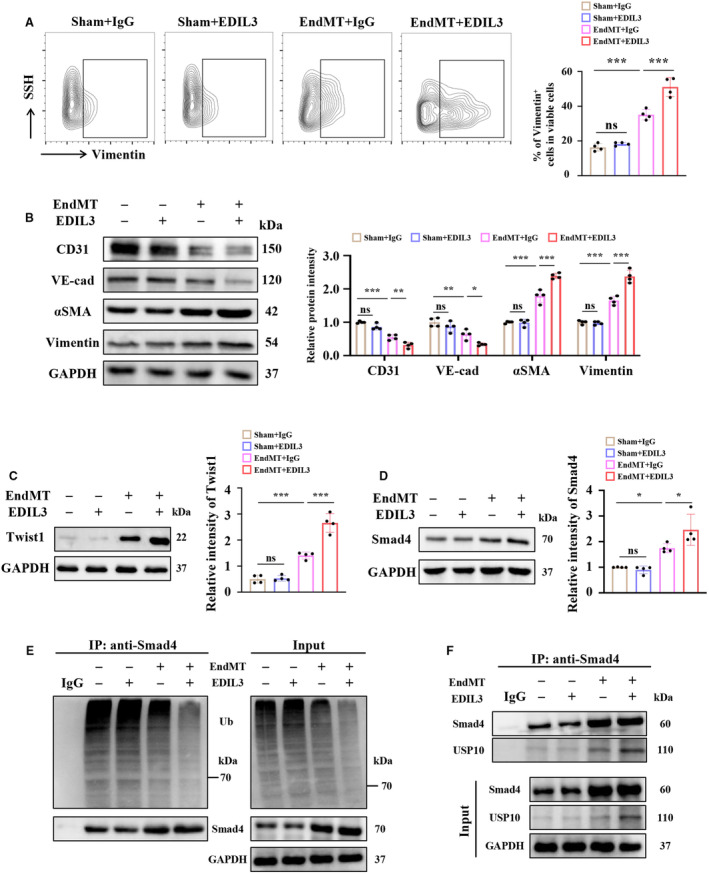
A, Representative flow cytometry images and corresponding quantification showing the ratio of Vimentin positive HUVECs (n=4). B, Representative immunoblots and corresponding quantification showing CD31, VE‐cadherin, αSMA and vimentin in HUVECs (n=4). C, Representative immunoblots and corresponding quantification showing Twist1 in HUVECs (n=4). D, Representative immunoblots and corresponding quantification showing Smad4 (n=4). E, Representative immunoblots showing ubiquitination of Smad4 in HUVECs (n=4). F, Representative immunoblots showing USP10 bound to Smad4 in HUVECs (n=4). Data are presented as mean±SD. In (A), (C) and (D), data were analyzed using 1‐way ANOVA followed by Tukey test. In (B), data were analyzed using 2‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. DCM indicates dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; HUVEC, human umbilical vein endothelial cell; SSH, suppression subtractive hybridization; Ub, ubiquitin; USP, ubiquitin specific peptidase; VE‐cadherin, vascular endothelial‐cadherin; and αSMA, α‐smooth muscle actin.
Inhibiting USP10 Abolished EndMT Exacerbated by EDIL3
Spautin 1, a specific USP10 inhibitor, was given to HUVECs to explore the role of USP10 in EndMT exacerbated by EDIL3. Spautin 1 significantly abolished EndMT exacerbated by EDIL3 (Figure 7A). Inhibiting USP10 also reduced the protein levels of Smad4 significantly (Figure 7B), which may be due to the reinforced ubiquitination and degradation of Smad4 (Figure 7C). These results indicated that inhibiting USP10 abolished EndMT exacerbated by EDIL3.
Figure 7. Inhibiting USP10 abolished EndMT exacerbated by EDIL3.
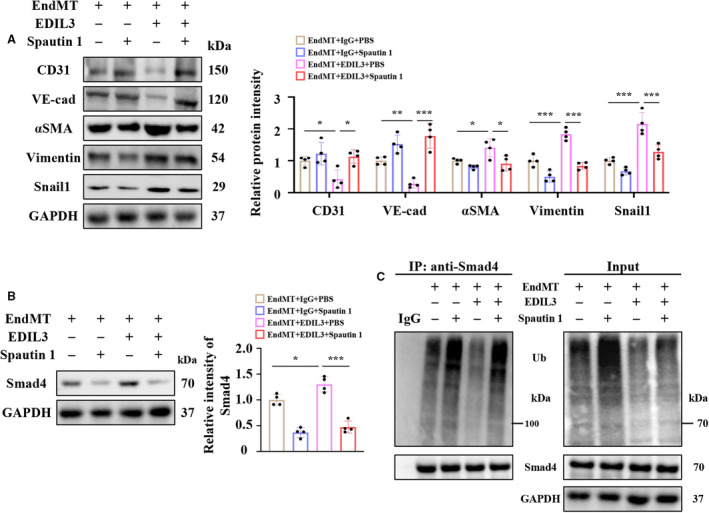
A, Representative immunoblots and corresponding quantification showing CD31, VE‐cadherin, αSMA, vimentin, and Snail1 in HUVECs (n=4). B, Representative immunoblots and corresponding quantification showing Smad4 (n=4). C, Representative immunoblots showing ubiquitination of Smad4 in HUVECs (n=4). Data are presented as mean±SD. In (A), data were analyzed using 2‐way ANOVA followed by Tukey test. In (B), data were analyzed using 1‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. DCM indicates dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; HUVEC, human umbilical vein endothelial cell; Ub, ubiquitin; VE‐cadherin, vascular endothelial‐cadherin; and αSMA, α‐smooth muscle actin.
Inhibiting β3 Integrin/ERK Signal Blocked USP10 Expression Promoted by EDIL3
EDIL3 is reported to promote epithelial‐mesenchymal transition by binding to ITGB3. 22 We speculate that ITGB3 mediates EDIL3‐promoted USP10 expression in HUVEC with EndMT. Knockdown of ITGB3 with siITGB3 significantly inhibited EDIL3‐mediated USP10 expression (Figure S2A and S2B). Furthermore, ITGB3 deficiency blocked EDIL3‐induced Smad4 expression in HUVEC with EndMT (Figure S2B). The classic downstream signaling of ITGB3 including ERK, AKT, and P65 was examined in HUVECs. 43 Recombinant EDIL3 promoted the phosphorylation of ERK and AKT but not P65 in HUVECs with EndMT (Figure S2C). We next treated HUVECs with siERK and siAKT to figure out whose downstream signaling is responsible for the expression of USP10. SiERK but not siAKT treatment inhibited the level of USP10 and Smad4 (Figure S2D). Taken together, EDIL3 may promote the expression of USP10 via regulating ITGB3/ERK signaling.
Recombinant EDIL3 Promotes Doxorubicin‐Induced EndMT Via Reinforcing Deubiquitination of Smad4 in HUVECs
To further explore the role of EDIL3 in doxorubicin‐induced DCM, we treated HUVECs with doxorubicin. Consistent with previous reports, 38 doxorubicin promoted EndMT, as shown by the reduced VE‐cadherin level and increased vimentin level (Figure 8A). In addition, doxorubicin treatment increased the level of Smad4 and USP10 in HUVECs (Figure 8A). EDIL3 deficiency attenuated doxorubicin‐induced EndMT by reducing the levels of Smad4 and USP10 (Figure 8A). Recombinant EDIL3 treatment aggravated doxorubicin‐induced EndMT by increasing the levels of Smad4 and USP10 (Figure 8B). In addition, EDIL3 also inhibited doxorubicin‐induced ubiquitination of SMAD4 (Figure 8C). Taken together, these results suggested that EDIL3 promotes doxorubicin‐induced EndMT via reinforcing deubiquitination of Smad4 in HUVECs.
Figure 8. Recombinant EDIL3 promotes doxorubicin‐induced EndMT via reinforcing deubiquitination of Smad4 in HUVECs.
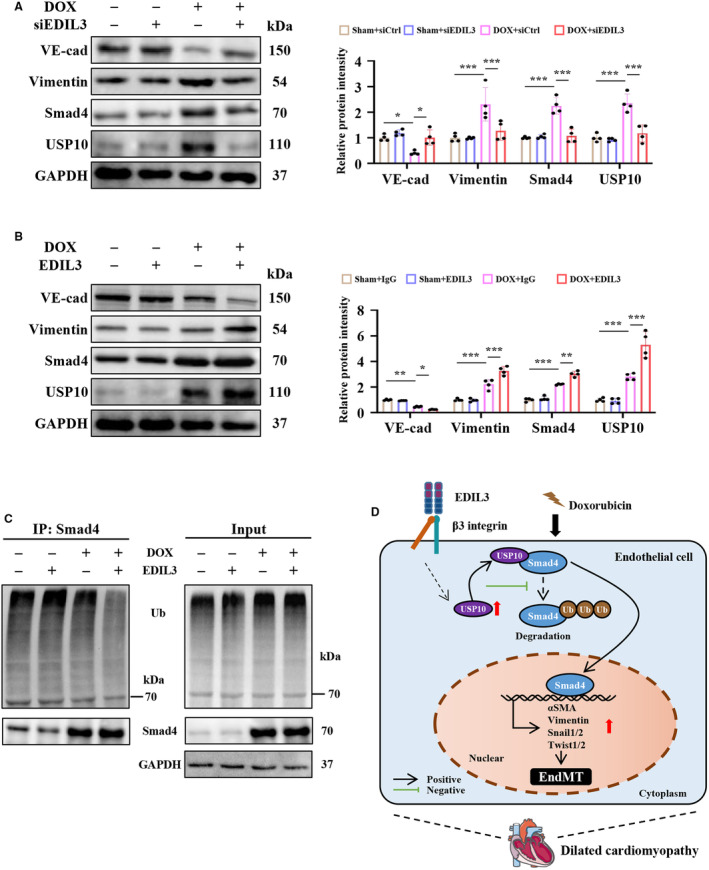
A, Representative immunoblots and corresponding quantification showing VE‐cadherin, vimentin, Smad4, and USP10 in HUVECs (n=4). B, Representative immunoblots and corresponding quantification showing VE‐cadherin, vimentin, Smad4, and USP10 in HUVECs (n=4). C, Representative immunoblots showing ubiquitination of Smad4 in HUVECs (n=4). D, EDIL3 aggravates EndMT by promoting USP10‐dependent deubiquitination of Smad4, thereby exacerbating DCM‐induced cardiac dysfunction and remodeling. Data are presented as mean±SD and were analyzed using 2‐way ANOVA followed by Tukey test. * indicates P<0.05, ** indicates P<0.01, *** indicates P<0.001, ns indicates P>0.05. DCM indicates dilated cardiomyopathy; EDIL3, epidermal growth factor‐like repeats and discoidin I‐like domains 3; EndMT, endothelial to mesenchymal transition; HUVEC, human umbilical vein endothelial cell; Ub, ubiquitin; USP, ubiquitin specific peptidase; and VE‐cadherin, vascular endothelial‐cadherin.
Discussion
In this study, we first found that EndMT signals and EDIL3 were activated in DCM mice. EDIL3 deficiency inhibited EndMT in vivo and in vitro. Mechanically, EDIL3 deficiency reinforced ubiquitination of Smad4 by inhibiting USP10. Recombinant EDIL3 promoted EndMT via reinforcing deubiquitination of Smad4 in HUVECs, which was abolished by USP10 inhibitor. Taken together, these results suggest that EDIL3 aggravates EndMT by promoting USP10‐dependent deubiquitination of Smad4, thereby exacerbating DCM‐induced cardiac dysfunction and remodeling (Figure 8D).
The role and mechanism of EndMT in DCM still lack sufficient evidence. But cardiac fibrosis is considered to be one of the important mechanisms of DCM progression. 44 Cardiac fibrosis occurs early in DCM progression, increases cardiac rigidity, reduces myocardial function, and increases the risk of sudden cardiac death and malignant arrhythmias. 45 , 46 Fibrogenesis is based on myofibroblasts that come from the activation of cardiac fibroblasts or from EMT. 44 EndMT‐mediated cardiac fibrosis plays an important role in diabetic cardiomyopathy and pressure overload‐induced cardiac hypertrophy. 10 Moreover, inhibition of EndMT can effectively alleviate the progression of various cardiovascular diseases. 10 In this study, we found that EndMT signaling was activated in the cardiac tissue of patients with DCM, providing new evidence for the role of EndMT in DCM.
In previous reports, EDIL3, as an endogenous anti‐inflammatory factor, was considered an important target for the treatment of cardiovascular diseases. 26 But in fact, the function and role of EDIL3 are still controversial. EDIL3 overexpression abolishes cardiovascular remodeling and hypertension progression through suppression of inflammation. 28 In contrast, we found that EDIL3 deletion attenuated DCM‐induced cardiac dysfunction and remodeling. EDIL3 loss has also been reported to improve cardiac remodeling after myocardial infarction by promoting the inflammatory response. 29 Although EDIL3‐mediated EndMT has not been discussed, it cannot be ignored. Previous studies have reported that the expression of EDIL3 is increased in various cancer tissues and induces cancer cell migration, invasion, and EMT. 21 , 22 , 24 , 25 EDIL3 depletion inhibits proliferation and EMT of human lens ECs and may be a potential therapeutic target for posterior capsule opacities. 23 We speculate that the different functions of EDIL3 may be due to the fact that EDIL3 is a representative of local tissue signals and exerts different regulatory functions in different expression regions. 15 EDIL3 accelerates the process of inflammation resolution in inflamed areas but not in noninflamed areas. 47 EDIL3 from vascular tissue or cancer tissue may mediate EndMT, thereby accelerating disease tension. EDIL3‐mediated EndMT may be involved in doxorubicin‐induced DCM.
Smad4 forms a heteromeric complex with other Smads, which translocates into the nucleus, binds regulatory elements, and induces the transcription of key genes related to EndMT. 40 Smad4 has also been reported to play critical roles in other cardiovascular diseases, including diabetic cardiomyopathy and cardiac hypertrophy. 48 , 49 Here, we propose that EDIL3 promotes the activation of Smad4 but not Smad2/3 in HUVECs, leading to EndMT. We also found that EDIL3 did not affect Smad4 transcription, which contradicts the finding of upregulation at the protein level. A previous study showed that USP10 contributes to Smad4 deubiquitination and cardiac fibrosis. 41 Subsequently, we found that USP10 directly interacts with Smad4 and causes Smad4 deubiquitination to maintain Smad4 activity in HUVECs. However, USP4 was also identified to regulate deubiquitination of Smad4 and was excluded in this study as the content of USP4 in HUVECs was much lower than USP10. 50 Previous study has shown that monoubiquitinated Smad4 interacts with and is rapidly cleaved by active USP4. 50 Here, we speculate that the specific binding domain may mediate the binding of USP10 to Smad4, which requires more experiments. USP10 is a highly conserved deubiquitinating enzyme, which contributes to the initiation and progression of multiple cancer types, DNA damage response, and signaling pathways. 37 , 51 , 52 USP10‐mediated deubiquitination plays important roles in multiple cardiovascular diseases, including diabetic cardiomyopathy, cardiac hypertrophy and ischemia/reperfusion‐induced cardiac remodeling. 41 , 53 , 54 In addition, knockdown of USP10 inhibits EndMT, whereas overexpression promotes EndMT. 55 Our study provides new evidence indicating the role of USP10 on EndMT. EDIL3 promoted the level of USP10 bound to Smad4, thereby promoting the deubiquitination of Smad4.
ITGB3, also known as CD61 or GP3A, is one of the most extensively studied members of the integrin family and plays an important role in EMT and EndMT. 43 However, the role of ITGB3 in cardiomyopathy seems to remain controversial. ITGB3 is necessary to maintain ventricular function after pressure overload in mice. 56 , 57 ITGB3 deficiency aggravated pressure overload‐induced cardiac hypertrophy and cardiomyocyte apoptosis. 56 , 57 , 58 However, ITGB3 deletion significantly reduced collagen accumulation in the left ventricle and alleviated cardiac fibrosis in mice with pressure overload. 59 In another study, annexin A2 directly binds to and activates ITGB3, promoting neovascularization and preventing myocardial infarction damage. 60 These reports suggested that ITGB3 mediates different functions in different cells and plays complex functions in cardiovascular diseases. EDIL3 has been reported to bind to ITGB3 and activate Tgf‐β signaling, thereby promoting EMT. 22 , 23 , 24 ITGB3 knockdown inhibited the EDIL3‐induced USP10 expression in HUVECs with EndMT. Inhibiting ERK, one of the classic downstream signalings of ITGB3, significantly blocked the expression of USP10 in HUVECs treated with EDIL3. EDIL3 may promote the expression of USP10 via regulating ITGB3/ERK signaling. Taken together, EDIL3 promotes EndMT by promoting USP10‐dependent deubiquitination of Smad4, thereby exacerbating DCM‐induced cardiac dysfunction and remodeling.
There are still several limitations in this study. First, EDIL3 gene systemic knockout mice were used in this study. Endothelial cell‐specific EDIL3 knockout mice may help us further understand the role and mechanism of EDIL3 in DCM. Second, the role of recombinant EDIL3 in DCM mice remains unknown, which needs further exploration.
Conclusions
In summary, we explored the role of EDIL3‐mediated EndMT in DCM‐induced cardiac dysfunction and remodeling and found that EDIL3 promotes USP10‐dependent Smad4 deubiquitination, thereby promoting EndMT.
Sources of Funding
This work was supported by grants from National Natural Science Foundation of China (82070436, 82270454, 82100292), Excellent Doctoral Program of Zhongnan Hospital of Wuhan University (ZNYB2022001), and Youth Interdisciplinary Special Fund of Zhongnan Hospital of Wuhan University (ZNQNJC2023001).
Disclosures
None.
Supporting information
Tables S1–S3.
Figures S1–S2.
Acknowledgments
Jun Wan, Menglong Wang, Juan‐Juan Qin, and Mengmeng Zhao were responsible for the experimental design and wrote the article. Zihui Zheng and Mengmeng Zhao contributed to the Western blots and quantitative polymerase chain reaction. Wei Pan and Jishou Zhang contributed to the animal models. Zheng Yin contributed to the graphical abstract. Yao Xu, Shuwan Xu, and Cheng Wei contributed to the acquisition and analysis of the data. Shanshan Peng contributed to the cell culture and in vitro experiments. Jianfang Liu reviewed this article.
This article was sent to Julie K. Freed, MD, PhD, Associate Editor, for review by expert referees, editorial decision, and final disposition.
Supplemental Material is available at https://www.ahajournals.org/doi/suppl/10.1161/JAHA.123.031283
For Sources of Funding and Disclosures, see page 16.
Contributor Information
Jun Wan, Email: wanjun@whu.edu.cn.
Juan‐Juan Qin, Email: qinjuanjuan@whu.edu.cn.
References
- 1. Merlo M, Cannatà A, Gobbo M, Stolfo D, Elliott PM, Sinagra G. Evolving concepts in dilated cardiomyopathy. Eur J Heart Fail. 2018;20:228–239. doi: 10.1002/ejhf.1103 [DOI] [PubMed] [Google Scholar]
- 2. McKenna WJ, Maron BJ, Thiene G. Classification, epidemiology, and global burden of cardiomyopathies. Circ Res. 2017;121:722–730. doi: 10.1161/circresaha.117.309711 [DOI] [PubMed] [Google Scholar]
- 3. Weintraub RG, Semsarian C, Macdonald P. Dilated cardiomyopathy. Lancet. 2017;390:400–414. doi: 10.1016/s0140-6736(16)31713-5 [DOI] [PubMed] [Google Scholar]
- 4. Hershberger RE, Hedges DJ, Morales A. Dilated cardiomyopathy: the complexity of a diverse genetic architecture. Nat Rev Cardiol. 2013;10:531–547. doi: 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- 5. Heidenreich PA, Bozkurt B, Aguilar D, Allen LA, Byun JJ, Colvin MM, Deswal A, Drazner MH, Dunlay SM, Evers LR, et al. 2022 AHA/ACC/HFSA guideline for the management of heart failure. Circulation. 2022;145:e876–e894. doi: 10.1161/CIR.0000000000001062 [DOI] [PubMed] [Google Scholar]
- 6. Zecchin M, Merlo M, Pivetta A, Barbati G, Lutman C, Gregori D, Serdoz LV, Bardari S, Magnani S, Di Lenarda A, et al. How can optimization of medical treatment avoid unnecessary implantable cardioverter‐defibrillator implantations in patients with idiopathic dilated cardiomyopathy presenting with "SCD‐HeFT criteria?". Am J Cardiol. 2012;109:729–735. doi: 10.1016/j.amjcard.2011.10.033 [DOI] [PubMed] [Google Scholar]
- 7. Peng Q, Shan D, Cui K, Li K, Zhu B, Wu H, Wang B, Wong S, Norton V, Dong Y, et al. The role of endothelial‐to‐mesenchymal transition in cardiovascular disease. Cell. 2022;11:1834. doi: 10.3390/cells11111834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Haynes BA, Yang LF, Huyck RW, Lehrer EJ, Turner JM, Barabutis N, Correll VL, Mathiesen A, McPheat W, Semmes OJ, et al. Endothelial‐to‐mesenchymal transition in human adipose tissue vasculature alters the particulate secretome and induces endothelial dysfunction. Arterioscler Thromb Vasc Biol. 2019;39:2168–2191. doi: 10.1161/atvbaha.119.312826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Baumann K. Mechanotransduction: kindlin' the fate of mesenchymal stem cells. Nat Rev Mol Cell Biol. 2018;19:278–279. doi: 10.1038/nrm.2018.21 [DOI] [PubMed] [Google Scholar]
- 10. Li Y, Lui KO, Zhou B. Reassessing endothelial‐to‐mesenchymal transition in cardiovascular diseases. Nat Rev Cardiol. 2018;15:445–456. doi: 10.1038/s41569-018-0023-y [DOI] [PubMed] [Google Scholar]
- 11. Xie Y, Liao J, Yu Y, Guo Q, Yang Y, Ge J, Chen H, Chen R. Endothelial‐to‐mesenchymal transition in human idiopathic dilated cardiomyopathy. Mol Med Rep. 2018;17:961–969. doi: 10.3892/mmr.2017.8013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang L, He J, Wang J, Liu J, Chen Z, Deng B, Wei L, Wu H, Liang B, Li H, et al. Knockout RAGE alleviates cardiac fibrosis through repressing endothelial‐to‐mesenchymal transition (EndMT) mediated by autophagy. Cell Death Dis. 2021;12:470. doi: 10.1038/s41419-021-03750-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Wang B, Ge Z, Wu Y, Zha Y, Zhang X, Yan Y, Xie Y. MFGE8 is down‐regulated in cardiac fibrosis and attenuates endothelial‐mesenchymal transition through Smad2/3‐snail signalling pathway. J Cell Mol Med. 2020;24:12799–12812. doi: 10.1111/jcmm.15871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ho HK, Jang JJ, Kaji S, Spektor G, Fong A, Yang P, Hu BS, Schatzman R, Quertermous T, Cooke JP. Developmental endothelial locus‐1 (Del‐1), a novel angiogenic protein: its role in ischemia. Circulation. 2004;109:1314–1319. doi: 10.1161/01.Cir.0000118465.36018.2d [DOI] [PubMed] [Google Scholar]
- 15. Li M, Zhong D, Li G. Regulatory role of local tissue signal Del‐1 in cancer and inflammation: a review. Cell Mol Biol Lett. 2021;26:31. doi: 10.1186/s11658-021-00274-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhong J, Eliceiri B, Stupack D, Penta K, Sakamoto G, Quertermous T, Coleman M, Boudreau N, Varner JA. Neovascularization of ischemic tissues by gene delivery of the extracellular matrix protein Del‐1. J Clin Invest. 2003;112:30–41. doi: 10.1172/jci17034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Niu X, Chang W, Liu R, Hou R, Li J, Wang C, Li X, Zhang K. mRNA and protein expression of the angiogenesis‐related genes EDIL3, AMOT and ECM1 in mesenchymal stem cells in psoriatic dermis. Clin Exp Dermatol. 2016;41:533–540. doi: 10.1111/ced.12783 [DOI] [PubMed] [Google Scholar]
- 18. Schürpf T, Chen Q, Liu JH, Wang R, Springer TA, Wang JH. The RGD finger of Del‐1 is a unique structural feature critical for integrin binding. FASEB J. 2012;26:3412–3420. doi: 10.1096/fj.11-202036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kun Z, Xin G, Tao W, Chenglong Z, Dongsheng W, Liang T, Tielong L, Jianru X. Tumor derived EDIL3 modulates the expansion and osteoclastogenesis of myeloid derived suppressor cells in murine breast cancer model. J Bone Oncol. 2019;16:100238. doi: 10.1016/j.jbo.2019.100238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fan Y, Zhu W, Yang M, Zhu Y, Shen F, Hao Q, Young WL, Yang GY, Chen Y. Del‐1 gene transfer induces cerebral angiogenesis in mice. Brain Res. 2008;1219:1–7. doi: 10.1016/j.brainres.2008.05.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xia H, Chen J, Shi M, Gao H, Sekar K, Seshachalam VP, Ooi LL, Hui KM. EDIL3 is a novel regulator of epithelial‐mesenchymal transition controlling early recurrence of hepatocellular carcinoma. J Hepatol. 2015;63:863–873. doi: 10.1016/j.jhep.2015.05.005 [DOI] [PubMed] [Google Scholar]
- 22. Gasca J, Flores ML, Jiménez‐Guerrero R, Sáez ME, Barragán I, Ruíz‐Borrego M, Tortolero M, Romero F, Sáez C, Japón MA. EDIL3 promotes epithelial‐mesenchymal transition and paclitaxel resistance through its interaction with integrin α(V)β(3) in cancer cells. Cell Death Dis. 2020;6:86. doi: 10.1038/s41420-020-00322-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Zhang R, Wei YH, Zhao CY, Song HY, Shen N, Cui X, Gao X, Qi ZT, Zhong M, Shen W. EDIL3 depletion suppress epithelial‐mesenchymal transition of lens epithelial cells via transforming growth factor β pathway. Int J Ophthalmol. 2018;11:18–24. doi: 10.18240/ijo.2018.01.04 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang L, Peng KW, Wang B, Yang XF, Zhang ZM. EDIL3 regulates gastric cancer cell migration, invasion and epithelial‐mesenchymal transition via TGF‐β1/XIST/miR‐137 feedback loop. Transl Cancer Res. 2020;9:6313–6330. doi: 10.21037/tcr-19-2967b [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lee SH, Kim DY, Jing F, Kim H, Yun CO, Han DJ, Choi EY. Del‐1 overexpression potentiates lung cancer cell proliferation and invasion. Biochem Biophys Res Commun. 2015;468:92–98. doi: 10.1016/j.bbrc.2015.10.159 [DOI] [PubMed] [Google Scholar]
- 26. Zhao M, Zheng Z, Li C, Wan J, Wang M. Developmental endothelial locus‐1 in cardiovascular and metabolic diseases: a promising biomarker and therapeutic target. Front Immunol. 2022;13:1053175. doi: 10.3389/fimmu.2022.1053175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhao M, Zheng Z, Yin Z, Zhang J, Peng S, Liu J, Pan W, Wei C, Xu Y, Qin JJ, et al. DEL‐1 deficiency aggravates pressure overload‐induced heart failure by promoting neutrophil infiltration and neutrophil extracellular traps formation. Biochem Pharmacol. 2023;218:115912. doi: 10.1016/j.bcp.2023.115912 [DOI] [PubMed] [Google Scholar]
- 28. Failer T, Amponsah‐Offeh M, Neuwirth A, Kourtzelis I, Subramanian P, Mirtschink P, Peitzsch M, Matschke K, Tugtekin SM, Kajikawa T, et al. Developmental endothelial locus‐1 protects from hypertension‐induced cardiovascular remodeling via immunomodulation. J Clin Invest. 2022;132:e126155. doi: 10.1172/jci126155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wei X, Zou S, Xie Z, Wang Z, Huang N, Cen Z, Hao Y, Zhang C, Chen Z, Zhao F, et al. EDIL3 deficiency ameliorates adverse cardiac remodelling by neutrophil extracellular traps (NET)‐mediated macrophage polarization. Cardiovasc Res. 2022;118:2179–2195. doi: 10.1093/cvr/cvab269 [DOI] [PubMed] [Google Scholar]
- 30. Chan KY, Xiang P, Zhou L, Li K, Ng PC, Wang CC, Zhang L, Deng HY, Pong NH, Zhao H, et al. Thrombopoietin protects against doxorubicin‐induced cardiomyopathy, improves cardiac function, and reversely alters specific signalling networks. Eur J Heart Fail. 2011;13:366–376. doi: 10.1093/eurjhf/hfr001 [DOI] [PubMed] [Google Scholar]
- 31. Wang M, Zhao M, Zheng Z, Pan W, Zhang J, Yin Z, Wei C, Xu Y, Wan J. TRPA1 deficiency aggravates dilated cardiomyopathy by promoting S100A8 expression to induce M1 macrophage polarization in rats. FASEB J. 2023;37:e22982. doi: 10.1096/fj.202202079RR [DOI] [PubMed] [Google Scholar]
- 32. Wang M, Zhao M, Yu J, Xu Y, Zhang J, Liu J, Zheng Z, Ye J, Wang Z, Ye D, et al. MCC950, a selective NLRP3 inhibitor, attenuates adverse cardiac remodeling following heart failure through improving the cardiometabolic dysfunction in obese mice. Front Cardiovasc Med. 2022;9:727474. doi: 10.3389/fcvm.2022.727474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wang Z, Xu Y, Wang M, Ye J, Liu J, Jiang H, Ye D, Wan J. TRPA1 inhibition ameliorates pressure overload‐induced cardiac hypertrophy and fibrosis in mice. EBioMedicine. 2018;36:54–62. doi: 10.1016/j.ebiom.2018.08.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Monteiro JP, Rodor J, Caudrillier A, Scanlon JP, Spiroski AM, Dudnakova T, Pflüger‐Müller B, Shmakova A, von Kriegsheim A, Deng L, et al. MIR503HG loss promotes endothelial‐to‐mesenchymal transition in vascular disease. Circ Res. 2021;128:1173–1190. doi: 10.1161/circresaha.120.318124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lee SH, Kim DY, Kang YY, Kim H, Jang J, Lee MN, Oh GT, Kang SW, Choi EY. Developmental endothelial locus‐1 inhibits MIF production through suppression of NF‐κB in macrophages. Int J Mol Med. 2014;33:919–924. doi: 10.3892/ijmm.2014.1645 [DOI] [PubMed] [Google Scholar]
- 36. Salazar G, Cullen A, Huang J, Zhao Y, Serino A, Hilenski L, Patrushev N, Forouzandeh F, Hwang HS. SQSTM1/p62 and PPARGC1A/PGC‐1alpha at the interface of autophagy and vascular senescence. Autophagy. 2020;16:1092–1110. doi: 10.1080/15548627.2019.1659612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang X, Xia S, Li H, Wang X, Li C, Chao Y, Zhang L, Han C. The deubiquitinase USP10 regulates KLF4 stability and suppresses lung tumorigenesis. Cell Death Differ. 2020;27:1747–1764. doi: 10.1038/s41418-019-0458-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pan JA, Zhang H, Lin H, Gao L, Zhang HL, Zhang JF, Wang CQ, Gu J. Irisin ameliorates doxorubicin‐induced cardiac perivascular fibrosis through inhibiting endothelial‐to‐mesenchymal transition by regulating ROS accumulation and autophagy disorder in endothelial cells. Redox Biol. 2021;46:102120. doi: 10.1016/j.redox.2021.102120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Zhao M, Zheng Z, Zhang P, Xu Y, Zhang J, Peng S, Liu J, Pan W, Yin Z, Xu S, et al. IL‐30 protects against sepsis‐induced myocardial dysfunction by inhibiting pro‐inflammatory macrophage polarization and pyroptosis. iScience. 2023;26:107544. doi: 10.1016/j.isci.2023.107544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gonzalez DM, Medici D. Signaling mechanisms of the epithelial‐mesenchymal transition. Sci Signal. 2014;7:re8. doi: 10.1126/scisignal.2005189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Xie S, Xing Y, Shi W, Zhang M, Chen M, Fang W, Liu S, Zhang T, Zeng X, Chen S, et al. Cardiac fibroblast heat shock protein 47 aggravates cardiac fibrosis post myocardial ischemia‐reperfusion injury by encouraging ubiquitin specific peptidase 10 dependent Smad4 deubiquitination. Acta Pharm Sin B. 2022;12:4138–4153. doi: 10.1016/j.apsb.2022.07.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yuan T, Chen Z, Yan F, Qian M, Luo H, Ye S, Cao J, Ying M, Dai X, Gai R, et al. Deubiquitinating enzyme USP10 promotes hepatocellular carcinoma metastasis through deubiquitinating and stabilizing Smad4 protein. Mol Oncol. 2020;14:197–210. doi: 10.1002/1878-0261.12596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhu C, Kong Z, Wang B, Cheng W, Wu A, Meng X. ITGB3/CD61: a hub modulator and target in the tumor microenvironment. Am J Transl Res. 2019;11:7195–7208. [PMC free article] [PubMed] [Google Scholar]
- 44. Cojan‐Minzat BO, Zlibut A, Agoston‐Coldea L. Non‐ischemic dilated cardiomyopathy and cardiac fibrosis. Heart Fail Rev. 2021;26:1081–1101. doi: 10.1007/s10741-020-09940-0 [DOI] [PubMed] [Google Scholar]
- 45. Morita H, Seidman J, Seidman CE. Genetic causes of human heart failure. J Clin Invest. 2005;115:518–526. doi: 10.1172/jci24351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Venero JV, Doyle M, Shah M, Rathi VK, Yamrozik JA, Williams RB, Vido DA, Rayarao G, Benza R, Murali S, et al. Mid wall fibrosis on CMR with late gadolinium enhancement may predict prognosis for LVAD and transplantation risk in patients with newly diagnosed dilated cardiomyopathy‐preliminary observations from a high‐volume transplant centre. ESC Heart Fail. 2015;2:150–159. doi: 10.1002/ehf2.12041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hajishengallis G, Chavakis T. DEL‐1‐regulated immune plasticity and inflammatory disorders. Trends Mol Med. 2019;25:444–459. doi: 10.1016/j.molmed.2019.02.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tian J, Zhang M, Suo M, Liu D, Wang X, Liu M, Pan J, Jin T, An F. Dapagliflozin alleviates cardiac fibrosis through suppressing EndMT and fibroblast activation via AMPKα/TGF‐β/Smad signalling in type 2 diabetic rats. J Cell Mol Med. 2021;25:7642–7659. doi: 10.1111/jcmm.16601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Umbarkar P, Singh AP, Gupte M, Verma VK, Galindo CL, Guo Y, Zhang Q, McNamara JW, Force T, Lal H. Cardiomyocyte SMAD4‐dependent TGF‐β signaling is essential to maintain adult heart homeostasis. JACC Basic Transl Sci. 2019;4:41–53. doi: 10.1016/j.jacbts.2018.10.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou F, Xie F, Jin K, Zhang Z, Clerici M, Gao R, van Dinther M, Sixma TK, Huang H, Zhang L, et al. USP4 inhibits SMAD4 monoubiquitination and promotes activin and BMP signaling. EMBO J. 2017;36:1623–1639. doi: 10.15252/embj.201695372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Zhu H, Yan F, Yuan T, Qian M, Zhou T, Dai X, Cao J, Ying M, Dong X, He Q, et al. USP10 promotes proliferation of hepatocellular carcinoma by deubiquitinating and stabilizing YAP/TAZ. Cancer Res. 2020;80:2204–2216. doi: 10.1158/0008-5472.Can-19-2388 [DOI] [PubMed] [Google Scholar]
- 52. Lim R, Sugino T, Nolte H, Andrade J, Zimmermann B, Shi C, Doddaballapur A, Ong YT, Wilhelm K, Fasse JWD, et al. Deubiquitinase USP10 regulates notch signaling in the endothelium. Science. 2019;364:188–193. doi: 10.1126/science.aat0778 [DOI] [PubMed] [Google Scholar]
- 53. Lu L, Ma J, Liu Y, Shao Y, Xiong X, Duan W, Gao E, Yang Q, Chen S, Yang J, et al. FSTL1‐USP10‐Notch1 signaling axis protects against cardiac dysfunction through inhibition of myocardial fibrosis in diabetic mice. Front Cell Dev Biol. 2021;9:757068. doi: 10.3389/fcell.2021.757068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Zhang DH, Zhang JL, Huang Z, Wu LM, Wang ZM, Li YP, Tian XY, Kong LY, Yao R, Zhang YZ. Deubiquitinase ubiquitin‐specific protease 10 deficiency regulates Sirt6 signaling and exacerbates cardiac hypertrophy. J Am Heart Assoc. 2020;9:e017751. doi: 10.1161/jaha.120.017751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Ouchida AT, Kacal M, Zheng A, Ambroise G, Zhang B, Norberg E, Vakifahmetoglu‐Norberg H. USP10 regulates the stability of the EMT‐transcription factor Slug/SNAI2. Biochem Biophys Res Commun. 2018;502:429–434. doi: 10.1016/j.bbrc.2018.05.156 [DOI] [PubMed] [Google Scholar]
- 56. Ren J, Avery J, Zhao H, Schneider JG, Ross FP, Muslin AJ. Beta3 integrin deficiency promotes cardiac hypertrophy and inflammation. J Mol Cell Cardiol. 2007;42:367–377. doi: 10.1016/j.yjmcc.2006.11.002 [DOI] [PubMed] [Google Scholar]
- 57. Johnston RK, Balasubramanian S, Kasiganesan H, Baicu CF, Zile MR, Kuppuswamy D. Beta3 integrin‐mediated ubiquitination activates survival signaling during myocardial hypertrophy. FASEB J. 2009;23:2759–2771. doi: 10.1096/fj.08-127480 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Suryakumar G, Kasiganesan H, Balasubramanian S, Kuppuswamy D. Lack of beta3 integrin signaling contributes to calpain‐mediated myocardial cell loss in pressure‐overloaded myocardium. J Cardiovasc Pharmacol. 2010;55:567–573. doi: 10.1097/FJC.0b013e3181d9f5d4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Balasubramanian S, Quinones L, Kasiganesan H, Zhang Y, Pleasant DL, Sundararaj KP, Zile MR, Bradshaw AD, Kuppuswamy D. β3 integrin in cardiac fibroblast is critical for extracellular matrix accumulation during pressure overload hypertrophy in mouse. PLoS One. 2012;7:e45076. doi: 10.1371/journal.pone.0045076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y, Wang Y, Li J, Li C, Liu W, Long X, Wang Z, Zhao R, Ge J, Shi B. ANNEXIN A2 facilitates neovascularization to protect against myocardial infarction injury via interacting with macrophage yap and endothelial integrin Β3. Shock. 2023;60:573–584. doi: 10.1097/shk.0000000000002198 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Tables S1–S3.
Figures S1–S2.