

Key words: Cutaneous leishmaniasis, in vitro experimental chemotherapy, in vivo, Leishmania amazonensis, shape-based virtual screening
Abstract
Cutaneous leishmaniasis (CL) is a spectrum of clinical manifestations characterized by severe skin ulcerations that leads to social stigma. There are limited treatment options for CL, and the available drugs are becoming less efficacious due to drug resistance. More efficacious and safer antileishmanial drugs are needed. In this study, the biological effect of seven synthetically accessible nitroaromatic compounds was evaluated in vitro against amastigotes of Leishmania amazonensis, followed by in vivo evaluation using mouse models of CL. Two compounds (6 and 7) were active against amastigotes in vitro [half-maximal effective concentration (EC50): 4.57 ± 0.08 and 9.19 ± 0.68 μm, respectively], with selectivity indexes >50, and the other compounds were not selective. In vivo, compounds 6 and 7 (10 mg kg−1, twice a day for 14 days) failed to reduce skin lesion sizes and parasite loads determined by light microscopy of lesion imprints and quantitative polymerase chain reaction. Nevertheless, the in vitro leishmanicidal efficacy sustained their use as templates for nitroimidazole-based antileishmanial drug discovery programmes focusing on analogues with more suitable properties.
Introduction
Cutaneous leishmaniasis (CL) comprises a broad spectrum of clinical manifestations caused by over 20 different kinetoplastid Leishmania species transmitted by female sandflies (WHO, 2020). In Asia and Africa, most CL cases are caused by Leishmania major and Leishmania tropica, leading to self-limiting ulcers. In the Americas, the disease is caused by several species, including Leishmania braziliensis, Leishmania mexicana, Leishmania guyanensis, Leishmania naiffi and Leishmania amazonensis. The clinical presentations range from self-healing localized skin lesions to disfiguring mucocutaneous ulcerations (Martins et al., 2014; de Vries et al., 2015). Annually, about 1.2 million new CL cases are reported, and a significant number of patients develop severely disfiguring permanent scars, which results in social stigma and loss of economic productivity (Alvar et al., 2012; Baileyid et al., 2019). CL is one of the most mistreated illnesses among neglected tropical diseases (NTDs). The large variation of drug susceptibility among the diverse parasite species makes the drug discovery process even more difficult (de Vries et al., 2015; Van Bocxlaer et al., 2019).
The first-line treatment for CL includes pentavalent antimonials: sodium stibogluconate and meglumine antimoniate. Antimonials present several drawbacks in terms of efficacy, safety (cardiotoxicity and hepatotoxicity) and protracted periods of drug administration (Weisz, 2006). Alternative systemic agents include miltefosine, amphotericin B, pentamidine isethionate, paromomycin and granulocyte-macrophage colony-stimulating factor, limited by severe adverse effects (de Vries et al., 2015). There is a need for safer, more selective and easily accessible therapies for CL.
A few new small molecules are currently in preclinical and clinical development against Leishmania. One example is the first-in-class kinetoplastid-selective proteasome inhibitor compound LXE408. LXE408 was discovered by Novartis with financial support from the Wellcome Trust. LXE408 is efficacious in the murine model of CL, and it is under clinical trial for the treatment of visceral leishmaniasis (Nagle et al., 2020). Partnerships between not-for-profit research and private pharmaceutical companies for the research and development of new treatments for NTDs such as leishmaniasis are increasing (Katsuno et al., 2015).
Some of the compounds in development include nitroaromatic compounds, which are historically used to treat several diseases caused by protozoans, for example, DNDI-8219, DNI-VL2098 and DNDI-0690 (Van Bocxlaer et al., 2019). In this study, we envision that scaffold-driven and easily synthesizable nitroaromatic compounds based on prior art can facilitate rapid phenotypic screenings using in vitro and in vivo experimental models. The scaffolds were selected based on our ongoing research on the use of natural product-derived motifs in the design and development of antiprotozoal agents (Zhang et al., 2018). The compounds were evaluated against the free amastigote forms (AFs) of L. amazonensis (strain LTB0016), purified from paw lesion, followed by evaluation against the intracellular forms as well as on peritoneal macrophages (PMMs). The most selective were evaluated in L. amazonensis-infected BALB/c mice. The methods and results are presented below.
Materials and methods
Compounds: Dimethyl sulphoxide (DMSO, Merck) solutions (30 mm) of the compounds (Fig. 1) and miltefosine ⩾98% (Sigma-Aldrich, St. Louis, USA) were diluted just before the in vitro experiments into RPMI-1640 medium (pH: 7.2–7.4) without phenol red (Sigma-Aldrich (St. Louis, USA), R7509) but supplemented with 1% l-alanyl-l-glutamine (GLUTAMAX I, Gilbco™), 1% penicillin–streptomycin 5000 U mL−1 (Gibco™, PEN-STR) and 10% heat-inactivated sterile-filtered fetal bovine serum (FBS), from Cultilab, Brazil. The concentration of DMSO was <0.6% for all in vitro experiments to prevent non-specific toxicity to host cells (Romanha et al., 2010). For in vivo assays, compounds 6 and 7 were prepared daily in a solution composed of 5% Gum Arabic from acacia tree (Sigma-Aldrich, St. Louis, USA), 6% DMSO and 3% Tween™ 80 Surfact-Amps™ detergent solution (Thermo Scientific™). The reference drug, Milteforan™ (Mt™), was dissolved in sterile deionized water.
Fig. 1.

Structures of the tested compounds.
Parasite strain and mammalian host cell cultures
Leishmania amazonensis (strain LTB0016) was used throughout the study. AFs were purified from male BALB/c mice. Briefly, the foot paws were inoculated (subcutaneously) with 20 μL containing 106 amastigotes, and the animal skin lesions were aseptically removed 30 days post-infection (dpi). The parasites in the lesions were mechanically dispersed by pipetting and used for the assays. The compounds were tested on (1) extracellular AFs; (2) intracellular amastigote forms (IA) in PMMs and (3) BALB/c mouse models infected with amastigotes as recently described (Cardoso Santos et al., 2021). PMMs were obtained from Swiss male mice (18–20 g) inoculated with 3% Brewer thioglycollate medium (Merck) previously diluted in water and autoclaved. After 4 days of stimulation, the cells were collected by rinsing the animals' peritoneum with RPMI 1640. The cells were subsequently seeded in 24- (3 × 105 cells well−1) and 96-well (5 × 104 cells well−1) plates for analysis of infection and cytotoxicity assays, respectively. Cell cultures were maintained at 37°C, 5% CO2 atmosphere in RPMI 1640 medium (pH: 7.2–7.4) without phenol red, but supplemented with 1% l-glutamine, 1% (PEN-STR) and 10% FBS. Assays using AF were maintained at 32°C using RPMI culture medium containing 5% FBS.
Cytotoxicity and in vitro leishmanicidal analysis
The toxicity of the compounds on host cells was evaluated 24 h after PMMs were seeded into culture plates. The compounds (0–500 μm) were added to the wells and incubated as described above for 48 h, and cellular viability was evaluated using the AlamarBlue test (Invitrogen) according to the manufacturer's recommendations. The effect of the compounds (0–20 μm) on AFs was evaluated using 106 parasites per well (0.2 mL) for 48 h, and parasite viability assessed with AlamarBlue tests as reported (Mikus and Steverding, 2000; Santos et al., 2020). Phenotypic screenings against IAs were performed by infecting 3 × 105 PMMs with 9 × 105 amastigotes, multiplicity of infection being 3. After 48 h of drug treatment, cultures were rinsed with saline, fixed 5 min with Bouin and stained for 15 min with Giemsa solution (Sigma-Aldrich (St. Louis, USA), 32884), filtered and diluted five times in distilled water (Santos et al., 2018; Feitosa et al., 2019). Samples were subsequently evaluated by light microscopy and photographs were obtained by using a Zeiss AxioObserver MI microscope (Oberkochen, Germany). The percentage of infected host cells, the number of parasites per infected cell and the corresponding infection index were obtained. Parasites with well-defined nuclei and kinetoplasts were counted as surviving since irregular structures could mean parasites undergoing death. The results were expressed as % reduction in parasite burden, and the half-maximal effective concentration (EC50) and 90% maximal effective concentration (EC90) were calculated by non-linear regression analysis using GraphPad Prism v.9.1.2. All assays were run at least twice with three independent replicates each time.
Hep G2 viability assay
Hep G2 (CRL-11997™) cells were grown in complete growth medium (Dulbecco's modified Eagle medium: F12 containing l-glutamine and sodium bicarbonate, 10% FBS and 1% penicillin/streptomycin) incubated at 37°C in a 5% CO2 environment. Cells were seeded into 96-well plates at 5 × 105 cells mL−1 and incubated for about 24 h. Cells were treated with the compounds prepared in DMSO for 72 h at a final concentration range of 160–1.2 μm in triplicates. Cell viability was assessed using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-2H-tetrazolium bromide assay as previously described (Zhang et al., 2018). Podophyllotoxin was used as cytotoxicity control, and sodium dodecyl sulphate (10%) was used as assay control.
In vivo efficacy analysis
Male BALB/c mice (18–20 g) obtained from the animal facilities of ICTB (Institute of Science and Biomodels Technology, Fiocruz, RJ, Brazil) were housed five per cage, kept in a room at 20–24°C under a 12-light and 12-h dark cycle and provided with sterile water and chow ad libitum. Animals were subcutaneously inoculated with 5 × 105 amastigotes on the left foot paw (Godinho et al., 2012). The treatment started 15 dpi, corresponding to the lesion onset (size diameter of 3–4 mm) (Van Bocxlaer et al., 2019). The compounds were given for 14 days by oral gavage (100 μL) at 10 mg kg−1, twice a day (q12h). Mt™ was administered at a standardized dose (40 mg kg−1) once a day (q24h). The lesions were measured regularly in three dimensions (height, width and depth), and animals were euthanized at 31 dpi. Skin lesions aseptically removed were used for molecular analysis of parasite load by quantitative polymerase chain reaction (qPCR) and light microscopy inspections of imprints (Ribeiro-Romão et al., 2016). All procedures were carried out in accordance with the guidelines established by the FIOCRUZ Committee of Ethics for the Use of Animals (CEUA L038/2017). Statistical analysis was conducted in GraphPad Prism v.9.1.2 by analysis of variance (ANOVA): (1) one-way Fisher's LSD test or (2) two-way unpaired t-test with Welch's correction, significance P < 0.05 [95% confidence interval (CI)].
Results
All experimental details are fully described in the Supplementary material. The compounds (Fig. 1) were synthesized as shown in Fig. S1 and diluted just before the in vitro experiments. First, the compounds were screened at 10 μm against extracellular amastigotes of L. amazonensis purified from male BALB/c paw lesions (Feitosa et al., 2019) using colorimetric assays (AlamarBlue test, Invitrogen) (Mikus and Steverding, 2000; Godinho et al., 2012). Two (6 and 7) out of the seven compounds caused a significant decline in the total number of live parasites (⩾50% decrease compared to untreated parasites) with only 48 h of drug exposition. Next, 6 and 7 were further analysed in dose–response assays. The EC50 values obtained were 10.78 ± 0.80 and 13.12 ± 1.70 μm, respectively (Table 1). Host cell viability showed that compounds did not exhibit any toxicity up to the highest tested concentration, leading to CC50 PMM values >500 μm. Also, when cellular viability was assessed using Hep G2 cultures, no toxicity was observed (CC50 Hep values >160 μm, Table 1). The selective antiparasitic activity was encouraging, and we decided to evaluate the compounds on intracellular amastigotes (IA) in PMMs (9 × 105 IA: 3 × 105 PMMs well−1), which mimics typical infection in mammalian cells. Light microscopy analysis showed that 6 was about 3-fold more active compared to the reference drug (miltefosine, P = 0.0273). Its EC50 was 4.57 μm, and it is quite selective [selective index (SI) > 100]. Compound 6 also displayed low EC90 (9.14 μm) and caused a 92% decrease in PMM infection at 10 μm. Compound 7 is slightly less potent (EC50 = 9.19 μm and EC90 = 17.2 μm) with an SI of >50 (Table 1). Both compounds displayed an impressive leishmanicidal effect due to a drastic decrease in the number of parasites per cell and the ratio of infected cells at 20 μm (Fig. S2). These favourable results prompted us to conduct preliminary in vivo proof of concepts using both hit compounds.
Table 1.
Phenotypic studies of compounds 6 and 7 against Leishmania amazonensis and their toxicity profile
| Compound | EC50 (μm)AF | EC50 (μm)IA | CC50 (μm)PMM | SIAF | SIIA | CC50 (μm) Hep G2 |
|---|---|---|---|---|---|---|
| 6 | 10.78 ± 0.80 | 4.57 ± 0.08 (P = 0.027)a | >500 | >46 | >109 | >160 |
| 7 | 13.12 ± 1.70 | 9.19 ± 0.68 | >500 | >38 | >54 | >160 |
| Miltefosine | 4.37 ± 0.11 | 13.30 ± 3.03a | 169.69 ± 5.07 | >38 | >12 | – |
AF, amastigote forms of L. amazonensis (LTB0016 strain) purified from animal lesions; IA, intracellular forms in peritoneal macrophages; SI, selective index.
EC50 is reported as mean and s.d. values.
Statistical analysis was performed with GraphPad prism v.9.1.2 by ordinary ANOVA test.
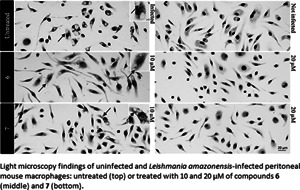
In the in vivo experiment, we observed a gradual increase in skin lesion size of mice treated with the vehicle alone, attaining 438.8 ± 42.05 mm3 at the endpoint (Fig. 2). Mice treated with compound 6 displayed similar lesions size (443.7 ± 47.76 mm3, P = 0.4798), whereas a slight increase was observed using 7 (550.9 ± 30.23 mm3, P = 0.2398), as compared to the vehicle-treated group. Mt™ achieved a 72% decrease in the size of skin lesions and significantly improved the clinical condition (P < 0.0001 (Fig. 2A and B). The qPCR standardization curves achieved all the desired efficiency and linearity coefficient criteria, ranging between 93.2–91.4% and 0.98–0.99% for the parasite kDNA amplification (Fig. S3A) and mice GAPDH target (Fig. S3B), respectively. At 31 dpi, qPCR readout expressed as DNA equivalents per mg of tissue (14) showed a 5–10-fold increase in the parasite load of mice treated with 6 (276 526 ± 40 519 eq. par mg−1 tissue, P = 0.0208) and 7 (153 765 ± 54 932 eq. par mg−1 tissue, P = 0.0829) as compared to the vehicle-treated group (27 753 ± 9539 eq. par mg−1 tissue) (Fig. 2B). Mt™ completely suppressed (99.99%) parasite load (0.256 ± 0.1022 eq. par mg−1 tissue, P = 0.0437). The qPCR showed that the hit compounds lacked in vivo efficacy, and it was corroborated by a qualitative light microscopy analysis of lesions imprints (Fig. 2C). The findings clearly showed that both 6- and 7-treated animal groups have high parasitaemia coupled with the presence of lymphomononuclear diffuse infiltrates and several parasite niches inside mononuclear host cells (Fig. 2).
Fig. 2.

Effect of 6 and 7 in CL experimental mouse models using BALB/c mice infected with Leishmania amazonensis. The graphics show: average lesion size during treatment (A), the correlation between parasite load by qPCR (black bars) and average lesion size measurements (grey bars) at 31 dpi (B), according to each experimental group. Light microscopy of lesion imprints of infected mice treated with vehicle and after administration of compounds 6 and 7 at 10 mg kg−1 q12h po (C). Arrow: intracellular parasites. Statistical analysis was performed with GraphPad prism v.9.1.2 by ANOVA test (95% CI). When P value ⩾0.05 = not significant (ns).
The lack of in vivo efficacy suggests low bioavailability of the compounds in mice. Low bioavailability is generally caused by impaired absorption or high clearance of compounds. Therefore, an in silico tool, pkCSM, was used to predict some critical absorption, distribution and metabolism parameters of 6 and 7, comparing to miltefosine (Gleeson, 2008; Pires et al., 2015). Compounds 6 and 7 do not violate Lipinski's rule. The cLog Ps of 6 and 7 are 2.97 and 2.13, respectively, while that of miltefosine is 5.68 (Table 2). Since miltefosine is zwitterionic, its relatively high cLog P is ideal for high intestinal absorption. Similarly, since compounds 6 and 7 have low molecular weights (324.34 and 297.27 g mol−1, respectively) and are neutral molecules at physiological pH with relatively low cLog P, sufficient intestinal adsorption was expected from oral gavage. The predicted high intestinal absorption further supports this in addition to the predicted moderate to high Caco-2 permeability (0.83 and 1.04, respectively) from in silico Absorption, Distribution, Metabolism, and Excretion (ADME) calculations. Oral gavage was selected for this study because oral administration is the preferred route for the clinical use of new antileishmanial drugs. The predicted volume of distribution (VDss) is high and ideal for tissue distribution for both compounds. However, the predicted unbound fraction for 6 is relatively low (5%) and ideal for 7 (21%). Both compounds are predicted as CYP3A4 substrates, which suggest significant first-pass hepatic clearance and low overall plasma and tissue concentration. Therefore, a comparative pharmacokinetics study (oral and intravenous) can provide insight into plasma concentrations and bioavailability of both compounds in addition to determining the appropriate dosage and structural changes necessary for in vivo efficacy. Also, in silico toxicological analysis of both compounds suggests probable mutagenicity (positive for Ames tests) and potential to inhibit hERG II and that 6 is potentially hepatotoxic. However, animals treated with compounds 6 and 7 did not show any observable side-effects. A cell proliferation assay using Hep G2 cells indicates that the compounds are not hepatotoxic at the tested concentrations (160–1.2 μm).
Table 2.
Predicted ADME properties of 6, 7 and miltefosine
| Compound | cLog P | Caco2-permeability | Intestinal absorptiona | VDssa | Fraction unbounda | CYP3A4 substrate |
|---|---|---|---|---|---|---|
| 6 | 2.97 | 0.83 | 90.23 | 1.28 | 0.05 | Yes |
| 7 | 2.13 | 1.043 | 90.03 | 0.81 | 0.21 | Yes |
| Miltefosine | 5.68 | 1.049 | 92.02 | 0.36 | 0.16 | Yes |
The predicted values are for humans.
Discussion
Leishmania amazonensis is one of the most common Leishmania species in Brazil, known to cause a wide spectrum of pathologies, including highly severe and diffuse CL (Lainson et al., 1994). Most CL patients live in impoverished communities with limited access to primary healthcare facilities (Okwor and Uzonna, 2016). The lack of access to primary healthcare typically means that the disease is usually at an advanced stage before medical intervention is sought, if available (Ruoti et al., 2013). In this study, we explored the in vitro and in vivo leishmanicidal effects of compounds derived from two nitroaromatics scaffolds (4-nitrophenylacetyl and 4-nitro-1H-imidazolyl). The in vitro data demonstrated that two 4-nitro-1H-imidazolyl compounds have acceptable selectivity indices (>50 for intracellular forms of L. amazonensis) as previously discussed by Caridha et al. (2019) as well as by Alcântara et al. (2018). Therefore, their antileishmanial activities were investigated further. The compounds were administered to BALB/c mouse models of CL for 14 days at a relatively low dose, starting at the onset of lesions (Godinho et al., 2012). The results showed that the in vitro potency of the hit compounds 6 and 7 did not translate to desired in vivo efficacy. The measurements of the mouse lesions (using a paquimeter) and the molecular analysis of animal parasitism (qPCR) demonstrated a lack of in vivo activity of both compounds, whereas Mt™ suppressed both parameters, as reported (Van Bocxlaer et al., 2019). The lack of in vivo efficacy of the tested compounds could be, at least in part, due to low bioavailability and/or high metabolic clearance. Nevertheless, as nitro-drugs are activated by Leishmania nitroreductases (NTR2) (Wyllie et al., 2016), it is possible that 6 and 7 may be also activated in a similar way, and thus, the in vitro leishmanicidal efficacy supports their use as templates for nitroimidazole-based antileishmanial drug discovery programmes focusing on analogues that have the appropriate Target Product Profile (TPP) for new CL drugs, as recommended (Drugs for Neglected Diseases Initiative, 2020).
Acknowledgements
The authors acknowledge the Fortalecimento dos Programas de Gestão Estratégica de Pesquisa da Fiocruz Rede de Plataformas Fiocruz (VPPLR-001-Fio 14) and Programa de Excelência Acadêmica (PROEX) from CAPES. They appreciate the excellent technical contribution of Roberson Donola Girão, Dr Ana Lia Mazzetti and Dr Cristiane França da Silva (Laboratory of Cellular Biology/IOC/Fiocruz) and Kingsley Bimpeh (Jackson State University).
Supplementary material
The supplementary material for this article can be found at https://doi.org/10.1017/S0031182021002079
Financial support
The current study was supported by grants from Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ), Conselho Nacional Desenvolvimento científico e Tecnológico (CNPq), Coordenação de Aperfeiçoamento de Pessoal de Nível Superior (CAPES) and Fundação Oswaldo Cruz PAEF/CNPq/Fiocruz. MNCS and OCM are research fellows of CNPq and CNE and JCNE researchers. The work at Jackson State University was supported, in part, by the US National Institutes of Health (SC3GM122629 and G12MD007581).
Ethical standards
All procedures were carried out in accordance with the guidelines established by the FIOCRUZ Committee of Ethics for the Use of Animals (CEUA L038/2017).
Conflict of interest
None.
References
- Alcântara LM, Ferreira TC, Gadelha FR and Miguel DC (2018) Challenges in drug discovery targeting TriTryp diseases with an emphasis on leishmaniasis. International Journal for Parasitology: Drugs and Drug Resistance 8, 430–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvar J, Vélez ID, Bern C, Herrero M, Desjeux P, Cano J, Jannin J and de Boer M (2012) Leishmaniasis worldwide and global estimates of its incidence. PLoS ONE 7, e35671. doi: 10.1371/journal.pone.0035671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baileyid F, Mondragon-Shem K, Haines LR, Olabi A, Alorfi A, Ruiz-Postigo JA, Alvar J, Hotez P, Adams ER, Vélez ID, Al-Salem W, Eaton J, Acosta-Serrano Á and Molyneux DH (2019) Cutaneous leishmaniasis and co-morbid major depressive disorder: a systematic review with burden estimates. PLoS Neglected Tropical Diseases 13, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso Santos C, Meuser Batista M, Inam Ullah A, Rama Krishna Reddy T and Soeiro MdNC (2021) Drug screening using shape-based virtual screening and in vitro experimental models of cutaneous leishmaniasis. Parasitology 148, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caridha D, Vesely B, Van Bocxlaer K, Arana B, Mowbray CE, Rafati S, Uliana S, Reguera R, Kreishman-Deitrick M, Sciotti R and Buffet P (2019) Route map for the discovery and pre-clinical development of new drugs and treatments for cutaneous leishmaniasis. International Journal for Parasitology: Drugs and Drug Resistance 11, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries HJC, Reedijk SH and Schallig HDFH (2015) Cutaneous leishmaniasis: recent developments in diagnosis and management. American Journal of Clinical Dermatology 16, 99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drugs for Neglected Diseases Initiative (2020) Target product profile for cutaneous leishmaniasis. Retrieved from DNDi website: https://dndi.org/diseases/cutaneous-leishmaniasis/target-product-profile/ (accessed 30 January 2021).
- Feitosa LM, da Silva ER, Hoelz LVB, Souza DL, Come JAASS, Cardoso-Santos C, Batista MM, Soeiro MDNC, Boechat N and Pinheiro LCS (2019) New pyrazolopyrimidine derivatives as Leishmania amazonensis arginase inhibitors. Bioorganic and Medicinal Chemistry 27, 3061–3069. doi: 10.1016/j.bmc.2019.05.026 [DOI] [PubMed] [Google Scholar]
- Gleeson MP (2008) Generation of a set of simple, interpretable ADMET rules of thumb. Journal of Medicinal Chemistry 51, 817–834. [DOI] [PubMed] [Google Scholar]
- Godinho JLP, Simas-Rodrigues C, Silva R, Ürmenyi TP, De Souza W and Rodrigues JCF (2012) Efficacy of miltefosine treatment in Leishmania amazonensis-infected BALB/c mice. International Journal of Antimicrobial Agents 39, 326–331. [DOI] [PubMed] [Google Scholar]
- Katsuno K, Burrows JN, Duncan K, van Huijsduijnen RH, Kaneko T, Kita K, Mowbray CE, Schmatz D, Warner P and Slingsby BT (2015) Hit and lead criteria in drug discovery for infectious diseases of the developing world. Nature Reviews Drug Discovery 14, 751–758. doi: 10.1038/nrd4683 [DOI] [PubMed] [Google Scholar]
- Lainson R, Shaw JJ, Silveira FT, de Souza AAA, Braga RR and Ishikawa EAY (1994) The dermal leishmaniases of Brazil, with special reference to the eco-epidemiology of the disease in Amazonia. Memorias do Instituto Oswaldo Cruz 89, 435–443. [DOI] [PubMed] [Google Scholar]
- Martins ALGP, Barreto JA, Lauris JRP and Martins ACGP (2014) American tegumentary leishmaniasis: correlations among immunological, histopathological and clinical parameters. Anais Brasileiros de Dermatologia 89, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikus J and Steverding D (2000) A simple colorimetric method to screen drug cytotoxicity against Leishmania using the dye Alamar Blue®. Parasitology International 48, 265–269. [DOI] [PubMed] [Google Scholar]
- Nagle A, Biggart A, Be C, Srinivas H, Hein A, Caridha D, Sciotti RJ, Pybus B, Kreishman-Deitrick M, Bursulaya B, Lai YH, Gao MY, Liang F, Mathison CJN, Liu X, Yeh V, Smith J, Lerario I, Xie Y, Chianelli D, Gibney M, Berman A, Chen YL, Jiricek J, Davis LC, Liu X, Ballard J, Khare S, Eggimann FK, Luneau A, Groessl T, Shapiro M, Richmond W, Johnson K, Rudewicz PJ, Rao SPS, Thompson C, Tuntland T, Spraggon G, Glynne RJ, Supek F, Wiesmann C and Molteni V (2020) Discovery and characterization of clinical candidate LXE408 as a kinetoplastid-selective proteasome inhibitor for the treatment of leishmaniases. Journal of Medicinal Chemistry 63, 10773–10781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okwor I and Uzonna J (2016) Social and economic burden of human leishmaniasis. American Journal of Tropical Medicine and Hygiene 94, 489–493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires DEV, Blundell TL and Ascher DB (2015) pkCSM: predicting small-molecule pharmacokinetic and toxicity properties using graph-based signatures. Journal of Medicinal Chemistry 58, 4066–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro-Romão RP, Saavedra AF, Da-Cruz AM, Pinto EF and Moreira OC (2016) Development of real-time PCR assays for evaluation of immune response and parasite load in golden hamster (Mesocricetus auratus) infected by Leishmania (Viannia) braziliensis. Parasites and Vectors 9, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanha AJ, de Castro SL, Soeiro M, Lannes-Vieira J, Ribeiro I, Talvani A, Bourdin B, Blum B, Olivieri B, Zani C, Spadafora C, Chiari E, Chatelain E, Chaves G, Calzada JE, Bustamante JM, Freitas-Junior LH, Romero LI, Bahia MT, Lotrowska M, Soares M, Andrade SG, Armstrong T, Degrave W and Andrade ZdA (2010) In vitro and in vivo experimental models for drug screening and development for Chagas disease. Memorias do Instituto Oswaldo Cruz 105, 233–238. [DOI] [PubMed] [Google Scholar]
- Ruoti M, Oddone R, Lampert N, Orué E, Miles MA, Alexander N, Rehman AM, Njord R, Shu S, Brice S, Sinclair B and Krentel A (2013) Mucocutaneous leishmaniasis: knowledge, attitudes, and practices among Paraguayan communities, patients, and health professionals. Journal of Tropical Medicine, ID 538629. doi: 10.1155/2013/538629 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CC, Lionel JR, Peres RB, Batista MM, da Silva PB, de Oliveira GM, da Silva CF, Batista DGJ, Souza SMO, Andrade CH, Neves BJ, Braga RC, Patrick DA, Bakunova SM, Tidwell RR and Soeiro MdNC (2018) In vitro, in silico, and in vivo analyses of novel aromatic amidines against Trypanosoma cruzi. Antimicrobial Agents and Chemotherapy 62, e02205-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos CC, Zhang H, Batista MM, de Oliveira GM, Demarque KC, da Silva-Gomes NL, Moreira OC, Ogungbe IV and Soeiro MdNC (2020) In vitro and in vivo evaluation of an adamantyl-based phenyl sulfonyl acetamide against cutaneous leishmaniasis models of Leishmania amazonensis. Antimicrobial Agents and Chemotherapy 64, e01188-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Bocxlaer K, Caridha D, Black C, Vesely B, Leed S, Sciotti RJ, Wijnant GJ, Yardley V, Braillard S, Mowbray CE, Ioset JR and Croft SL (2019) Novel benzoxaborole, nitroimidazole and aminopyrazoles with activity against experimental cutaneous leishmaniasis. International Journal for Parasitology: Drugs and Drug Resistance 11, 129–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisz G (2006) Making medical history. Bulletin of the History of Medicine 80, 153–159. [Google Scholar]
- World Health Organization, Regional Office for the Eastern Mediterranean (2020) Infectious agent (s) WHO case definition incubation period communicability period epidemiology and risk factors Situation in countries affected by crisis in Syria. pp. 1–7.
- Wyllie S, Roberts AJ, Norval S, Patterson S, Foth BJ, Berriman M, Read KD and Fairlamb AH (2016) Activation of bicyclic nitro-drugs by a novel nitroreductase (NTR2) in Leishmania. PLoS Pathogens 12, e1005971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Collins J, Nyamwihura R, Ware S, Kaiser M and Ogungbe IV (2018) Discovery of a quinoline-based phenyl sulfone derivative as an antitrypanosomal agent. Bioorganic and Medicinal Chemistry Letters 28, 1647–1651. [DOI] [PMC free article] [PubMed] [Google Scholar]