

Abstract
Scope
Fucosylated human milk oligosaccharides (fHMOs) are metabolized by Bifidobacterium infantis and promote syntrophic interactions between microbiota that colonize the infant gut. The role of fHMO structure on syntrophic interactions and net microbiome function is not yet fully understood.
Methods and Results
Metabolite production and microbial populations were tracked during mono- and co-culture fermentations of 2′fucosyllactose (2′FL) and difucosyllactose (DFL) by two B. infantis strains and Eubacterium hallii. This was also conducted in an in vitro modeled microbiome supplemented by B. infantis and/or E. hallii. Metabolites were quantified by high performance liquid chromatography. Total B. infantis and E. hallii populations were quantified through qRT-PCR and community composition through 16S amplicon sequencing. Differential metabolism of 2′FL and DFL by B. infantis strains gave rise to strain- and fHMO structure-specific syntrophy with E. hallii. Within the modeled microbial community, fHMO structure did not strongly alter metabolite production in aggregate, potentially due to functional redundancy within the modeled community. In contrast, community composition was dependent on fHMO structure.
Conclusion
Whereas short chain fatty acid production is not significantly altered by the specific fHMO structure introduced to the modeled community, fHMOs influences the composition of the gut microbiome.
Keywords: Bifidobacterium, Eubacterium hallii, fucose, human milk oligosaccharide, microbiome, trophic interactions
Graphical Abstract

Human milk oligosaccharides (HMO) are metabolized by bifidobacteria in the infant gut, and the impact of specific HMO structures on interactions between bifidobacteria and microbiota are incompletely characterized. To investigate this, in vitro approaches including modeled microbiomes determined the influence of HMO structure on interactions between Bifidobacterium infantis and Eubacterium hallii within a modeled infant gut microbiome. It was determined that specific HMO structure dictated the composition of the modeled community, yet the microbiome function remained similar regardless of HMO.
1. Introduction
Human milk contains host-indigestible oligosaccharides that shift the composition of the breastfed infant gut microbiome towards specific beneficial microbial populations, including enriching for bifidobacteria.[1–5] These human milk oligosaccharides (HMOs) generally consist of a neutral parent structure composed of glucose, galactose, and N-acetylglucosamine, giving rise to myriad variants based on degree of polymerization, branching, and stereochemistry. These parent structures are often further diversified through the addition of terminal sialic acid or fucose.[3,6] Fucosylated HMOs (fHMOs) are highly abundant in secretor women and are dominated by 2′fucosyllactose (2′FL), which can account for up to 30% of all HMOs.[6,7]
Bifidobacterium longum subsp. infantis (Bifidobacterium infantis) is often a predominant colonizer of the breastfed infant gut where it metabolizes fHMOs such as 2′FL.[8–11] Bifidobacterial metabolism of fHMOs results in the production of 1,2-propanediol (1,2-PD) in addition to organic acids such as acetic acid and lactic acid, as well as formic acid and ethanol that are produced from carbohydrate metabolism. These end-products contribute to infant development and homeostasis, and are available for syntrophic interactions with other gut microbes (i.e. cross-feeding). Of particular relevance, 1,2-PD can be metabolized by several gut commensals to result in propionate,[12–15] which supports host gluconeogenesis and may be protective against pathogens.[16–18] Similarly, acetic acid and lactic acid can be metabolized to butyrate, which suppresses inflammation and is an energy source for colonocytes.[16,19,20] In addition to inter-genus relationships, fHMOs could mediate interactions between bifidobacterial taxa.[21–23]
Eubacterium hallii is postulated as one of the first butyrate producers to colonize in the infant gut, although the timing of emergence is not yet fully understood. It is, however, generally accepted that E. hallii populations increase through weaning as solid food is introduced and persists throughout adulthood.[13,24–26] E. hallii can metabolize 1,2-PD generated during bifidobacterial fucose utilization to produce propionate. E. hallii secretes butyrate produced from exogenous acetic and lactic acid substrates. Recently, E. hallii was reclassified based on 16S rRNA phylogenic analysis to Anaerobutyricum hallii within the family Lachnospiraceae.[27] Unlike bifidobacteria, E. hallii cannot metabolize complex oligosaccharides such as HMOs.[14,15,28] Furthermore, cross-feeding of E. hallii on 1,2-PD produced by bifidobacterial metabolism of 2′FL has been previously observed in pure culture,[13] although this syntrophic interaction has not been characterized in a competitive environment. A schematic of metabolic pathways of both E. hallii and B. infantis during syntrophic 2′FL metabolism is depicted in Fig 1. Since E. hallii can produce both propionate and butyrate from bifidobacterial metabolic end-products, the relative abundance of microbes capable of producing propionate from 1,2-PD such as Rosburia spp., Ruminococcus spp., and Lactobacillus spp.,[15,29] may shift E. hallii metabolism to favor butyrate production as a means of relieving substrate competition. This study investigates the role of fHMO structure on B. infantis and E. hallii syntrophy within a modeled microbial community. The experimental system quantifies B. infantis growth kinetics, net secreted end-products, B. infantis and E. hallii populations, and the phylogenetic composition of the community throughout fermentation. We hypothesize that fHMO structure mediates syntrophic interactions between the infant gut commensals B. infantis and E. hallii to directly and indirectly influence the composition and function of a modeled microbial community.
Figure 1.

Schematic of B. infantis and E. hallii metabolic pathways to produce short chain fatty acids during bifidobacterial 2′FL metabolism.
2. Experimental Section
Sample collection and processing
A discarded stool sample was collected from a healthy one-year-old infant who had not received antibiotics in the previous three months and was consuming both solid foods and human milk. The diaper containing freshly voided feces was frozen at −20°C for transport and subsequently thawed to 4°C upon receipt. Thawed feces were immediately scraped from the diaper and transferred in ~8 g aliquots to sterile tubes and stored at −80°C until subsequent use.
Bacterial strains and propagation
B. longum subsp. infantis strain ATCC 15697T (B. infantisT) and E. hallii strain ATCC 27751T (E. halliiT) were used in this study. A B. longum subsp. infantis strain was isolated from the infant stool that seeded the modeled community (UMA0169, intrinsic or B. infantisi). B. infantis propagation was performed in De Man Rogosa-Sharpe medium (MRS; BD Difco) supplemented with 0.5 gL−1 L-cysteine hydrochloride (Sigma-Aldrich). E. hallii propagation was performed in anoxic media pre-reduced with 100% O2-free CO2 following a vacuum-vortex method.[30] Plating and roll tube enumeration[31] were performed on reinforced clostridial agar (MillliporeSigma) with 2% (wt/vol) agar. Infant colon medium (ICM) for the modeled microbiome was modified from Cinquin et al [32] and contained (gL−1): yeast extract (2.5), Bacto tryptone (0.5), peptone (0.5), casein hydrolysate (8.4), soluble starch (0.6), NaHCO3 (1.5), FeSO4·7H2O (5 mg), KCl (4.5), NaCl (4.5), MgSO4·7H2O (1.25), CaCl2·6H2O (0.225), hemin (10mg), L-cysteine hydrochloride (1), K2HPO4 (0.5), bile salts (50 mg). Carbohydrate (10 gL−1) and vitamin solution (100 μL per L) were dissolved into a portion of autoclaved media (15 min, 121°C) and subsequently filter- sterilized. The vitamin solution was adapted from Gibson and Wang [33] to contain (mgL−1): biotin (20), calcium pantothenate (100), nicotinamide (50), p-aminobenzoic acid (50), thiamine hydrochloride (40), cobalamin (5), vitamin K1 (10 μL). All cultures were incubated at 37°C under anaerobic conditions (7% H2, 10% CO2, N2 balance) (Coy Lab). A schematic of the various culture work performed in this study is given in Supplemental Fig S1.
Isolation of B. infantis from infant stool
Infant stool was thawed and subsequently homogenized in peptone water, and the resultant 1% (vol/vol) fecal slurry used to inoculate Bifidobacterium-selective MRS broth supplemented with 0.5gL−1 L-cysteine hydrochloride (Sigma-Aldrich) and 100 mgL−1 mupirocin[34,35] (PanReac Applichem) and incubated overnight at 37°C under anaerobic conditions. 100 μL overnight culture was plated on Bifidobacterium- selective MRS agar with presumptive colonies selected and re-plated. DNA was extracted from overnight cultures using Qiagen DNeasy UltraClean Microbial kit (Qiagen) according to manufacturer instructions using a bead beating step to disrupt cells. Powerlyzer tubes were beat at 6 m/s for 30 sec 2X using a FastPrep 24 bead beater (MP Biomedicals) with an incubation step on ice for 3 min between each step. Amplification of the 16S-internal transcribed spacer (ITS) sequence was performed with previously developed primers BIF-specific, 23S_bif, and the Sanger sequencing primers bif_secF and bif_secR (sequences given in Table 1).[36] PCR products were purified from agarose gel and using the Zymoclean Gel DNA Recovery Kit (Zymo Research) according to the manufacturer’s instructions and subsequently sequenced (Genewiz).
Table 1:
Primers used in this study.
| Primer name | Primer sequence (5’−3’) | Target gene | Primer designed |
|---|---|---|---|
| BIF-specific | GGT GTG AAA GTC CAT CGC T | 16S-internal transcribed spacer | Turroni et al., 2009[36] |
| 23S_bif | GTC TGC CAA GGC ATC CAC CA | ||
| bif_secF | GGT GTG AAA GTC CAT CGC T | ||
| bif-secR | CAT GCC CCT ACG TCC AG | ||
| Ehal_2FP | ATG CAA GTC GAA CGA AGC AC | 16S rRNA | This study |
| Ehal_2RP | ACC CTG CCA ACC AGC TAA TC | ||
| NanH2_2FP | TGG CCG TGT GAT GCT GAA | Sialidase | Sela et al., 2011[38] |
| NanH2_2RP | CCG GGA GAT GGC GAC ATA |
Microplate bacterial growth assay
B. infantis growth phenotypes were analyzed in a 96-well format using a PowerWave HT Microplate Spectrophotometer (BioTek). Overnight cultures were inoculated 1% (vol/vol) in modified MRS media (mMRS) that contains a defined carbohydrate substrate without acetate and Tween80. Carbohydrates include lactose (Sigma-Aldrich), 2′fucosyllactose (2′FL), 3′fucosyllactose (3′FL), and difucosyllactose (DFL) (provided by DSM) at 20 gL−1 as the sole carbohydrate source(s). The growth assay was conducted anaerobically at 37°C by shaking for 5 sec and assessing OD600 nm at 15-minute intervals until bacterial growth reached stationary phase. Each carbohydrate source was evaluated in biological triplicate with three technical replicates. Inoculated mMRS without carbohydrate served as the negative control, and inoculated mMRS with 20 gL−1 lactose served as the positive control. Bacterial growth kinetics were calculated using Wolfram Mathematica with the equation described by Dai et al,
| (1) |
where corresponds to the adjusted optical density, the optical density at stationary phase, the growth rate, and the time to reach maximum growth rate.[37] After reaching stationary phase, technical replicates were pooled, pelleted, and the resulting cell-free spent growth medium reserved for extracellular metabolite analysis. Growth kinetics were subjected to one-way analysis of variance and Tukey’s HSD test for multiple comparisons between carbohydrate source and B. infantis strain.
in vitro batch culture modeled fecal community fermentation
Coculture and modeled fecal community fermentation was performed in ICM within a DASGIP bioreactor system (Eppendorf). Carbohydrate-free ICM for in vitro batch culture fermentations was autoclaved without degassing and cooled prior to the addition of filter-sterilized vitamin solution and filling of sterilized 1L vessels. Filled vessels were degassed by sparging overnight with CO2 (100% O2- free) at 37°C in a DASGIP Bioblock under constant agitation (100 rpm). Immediately prior to inoculation, carbohydrate (10 gL−1) was added to 20 mL aliquots of media that were aseptically removed, filter- sterilized, and returned to their respective vessels. B. infantis and E. hallii inocula were conditioned on pre-reduced ICM containing lactose (10 gL−1) for 12 and 24 hours, respectively. Cultures were pelleted, washed, and resuspended in carbohydrate-free pre-reduced ICM to minimize carryover of residual sugars or metabolites. Vessels were inoculated to 1% (vol/vol) with each strain. The inoculum volume was constant for all vessels regardless of bacterial supplementation using carbohydrate-free pre-reduced ICM as balance. Infant stool was thawed to 4°C, suspended in carbohydrate-free pre-reduced ICM at 50 gL−1 in a sterile stomacher bag (Thermo Fisher Scientific), and homogenized for 1 minute. Vessels were inoculated with the fecal slurry to 5 gL-1.
Fermentations proceeded for up to 52 hours at 37°C and pH 6.8 with constant sparging of 100% O2-free CO2 and agitation (100 rmp). pH was maintained through the addition of either 2 M HCl or 4 N NaOH as controlled by pH sensors in each vessel. Throughout fermentation, (1 cm path length; Eppendorf), redox potential (Metler Toledo), and pH (EasyFerm Plus) were monitored. Throughout fermentation, 3 mL (4 mL for samples before 10 hours) samples were removed aseptically from bioreactor vessels and divided into 1 mL aliquots. Cells were pelleted, and supernatants were reserved separately with both stored at −80°C until analysis. Sampling ports were flushed several times prior to sample collection.
Characterization of microbial metabolic end-products
Extracellular metabolites were quantitated by HPLC. Cell-free spent growth medium was thawed and filtered through a 0.22-μm filter (Sartorius) and stored at 4°C during analysis. Metabolite (2′fucosyllactose, 3′fucosyllactose, difucosyllactose, fucose, lactose, glucose, galactose, 1,2-propanediol, acetic acid, lactic, acid, formic acid, butyric acid, propionic acid, and ethanol) concentrations were quantified using an 1100 series HPLC system equipped with a refractive-index detector (Agilent Technologies). Separation was performed using an Aminex HPX-87H column (ID 7.8 mm by 300 mm; Bio-Rad Laboratories) at 30°C in a mobile phase of 5 mM H2SO4 with a flow rate of 0.6 mL/min and an injection volume of 20 μL. Analytical standards of 2′FL, 3′FL, DFL, LNT, LNnT (DSM), fucose, lactose, glucose, several organic acids (Sigma-Aldrich), and 1,2-propanediol (Alfa Aesar) were used to generate a standard curve from six concentrations (0.5, 1, 5, 10, 20, and 40 mM). Each measurement was performed in duplicate. Metabolite concentrations were subjected to one-way analysis of variance and Tukey’s HSD test for multiple comparisons between carbohydrate source, pure culture and co-culture B. infantis strain, and in vitro batch culture B. infantis and/or E. hallii supplementation.
DNA extraction from in vitro batch culture fermentation
Total DNA from pure and coculture pellets was extracted using Qiagen DNeasy UltraClean Microbial kit according to manufacturer instructions with a bead beating step to disrupt cell envelopes. Powerlyzer tubes were subjected to bead-beating at 6 m/s for 30 sec 2X on a MP Biomedicals FastPrep 24 bead beater with tubes cooled on ice for 3 min between steps. Total DNA from modeled fecal community pellets was extracted using Zymo Research Quick-DNA Fecal/Soil Microbe MiniPrep kit according to manufacturer instructions using a bead beating step to disrupt cells. BashingBead tubes were beat at 6 m/s for 1 min 3X using a FastPrep 24 bead beater and tubes were cooled on ice for 3 min between each bead- beating step. DNA concentration (between 5 and 750 ng μL−1 from mono- and co-culture pellets and 5 and 25 ng μL−1 for modeled fecal community pellets) and quality was measured by a NanoDrop spectrophotometer and stored at −80°C until analysis.
Primer design for qRT-PCR quantification of E. hallii and B. infantis
E. hallii (ATCC 27751) specific 16S rRNA gene primers were designed using NCBI/BLAST (https://blast.ncbi.nlm.nih.gov/Blast.cgi). To test primer pair specificity, each pair was compared in silico against the nucleotide collection database from NCBI using Primer BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast). Potential primer dimers and hairpins were evaluated using OligoAnalyzer™ Tool (IDT). B. infantis sialidase gene specific primers were used as described previously.[38] Primers were synthesized by Invitrogen (Thermo Fisher Scientific). The primer concentration and PCR conditions were optimized for each primer set. The list of primers used in this study is summarized in Table 1.
B. infantis and E. hallii quantification determined by qRT-PCR
Quantitative real time PCR (qRT-PCR) was performed targeting the E. halliiT (ATCC 27751) 16S rRNA gene and B. infantisT (ATCC 15697) sialidase gene with specific primers (Table 1). Genomic DNA from E. halliiT and B. infantisT was used as the qRT-PCR standard for quantification. An overnight culture of B. infantisT and E. halliiT (109 and 108 CFU/ml respectively) was plated and colonies were counted on solid media following serial dilutions. E. halliiT serial dilutions were plated using reinforced clostridial agar (2% w/v agar) (MilliporeSigma,) in roll tubes.[31] Genomic DNA was extracted from 1 ml of overnight culture from each reference strain (DNeasy UltraClean Microbial Kit, Qiagen). Serial 10-fold dilutions of genomic DNA (107 - 101 genome copies/reaction) were prepared using qRT-PCR grade water (Invitrogen). For quantitative analysis bacterial genome copy number was computed as follows:
| (2) |
Reaction mixtures were prepared to a total volume of 20μL, with 10 μL PowerUp SYBR Green 2X Master Mix (Applied Biosystems), 0.5 μL of each primer (10 μM), 7.0 μL nuclease free water, and 2 μL of DNA template (2.5ng/μL). Thermocycling was performed on an ABI 7500 Fast Real-Time PCR System (Applied Biosystems, Life technologies) with the following protocol: 50°C for 2 minutes, 95°C for 10 minutes followed by 40 cycles of 95°C for 30 seconds, 55°C for 60 seconds, 72°C for 60 seconds, and final extension at 72°C for 7 minutes. A post-amplification melting curve was generated with a temperature gradient from 60°C to 95°C to confirm product specific amplification. Standards were included on each plate along with technical replicates for every dilution and no template controls in duplicate. A total of two independent runs on separate plates were analyzed and average values were calculated. Based on the equation for the linear regression line (R2 > 0.99, E > 94%) initial quantity in the samples were determined. The PCR efficiency (E) was calculated by the slope of the standard curve as follows:
| (3) |
16S amplicon sequencing library construction
Modeled fecal community DNA was diluted to 5 ng uL−1 and 16S libraries were prepared with 30 ng total DNA per sample using Quick-16S Plus NGS Library Prep kit (V3-V4) (Zymo) following manufacturer instructions. Library size (611 bp) was measured by DNA analysis ScreenTape Assay on a Tape Station 2200 (Agilent Technologies). Sequencing was performed on an Illumina MiSeq platform (paired-end, MiSeq Reagent Kit V3 (600 cycle), 20% PhiX) (Illumina).
16S sequence analysis
Demultiplexed fastQ files were imported into the QIIME 2 pipeline (version 2018.8).[39] The DADA2 plugin was used to denoise reads and remove chimeras using the consensus method.[40] Forward and reverse reads were trimmed at positions 17, and 21 respectively, and truncated at position 251. All other parameters were set to default. Samples from 2′FL and DFL modeled fecal community fermentations were rarefied to a sampling depth of 48,000 and 45,000 respectively prior to alpha and beta diversity analyses. The contribution of exogenous bacteria supplementation to overall bacterial community structure was analyzed using principal coordinate analysis (PCoA) with matrices for ordinations computed using weighted UniFrac distances.[41] The phylogeny was inferred using the align-to-tree-mafft- fasttree pipeline in QIIME2. Taxonomy was assigned with the pre-trained naïve Bayesian classifier based on the SILVA reference database v 132[42] using the q2-feature-classifier plugin.[43,44]
Alpha and beta diversity metrics
We reported the relative abundances of microbial taxa at the genus level (Fig 5). Statistical analyses of alpha diversity were conducted in QIIME 2 [39] using the Kruskal–Wallis test. Alpha diversity results generated in QIIME 2 were plotted in R[45] and RStudio version 2022.07.1[46] with post hoc pairwise comparisons to examine differences in alpha diversity between supplementation regimes. Post hoc test P values were adjusted for multiple comparisons using the Benjamini–Hochberg method[47]. Statistical analyses of beta diversity were conducted in QIIME 2 using the permutational multivariate analysis of variance (PERMANOVA) test with 999 permutations[48]. PCoA plots of beta diversity with 95% confidence intervals (stat_ellipse) were generated using the ggplot2[49], tidyverse [43], and qiime2R [44] packages in R. In addition, alpha diversity plots used the ggpubr[50] package in R. All further visualization was done using OriginPro (version 2022, OriginLab Corporation).
Figure 5.
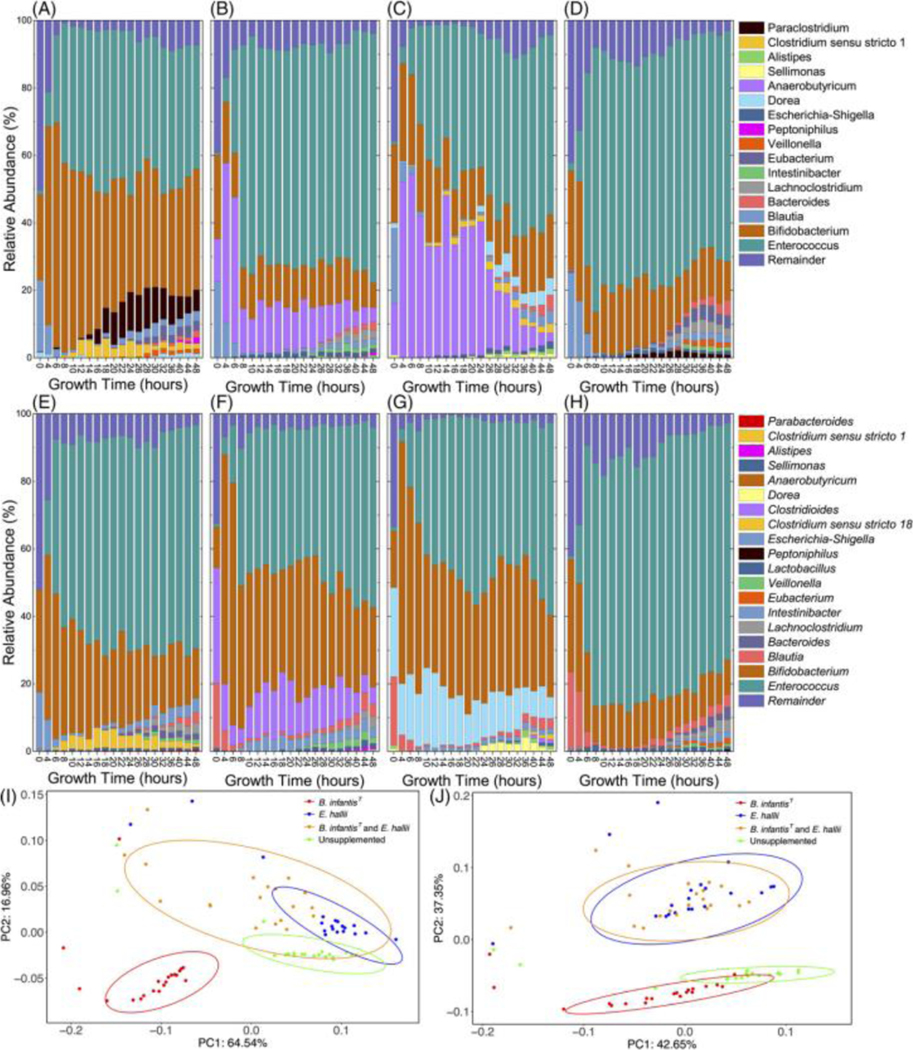
Relative abundance of the top 10 most abundant taxa from 16S analysis of modeled fecal community fermentations of 2′FL (A-D) and DFL (E-H) supplemented with B. infantisT (A, E), E. halliiT (B, F), and B. infantisT & E. halliiT (C, G), or unsupplemented (D, H). Remainder was calculated as the sum of all other taxa abundance. Principal-coordinate analysis based on weighted UniFrac distances (I, J). Each point represents a sample colored by supplementation status of modeled fecal community fermentations of 2′FL (I) or DFL (J). There are significant differences across supplementation regimes based on PERMANOVA (2′FL: F = 27.2889, P = 0.001 and DFL: F = 20.763, P = 0.001). Ellipses indicate a 95% confidence interval.
Data availability statement
Raw sequencing reads from the 16S amplicon sequencing are available via the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) under BioProject PRJNA897896 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA897896).
3. Results and Discussion
Intrinsic B. infantisi strain metabolizes fHMOs more efficiently than the type strain
Given that bacterial strains of the same species can exhibit divergent physiology and that multiple strains can coexist within a specific niche,[51–54] the metabolic phenotypes of both B. infantis strains were investigated. B. infantis strains were grown on 2′FL and DFL in pure culture to determine the variation in metabolite production and growth phenotype between the type strain (B. infantisT, ATCC 15697) and the endogenous strain present in the infant stool used to seed the bioreactor, herein termed intrinsic strain (B. infantisi, UMA0169). These strains exhibited growth on both 2′FL and DFL as sole carbohydrate sources (Supplemental Table S1 and Fig S2). The intrinsic strain B. infantisi exhibits higher growth rates and increased biomass as characterized by asymptotic OD600 than the type strain B. infantisT during fermentation of both 2′FL () and DFL () (Supplemental Table S1). Metabolic efficiency is described by several factors including biomass, growth rate, and metabolite production as it cannot be directly measured and is dependent on the culture environment and microbial growth regime.[55,56] A more predictive measure of metabolic efficiency is provided by ratios of metabolic end-products. The theoretical yield of bifidobacterial metabolism results in the secretion of 3 moles of acetic acid to 2 moles of lactic acid for every 2 moles of consumed hexose through the fructose-6- phosphate phosphoketolase (F6PPK) pathway.[57] An acetic acid:lactic acid ratio that is further from the theoretical yield 1.5 may indicate that cellular metabolism has shifted to convert pyruvate to formic acid and acetic acid at the expense of lactic acid.[57–59] Consequently, the formic acid:acetic acid ratio is a measure of metabolic efficiency with a lower ratio indicating a more efficient metabolism. These ratios are modified by metabolism of fucose outside the F6PPK pathway, which results in the production of 1 mole 1,2-PD, 1 mole lactic acid, and 1 mole pyruvate per mole of consumed fucose.[8,9,11,60] Taking the F6PPK and fucose metabolism pathways into consideration, the theoretical acetic acid:lactic acid ratio is effectively reduced by every added fucose, although the exact ratio depends on the metabolic fate of the pyruvate intermediate.
B. infantisi isolated from infant feces used to seed the bioreactor exhibits more efficient metabolism of both 2′FL and DFL as measured by significantly lower acetic acid:lactic acid and formic acid:lactic acid ratios. B. infantisi exhibits an acetic acid:lactic acid ratio of 2.14 and 1.77 during metabolism of 2′FL and DFL, respectively (Supplemental Table S1). These ratios are significantly lower than those of the type strain B. infantisT (3.20, p ≤ 0.01 and 3.80, p ≤ 0.00001 respectively, Supplemental Table S1) suggesting that the intrinsic strain more efficiently metabolizes both 2′FL and DFL under experimental conditions. Drastically reduced formic acid:acetic acid ratios (0.06 and 0.08 for 2′FL and DFL, respectively) further support this claim. Despite the seemingly more efficient metabolism of both 2′FL and DFL, the intrinsic strain produces significantly less 1,2-PD as compared to B. infantisT (p ≤ 0.0001 for both net and biomass-normalized 1,2-PD production during 2′FL and DFL fermentation, Supplemental Table S1). The difference in 1,2-PD production is particularly evident when accounting for biomass accumulation by normalizing 1,2-PD concentration to asymptotic (p ≤ 0.00001 for both 2′FL and DFL fermentation). Assuming that fucose is fully metabolized to 1,2-PD and associated end-products, the theoretical ratio of consumed fucose:secreted1,2-PD is hypothesized to be 1. Thus, a ratio of consumed HMO:secreted 1,2-PD can be predicted for fHMOs based on degrees of fucosylation. Singly fucosylated 2′FL would give a ratio of 1 and the di-fucosylated DFL would have a ratio of 0.5 (i.e. 1 mole DFL promotes secretion of 2 moles 1,2-PD). 2′FL and DFL fermentation by B. infantisi resulted in ratios of 2.2 and 2.3, respectively, indicating that not all fHMO-derived fucose was metabolized to 1,2-PD. The type strain B. infantisT exhibited ratios of 0.92 and 0.65 for 2′FL and DFL fermentation (Supplemental Table S1), suggesting that the type strain metabolizes fHMO-derived fucose more completely than the intrinsic strain. This agrees with the intrinsic strain’s decreased acetic acid:lactic acid ratio, as increased fucose metabolism would decrease overall acetic acid:lactic acid.[8,9] A fraction of the unmetabolized fucose may be excreted, as low amounts of fucose were found in the spent growth medium (Supplemental Table S1) but the remaining fucose is not accounted for.
E. hallii syntrophic interactions with B. infantis is dependent on 2′FL or DFL utilization phenotypes
To investigate the role of strain-dependent bifidobacterial 2′FL and DFL metabolism on syntrophic interactions between B. infantis and E. hallii, both B. infantisi and B. infantisT were grown in coculture with E. hallii ATCC27751T on 2′FL or DFL as the sole carbohydrate sources in an in vitro batch culture fermentation. qRT-PCR was used to monitor growth of B. infantis strains with E. halliiT independently. Fermentation of 2′FL by either B. infantis strain in the presence of E. halliiT resulted in metabolic end- products that are consistent with strain-dependent bifidobacterial 2′FL metabolism (Fig 2 and Supplemental Table S2). B. infantis fHMO utilization phenotypes were consistent between the microplate assay and bioreactor in vitro batch culture environments. B. infantisT 2′FL fermentation in the batch culture fermentation environment produced concentrations and ratios of metabolic end-products comparable to those observed in the microplate assay (data not shown).
Figure 2.

Total B. infantis and E. hallii populations during coculture of B. infantisT (A, C) and B. infantisi (B, D) with E. hallii during bifidobacterial fermentation of 2′FL (A,B) and DFL (C, D) as sole carbohydrate sources.
B. infantisT and E. halliiT produced more propionate on 2′FL cooperatively than in coculture of intrinsic B. infantisi and E. halliiT (12.38±0.04 mM and 10.1±0.2 mM, respectively, Supplemental Table S2). This aligns with pure-culture 1,2-PD production, as B. infantisT produces more 1,2-PD per mole of consumed 2′FL than B. infantisi (Supplemental Table S1). Formic acid production on 2’FL was consistent with pure- culture fermentation of 2′FL and the metabolic phenotype variation of the two B. infantis strains (Supplemental Tables S1 and S2).
B. infantisT and E. halliiT cooperatively produced less butyrate and more acetic acid than cocultures of intrinsic B. infantisi and E. halliiT during fermentation of both 2′FL (23.4±0.4 mM as compared to 31.0±0.2 mM, Supplemental Table S2) and DFL (70.3±0.3 mM as compared to 61.5±0.8 mM, Supplemental Table S2). In both cocultures, lactic acid is below the limits of detection (< 0.5mM), which suggests that E. halliiT production of butyrate from acetic and lactic acids is limited by bifidobacterial lactic acid production. From pure-culture fermentation of 2′FL, B. infantisT produces significantly less lactic acid than B. infantisi (10.5±0.4 mM as compared to 16.7±0.1 mM, p ≤ 0.00001, Supplemental Table S1). This is in line with the reduced butyrate and increased acetic acid concentrations in the coculture with B. infantisT as compared to coculturing E. halliiT with the intrinsic B. infantisi. When bifidobacterial lactic acid production is considered as the rate-limiting factor for butyrate production and acetic acid consumption, the coculture results are consistent with pure-culture fermentation of 2′FL.
As expected, the additional fucosyl moiety in DFL increases propionate secretion due to additional 1,2- PD produced by bifidobacterial fucose metabolism. The coculture of B. infantisT and E. halliiT (Fig 2C) produces more propionate, acetic acid, and formic acid but less butyrate than the B. infantisi coculture (Fig 2D). The variation in butyrate production may be explained in the context of bifidobacterial lactic acid production as the type strain produced less lactic acid in pure culture (4.1±0.3 and 17.2±0.2 mM), Supplemental Table S1). B. infantisT coculture of DFL showed a B. infantis population increase of 1.27 log copies mL−1 (5.99 to 7.26 log copies mL−1, Fig 2C) and an E. hallii population increase of 1.33 log copies mL−1 (6.21 to 7.54 log copies mL−1, Fig 2C). DFL coculture by B. infantisi and E. halliiT increased B. infantis and E. hallii populations by 1.87 log copies mL−1 (5.43 to 7.30 log copies mL−1, Fig 2D) and 1.55 log copies mL−1 (5.86 to 7.41 log copies mL−1, Fig 2D), respectively. It is interesting that the change in E. hallii population is similar between the two cocultures. Divergent E. hallii population growth may be anticipated given that the B. infantisT and B. infantisi metabolize DFL with varying efficiency and, thus, produce differing relative amounts of metabolic end-products. While there may be a difference in substrates produced by bifidobacterial metabolism, E. halliiT can utilize 1,2-PD, acetic acid, and lactic acid as energy sources. There is a potential for substrate preference by E. halliiT, which is consistent with the relative concentrations of endproducts where the B. infantisT coculture exhibited higher propionate and lower butyrate than what occurred in the B. infantisi coculture.
Given that both B. infantis and E. hallii are not predicted to be uniformly present in a strict 1:1 population ratio, a coculture of 1% (vol/vol) B. infantisT and 0.1% (vol/vol) E. halliiT was performed to determine whether trophic interactions were dependent on the initial population ratio. The final B. infantis and E. hallii populations were comparable to the 1:1 inoculated coculture fermentation (Supplemental Table S3), suggesting that E. hallii growth is limited by B. infantis metabolism of 2′FL. This is supported by the control fermentation of 2′FL inoculated with only E. hallii (1% by volume), which exhibited no detectable change in 2′FL concentrations, consistent with literature reports that E. hallii cannot consume 2′FL.[9,28]
fHMO-mediated syntrophy is impacted by the microbiome background in which it occurs
An in vitro microbiome model was used to determine the extent to which fHMO-mediated syntrophy between B. infantis and E. hallii is influenced within a community context. This in vitro batch culture fermentation model was initially seeded with a fecal inoculum. Consistent with the 2′FL coculture, propionate production is limited by bifidobacterial 1,2-PD production in the modeled microbiome. Accordingly, propionate concentrations were consistent across all four modeled community fermentations of 2′FL regardless of B. infantisT or E. halliiT supplementation and population (Fig 3 and Supplemental Table S4). Despite varying B. infantis populations, a consistent amount of 1,2-PD is produced during fHMO fermentation in agreement with a consistent rate of substrate disappearance in all fermentations (Supplemental Fig S3, S4). The stable propionate concentrations suggest that other microbes are producing propionate when the total E. hallii population is low. This is expected as other gut commensals are capable of producing propionate from 1,2-PD.[15,18] While the final propionate concentration does not appear to be dependent on E. hallii population concentrations, the rate of propionate production is much higher when E. hallii is supplemented into the modeled community fermentation (Fig 3 B,C). Again, this may be related to varying preference and metabolic efficiency across different substrates of the microbes that comprise the modeled microbiome and supplemented E. halliiT.
Figure 3.

Total B. infantis and E. hallii populations and metabolite production during modeled fecal community fermentation of 2′FL supplemented with A) B. infantisT, B) E. halliiT, C) B. infantisT & E. halliiT, and D) no supplementation.
Butyrate production, in contrast, appears to be dependent on both total E. hallii population and B. infantisT supplementation. Higher butyrate production is observed in the modeled community supplemented with E. halliiT alone. This supports the hypothesis that E. hallii butyrate production is limited by bifidobacterial lactate production. B. infantisi is endogenous to the fecal inoculum at approximately 4 log copies mL−1 and produces a higher ratio of lactic acid to acetic acid than B. infantisT (Supplemental Table S1).
As with 2′FL, modeled community fermentations of DFL show a consistent final propionate concentration but varying rate of propionate production dependent on E. halliiT supplementation (Fig 4). The amount of propionate produced during 2′FL and DFL fermentations is very similar (Supplemental Table S4). In the modeled community fermentations, a fucosyl group is cleaved from DFL to produce free fucose and 3′FL (Supplemental Fig S4). Due to this cleavage, a direct comparison between coculture and modeled community fermentation of DFL is difficult to make. The cleaved free fucose is likely not metabolized by B. infantis to 1,2-PD due to low efficiency by which this substrate is translocated and metabolized in vitro.[8] It is of note that in pure culture, both B. infantisT and B. infantisi metabolize the fucosyl moiety of 3′FL to a lesser extent than during 2′FL fermentation as evidenced by a much higher ratio of consumed HMO to produced 1,2-PD (Supplemental Table S1). Modeled community fermentations of 2′FL do not appear to result in obvious fucosyl group cleavage from 2′FL (Supplemental Fig S3).
Figure 4.

Total B. infantis and E. hallii populations and metabolite production during modeled fecal community fermentation of DFL supplemented with A) B. infantisT, B) E. halliiT, C) B. infantisT & E. halliiT, and D) no supplementation
fHMO structure drives microbial community composition
While the organic acid profiles of the modeled microbiomes are similar between 2′FL and DFL fermentations, the community population responds to fHMO structure specificity. Modeled community fermentations of 2′FL and DFL exhibit a net increase in total B. infantis population regardless of supplementation (Fig 3 and 4). Both E. hallii and B. infantis populations are less stable during DFL fermentation than 2′FL fermentation and peak much later in fermentation. In both 2′FL and DFL fermentations, total B. infantis population growth is highest when only B. infantisT is supplemented into the modeled fecal community. In contrast, 2′FL fermentation drives an increase in total E. hallii population in the absence of E. hallii supplementation whereas DFL fermentation drives a decrease. When both B. infantisT and E. halliiT are supplemented into the modeled community, DFL fermentation drives an increase in total E. hallii population whereas 2′FL fermentation does not result in a net change in total E. hallii population.
All modeled communities exhibited a relatively consistent initial composition, due to the common fecal inoculum (Fig 5). Principal-coordinate analysis based on weighted UniFrac distances (Fig 5I) indicates that 2′FL supplemented with only E. hallii drives the community composition to most closely cluster with unsupplemented 2′FL fermentation (F = 3.48, P = 0.055). All other supplementations significantly differ from each other (P = 0.001), where varying effect sizes show B. infantisT supplementation alone has a larger impact on overall community structure (F = 46.76) than dual supplemented B. infantisT and E. halliiT (F = 11.18) during 2′FL fermentation. B. infantisT supplemented fermentations of 2′FL (P = 0.001, F = 35.23) and DFL (P = 0.001, F = 33.29; Fig 5 I, J) significantly differ from each other.
During DFL fermentation, all supplementation regimes significantly differed from each other (P = 0.001) except E. halliiT supplemented and dual supplemented B. infantisT and E. halliiT (F = 1.39, P = 0.240; Fig 5J). Relative effect sizes indicate that E. halliiT-containing supplementations have the largest impact on unsupplemented community structure during DFL fermentation (E. halliiT F= 22.07, dual supplemented F = 24.59) compared to B. infantisT alone (F = 11.18). In addition, Bray-Curtis dissimilarity of the communities cluster according to E. halliiT supplementation during DFL fermentation, where there was not a significant difference between the two E. halliiT-containing supplementations (F = 1.58, P = 0.14; Supplemental Fig S5E). Although communities cluster according to B. infantisT supplementation during 2′FL fermentation (Supplemental Fig S5B), there were significant differences based on the two B. infantisT-containing supplementations (PERMANOVA; F = 19.26, P = 0.001).
The alpha diversity was increased in DFL fermentations during B. infantisT supplemented fermentations relative to dual supplemented (P ≤ 0.01, Supplemental Fig S6) and unsupplemented fermentations (P ≤ 0.05; Supplemental Fig S6). In addition, Faith’s Phylogenetic Diversity increased during B. infantisT supplementation relative to dual supplementation (P ≤ 0.01), but not relative to unsupplemented fermentations (Supplemental Fig S6C). Shannon diversity decreased with the addition of B. infantisT relative to unsupplemented DFL fermentations (P ≤ 0.01, Supplemental Fig S6A). Interestingly, dual supplementation also decreased the Shannon diversity compared to the unsupplemented (P ≤ 0.001, Supplemental Fig S6A) and E. halliiT supplementation alone (P ≤ 0.01, Supplemental Fig S6A) but the Shannon diversity of B. infantisT supplementation was not significantly different than any of the other supplementation regimes. During 2′FL fermentations Shannon diversity decreased with B. infantisT supplementation (P ≤ 0.0001, Supplemental Fig S6A). Interestingly, E. halliiT supplementation increased Shannon diversity (P ≤ 0.05, Supplemental Fig S6A), and dual supplementation did not significantly differ from unsupplemented fermentations.
Community fermentation of 2′FL supplemented solely with B. infantisT shows a rapid expansion of Bifidobacterium in the first 6 hours consistent with the qRT-PCR results (Fig 3, 5). Enterococcus abundance mirrors the expansion of Bifidobacterium, reaching a relative abundance of ~45% at around 8 hours and remaining at this fraction for the remainder of the fermentation. Anaerobutyricum (the genus containing E. hallii) relative abundance declines rapidly from an initial 8% to 1% at hour 6 and then remains less than 1% for the remainder of the fermentation. Modeled community fermentation of 2′FL supplemented with only E. halliiT exhibits an initial expansion of Anaerobutyricum within the first 6 hours. Enterococcus relative abundance mirrors this early expansion of Anaerobutyricum and stabilizes at a relative abundance of ~65%, much higher than the B. infantisT supplemented fermentation. Anaerobutyricum is enriched in relative abundance in the dual-supplemented 2′FL fermentation from an initial 15% to a maximum of 54% at hour 6 followed by a steady decline to a final 3% at hour 48. Dual supplementation gives rise to the highest relative abundance of Anaerobutyricum of all 2′FL fermentations. Dual-supplemented Bifidobacterium relative abundance does not increase as much as in the B. infantisT supplemented fermentation despite starting at a comparable initial relative abundance. Unsupplemented 2′FL fermentation exhibits low Anaerobutyricum relative abundance declining from an initial 7% to less than 1% by hour 8. Bifidobacterium relative abundance is relatively constant throughout fermentation and resembles the abundance trends across time in the dual-supplemented fermentation.
In B. infantisT supplemented modeled community fermentation of DFL, the relative abundance of Bifidobacterium increases from an initial 30% to a maximum of 49% in the first 4 hours before slowly decreasing to a final abundance of 15%. Contrary to 2′FL fermentations, Enterococcus is below the limits of detection at inoculation and steadily increases across fermentation to a final abundance of 66%. Similar to the B. infantisT supplemented 2′FL fermentation, Anaerobutyricum falls below 1% by hour 8 and remains below 1% throughout the remainder. In the E. halliiT supplemented DFL fermentation, Anaerobutyricum relative abundance shows an overall downward trend from a maximum of 72% at hour 6 but has a secondary maximum of 42% around hour 22 before decreasing to a final 24%. Bifidobacterium populations indicate an overall downward trend with a secondary maximum around hour 18, preceding the Anaerobutyricum abundance. Enterococcus again increases with time but settles at a lower relative abundance than B. infantisT supplemented fermentation. In the dual-supplemented DFL fermentation, Anaerobutyricum again shows an overall downward trend from maximum of 72% at hour 4 with a local maximum of 40% at hour 28 to a final 24%. Bifidobacterium mirrors this trend to a lesser extent. In unsupplemented DFL fermentation, Bifidobacterium shows an overall downward trend with a very slow decay after hour 8 to a final abundance of 10%. Anaerobutyricum decays from a starting abundance of 8% below limits of detection by hour 12, consistent with qRT-PCR. Enterococcus steadily increases from less than 1% initially to a maximum of 78% at hour 16 before declining until hour 24 where it increases.
Dorea relative abundance is correlated to B. infantisT supplementation during 2′FL fermentation, as it is below the limits of detection at the end of fermentations without supplemental B. infantisT but is in the top ten most abundance genera in both B. infantisT and the dual-supplemented fermentations (1.2% and 5%, respectively). E. halliiT supplementation is associated with decreased relative abundance of Lachnoclostridium and Intestinibacter during DFL fermentation. Lachnoclostridium reaches a comparable final abundance in the unsupplemented and B. infantisT supplemented modeled fecal community fermentations of DFL (3.6% and 3%, respectively). In 2′FL fermentations, Intestinibacter abundance is negatively correlated with B. infantisT supplementation, but Intestinibacter abundance is negatively correlated with E. halliiT supplementation in DFL fermentations. Intestinibacter is a relatively recent delineated genus[61] and includes I. bartletti (formerly Clostridium bartlettii). While little is characterized about this genus, cursory analysis of functional annotations in public genomic databases (i.e. SEED and gut microbial modules) suggest Intestinibacter species may metabolize fucose as it contains requisite gene sequences.[62] If Intestinibacter is fucose-consuming microbe the community dynamics are not understood to how this taxon is not more prevalent in the DFL fermentations where free fucose is cleaved from DFL, as it is not predicted that E. hallii metabolizes fucose.[13] It is possible that the free fucose is sequestered more rapidly by heterologous microbes, is covalently modified, or access to fucose does not confer a fitness advantage to Intestinibacter in this experimental context or in general.
The modeled microbiome exhibits functional redundancy in both butyrate and propionate production during fHMO fermentation
Considering the representation of the ten most abundant taxa, along with the secreted metabolic end-products, the potential for additional cross-feeding relationships are likely to occur in the microbiome, with divergent microbial communities performing the same net function. In all fermentations of 2′FL and DFL, Blautia appears a top five abundant genus at the end of the fermentation with a relative abundance of <5% after starting between 15 and 23% at inoculation. Of particular interest, Blautia species produce propionate from 1,2-PD.[15] In fermentations that are not supplemented with E. hallii, propionate production and Blautia relative abundance increases coinciding with fermentation time. Propionate production, however, begins earlier in the fermentation when E. halliiT is added to the modeled community. Lachnoclostridium may also contribute, albeit indirectly, to aggregate propionate production. In a previous study, Lachnoclostridium clostridioforme exhibited a positive relationship with propionate production during fermentation of inulin within a modeled microbiome despite not encoding for propionate production within its genome.[63] While Lachnoclostridium is not consistently within the most abundant genera, there may be indirect interactions that promote propionate production regardless of taxon representation.
Butyrate production is somewhat low in the dual-supplemented fermentations with both 2′FL and DFL substrates given that E. hallii populations are very similar across time for the E. halliiT supplemented and dual-supplemented fermentations. Butyrate-producing Lachnoclostridium is found at higher relative abundance with E. halliiT supplementations as compared to dual-supplemented fermentations, which may contribute to the differing butyrate concentrations despite consistent E. hallii populations. Lachnoclostridium is a butyrate producer as it utilizes a different anabolic pathway than E. hallii that relies on γ-aminobutyrate or succinate degradation.[64,65] This variant butyrate pathway may relieve potential competition for lactic acid, which appears to be the limiting factor for E. halliiT butyrate production in coculture. In a previously reported model gut microbiome, L. symbiosum significantly increased butyrate production when included in a fermentation of inulin.[63] In DFL fermentations without E. halliiT supplementation, Lachnoclostridium and Eubacterium are present at high relative abundances, which may partially explain the modest butyrate production in these fermentations despite the low total population and relative abundance of E. hallii. A similar phenomenon is observed during 2′FL fermentation, though the associated genera are Paraclostridium and Eubacterium. Eubacterium species, in particular E. rectale, are known to produce butyrate but not propionate.[15] Paraclostridium exhibits an inverse relationship with E. halliiT supplementation during 2′FL fermentation. The Paraclostridium genus is characterized to produce butyrate through the degradation of lysine and succinate, similar to Lachnoclostridium.[66] The mechanisms and community dynamics that promote expansion of Paraclostridium during 2′FL fermentation and Lachnoclostridium during DFL fermentation are unclear as they both appear to expand into the butyrate-producing niche left vacated by E. hallii.
4. Concluding Remarks
The modeled microbial community background in which cocultures interact engenders functional differences in model outcomes. This microbiome background allows for competition between microbes that are not the primary subjects of experimental investigation (i.e. B. infantis and E. hallii). Similarly, modification of available HMO substrates may occur independently. It is established that substrate modification by a particular species can support heterologous microbial species in the gut microbiome to increase diversity. The interplay between extracellular cleavage and metabolism of intact fHMOs promotes competition that is expected to shift the production of short chain fatty acids (SCFAs) and may not be fully predicted by a mono-culture or coculture model. Microbial interactions during modeled community fermentation, however, cannot be fully characterized without reducing the complexity of the system in a systematic manner. The predictive value of each model (i.e. pure-culture, coculture, and modeled community culture) is increased when examined together, providing a basic understanding to inform future directions. The in vitro batch culture model allows for a direct comparison with pure culture characterization of bifidobacterial fHMO metabolism. In addition, this model adds an additional layer of complexity with a competitive modeled community background. Although, and with similar studies, does not fully capture community interactions that may occur in a steady-state in vivo model. A natural progression from the in vitro batch culture model would be to further increase similarity to an in vivo model through the addition of pulsed feeding, which would mimic another dimension of substrate access by interacting microbes and allow the community to potentially achieve a stable state. It is likely that this may shift relative community population fluxes and alter aggregate community function.
Ultimately, every trophic interaction between two microbes may not be a major driver of the overall function of a microbiome. The emergent physiology of the microbiome exhibits flexibility and resilience in maintaining homeostasis and perturbations to syntrophic partners may be limited in context of overall community function. Accordingly, consistent SCFA profiles during non-digestible oligosaccharide fermentation by microbial communities have been previously ascribed to functional redundancy.[67] Herein, populations of a consistent modeled microbiome background changed with microbial supplementation and fHMO structure and produced similar metabolic profiles despite changes in community structure. Coculture fermentations in the absence of the modeled community background consistently increased total B. infantis and E. hallii populations during metabolism of both 2′FL and DFL, and the addition of the modeled community background exhibited fHMO-dependent population changes. E. hallii populations were relatively static during dual-supplemented community fermentation of 2′FL, in contrast to a strong increase in overall E. hallii population during dual-supplemented community fermentation of DFL.
fHMOs direct the composition and function of the infant gut microbiome well into the weaning period and, unsurprisingly, promote the expansion of fHMO-utilizing B. infantis.[68,69] While there is no universal microbiome community composition, this work suggests that bifidobacterial metabolism of 2′FL and DFL supports interactions between infant-associated microbes and adult-associated microbes. The modulation of SCFA production may also impact host health and development, as SCFAs are absorbed by the infant host as well as serving as substrates for microbial metabolism.
Supplementary Material
Acknowledgements
The authors thank Cindy Kane for technical assistance and Dr. Ezgi Özcan and Michelle Rozycki for helpful discussions. Dr. Ravi Ranjan is acknowledged for technical support at the UMass Amherst Genome Resource Laboratory. DSM (formerly Glycom) is acknowledged for their generous donation of HMOs. This work was supported by NIH-NICHD grant R01 HD106554 (DAS). LRD acknowledges fellowship support from USDA-NIFA (2020-67034-31717).
Abbreviations:
- HMO
Human milk oligosaccharide
- fHMO
Fucosylated human milk oligosaccharide
- 2′FL
2′fucosyllactose
- 3′FL
3′fucosyllactose
- DFL
Difucosyllactose
- 1,2-PD
1,2-propanediol
- SCFA
Short chain fatty acid
- ATCC
American Type Culture Collection
- MRS
De Man Rogosa-Sharpe medium
- ICM
Infant colon medium
- mMRS
Modified De Man Rogosa-Sharpe medium
- qRT-PCR
quantitative real time polymerase chain reaction
- PCoA
Principal coordinate analysis
- PERMANOVA
permutational multivariate analysis of variance
- F6PPK
Fructose-6-phosphate phosphoketolase
- OTU
Operational taxonomic unit
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest.
5 References
- [1].Lewis ZT, Totten SM, Smilowitz JT, Popovic M, Parker E, Lemay DG, Van Tassell ML, Miller MJ, Jin Y-S, German JB, Lebrilla CB, Mills DA, Microbiome 2015, 3, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Wang M, Li M, Wu S, Lebrilla CB, Chapkin RS, Ivanov I, Donovan SM, J Pediatr Gastroenterol Nutr 2015, 60, 825–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Bode L, Early Hum Dev 2015, 91, 619–622. [DOI] [PubMed] [Google Scholar]
- [4].Sakanaka M, Hansen ME, Gotoh A, Katoh T, Yoshida K, Odamaki T, Yachi H, Sugiyama Y, Kurihara S, Hirose J, Urashima T, zhong Xiao J, Kitaoka M, Fukiya S, Yokota A, Lo Leggio L, Hachem MA, Katayama T, Sci Adv 2019, 5, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Borewicz K, Gu F, Saccenti E, Hechler C, Sci Rep 2020, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bode L, Glycobiology 2012, 22, 1147–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Chaturvedi P, Warren CD, Altaye M, Morrow AL, Ruiz-Palacios G, Pickering LK, Newburg DS, Glycobiology 2001, 11, 365–372. [DOI] [PubMed] [Google Scholar]
- [8].Dedon LR, Özcan E, Rani A, Sela DA, Front Nutr 2020, 7, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Bunesova V, Lacroix C, Schwab C, BMC Microbiol 2016, 16, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Sela DA, Garrido D, Lerno L, Wu S, Tan K, Eom H-J, Joachimiak A, Lebrilla CB, Mills DA, Appl Environ Microbiol 2012, 78, 795–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Zabel B, Yde CC, Roos P, Marcussen J, Jensen HM, Salli K, Hirvonen J, Ouwehand AC, Morovic W, Sci Rep 2019, 9, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Crost EH, Tailford LE, Le Gall G, Fons M, Henrissat B, Juge N, PLoS One 2013, 8, DOI 10.1371/journal.pone.0076341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Schwab C, Ruscheweyh H-J, Bunesova V, Pham VT, Beerenwinkel N, Lacroix C, Front Microbiol 2017, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Engels C, Ruscheweyh HJ, Beerenwinkel N, Lacroix C, Schwab C, Front Microbiol 2016, 7, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Louis P, Flint HJ, Environ Microbiol 2017, 19, 29–41. [DOI] [PubMed] [Google Scholar]
- [16].Gentile CL, Weir TL, Science (1979) 2018, 362, 776–780. [DOI] [PubMed] [Google Scholar]
- [17].Hung CC, Garner CD, Slauch JM, Dwyer ZW, Lawhon SD, Frye JG, Mcclelland M, Ahmer BMM, Altier C, Mol Microbiol 2013, 87, 1045–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Reichardt N, Duncan SH, Young P, Belenguer A, McWilliam Leitch C, Scott KP, Flint HJ, Louis P, ISME J 2014, 8, 1323–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Plöger S, Stumpff F, Penner GB, Schulzke J-D, Gäbel G, Martens H, Shen Z, Günzel D, Aschenbach JR, Ann NY Acad Sci 2012, 1258, 52–59. [DOI] [PubMed] [Google Scholar]
- [20].Morrison DJ, Preston T, Gut Microbes 2016, 7, 189–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Walsh C, Lane JA, van Sinderen D, Hickey RM, Sci Rep 2022, 12, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Lawson MAE, O’Neill IJ, Kujawska M, Gowrinadh Javvadi S, Wijeyesekera A, Flegg Z, Chalklen L, Hall LJ, ISME Journal 2020, 14, 635–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Turroni F, Milani C, Duranti S, Mahony J, van Sinderen D, Ventura M, Trends Microbiol 2018, 26, 339–350. [DOI] [PubMed] [Google Scholar]
- [24].Pham VT, Lacroix C, Braegger CP, Chassard C, Environ Microbiol 2016, 18, 2246–2258. [DOI] [PubMed] [Google Scholar]
- [25].Yatsunenko T, Rey FE, Manary MJ, Trehan I, Dominguez-Bello MG, Contreras M, Magris M, Hidalgo G, Baldassano RN, Anokhin AP, Heath AC, Warner B, Reeder J, Kuczynski J, Caporaso JG, Lozupone CA, Lauber C, Clemente JC, Knights D, Knight R, Gordon JI, Nature 2012, 486, 222–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bergström A, Skov TH, Bahl MI, Roager HM, Christensen LB, Ejlerskov KT, Mølgaard C, Michaelsen KF, Licht TR, Appl Environ Microbiol 2014, 80, 2889–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Shetty SA, Zuffa S, Bui TPN, Aalvink S, Smidt H, De Vos WM, Int J Syst Evol Microbiol 2018, 68, 3741–3746. [DOI] [PubMed] [Google Scholar]
- [28].Scott KP, Martin JC, Duncan SH, Flint HJ, FEMS Microbiol Ecol 2014, 87, 30–40. [DOI] [PubMed] [Google Scholar]
- [29].Barcenilla A, Pryde SE, Martin JC, Duncan SH, Stewart CS, Henderson C, Flint HJ, Appl Environ Microbiol 2000, 66, 1654–1661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Wolfe RS, Metcalf WW, Anaerobe 2010, 16, 216–219. [DOI] [PubMed] [Google Scholar]
- [31].Hungate RE, Methods in Microbiology 1969, 3, 117–132. [Google Scholar]
- [32].Cinquin C, le Blay G, Fliss I, Lacroix C, FEMS Microbiol Ecol 2006, 57, 324–336. [DOI] [PubMed] [Google Scholar]
- [33].Gibson GR, Wang X, FEMS Microbiol Lett 1994, 118, 121–127. [DOI] [PubMed] [Google Scholar]
- [34].Simpson PJ, Fitzgerald GF, Stanton C, Ross RP, J Microbiol Methods 2004, 57, 9–16. [DOI] [PubMed] [Google Scholar]
- [35].Leuschner RGK, Bew J, Simpson P, Ross PR, Stanton C, Int J Food Microbiol 2003, 83, 161–170. [DOI] [PubMed] [Google Scholar]
- [36].Turroni F, Foroni E, Pizzetti P, Giubellini V, Ribbera A, Merusi P, Cagnasso P, Bizzarri B, De’Angelis GL, Shanahan F, van Sinderen D, Ventura M, Appl Environ Microbiol 2009, 75, 1534–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Dai Y, McLandsborough LA, Weiss J, Peleg M, J Food Sci 2010, 75, M482–M488. [DOI] [PubMed] [Google Scholar]
- [38].Sela DA, Li Y, Lerno L, Wu S, Marcobal AM, German JB, Chen X, Lebrilla CB, Mills DA, Journal of Biological Chemistry 2011, 286, 11909–11918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, Bai Y, Bisanz JE, Bittinger K, Brejnrod A, Brislawn CJ, Brown CT, Callahan BJ, Caraballo-Rodríguez AM, Chase J, Cope EK, Da Silva R, Diener C, Dorrestein PC, Douglas GM, Durall DM, Duvallet C, Edwardson CF, Ernst M, Estaki M, Fouquier J, Gauglitz JM, Gibbons SM, Gibson DL, Gonzalez A, Gorlick K, Guo J, Hillmann B, Holmes S, Holste H, Huttenhower C, Huttley GA, Janssen S, Jarmusch AK, Jiang L, Kaehler BD, Bin Kang K, Keefe CR, Keim P, Kelley ST, Knights D, Koester I, Kosciolek T, Kreps J, Langille MGI, Lee J, Ley R, Liu YX, Loftfield E, Lozupone C, Maher M, Marotz C, Martin BD, McDonald D, McIver LJ, Melnik AV, Metcalf JL, Morgan SC, Morton JT, Naimey AT, Navas-Molina JA, Nothias LF, Orchanian SB, Pearson T, Peoples SL, Petras D, Preuss ML, Pruesse E, Rasmussen LB, Rivers A, Robeson MS, Rosenthal P, Segata N, Shaffer M, Shiffer A, Sinha R, Song SJ, Spear JR, Swafford AD, Thompson LR, Torres PJ, Trinh P, Tripathi A, Turnbaugh PJ, Ul-Hasan S, van der Hooft JJJ, Vargas F, Vázquez-Baeza Y, Vogtmann E, von Hippel M, Walters W, Wan Y, Wang M, Warren J, Weber KC, Williamson CHD, Willis AD, Xu ZZ, Zaneveld JR, Zhang Y, Zhu Q, Knight R, Caporaso JG, Nat Biotechnol 2019, 37, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, Holmes SP, Nat Methods 2016, 13, 581–583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Lozupone C, Lladser ME, Knights D, Stombaugh J, Knight R, ISME Journal 2011, 5, 169–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO, Nucleic Acids Res 2013, 41, 590–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Wickham H, Averick M, Bryan J, Chang W, McGowan L, François R, Grolemund G, Hayes A, Henry L, Hester J, Kuhn M, Pedersen T, Miller E, Bache S, Müller K, Ooms J, Robinson D, Seidel D, Spinu V, Takahashi K, Vaughan D, Wilke C, Woo K, Yutani H, J Open Source Softw 2019, 4, 1686. [Google Scholar]
- [44].Bisanz JE, 2018. [Google Scholar]
- [45].R Core Team (2022), n.d. [Google Scholar]
- [46].Team RStudio (2020), n.d. [Google Scholar]
- [47].Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing, 1995. [Google Scholar]
- [48].Anderson MJ, Austral Ecol 2001, 26, 32–46. [Google Scholar]
- [49].Wickham H, Ggplot2: Elegant Graphics for Data Analysis, Springer International Publishing; 2016. [Google Scholar]
- [50].Kassambara A, 2020. [Google Scholar]
- [51].Fridman Y, Wang Z, Maslov Id S, Goyal Id A, PLoS Comput Biol 2022, 18, e1010244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Goyal A, Bittleston LS, Leventhal GE, Lu L, Cordero OX, Elife 2022, 11, DOI 10.7554/ELIFE.74987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Bittleston LS, Gralka M, Leventhal GE, Mizrahi I, Cordero OX, Nature Communications 2020 11:1 2020, 11, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Wolff R, Shoemaker W, Garud N, bioRxiv 2022, 2021.09.30.462616. [Google Scholar]
- [55].Linton JD, FEMS Microbiol Lett 1990, 75, 1–18. [Google Scholar]
- [56].Linton JD, Antonie Van Leeuwenhoek 1991, 60, 293–311. [DOI] [PubMed] [Google Scholar]
- [57].Palframan RJ, Gibson GR, Rastall RA, Curr Issues Intest Microbiol 2003, 4, 71–5. [PubMed] [Google Scholar]
- [58].Özcan E, Sela DA, Front Nutr 2018, 5, 1–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Van Der Meulen R, Adriany T, Verbrugghe K, De Vuyst L, Appl Environ Microbiol 2006, 72, 5204–5210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].James K, Bottacini F, Contreras JIS, Vigoureux M, Egan M, Motherway MO, Holmes E, van Sinderen D, Sci Rep 2019, 9, 15427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Gerritsen J, Fuentes S, Grievink W, van Niftrik L, Tindall BJ, Timmerman HM, Rijkers GT, Smidt H, Int J Syst Evol Microbiol 2014, 64, 1600–1616. [DOI] [PubMed] [Google Scholar]
- [62].Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira- Silva S, Gudmundsdottir V, Krogh Pedersen H, Arumugam M, Kristiansen K, Yvonne Voigt A, Vestergaard H, Hercog R, Igor Costea P, Roat Kultima J, Li J, Jørgensen T, Levenez F, Dore J, Bjørn Nielsen H, Brunak S, Raes J, Hansen T, Wang J, Dusko Ehrlich S, Bork P, Pedersen O, Nature 2015, 528, 262–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gutiérrez N, Garrido D, mSystems 2019, 4, DOI 10.1128/msystems.00185-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Muñoz-Tamayo R, Laroche B, Walter É, Doré J, Duncan SH, Flint HJ, Leclerc M, FEMS Microbiol Ecol 2011, 76, 615–624. [DOI] [PubMed] [Google Scholar]
- [65].Vital M, Howe AC, Tiedje JM, mBio 2014, 5, DOI 10.1128/mBio.00889-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Frolova MS, Suvorova IA, Iablokov SN, Petrov SN, Rodionov DA, Front Mol Biosci 2022, 9, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Reichardt N, Vollmer M, Holtrop G, Farquharson FM, Wefers D, Bunzel M, Duncan SH, Drew JE, Williams LM, Milligan G, Preston T, Morrison D, Flint HJ, Louis P, ISME Journal 2018, 12, 610–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Bai Y, Tao J, Zhou J, Fan Q, Liu M, Hu Y, Xu Y, Zhang L, Yuan J, Li W, Ze X, Malard P, Guo Z, Yan J, Li M, mSystems 2018, 3, DOI 10.1128/msystems.00206-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Smith-Brown P, Morrison M, Krause L, Davies PSW, PLoS One 2016, 11, e0161211. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw sequencing reads from the 16S amplicon sequencing are available via the National Center for Biotechnology Information (NCBI) Short Read Archive (SRA) under BioProject PRJNA897896 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA897896).