

Abstract
The Cone-rod homeobox (CRX) protein is a key transcription factor essential for photoreceptor function and survival. Mutations in human CRX gene are linked to a wide spectrum of blinding diseases ranging from mild macular dystrophy to severe Leber Congenital Amaurosis (LCA), Cone-Rod Dystrophy (CRD) and Retinitis Pigmentosa (RP). These diseases are still incurable and mostly inherited in an autosomal dominant form. Dysfunctional mutant CRX protein interferes with the function of wildtype CRX protein, demonstrating the dominant negative effect. At present, gene augmentation is the most promising treatment strategy for hereditary diseases. This study aims to review the pathogenic mechanisms of various CRX mutations and propose two therapeutic strategies to rescue sick photoreceptors in CRX-associated retinopathies, namely, Tet-On-hCRX system and Adeno-associated Virus (AAV)-mediated gene augmentation. The outcome of proposed studies will guide future translational research and suggest guidelines for therapy evaluation in terms of treatment safety and efficacy.
Keywords: CRX mutations, dominant negative effects, gene augmentation, Tet-On, AAV gene therapy
1. Pathogenic mechanisms of CRX mutations
The human cone-rod homeobox gene CRX (OMIM #602225) is located on chromosome 19q13.33. There are four exons in CRX, specifically, the first noncoding and three coding exons. CRX codes for a 299 amino acid transcription factor which is predominantly expressed in photoreceptors and the pineal gland, regulating photoreceptor development and maintenance [1, 2]. The protein consists of three major domains: the homeodomain at residues 39-99 facilitates the DNA binding; the activation domain at residues 113-284, including a WSP motif at residues 158-170, contains binding sites for other transcription coregulators; a conserved carboxyl terminus motif, also known as the OTX tail, is found at resides 284-295 [1, 3].
To date, 93 disease-causing CRX mutations have been identified in human patients (www.hgmd.cf.ac.uk). Pathogenic CRX variants are associated with a complex group of inherited retinal diseases (IRDs) such as macular dystrophy [4], cone-rod dystrophy (CRD) [1], retinitis pigmentosa (RP) [5], and Leber congenital amaurosis (LCA) [6, 7]. Mutations within CRX are known to occur de novo or to be inherited mostly in an autosomal dominant pattern, consisting of substitution (nonsense and missense mutations) and frameshift mutations (deletions and insertions) [8]. Our understanding of these mutations has significantly expanded, as a great variety are being analyzed with the advent of cutting-edge tools from the sequencing and analytic technologies. However, a tremendous challenge for the pathogenic analysis of CRX-associated retinopathies, particularly of the autosomal dominant disorders, is to explore the phenotype-genotype correlations between disease severity and CRX mutations.
CRX mutations could cause dominant disorders by two possible mechanisms, namely, the haploinsufficiency of the functional CRX, and/or various dominant negative or gain-of-function effects of the mutant CRX. To the extent of data published, haploinsufficiency may not cause severe phenotypes. The study on Crx+/− mice corroborates this claim, as they do not develop any detectable functional defects up to 6 months [9]. A rare case of human patients with the heterozygosity of CRX also supports these observations, as only the patients with the nullizygosity of CRX develop LCA [10]. However, in the autosomal dominant disorders, it remains unknown if the mutant CRX allele could partially abrogate the production of a functional CRX from the normal allele. Further studies are needed to address this question in detail. Alternatively, dominant negative activities of various CRX mutant proteins have been demonstrated in animal models [9, 11]. The reported dominant-negative mutations that arise in the homeodomain are mostly missense mutations, and those identified in the activation domain are largely frameshifts [7, 9]. Pathogenic mechanisms thus are constituted by altered DNA binding properties and/or transactivation activities of CRX mutant proteins.
CRX R90W mutation presents a hypomorphic missense mutation located in the homeodomain [3, 12], and is associated with a dominant late-onset mild CRD and recessive LCA. The mutant protein has reduced DNA binding activity, and thus malfunctions to transactivate CRX-downstream target genes such as Rhodopsin [3, 12]. The transactivation activity of wild-type (WT) CRX seems normal in the heterozygous model [3], which proves the limited dominant negative effect of the CRX R90W mutation through the process of retinal development. On the other hand, CRX E80A and K88N mutations represent distinct antimorphic missense mutations located in the homeodomain [13–15]. Both mutations manifest LCA in human patients [1, 14]. Interestingly, some LCA-associated CRX homeodomain mutations are located at non-conserved residues in human homeodomain paralogues [4]. These mutant proteins are predicted to bind discrete DNA sequences and show different transactivation activities from the WT protein, although this has not yet been studied in mammalian models. Hence the dominant negative effects of the mutant proteins, if any, remain to be proven.
The frameshift deletion CRX E168d2 presents an antimorphic mutation located in the activation domain [3], and is associated with dominant LCA in human patients [6, 16]. This mutation results in the early truncation of the activation domain, producing a protein that retains the ability to bind target DNA but fails to transactivate CRX-downstream target genes [3]. In addition, CRX E168d2 allele overproduces the mutant protein at about 4 times more than the WT protein in heterozygous mice, which exacerbates the dominant-negative effect on the binding competition [3]. As a result, the heterozygous mice have impaired visual functions at 1 month-old (MO) [3] and limited electroretinography (ERG) response at 3MO (data not shown). Cone degeneration occurs prior to rod degeneration in the heterozygous mice. As compared to the WT samples, the heterozygous mice have only about 30% survived cones at 1 month-old (MO) and no detectable cones at 3MO (data not shown); mutants rods are functional with shorter outer segments (OS) at 1MO but undergo progressive degeneration till complete lost at 6MO [3]. Interestingly, the ratio of mutant to WT CRX proteins directly correlates with the disease phenotype severity and age of onset [3]. In addition, truncation at the last exon by frameshift results in premature terminations [7, 8], hence the production of shortened but stable mutant mRNA that can avoid nonsense-mediated decay [17], which partially contributes to the dominant negative effect. On the other hand, CrxRip presents the c.763del1 mutation located in the last exon, which results in a skipping of the OTX tail and a non-homologous extension of 133 residues [18]. The mutant protein does not transactivate CRX-downstream target genes, suggesting its antimorphic activity [18]. More notably, the mutant protein does not bind target DNA [18]. Similar to the retinal manifestation in patients, CrxRip/+ mice show a LCA-like phenotype [18]. Photoreceptors in CrxRip/+ mice do not form outer segments, due to impaired photoreceptor gene expression and incomplete differentiation at early development [18]. In addition, the mutant protein is not overexpressed in CrxRip/+ mice. Taken together, the dominant negative effect of CrxRip mutation does not signify a competition between the mutant and WT proteins, but likely arises from the disruption of the photoreceptor transcription factor network.
2. Gene augmentation as a strategy to treat CRX-associated retinopathies
There are currently limited therapeutic options for CRX-associated retinopathies. Fortunately, retinal gene therapy recently shines promise for treatment of various inherited blindness [19]. In particular, the gene augmentation approach has been developed to deliver a healthy gene to a diseased retina to rescue the defective phenotypes and visual functions without replacing the mutant allele. At present, successes of gene augmentation are largely restricted to recessive mutations [20]. Five important questions need to be answered in order to apply gene augmentation to treat CRX-associated autosomal dominant disorders. Firstly, how much of augmented CRX is needed to overcome the dominant negative effect of a given mutation? If a mutant protein competes with WT CRX proteins for binding sites, how much is augmented CRX needed to balance the ratio of mutant to WT CRX proteins in each photoreceptor? Can altering this ratio rescue the mutant phenotypes? Secondly, considering CRX is an early photoreceptor transcription factor that activates the downstream gene regulatory network in the retinal development, could gene augmentation impose overexpression toxicity in a treated retina? Thirdly, when is augmented CRX needed in the treatment? Fourthly, as human patients with CRX-associated retinopathies are often diagnosed at different stages of disease progression, what are the windows of opportunities for gene augmentation to a given disease? Do diseased photoreceptors possess the neuroplasticity for gene augmentation outside the development window? Lastly, can the treated photoreceptors retain their fated cell identity and functional capability upon gene augmentation?
Answers to these questions could inform the feasibility and strategies to treat CRX-associated retinopathies. To address these questions at least partially, a new transgenic mouse model, Tet-On-hCRX, has been developed to allow both quantitative and temporal control of augmented human CRX (hCRX) gene expression under a given CRX mutant background (Figure 1). Upon binding of doxycycline, a conformation change of the reverse tetracycline-controlled transcriptional activator (rtTA) is triggered [21], which allows the resulted complex bind to tetracycline-responsive element (TRE) (Tet-On) [22]. FLAG-tagged human CRX protein will hence be produced (Figure 1A). Moreover, the photoreceptor-specific induction can be achieved by the combination of two transgenes, namely, a Cre-dependent pCAG-LSL-rtTA (Figure 1B) and a Crx promoter-driven Cre, pCrx-Cre [23] (Figure 1C). Proof-of-concept validation of gene augmentation by Tet-On-hCRX system shows that CRX expression can be induced and detected in Crx−/− mouse retinae at all tested ages throughout the entire developmental window (postnatal (P) day 5, 14, 21 and 35) (Figure 2), confirming the success of Tet-On-hCRX system. Doxycycline-treated samples also display significant upregulation of CRX-downstream Rho expression. Furthermore, the comparisons between treated mice with different starts of the treatments suggests the ‘early the better’ effect on induced CRX and Rho expression at P35, as samples with P0-35 treatment display significantly higher induced CRX and Rho expression than those with P14-35 or P21-35 treatments (Figure 2A, 2B). However, as compared to the WT samples, treated mice still have significantly lower CRX and Rho expression at P35 under current treatment schemes, implying a partial rescue effect. These results suggest that Crx-null photoreceptors retain the machinery for CRX-mediated gene expression and neuroplasticity for therapeutic intervention.
Figure 1.
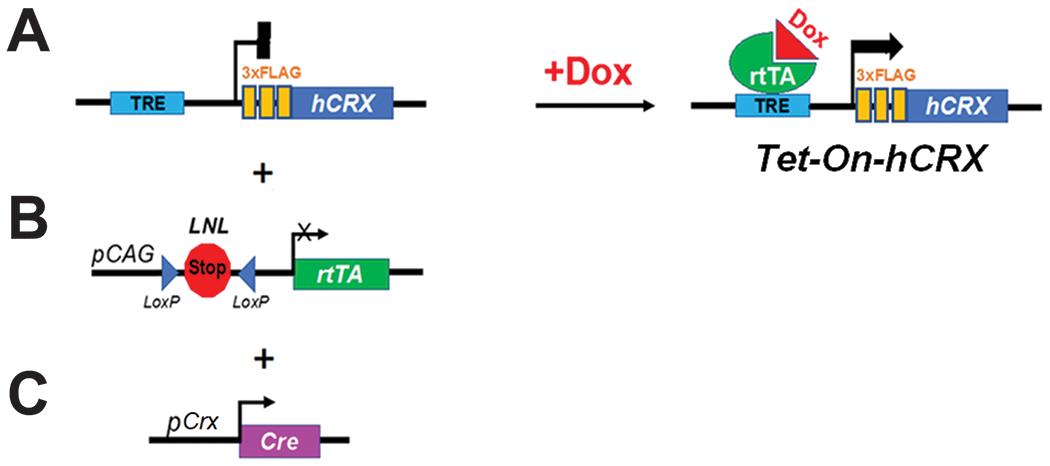
Components of Tet-On-hCRX system. (A) TRE-hCRX (B) pCAG-LSL-rtTA (C) pCrx-Cre.
Figure 2.
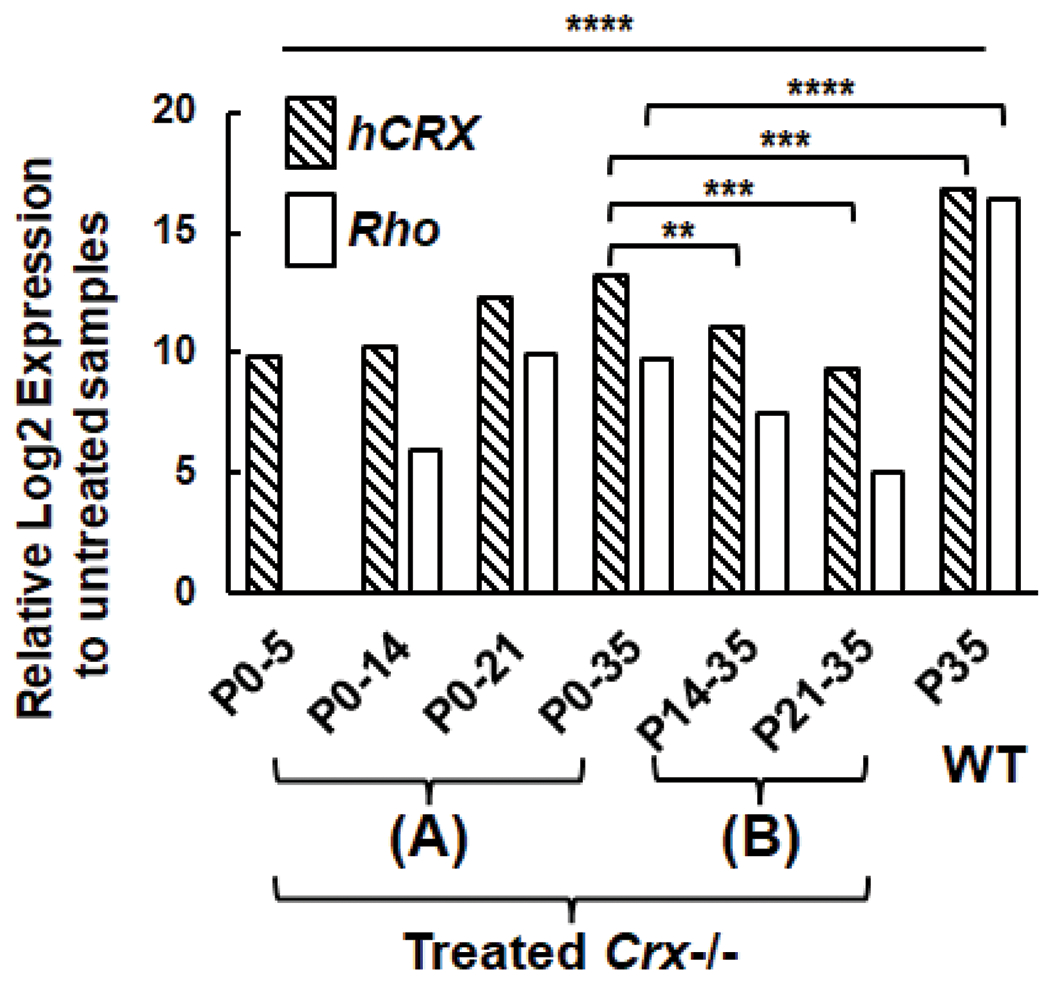
Quantitative PCR (qPCR) analysis of hCrx and Rho expression in untreated Crx−/−, Crx-BAC-Cre+, Tet-On-hCRX+; doxycycline-treated Crx−/−, Crx-BAC-Cre+, Tet-On-hCRX+; WT mice. (A) Treatments started at P0, samples were harvested at P5, P14, P21 or P35. (B) Treatments started at P14 or P21, samples were harvested at P35. Results are plotted as relative Log2 expression to untreated samples (n≥4). Asterisks (**, ***, ****) denote p ≤ 0.01, p ≤ 0.001 and p ≤ 0.0001 respectively by T-test.
As compared to Tet-On-hCRX system, a clinically applicable approach is Adeno-associated Virus (AAV)-mediated gene augmentation. The retina is surgically accessible, which facilitates intravitreal or subretinal administration of the viral vector [24, 25]. Since the retina is a relatively immune privileged tissue [24] and photoreceptors are nondividing and differentiated [26], a small amount of the non-integrating AAV viral vector can be tolerated to generate a therapeutic response [24, 25] with minimal risk of stimulating a severe inflammatory immune response [25]. Yet, two immediate challenges emerge during testing AAV-mediated CRX gene augmentation with animal models, i.e., choice of AAV vectors and cell-type specificity. To date, the AAV2/5 vector yields efficient transduction and good tropism for photoreceptors in P0 mice [27]. And the size of human or mouse CRX cDNA well fits into the AAV packaging capacity. Also, photoreceptor-specific promoters, including CRX or GRK, can be employed to achieve desirable sensitivity and precision.
However, there are still several safety precautions of applying AAV-mediated gene augmentation. Although subretinal administration is currently the most efficient route for targeting photoreceptors [28], it causes a transient detachment of the RPE from photoreceptors, which might damage photoreceptors at the injection site. A possible solution is the use of low-volume and low-concentration multiple blebs to reduce the danger of the RPE detachment and systemic exposure of high concentration by a single bleb, despite of associated minor risks [29, 30]. In addition, immunity and tolerance to AAV is untested in human photoreceptors at early and middle childhood [31], and multiplicity of infection (MOI) of viral vectors per cell may vary at different stages of the disease, all of which require further trials on human patients with early-onset diseases.
3. Unanswered questions and challenges
One-step gene augmentation may not rescue all CRX-associated autosomal dominant disorders. For examples, gain-of-function activities of a mutant protein with altered DNA binding specificity, such as CRX K88N, may generate the irreversible but detrimental effects on the developmental program. In such cases, expression from the mutant allele needs to be silenced to prevent the disease progression. Similar approaches of treating RHO-associated autosomal dominant RP in the mouse and canine models have shown encouraging outcomes [32, 33]. In addition, the CRISPR-Cas9/gRNA technique [34–37] can also be used to knockout the mutant allele.
Since gene augmentation utilizes the transcription machinery of the host cells to express the functional protein, cellular conditions of the host cells require close monitoring. In a developing retina, CRX gene augmentation may not fully rescue mutations that block early differentiation in photoreceptors, due to an insufficient but early window of opportunity. On the contrary, when the host cells are at the late stage of degeneration or the degeneration tends to progress too fast, gene augmentation may not succeed. Prerequisite anti-apoptotic or neuroprotective therapies can be combined to target the defective photoreceptors in order to gain a required time window for gene augmentation [38, 39].
Maculopathy has been reported in human patients of different CRX mutations with large variability in disease progression and symptoms [4, 40–42]. In addition, CRX may be involved in foveal development, as the haploinsufficiency of CRX results in subclinical foveal abnormalities in human patients [10]. None of these observations can be validated and studied in non-primate models.
The phenotypic and onset variability between patients sharing the same CRX mutant allele has constantly been reported [4, 7, 10, 43, 44]. It may be due to, firstly, possible polymorphisms in the CRX promoter region; secondly, the impacts of possible epigenetic modifications in coding regions; thirdly, any differential expression of other transcription factors such as NRL and NR2E3; fourthly, variable expression levels of the WT and/or mutant alleles; lastly, sexual dimorphisms [42], suggesting CRX may interact with specific targets on the sex chromosomes. None of these hypotheses has systematically been tested in animal models. In addition, the stochastic effects of CRX mutations and overexpression toxicity on the retinal degeneration are currently unknown, which appends uncertainty on the application of gene augmentation.
4. Methods
4.1. Transgenic mouse lines.
All male and female mice in this study were on the genetic background of C57BL/6J. TRE-hCRX line was generated for this study. The transgene had a TRE promoter upstream of full-length human CRX cDNA sequence, all inserted into the H11 locus. pCAG-LSL-rtTA (Rosa26:pCAG-LSL-rtTA) line was obtained from the Jackson Laboratory (Stock No: 029617). pCrx-Cre (BAC-Tg Crx-Cre) line was obtained from a published colony [23]. Doxycycline diets (200mg/kg, Bio-Serv, NJ) were provided to mice (> P14) and breeding dams. Intraperitoneal (IP) injection of 100μL of 20mg/mL doxycycline solution was provided to mice (> P21) once a week to maximize the treatment effect. All animal procedures were conducted according to the Guide for the Care and Use of Laboratory Animals of the National Institute of Health, and were approved by the Washington University in St. Louis Institutional Animal Care and Use Committee.
4.2. Quantitative PCR (qPCR).
Each RNA sample was extracted from 2 retinae of a mouse using the NucleoSpin RNA Plus kit (Macherey-Nagel, PA). 1μg of RNA was used to produce cDNA using First Strand cDNA Synthesis kit (Roche, IN). Technical triplicates were run for each gene. Primers (5’ to 3’) used in this study were Crx (F) TGTCCCATACTCAAGTGCCC, (R) TGCTGTTTCTGCTGCTGTCG; Rho (F) GCTTCCCTACGCCAGTGTG, (R) CAGTGGATTCTTGCCGCAG; Ubb (F) CAACATCCAGAAAGAGTCAACC, (R) ATGTTGTAATCAGAGAGGGTGC. The reaction master mix consisted of EvaGreen polymerase (Bio-Rad Laboratories, CA), 1μM primer mix, and diluted cDNA samples. Samples were run using a two-step 40-cycle protocol on a Bio-Rad CFX96 Thermal Cycler (Bio-Rad Laboratories, CA). The statistical analysis is done by Student’s t-test with p<0.05, CI:95%.
Acknowledgements
The authors are grateful to Dr. Tom Glaser for pCrx-Cre mice as a gift, to Dr. Ke Jiang for critical evaluation of the manuscript. Funding was provided by R01 EY012543 and R01 EY032136 (SC), RPB unrestricted fund (WU-DOVS), EY002687 (WU-DOVS), and McDonnell Center for Cellular and Molecular Neurobiology Fellowship (CS).
Reference
- 1.Freund CL, et al. , Cone-Rod Dystrophy Due to Mutations in a Novel Photoreceptor-Specific Homeobox Gene (CRX) Essential for Maintenance of the Photoreceptor. Cell, 1997. 91(4): p. 543–553. [DOI] [PubMed] [Google Scholar]
- 2.Chen S, et al. , Crx, a Novel Otx-like Paired-Homeodomain Protein, Binds to and Transactivates Photoreceptor Cell-Specific Genes. Neuron, 1997. 19(5): p. 1017–1030. [DOI] [PubMed] [Google Scholar]
- 3.Tran NM, et al. , Mechanistically distinct mouse models for CRX-associated retinopathy. PLoS genetics, 2014. 10(2): p. e1004111–e1004111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hull S, et al. , The Phenotypic Variability of Retinal Dystrophies Associated With Mutations in CRX, With Report of a Novel Macular Dystrophy Phenotype. Investigative Ophthalmology & Visual Science, 2014. 55(10): p. 6934–6944. [DOI] [PubMed] [Google Scholar]
- 5.Sohocki MM, et al. , Prevalence of mutations causing retinitis pigmentosa and other inherited retinopathies. Human mutation, 2001. 17(1): p. 42–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Freund CL, et al. , De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nature Genetics, 1998. 18(4): p. 311–312. [DOI] [PubMed] [Google Scholar]
- 7.Rivolta C, et al. , Novel frameshift mutations in CRX associated with Leber congenital amaurosis. Human Mutation, 2001. 18(6): p. 550–551. [DOI] [PubMed] [Google Scholar]
- 8.Stenson PD, et al. , The Human Gene Mutation Database: building a comprehensive mutation repository for clinical and molecular genetics, diagnostic testing and personalized genomic medicine. Human genetics, 2014. 133(1): p. 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tran NM and Chen S, Mechanisms of blindness: animal models provide insight into distinct CRX-associated retinopathies. Developmental dynamics : an official publication of the American Association of Anatomists, 2014. 243(10): p. 1153–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ibrahim MT, et al. , A complete, homozygous CRX deletion causing nullizygosity is a new genetic mechanism for Leber congenital amaurosis. Scientific Reports, 2018. 8(1): p. 5034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ruzycki PA, et al. , Crx-L253X Mutation Produces Dominant Photoreceptor Defects in TVRM65 Mice. Investigative ophthalmology & visual science, 2017. 58(11): p. 4644–4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Swaroop A, et al. , Leber Congenital Amaurosis Caused by a Homozygous Mutation (R90W) in the Homeodomain of the Retinal Transcription Factor CRX: Direct Evidence for the Involvement of CRX in the Development of Photoreceptor Function. Human Molecular Genetics, 1999. 8(2): p. 299–305. [DOI] [PubMed] [Google Scholar]
- 13.Chen S, et al. , Functional analysis of cone–rod homeobox (CRX) mutations associated with retinal dystrophy. Human Molecular Genetics, 2002. 11(8): p. 873–884. [DOI] [PubMed] [Google Scholar]
- 14.Nichols LL II, et al. , Two novel CRX mutant proteins causing autosomal dominant Leber congenital amaurosis interact differently with NRL. Human Mutation, 2010. 31(6): p. E1472–E1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Terrell D, et al. , OTX2 and CRX rescue overlapping and photoreceptor-specific functions in the Drosophila eye. Developmental Dynamics, 2012. 241(1): p. 215–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jacobson SG, et al. , Retinal degenerations with truncation mutations in the cone-rod homeobox (CRX) gene. Investigative Ophthalmology & Visual Science, 1998. 39(12): p. 2417–2426. [PubMed] [Google Scholar]
- 17.Lejeune F and Maquat LE. Mechanistic links between nonsense-mediated mRNA decay and pre-mRNA splicing in mammalian cells. Current Opinion in Cell Biology, 2005. 17(3): p. 309–315. [DOI] [PubMed] [Google Scholar]
- 18.Roger JE, et al. , OTX2 loss causes rod differentiation defect in CRX-associated congenital blindness. The Journal of clinical investigation, 2014. 124(2): p. 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Campa* C, et al. , The Role of Gene Therapy in the Treatment of Retinal Diseases: A Review. Current Gene Therapy, 2017. 17(3): p. 194–213. [DOI] [PubMed] [Google Scholar]
- 20.Vazquez-Dominguez I, Garanto A, and Collin RWJ, Molecular Therapies for Inherited Retinal Diseases-Current Standing, Opportunities and Challenges. Genes (Basel), 2019. 10(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Das AT, Tenenbaum L, and Berkhout B, Tet-On Systems For Doxycycline-inducible Gene Expression. Current gene therapy, 2016. 16(3): p. 156–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gossen M, et al. , Transcriptional activation by tetracyclines in mammalian cells. Science, 1995. 268(5218): p. 1766–1769. [DOI] [PubMed] [Google Scholar]
- 23.Prasov L and Glaser T, Pushing the envelope of retinal ganglion cell genesis: context dependent function of Math5 (Atoh7). Developmental biology, 2012. 368(2): p. 214–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Öner A, Recent Advancements in Gene Therapy for Hereditary Retinal Dystrophies. Turkish journal of ophthalmology, 2017. 47(6): p. 338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gupta PR and Huckfeldt RM, Gene therapy for inherited retinal degenerations: initial successes and future challenges. Journal of Neural Engineering, 2017. 14(5): p. 051002. [DOI] [PubMed] [Google Scholar]
- 26.Patrício MI, et al. , Inclusion of the Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element Enhances AAV2-Driven Transduction of Mouse and Human Retina. Molecular therapy. Nucleic acids, 2017. 6: p. 198–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Watanabe S, et al. , Tropisms of AAV for Subretinal Delivery to the Neonatal Mouse Retina and Its Application for In Vivo Rescue of Developmental Photoreceptor Disorders. PLOS ONE, 2013. 8(1): p. e54146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Planul A and Dalkara D, Vectors and Gene Delivery to the Retina. Annual Review of Vision Science, 2017. 3(1): p. 121–140. [DOI] [PubMed] [Google Scholar]
- 29.Peng Y, Tang L, and Zhou Y, Subretinal Injection: A Review on the Novel Route of Therapeutic Delivery for Vitreoretinal Diseases. Ophthalmic Research, 2017. 58(4): p. 217–226. [DOI] [PubMed] [Google Scholar]
- 30.Xue K, et al. , Technique of retinal gene therapy: delivery of viral vector into the subretinal space. Eye, 2017. 31(9): p. 1308–1316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Verdera HC, Kuranda K, and Mingozzi F, AAV Vector Immunogenicity in Humans: A Long Journey to Successful Gene Transfer. Molecular Therapy, 2020. 28(3): p. 723–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Millington-Ward S, et al. , Suppression and Replacement Gene Therapy for Autosomal Dominant Disease in a Murine Model of Dominant Retinitis Pigmentosa. Molecular Therapy, 2011. 19(4): p. 642–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cideciyan AV, et al. , Mutation-independent rhodopsin gene therapy by knockdown and replacement with a single AAV vector. Proceedings of the National Academy of Sciences, 2018. 115(36): p. E8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu W and Wu Z, In Vivo Applications of CRISPR-Based Genome Editing in the Retina. Frontiers in cell and developmental biology, 2018. 6: p. 53–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hung SSC, et al. , AAV-Mediated CRISPR/Cas Gene Editing of Retinal Cells In Vivo. Investigative Ophthalmology & Visual Science, 2016. 57(7): p. 3470–3476. [DOI] [PubMed] [Google Scholar]
- 36.Trapani I and Auricchio A, Seeing the Light after 25 Years of Retinal Gene Therapy. Trends in Molecular Medicine, 2018. 24(8): p. 669–681. [DOI] [PubMed] [Google Scholar]
- 37.Suzuki K, et al. , In vivo genome editing via CRISPR/Cas9 mediated homology-independent targeted integration. Nature, 2016. 540(7631): p. 144–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Petit L and Punzo C, Gene therapy approaches for the treatment of retinal disorders. Discovery medicine, 2016. 22(121): p. 221–229. [PMC free article] [PubMed] [Google Scholar]
- 39.Chinskey ND, Besirli CG, and Zacks DN, Retinal cell death and current strategies in retinal neuroprotection. Current Opinion in Ophthalmology, 2014. 25(3). [DOI] [PubMed] [Google Scholar]
- 40.Ozdek S, et al. , Bilateral Disc Edema and Unilateral Macular Hole in a Patient with Retinitis Pigmentosa. European Journal of Ophthalmology, 2006. 16(3): p. 487–490. [DOI] [PubMed] [Google Scholar]
- 41.Griffith JF, DeBenedictis MJ, and Traboulsi EI, A novel dominant CRX mutation causes adult-onset macular dystrophy. Ophthalmic Genetics, 2018. 39(1): p. 120–124. [DOI] [PubMed] [Google Scholar]
- 42.Gonzalez-Gonzalez LA, et al. , Novel clinical presentation of a <em>CRX</em> rod-cone dystrophy. BMJ Case Reports, 2021. 14(4): p. e233711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Paunescu K, et al. , Genotype–Phenotype Correlation in a German Family with a Novel Complex CRX Mutation Extending the Open Reading Frame. Ophthalmology, 2007. 114(7): p. 1348–1357.e1. [DOI] [PubMed] [Google Scholar]
- 44.Lu Q-K, et al. , A novel CRX mutation by whole-exome sequencing in an autosomal dominant cone-rod dystrophy pedigree. International journal of ophthalmology, 2015. 8(6): p. 1112–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]