

Abstract
We present the results of a 6-year study of 131 human immunodeficiency virus (HIV) type 2 (HIV-2)-infected individuals from a rural population in Guinea-Bissau. Proviral DNA sequences 1.3 kb in length were obtained from each individual and, together with clinical data, including proviral load and CD4 and CD8 levels, were used to assess whether viral genotype influences clinical outcome. With a phylogenetic model, a correlation was found between viral genotype and mortality; this correlation was not due to confounding factors, such as age-specific viral strains or cohabitation of patients. The data provide strong evidence for the involvement of viral genetic factors in determining HIV disease progression in vivo. The pattern of association found suggests that virulence factors are multiple and scattered throughout the HIV-2 genome and can be rapidly gained or lost by the virus through a combination of mutation and recombination. These findings may lead to the identification of viral determinants of HIV disease progression.
The majority of individuals infected with human immunodeficiency virus (HIV) type 1 (HIV-1) develop AIDS within 10 years. However, approximately 10% of cases progress rapidly (2 to 3 years) to AIDS, while 5 to 10% of cases are slow or nonprogressive, with patients remaining clinically asymptomatic after 10 years (20, 34). HIV type 2 (HIV-2) is claimed to have a longer incubation period between infection and overt AIDS than HIV-1 (29) but has recently been clearly characterized as having a similar diversity of clinical outcome (36). Among adults under the age of 45 years, the ratio of mortality in HIV-2-infected to uninfected individuals has been shown to be 5:1. However, unlike mortality with HIV-1, which rises with age (6), the mortality ratio for older people with HIV-2 infection is close to 1:1 (36), suggesting a dichotomy between rapid progression to AIDS and slow progression or even nonprogression.
Susceptibility to HIV-1 infection and the rate of progression of disease have been strongly associated with polymorphisms in host genes coding for the CCR5 (11, 32) and CCR2 (39) chemokine receptors, which also function as HIV coreceptors. In addition, there is some effect from host immunoregulatory factors, including the major histocompatibility complex (30). In contrast, although HIV-1 and HIV-2 have been shown to differ in biological properties in vitro (8, 38), little evidence has accumulated concerning the importance of viral genetic factors in determining disease progression in vivo (33, 40, 41). A reason for the current lack of certainty about the existence and role of viral virulence factors is the difficulty in classifying clinical outcome for HIV-1 infection, due to the continuum in the lengths of incubation period between infection and full-blown AIDS (7, 34). In contrast, the clear dichotomy in clinical outcome for HIV-2 infection (36) provides an opportunity to carry out systematic investigations into the relative roles of host versus viral genetic factors in determining the rate of progression to AIDS. Such investigations not only are important for the understanding and control of HIV-2 but also may reveal important features concerning the mechanisms leading to AIDS in chronic retroviral infections. In addition, there is some evidence that HIV-2 infection may protect against HIV-1 infection (2, 19, 23, 42). If viral genetic determinants of nonprogressive clinical outcome could be found for HIV-2, then this may have implications for the development of vaccines against both HIV-1 and HIV-2.
This paper presents results from a prospective study of HIV-2-infected residents of a village in rural Guinea-Bissau who, together with uninfected controls, have been monitored with clinical, virological, and immunological investigations for over 6 years (37, 43). These HIV-2-infected residents have suffered approximately three times the mortality rate of uninfected controls (37) and show a dichotomy in the rate of disease progression (1), in accordance with the findings for a similar cohort of HIV-2-infected individuals monitored in Bissau, the capital of Guinea-Bissau (36). Using novel phylogenetic methods, we investigated whether the mortality rate of HIV-2-infected individuals within this cohort depends upon viral genotype.
MATERIALS AND METHODS
Subjects.
A total of 134 HIV-2-positive subjects were originally entered into a cohort study between March and June 1991 and followed up with annual censuses until 1997 (37). All subjects were of the same ethnic origin, and none had received blood transfusions or blood products. Subjects were examined clinically, and blood samples were taken in both 1991 and 1997 and cryopreserved. The CD4 and CD8 levels in the cryopreserved blood samples taken in 1991 were measured previously (37, 43). In addition, a quantitative PCR was used to assess proviral load (4). Postmortem interviews were conducted with the relatives of all those who died. Lymphocyte DNA from 131 of the 134 HIV-2-positive subjects from whom samples were obtained in 1991 was available for analysis.
PCR amplification and sequencing.
Proviral DNA was extracted from one 200-μl aliquot of frozen lymphocytes obtained in 1991, and the gag, pol, env, and long terminal repeat (LTR) regions were amplified by the PCR. Primer sequences (with International Union of Pure and Applied Chemistry mixed base codes) and positions (in nucleotides) (numbered according to HIV-2ROD) were as follows: Gag 1, AGGTTACGGCCCGGCGGAAAGAAAA (603 to 627); Gag 2, CCTACTCCCTGACAGGCCGTCAGCATTTCTTC (1612 to 1581); Gag 3, GGATTAGCAGAGAGCCTGTTGGA (677 to 700); Gag 4 (44), CCTTAAGCTTTTGTAGAATCTATCTACATA (1466 to 1437); Pol 1, AATATTATAGTAGAYTCACARTATGT (603 to 627); Pol 2, CCWAYYCCYTGRCADGYXGTYARCATYTCYTC (1612 to 1581); Pol 3, TATGTTGCATGGGTCCCAGCCC (3397 to 3992); Pol 4, TTCATTGCTTCCACTACTCC (1279 to 1258); Env 1, TRCTATDARRTTTAGRTAYTG (6807 to 6851); Env 2, TCYCTDGGAGGCAAATAHAC (7340 to 7316); Env 3, TAGRTAYTGTGCACCRCCRGG (6911 to 6935); Env 4, TSCCTACYTTRTGCCAVGTRT (9301 to 9277); LTR 1, TAACCAAGGGAGGGACATGGG (9411 to 9431); LTR 2, TGGTGAGAGTCTAGCAGGG (9592 to 9574); LTR 3, AGGAGCTGGTGGGGAACGCCCT (9432 to 9453); and LTR 4 (5), AACACCCAGGCTCTACCTGCT (9573 to 9553).
A reaction mixture (50 μl) containing 200 to 400 ng of extracted proviral DNA, 1.5 mM MgCl2, 5 μl of 10× reaction buffer (Perkin Elmer), 200 mM each deoxynucleoside triphosphate, 50 pmol of each primer, and 1 U of Taq polymerase (Perkin Elmer) was subjected to hot-start denaturation at 95°C for 3 min followed by 35 cycles of 94°C for 1 min, 40°C for 2 min, and 72°C for 1.5 min and a final cycle of 94°C for 1 min, 40°C for 2 min, and 72°C for 2 min. Nested PCRs were carried out under the same conditions, except that 0.5 μl of the first-round product was added to the reaction mixture and the annealing temperature was raised to 55°C. Nested PCR products were electrophoresed on a 1.5% low-melting-temperature agarose gel, and a product of the correct size was extracted with the Wizard DNA purification system (Promega). The mixture was ethanol precipitated and directly cycle sequenced in both directions on an ABI 377 sequencer with fluorescent dye terminators. The env and LTR products were sequenced with the PCR primers. Additional primers used to sequence gag and pol were as follows: Gag 5, CACGCAGAAGAGAAAGTGAAA (810 to 830); Gag 6, TCTACTGTGCTTGTTGTTCCTG (1279 to 1258); Pol 5, TATGTTGCATGGGTCCCAGCCC (3971 to 3992); and Pol 6, TTCATTGCTTCCACTACTCC (4521 to 4502). In the majority of cases, the consensus sequence was verified by sequencing of products from two separate PCRs.
Phylogenetic trees.
The consensus sequences were aligned with ClustalV (21). Maximum-likelihood (ML) phylogenetic trees were constructed with a test version of PAUP* (d54) made available by David Swofford. Trees were constructed for all sequences independently and for a joint alignment of gag, LTR, and pol sequences. These trees were used when assessing the correlation between viral phenotype (see below) and various clinical features. In the case of any missing individual clinical data, trees were constructed with the relevant number of taxa. The env sequences were not included in a joint alignment, as in only 79 subjects were sequences available for all four regions. The substitution model used to construct the ML phylogenetic trees was F84 (for a description, see reference 16). ML estimates of the transition/transversion ratio were obtained for each tree. In the case of the joint gag, LTR, and pol alignment, different relative substitution rates were allowed for each gene.
Phylogenetic models.
To assess whether there are viral genetic determinants of clinical aspects of HIV-2 infection (which we term the viral phenotype), models of how viral phenotype changes with underlying viral genotype are required. The phenotypic characters modelled in this paper are the status of the infected individual (dead or alive), CD4 and CD8 levels (and their ratio), and viral load. The genotype of the virus is the consensus sequence obtained by direct sequencing from the infected individual and is represented by its position in the phylogenetic tree.
In a very simple model, an HIV-2 isolate can be said to be either benign (found in nonprogressors) or fatal. If the virus changes its state at rate α, then the probability (P) of change between benign and fatal states over any time t is given by
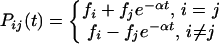 |
where f is the equilibrium frequency of benign or fatal isolates in the viral population. Given that a virus is in state i at time 0 and state j at time t, the likelihood (L) of this situation for a given rate α is calculated simply as L(α,t|ij) = Pij (t).
A measure of the time of separation of two viral isolates is simply the number of substitutions per site that have occurred in their nucleotide sequences, estimated with an appropriate substitution model. Thus, given two viral nucleotide sequences (e.g., the gag gene) and their viral phenotypes, the likelihood for this simple model of phenotypic change for a given rate α can be calculated. If substitutional information about the time of separation of the two viral isolates is discarded and the rate of phenotypic change and time are treated as a compound parameter αt, the ML estimate of this parameter for two viral isolates given their viral phenotypes can be found. For more than two isolates, substitutional information can be used to construct a phylogenetic tree which best represents their relatedness (as is done with PAUP*). The ML for a distribution of benign and fatal isolates on this phylogenetic tree can be found with a “pruning” algorithm (13–15), which relies only upon the accuracy of the reconstructed phylogenetic topology (not branch lengths).
With a similar procedure, the ML for a distribution of continuous phenotypic characters (e.g., CD4 and CD8 levels and viral load) on a given phylogeny can be calculated. The model implemented in this paper is that of Brownian motion (see, e.g., reference 14). In other words, the mean phenotype undergoes a random diffusion on an infinite linear scale. Thus, the probability (P) that a character with value x1 has value x2 after time t follows the normal density function
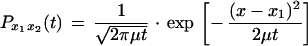 |
where μ is the rate parameter. As for the discrete model, this probability is equivalent to the likelihood L(μ,t|x1,x2) = Px1x2(t). Again, the compound parameter μt is considered and substitutional information is used only to construct a phylogenetic tree relating the sequences. The ML for a distribution of continuous characters on a phylogeny can be calculated with a pruning algorithm similar to that used for discrete characters but in which the length of the internal branch is altered following pruning (14).
The significance of each ML value calculated for the phylogenetic model relating viral genotype to clinical features was assessed by first calculating the MLs for 100 randomizations of the clinical data. The observed ML was then ranked among these likelihoods to obtain a P value. If the observed ML fell within or was better than the best five randomized likelihoods, then it was concluded that the data fit the model significantly better than would be expected by chance at the 5% level.
ML values have limited statistical power in differentiating hypotheses, since even randomized data may have an ML value that is quite high (12). Tests for significance based on ML values therefore tend to be conservative. The likelihood ratio is a far more powerful test statistic but relies on the existence of two competing hypotheses for which ML values can be calculated. An alternative hypothesis against which to test the models of genotype-phenotype correlation described here is difficult to develop. It is possible to use the normal distribution as an alternative hypothesis for the observed continuous characters and the binomial distribution for the discrete characters. However, care would need to be taken in interpreting nonsignificant likelihood ratios (i.e., ratios for which the hypothesis of genotype-phenotype correlation cannot be rejected), since the validity of such results would depend upon the validity of the alternative hypothesis. Thus, at present, only the ML statistic is used.
Investigation of other correlates of mortality.
Possible correlates of mortality, other than viral genotype, were investigated by survival analysis because of their suggested importance in a previous study (36). Factors investigated included subject age, gender, area of residence, spouse HIV-2 status, and activity as a prostitute. A Cox proportional-hazards model was used (9), with time since blood sampling as the underlying time scale. For analyses other than that of age of subject, age was controlled for as a background variable.
Identification of motifs and base changes associated with pathogenic viruses.
For each site in the gag, pol, LTR, and V3 alignments, the minimum number of changes in the respective ML phylogenetic trees necessary to explain the distribution of sequence diversity was calculated with MacClade (28). These changes were then subdivided into those occurring in viral lineages whose descendants were exclusively associated with mortality and those occurring in all other lineages. By use of a program written in C it was possible to obtain plots of the frequencies of nucleotide changes along the aligned sequences similarly subdivided. These plots displayed the site-specific changes associated with mortality against the background of changes occurring in all other lineages and hence allowed identification of any changes and/or motifs associated with mortality. A nonphylogenetic method for detecting “signature patterns” of query sequences was also used to assess whether there is a correlation of certain nucleotides with mortality (27). A signature pattern is comprised of nucleotides for which the ratio of their frequencies in the query sequence alignment and the background alignment is above a certain threshold. A computer program, VESPA (27), implementing this method was used to determine the frequencies of the nucleotides comprising the putative signature of sequences associated with mortality and their frequencies in the alignment of sequences isolated from individuals alive at the end of the follow-up period.
Nucleotide sequence accession numbers.
The following sequences were deposited in the EMBL database under accession no. AJ008441 and AJ011191, 539 and 222 for gag, respectively; AJ008540, 667 for pol; AJ008283 and AJ011223, 316 and 272 for env, respectively; and AJ008317, 440 for the LTR.
RESULTS
Subjects and nucleotide sequences.
During the study, 26 people died. Proviral gag (350 bp), pol (415 bp), env (V3; 357 bp), and LTR (164 bp) sequences were amplified from 123, 127, 84, and 124 patients, respectively. In 117 of the 131 subjects analyzed, including 25 of the 26 patients who died, at least three regions (gag, pol, and LTR) were amplified. The other patient who died and from whom sequences of the gag and V3 regions only were available was a 32-year-old woman who had a CD4 count of 32% and who died 4 days postpartum. Postmortem interviews conducted with the relatives of the other 25 subjects who died could not exclude HIV as a cause of death in all cases, with the possible exception of a 44-year-old man. However, all 25 patients who died were included in the analyses.
All sequences were found to be HIV-2 subtype A, in agreement with a previous analysis of a subset of the gag gene data set (44). The sequences have been deposited in the EMBL sequence database.
Phylogenetic trees.
Figure 1 shows an ML phylogenetic tree for the 117 sequences of the joint alignment of gag, pol, and LTR sequences; sequences isolated from people who subsequently died are marked.
FIG. 1.

ML phylogenetic tree of the HIV-2 isolates for the joint alignment of gag, pol, and LTR sequences, showing sequences isolated from people who subsequently died (marked by black circles). The estimated relative rates of substitution for the three regions of the HIV-2 genome were 1.19, 0.77, and 1.02, respectively. The estimated transition/transversion ratio was 1.65. The scale bar indicates the length of a branch with one substitution per 10 nucleotide sites.
Phylogenetic models.
A correlation was found between the death of HIV-2-infected individuals and viral genetic identity (Table 1). This correlation was highly significant for the joint alignment of LTR, gag, and pol sequences (P, <0.01) but became nonsignificant when the gag, pol, and LTR genes were examined individually (P, 0.45, 0.07, and 0.07, respectively). This result may reflect the increased phylogenetic resolution provided by use of a joint alignment with appropriate models for each gene (22) (although this effect may be counteracted by recombination occurring between the genes). A previous cohort study of HIV-2-infected individuals showed that deaths early in the follow-up period are more likely to involve rapid progressors (36). This trend was recently confirmed for the cohort studied here, with no excess mortality occurring in the HIV-2-positive cohort after 1995 (mortality rate ratio, 0.94, versus 3.38 before 1995 [1]). Analysis of only those deaths occurring prior to 1995 revealed a significant correlation with viral genotype for all but the gag sequences (Table 1).
TABLE 1.
Log likelihoods for a model of clinical outcome dependent upon viral genotype represented by different genes for a range of clinical features, together with P values denoting the probability of obtaining a likelihood this good by chancea
| Gene or region | Log likelihood (P) for:
|
||||||
|---|---|---|---|---|---|---|---|
| All deaths (n = 25) | Pre-1995 deaths (n = 15) | Deaths of those less than 65 yr old (n = 16) | % CD4 level | % CD8 level | Ratio of % CD4 level to % CD8 level | Viral load | |
| gag | −35.95 (0.45) | −26.07 (0.06) | −27.53 (0.07) | −141.92 (0.31) | −220.23 (0.41) | −244.59 (0.91) | −457.23 (0.55) |
| pol | −32.36 (0.07) | −21.69 (<0.01)b | −27.17 (0.10) | −175.62 (0.96) | −250.15 (0.91) | −142.85 (0.08) | −621.47 (0.10) |
| LTR | −30.36 (0.07) | −25.60 (0.05)b | −28.94 (0.35) | −144.78 (0.24) | −120.01 (0.17) | −256.39 (0.32) | −621.53 (0.36) |
| Joint alignment | −23.00 (<0.01)b | −17.26 (<0.01)b | −22.33 (<0.01)b | −138.61 (0.36) | −177.68 (0.26) | −143.07 (0.33) | −584.38 (0.89) |
P values were obtained by ranking the actual likelihood among the likelihoods of 100 randomizations of the clinical data for each analysis (see Materials and Methods for details). A less negative log likelihood indicates a better model fit. However, the likelihood values are not comparable across the table owing to differences in phylogenetic topology and slight variations in the number of people included in the analysis due to missing data (see the text). n, number of deaths included in the analysis of mortality.
The P value was considered significant at the 5% level.
No correlation was found between mortality and the V3 region of the envelope gene (P was 0.5 for both the analysis of all deaths and the analysis of deaths prior to 1995). This result may reflect the smaller number of V3 sequences than of gag, pol, and LTR sequences available for analysis. It may also reflect a lack of statistical power of the ML statistic.
It was possible to use these sequences to predict the in vitro replication phenotype of the HIV-2 isolates, since this phenotype was recently shown to correlate with certain amino acid changes in the V3 loop (3) as has been previously demonstrated for HIV-1 (18). The syncytium-inducing phenotype was, however, suggested by amino acid changes in only two subjects and was not found to correlate with mortality (χ2 test of independence: χ2, 0.683; P, 0.41). These results suggest the importance of some other viral virulence factor(s) and confirm the observation that the syncytium-inducing phenotype tends to be more prevalent only close to the onset of AIDS (26, 38).
No correlation was found between viral genotype and high proviral load or low CD4 count (Table 1), despite previous findings of a relationship between the latter two and progression to AIDS in HIV-2-infected people (4). This result may be explained by the fact that of the 25 deaths included in our analysis, only 15 were associated with a high viral load (>100 copies/105 CD4 cells) and low CD4 levels (<29%) at the time of blood sampling (in accordance with previous definitions for these categories [37]). Furthermore, the levels of HIV-2 provirus varied considerably, and the mean level was high even in subjects with high CD4 levels. Thus, it appears that these markers for disease progression are not consistent if the onset of AIDS is not imminent. A more suitable marker allowing early prognosis of the rate of progression to death in HIV-2-positive individuals may therefore be plasma viral load, as is the case for HIV-1 (31).
Investigation of other correlates of mortality.
Using the Cox proportional-hazards model (9), we found no correlation between mortality and subject gender, area of residence, spouse HIV-2 status, or prostitution (P, 0.385, 0.984, 0.056, and 0.433, respectively). A significant correlation was found, however, between mortality and the age of HIV-2-positive individuals when the latter was treated as a continuous variable (mortality rate ratio, 1.06 per year [95% confidence interval, 1.03 to 1.09]; P, <0.001). Further analysis revealed that, although mortality was higher in those over 65 years old, there was no increase in mortality with age in those under 65 years old (test for trend: P, 0.593) (Table 2). This finding agrees with the dichotomy in clinical progression for HIV-2 infection (36) and previous observations for the same community (37).
TABLE 2.
Mortality of HIV-2-positive individuals subdivided according to age group relative to the oldest age group
| Age (yr) | Mortality rate ratio | 95% Confidence interval | P |
|---|---|---|---|
| 15–34 | 0.27 | 0.08–0.93 | 0.039 |
| 35–54 | 0.15 | 0.04–0.52 | 0.003 |
| 55–64 | 0.17 | 0.05–0.58 | 0.005 |
| ≥65 | 1 |
Identification of motifs and base changes associated with pathogenic viruses.
Consistent patterns of base changes in lineages leading to the pathogenic viruses were not found by use of the phylogenetic approach described. Similarly, no signature patterns were found to define the sequences associated with mortality. Table 3 shows the putative signature nucleotides of pathogenic sequences and their frequencies in both these pathogenic sequences and sequences isolated from patients alive at the end of the follow-up period. These frequencies are not very different, indicating no clear correlation between a particular viral motif or single nucleotide change and mortality in the regions sequenced. This finding probably reflects the fact that a relatively small proportion (1,300 bases) of the entire HIV-2 genome (∼10,000 bases) was sequenced.
TABLE 3.
Putative signature patterns for the V3, gag, LTR, and pol sequences associated with mortality, as detected with VESPA (27)
| Signature | Position | Proportion of individuals
|
|
|---|---|---|---|
| Dead | Alive | ||
| V3 | |||
| T | 81 | 0.52 | 0.48 |
| A | 208 | 0.52 | 0.30 |
| G | 216 | 0.33 | 0.14 |
| G | 330 | 0.57 | 0.48 |
| A | 336 | 0.57 | 0.43 |
| A | 355 | 0.62 | 0.43 |
| gag | |||
| G | 114 | 0.56 | 0.45 |
| A | 117 | 0.52 | 0.35 |
| A | 143 | 0.48 | 0.44 |
| T | 190 | 0.64 | 0.49 |
| A | 444 | 0.44 | 0.41 |
| LTR | |||
| T | 34 | 0.56 | 0.44 |
| T | 71 | 0.52 | 0.24 |
| pol | |||
| G | 140 | 0.52 | 0.39 |
| A | 192 | 0.56 | 0.33 |
DISCUSSION
The data reported here reveal a correlation between viral genotype and mortality in HIV-2 infection in vivo. Furthermore, the fact that no distinct viral phylogenetic group (clade) is associated with mortality (Fig. 1) suggests that several viral virulence factors found within or linked to the gag, pol, and LTR regions sequenced can be gained or lost by the virus. Because such motifs were not detected within the sequences analyzed with the methods described here, linkage seems the most likely explanation.
It is possible that the correlation was confounded by other factors, which may covary in some way with both viral genotype and mortality. Gender, area of residence, spouse HIV-2 status, and prostitution, previously suggested as risk factors (36), were all found not to correlate with mortality. However, age was found to correlate with the mortality of HIV-2-positive individuals when treated as a continuous variable (P, <0.001). In addition, age was found to correlate with viral genotype for the gag and LTR sequences when assessed with the likelihood methods described previously (P, 0.02 and 0.03, respectively). It is therefore possible that certain viral strains were circulating at certain times, infecting specific age groups when they were sexually active, and that the death of older subjects caused a correlation between mortality and viral genotype. However, although mortality was higher in those over 65 years old, there was no correlation between mortality and age in those under 65 years old (Table 2). Since consideration of the deaths of only people under 65 years old still resulted in a significant correlation with viral genotype (Table 1), age could not be confounding the in vivo correlation between viral genotype and mortality.
The possibility that discrete viral genetic elements are associated with mortality in HIV-2 infections is supported by experimental evidence derived from strains of simian immunodeficiency virus. Acquisition of virulence by some strains of simian immunodeficiency virus has been associated with changes occurring in the env (17, 24, 35), gag (25, 35), tat (17), and nef (35) genes and the LTR (24, 35). Furthermore, deletions in the HIV-1 nef gene have been described for a cohort of unrelated long-term nonprogressors who received transfusions from a single donor, supporting the notion that viral determinants may influence disease progression (10).
In conclusion, this paper provides evidence for heritable viral virulence factors important in determining the rate of disease progression in vivo. These results are compatible with the role of other determinants of virulence, including both host factors and viral genetic changes that may occur within individual hosts (intrahost evolution). Given the rapid mutation rate and turnover of HIV-2 within humans, the latter may be of importance, in particular with regard to escape from immune recognition or the use of a broader range of receptors. However, the recognition of heritable virulence factors now prompts the need for their precise identification and further research toward an understanding of the mechanisms by which they are acquired and interact with host determinants of disease progression.
ACKNOWLEDGMENTS
We are indebted to the people who took part in the community-based study and to Andrew Wilkins for his contribution to the design of the study. We also thank Pedro Biri Gomes for additional clinical data, Thiru Surentheran for technical help, and Eddie Holmes, Paul Harvey, and Robin Weiss for constructive criticism regarding the manuscript.
This work was supported by the MRC (J.B.) and BBSRC (grants to N.C.G.).
REFERENCES
- 1.Aaby, P., H. Jensen, and M. Schim van der Loeff. Unpublished data.
- 2.Aaby P, Poulsen A G, Larsen O, Christiansen C B, Jensen H, Melbye M, Naucler A, Dias F. Does HIV-2 protect against HIV-1 infection? AIDS. 1997;11:939–940. [PubMed] [Google Scholar]
- 3.Albert J, Stalhandske P, Marquina S, Karis J, Fouchier R A M, Norrby E, Chiodi F. Biological phenotype of HIV type-2 isolates correlates with V3 genotype. AIDS Res Hum Retroviruses. 1996;12:821–828. doi: 10.1089/aid.1996.12.821. [DOI] [PubMed] [Google Scholar]
- 4.Ariyoshi K, Berry N, Wilkins A, Ricard D, Aaby P, Naucler A, N’Gum P T, Jobe O, Jaffar S, Dias F, Tedder R S, Whittle H. A community-based study of human immunodeficiency virus type 2 provirus load in a rural village in West Africa. J Infect Dis. 1996;173:245–248. doi: 10.1093/infdis/173.1.245. [DOI] [PubMed] [Google Scholar]
- 5.Berry N, Ariyoshi K, Jobe O, N’Gum P T, Corrah T, Wilkins A, Whittle H, Tedder R. HIV type-2 proviral load measured by quantitative polymerase chain reaction correlates with CD4+ lymphopenia in HIV type 2 infected individuals. AIDS Res Hum Retroviruses. 1994;10:1031–1037. doi: 10.1089/aid.1994.10.1031. [DOI] [PubMed] [Google Scholar]
- 6.Biggar R J. AIDS incubation in 1891 HIV seroconverters from different exposure groups. AIDS. 1990;4:1059–1066. doi: 10.1097/00002030-199011000-00002. [DOI] [PubMed] [Google Scholar]
- 7.Cao Y Z, Qin L M, Zhang L Q, Safrit J, Ho D D. Virological and immunological characterization of long-term survivors of human immunodeficiency virus type-1 infection. N Engl J Med. 1995;332:201–208. doi: 10.1056/NEJM199501263320401. [DOI] [PubMed] [Google Scholar]
- 8.Cheng-Mayer C, Seto D, Tateno M, Levy J A. Biological features of HIV-1 that correlate with virulence in the host. Science. 1988;240:80–82. doi: 10.1126/science.2832945. [DOI] [PubMed] [Google Scholar]
- 9.Cox D R, Oakes D. Analysis of survival data. London, England: Chapman & Hall, Ltd.; 1984. [Google Scholar]
- 10.Deacon N J, Tsykin A, Solomon A, Smith K, Ludfordmenting M, Hooker D J, McPhee D A, Greenway A L, Ellett A, Chatfield C, Lawson V A, Crowe S, Maerz A, Sonza S, Learmont J, Sullivan J S, Cunningham A, Dwyer D, Dowton D, Mills J. Genomic structure of an attenuated quasi-species of HIV-1 from a blood transfusion donor and recipients. Science. 1995;270:988–991. doi: 10.1126/science.270.5238.988. [DOI] [PubMed] [Google Scholar]
- 11.Dean M, Carrington M, Winkler C, Huttley G A, Smith M W, Allikmets R, Goedert J J, Buchbinder S P, Vittinghoff E, Gomperts E, Donfield S, Vlahov D, Kaslow R, Saah A, Rinaldo C, Detels R, O’Brien S J. Genetic restriction of HIV-1 infection and progression to AIDS by a deletion allele of the CKR5 structural gene. Science. 1996;273:1856–1862. doi: 10.1126/science.273.5283.1856. [DOI] [PubMed] [Google Scholar]
- 12.Edwards A W F. Likelihood (extended ed.). London, England: The John Hopkins University Press; 1992. [Google Scholar]
- 13.Felsenstein J. Evolutionary trees from DNA sequences: a maximum likelihood approach. J Mol Evol. 1981;17:368–376. doi: 10.1007/BF01734359. [DOI] [PubMed] [Google Scholar]
- 14.Felsenstein J. Evolutionary trees from gene frequencies and quantitative characters: finding maximum likelihood estimates. Evolution. 1981;35:1229–1242. doi: 10.1111/j.1558-5646.1981.tb04991.x. [DOI] [PubMed] [Google Scholar]
- 15.Felsenstein J. Maximum-likelihood estimation of evolutionary trees from continuous characters. Am J Hum Genet. 1973;25:471–492. [PMC free article] [PubMed] [Google Scholar]
- 16.Felsenstein J, Churchill G A. A hidden Markov model approach to variation among sites in rate of evolution. Mol Biol Evol. 1996;13:93–104. doi: 10.1093/oxfordjournals.molbev.a025575. [DOI] [PubMed] [Google Scholar]
- 17.Flaherty M T, Hauer D A, Mankowski J L, Zink M C, Clements J E. Molecular and biological characterization of a neurovirulent molecular clone of simian immunodeficiency virus. J Virol. 1997;71:5790–5798. doi: 10.1128/jvi.71.8.5790-5798.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fouchier R A M, Groenink M, Kootstra N A, Tersmette M, Huisman H G, Miedema F, Schuitemaker H. Phenotype-associated sequence variation in the third variable domain of the human immunodeficiency virus type 1 gp120 molecule. J Virol. 1992;66:3183–3187. doi: 10.1128/jvi.66.5.3183-3187.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Greenberg A E, Wiktor S Z, Decock K M, Smith P, Jaffe H W, Dondero T J. HIV-2 and natural protection against HIV-1 infection. Science. 1996;272:1959. [PubMed] [Google Scholar]
- 20.Haynes B F, Pantaleo G, Fauci A S. Towards an understanding of the correlates of protective immunity to HIV infection. Science. 1996;271:324–328. doi: 10.1126/science.271.5247.324. [DOI] [PubMed] [Google Scholar]
- 21.Higgins D G, Bleasby A J, Fuchs R. ClustalV: improved software for multiple sequence alignment. Comput Appl Biosci. 1992;8:189–191. doi: 10.1093/bioinformatics/8.2.189. [DOI] [PubMed] [Google Scholar]
- 22.Huelsenbeck J P, Bull J J, Cunningham C W. Combining data in phylogenetic analysis. Trends Ecol Evol. 1996;11:152–158. doi: 10.1016/0169-5347(96)10006-9. [DOI] [PubMed] [Google Scholar]
- 23.Kanki P J, Eisen G, Travers K U, Marlink R G, Essex M E, Hsieh C C, Mboup S. HIV-2 and natural protection against HIV-1 infection—response. Science. 1996;272:1959–1960. doi: 10.1126/science.272.5270.1959b. [DOI] [PubMed] [Google Scholar]
- 24.Karlsson G B, Halloran M, Li J, Park I W, Gomila R, Reimann K A, Axthelm M K, Iliff S A, Letvin N L, Sodroski J. Characterization of molecularly cloned simian human immunodeficiency viruses causing rapid CD4+ lymphocyte depletion in rhesus monkeys. J Virol. 1997;71:4218–4225. doi: 10.1128/jvi.71.6.4218-4225.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kimata J T, Overbaugh J. The cytopathicity of a simian immunodeficiency virus Mne variant is determined by mutations in gag and env. J Virol. 1997;71:7629–7639. doi: 10.1128/jvi.71.10.7629-7639.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Koot M, Keet I P M, Vos A H V, Degoede R E Y, Roos M T L, Coutinho R A, Miedema F, Schellekens P T A, Tersmette M. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4+ cell depletion and progression to AIDS. Ann Intern Med. 1993;118:681–688. doi: 10.7326/0003-4819-118-9-199305010-00004. [DOI] [PubMed] [Google Scholar]
- 27.Korber B, Myers G. Signature pattern analysis: a method for assessing viral sequence relatedness. AIDS Res Hum Retroviruses. 1992;8:1549–1560. doi: 10.1089/aid.1992.8.1549. [DOI] [PubMed] [Google Scholar]
- 28.Maddison W P, Maddison D R. MacClade, 3.05 ed. Sunderland, Mass: Sinauer Associates, Inc.; 1995. [Google Scholar]
- 29.Marlink R, Kanki P, Thior I, Travers K, Eisen G, Siby T, Traore I, Hsieh C C, Dia M C, Gueye E, Hellinger J, Gueyendiaye A, Sankale J L, Ndoye I, Mboup S, Essex M. Reduced rate of disease development after HIV-2 infection as compared to HIV-1. Science. 1994;265:1587–1590. doi: 10.1126/science.7915856. [DOI] [PubMed] [Google Scholar]
- 30.McNeil A J, Yap P L, Gore S M, Brettle R P, McColl M, Wyld R, Davidson S, Weightman R, Richardson A M, Robertson J R. Association of HLA types A1-B8-Dr3 and B27 with rapid and slow progression of HIV disease. QJM Monthly J Assoc Physicians. 1996;89:177–185. doi: 10.1093/qjmed/89.3.177. [DOI] [PubMed] [Google Scholar]
- 31.Mellors J W, Rinaldo C R, Gupta P, White R M, Todd J A, Kingsley L A. Prognosis in HIV-1 infection predicted by the quantity of virus in plasma. Science. 1996;272:1167–1170. doi: 10.1126/science.272.5265.1167. [DOI] [PubMed] [Google Scholar]
- 32.Michael N L, Chang G, Louie L G, Mascola J R, Dondero D, Birx D L, Sheppard H W. The role of viral phenotype and CCR-5 gene defects in HIV-1 transmission and disease progression. Nat Med. 1997;3:338–340. doi: 10.1038/nm0397-338. [DOI] [PubMed] [Google Scholar]
- 33.Mocroft A, Sabin C A, Phillips A N. Lower prevalence and incidence of HIV-1 syncytium-inducing phenotype among injecting drug users relative to homosexual men—a response. AIDS. 1996;10:344–345. doi: 10.1097/00002030-199603000-00020. [DOI] [PubMed] [Google Scholar]
- 34.Munoz A, Kirby A J, He Y D, Margolick J B, Visscher B R, Rinaldo C R, Kaslow R A, Phair J P. Long-term survivors with HIV-1 infection: incubation period and longitudinal patterns of CD4+ lymphocytes. J AIDS Hum Retrovirol. 1995;8:496–505. doi: 10.1097/00042560-199504120-00010. [DOI] [PubMed] [Google Scholar]
- 35.Novembre F J, Johnson P R, Lewis M G, Anderson D C, Klumpp S, McClure H M, Hirsch V M. Multiple viral determinants contribute to pathogenicity of the acutely lethal simian immunodeficiency virus SIVsmmPBj variant. J Virol. 1993;67:2466–2474. doi: 10.1128/jvi.67.5.2466-2474.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Poulsen A-G, Aaby P, Larsen O, Jensen H, Naucler A, Lisse I M, Christiansen C B, Dias F, Melbye M. 9-year HIV-2-associated mortality in an urban community in Bissau, West Africa. Lancet. 1997;349:911–914. doi: 10.1016/S0140-6736(96)04402-9. [DOI] [PubMed] [Google Scholar]
- 37.Ricard D, Wilkins A, N’Gum P T, Hayes R, Morgan G, Da Silva A P, Whittle H. The effects of HIV-2 infection in a rural area of Guinea-Bissau. AIDS. 1994;8:977–982. doi: 10.1097/00002030-199407000-00016. [DOI] [PubMed] [Google Scholar]
- 38.Schulz T F, Whitby D, Hoad J G, Corrah T, Whittle H, Weiss R A. Biological and molecular variability of human immunodeficiency virus type 2 isolates from The Gambia. J Virol. 1990;64:5177–5182. doi: 10.1128/jvi.64.10.5177-5182.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Smith M W, Dean M, Carrington M, Winkler C, Huttley G A, Lomb D A, Goedert J J, O’Brien T R, Jacobson L P, Kaslow R, Buchbinder S, Vittinghoff E, Vlahov D, Hoots K, Hilgartner M W, O’Brien S J. Contrasting genetic influence of CCR2 and CCR5 variants on HIV-1 infection and disease progression. Science. 1997;277:959–965. doi: 10.1126/science.277.5328.959. [DOI] [PubMed] [Google Scholar]
- 40.Soto-Ramirez L E, Renjifo B, McLane M F, Marlink R, O’Hara C, Sutthent R, Wasi C, Vithayasai P, Vithayasai V, Apichartpiyakul C, Auewarakul P, Cruz V P, Chui D S, Osathanondh R, Mayer K, Lee T H, Essex M. HIV-1 Langerhans’ cell tropism associated with heterosexual transmission of HIV. Science. 1996;271:1291–1293. doi: 10.1126/science.271.5253.1291. [DOI] [PubMed] [Google Scholar]
- 41.Spijkerman I J B, Koot M, Prins M, Keet I P M, Vandenhoek A, Miedema F, Coutinho R A. Lower prevalence and incidence of HIV-1 syncytium-inducing phenotype among injecting drug users compared with homosexual men. AIDS. 1995;9:1085–1092. doi: 10.1097/00002030-199509000-00016. [DOI] [PubMed] [Google Scholar]
- 42.Travers K, Mboup S, Marlink R, Gueyendiaye A, Siby T, Thior I, Traore I, Diengsarr A, Sankale J L, Mullins C, Ndoye I, Hsieh C C, Essex M, Kanki P. Natural protection against HIV-1 infection provided by HIV-2. Science. 1995;268:1612–1615. doi: 10.1126/science.7539936. [DOI] [PubMed] [Google Scholar]
- 43.Wilkins A, Ricard D, Todd J, Whittle H, Dias F, Da Silva A P. The epidemiology of HIV infection in a rural area of Guinea-Bissau. AIDS. 1993;7:1119–1122. doi: 10.1097/00002030-199308000-00015. [DOI] [PubMed] [Google Scholar]
- 44.Xiang Z, Ariyoshi K, Wilkins A, Dias F, Whittle H, Breuer J. HIV type 2 pathogenicity is not related to subtype in rural Guinea Bissau. AIDS Res Hum Retroviruses. 1997;13:501–505. doi: 10.1089/aid.1997.13.501. [DOI] [PubMed] [Google Scholar]