

Abstract
Type I positive allosteric modulators (PAMs) of the alpha7-nicotinic receptor enhance its cholinergic activation, while preserving the spatiotemporal features of synaptic transmission and the receptor’s characteristic rapid desensitization kinetics. Alpha7-nicotinic receptor agonists have shown promise for improving cognition in schizophrenia, but longer term trials have been disappointing. Therefore the type I PAM, AVL-3288, was evaluated for safety and preliminary evidence of neurocognitive effect in healthy human subjects as an alternative to direct agonists and a foundation for possible studies in schizophrenia. Single dose oral administration in ascending doses was conducted in a double-blind, placebo-controlled Phase 1 trial in non-smokers. To determine whether nicotine would alter the safety or effect of AVL-3288 a subsequent trial in smokers with concurrent nicotine replacement was conducted. The trial found evidence of effect on neurocognition at 10 and 30 mg, two doses that produced overlapping peak levels. There was also some evidence for effect on inhibition of the P50 auditory evoked potential to repeated stimuli, a biomarker that responds to alpha7-nicotinic receptor activation. A lower dose 3 mg had no effect. The pharmacokinetic characteristics were consistent between subjects, and there were no safety concerns. The effects and safety profile were also assessed at 3 mg in a cohort of smokers. Concurrent nicotine administration did not alter either effect or safety, but did modestly increase plasma levels compared to non-smokers. The trial demonstrates that a type I PAM can be safely administered to humans and that it may have positive neurocognitive effects in CNS disorders.
Keywords: receptors, nicotinic, allosteric modulators, cognition, drug evaluation, schizophrenia
Introduction
The alpha7-nicotinic cholinergic receptor has received increasing interest as a target for drug development in schizophrenia, because of its genetic association with sensory gating deficits and neurocognitive deficits (Freedman 2014; Dempster et al. 2006). Nicotine itself has neurocognitive effects in schizophrenia, but they are severely limited by desensitization, particularly in smokers with high chronic levels of nicotine (Harris et al. 1994). The alpha7-nicotinic receptor is a difficult target because it is readily desensitized by exposure to agonists (Peng et al. 1994). Three agonists in commercial development have had successful Phase 2 trials, but Phase 3 results have been equivocal (Lieberman et al. 2013, Haig et al. 2016; Keefe et al. 2015). We have demonstrated neurocognitive effects of the partial agonist 3-(2,4-dimethoxybenzylidene)-anabaseine after single-dose administration using the Repeatable Battery for the Assessment of Neurocognitive Status (RBANS) and with 1-month administration in the Attention and Executive Function Domains of the MATRICS Consensus Cognitive Battery (Olincy et al. 2006; Freedman et al. 2008). Compared to half-lives of many hours in commercially developed drugs, DMXB-A has an exceptionally short half-life, less than 90 minutes, which may minimize the impact of receptor desensitization (Kitigawa et al. 2003).
Unlike direct receptor agonists, allosteric modulators preserve the spatiotemporal integrity of neurotransmission and thus reduce the influence of this factor as potential confounds (Grolien et al. 2007). This paper reports the first-in-human Phase 1 study of the type I positive allosteric modulator (PAM) AVL-3288 in healthy subjects. The type II PAM JNJ39393406 was not effective in its initial trial, using similar assessment to that reported here (Winterer et al. 2013). Unlike type II PAMs, type I PAMs do not disrupt the kinetics of desensitization of the alpha7-nicotinic acetylcholine receptor (Ng et al. 2007). The desensitization of this receptor is believed to be a use-dependent, readily reversible type of signal plasticity that may shape synaptic efficacy in various brain regions and protects neurons from excessive excitation by tight control of cytosolic calcium homeostasis (Quick and Lester 2002: Giniatullin et al. 2005). Thus any disruption of desensitization may also cloud the observation of any potential therapeutic response to alpha7-nicotinic receptor modulation. The unique attributes of AVL-3288 as a type I PAM, including evidence for efficacy in multiple preclinical models of cognitive dysfunction (Thomsen et al., 2011; Nikiforuk et al., 2015, 2016) and a benign side-effect profile, render it an ideal PAM for clinical evaluation of the potential of the alpha7-nicotnic receptor as a drug target in CNS disorders that may stem in part from loss of function related to this receptor.
Methods & Materials
Active pharmaceutical ingredient
The active pharmaceutical ingredient is N-(4-Chlorophenyl)-a-[[(4-chlorophenyl) amino]methylene]-3-methyl-5 isoxazoleacetamide, also known as XY-4083 (Xytis, Inc., Irvine, CA) and then as AVL-3288 (Anvyl LLC, Irvine, CA). It was designated compound 6 in Ng et al. (2007) or CCMI (Hu et al., 2009; Nikiforuk et al., 2015, 2016). The preparation of GMP AVL-3288 started with commercially available 3-methyl-5-isoxazoleacetic acid. Reaction with oxalyl chloride afforded the corresponding acid chloride which was treated with 4-chloroaniline and base. The resulting amide was treated with base and then with ethyl formate. The hydroxymethylene intermediate was condensed with 4-chloroaniline to afford crude AVL-3288 as a mixture of geometrical isomers. The crude material was purified by recrystallization from acetonitrile and from ethanol to yield the active pharmaceutical ingredient.
Drug formulation
Because of the low aqueous solubility of AVL-3288, it was dissolved with heating in 91% polyethylene glycol and 9% Solutol HS15 (Polyethylene glycol 15hyroxysterate; Mutchler, Inc. Harrington, PA), with Red #40 coloring, bitterness masking (Fona International, Geneve, IL), and peppermint oil. The administered amount was 3 ml for each dose.
Additional preclinical study
PAMs have the theoretical potential of increasing the toxicity of nicotine if the channel were to remain open longer than expected. While that possibility is minimized with a type I PAM, the initial preclinical testing by Xytis as XY-4083 had not verified toxicity with combined nicotine and AVL-3288 administration. Therefore 6 rats, 3 males and 3 females, received AVL-3288 o.d. at 0.3 mg/kg i.p. with 100 mcg/ml nicotine in drinking water for 30 days. The research was prospectively reviewed by a duly constituted institutional animal care and use committee and adheres to the Guide for the Care and Use of Laboratory Animals, 8th edition, National Academic Press, Washington, D.C. http://oacu.od.nih.gov/guide/guide.pdf. Nicotine plasma levels were 25.7 ± 9.0 ng/ml (sem) in males and 14.7 ± 4.4 ng/ml in females; cotinine levels were 419 ± 21 ng/ml in males and 270 ± 15 ng/ml in females. No cerebral pathology or other pathology ascribable to drug administration was found (Pathology Service, Division of Laboratory Animal Medicine, UCLA). Control groups received vehicle, nicotine alone, and AVL-3288 alone.
Subjects
Normal healthy males and females age 18–50 years in equal numbers for each dose cohort were recruited from the community. After informed consent, they were screened for the absence of significant psychiatric or medical illness. Females had an assured method of birth control. All subjects had negative urine toxicology, normal clinical laboratory values, and a normal electrocardiogram. Non-smoking was verified by carbon monoxide levels. Smokers attested to regular use of tobacco cigarettes.
Assessments
Subjects received baseline RBANS and P50 auditory evoked-potential recording, and a side effect questionnaire prior to admission and then during AVL-3288 administration. The validity of the RBANS for the assessment of neuropsychological status in schizophrenia has been previously established (Gold et al. 1999). Methods for these assessments have been previously reported (Gold et al., 1999, Olincy et al. 2006, 2010). The inhibition of the P50 auditory evoked-potential to a repeated stimulus is measured as the ratio of the second response amplitude to the first. Alpha7-nicotinic receptors activate cerebral interneurons which increase inhibition of the response to stimuli and thereby decrease the P50 ratio (Freedman, 2014).
Clinical trial
The trial was conducted at the University of Colorado under FDA IND 116,103 and sponsored by the National Institute of Mental Health (NIMH U01MH094247) June 1, 2013-November 1, 2015. The trial was registered at ClinicalTrials.gov (NCT01851603) and prospectively reviewed and approved by the Western IRB (20121598) under the authority of the Colorado Multi-Institutional Review Board. The Clinical Trials Operation Branch of NIMH (DSMB-C) monitored the trial quarterly. During drug administration, subjects were admitted for 12 hr to the Clinical Trials Research Center, University of Colorado Hospital (NCATS UL1TR001082).
The subjects were administered the formulated AVL-3288. Venous blood specimens, vital signs, and ECGs were obtained at the intervals shown in Figure 1. At 3 hours, the predicted time of maximum dose, the subjects received the RBANS, followed by P50 recording at 4 hours. The subjects were monitored for side effects for 4 weeks after drug administration.
Figure 1.
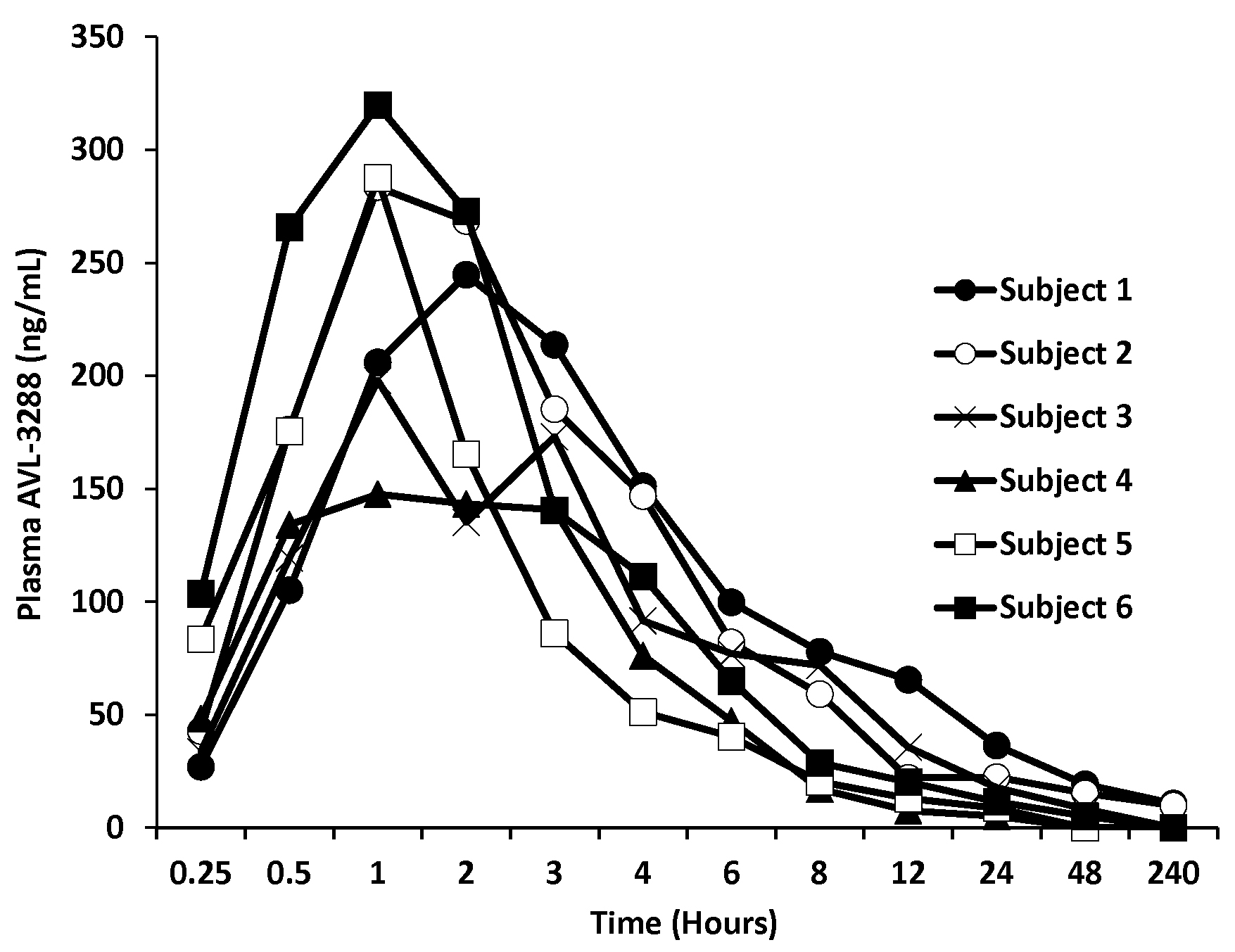
AVL-3288 plasma levels for individual subjects after 10 mg dose.
In preclinical studies, the rat was the most sensitive species, with a no observed adverse effect level (NOAEL) at 40 mg/kg/day, equivalent to 384 mg in humans. Based on animal model experiments, the predicted effective dose was 3 mg, which was therefore the starting dose. The dose was increased in successive cohorts (3 mg, 10 mg, 30 mg) because of the absence of significant adverse effects or toxicity. The trial was stopped in non-smoking subjects at the 30 mg dose, because the predicted plasma level at 90 mg would have approached the level found for inhibition of hERG IC50 in vitro (3 mcM) as well as exceeded the anticipated levels required for efficacy in preclinical models by over an order of magnitude (Ng et al., 2007). A single cohort of smoking subjects received AVL-3288 at 3 mg/kg. During the 12 hr in the Hospital they received transdermal nicotine as needed.
Each cohort was initiated by 2 subjects, one receiving drug and one placebo. If there were no adverse effects, the cohort was extended to 8 subjects total, 6 drug and 2 placebo. Randomization was conducted by a statistician, who along with the pharmacist were the only study personnel aware of treatment assignment.
Bioanalytical Analysis
Plasma samples were analyzed using LCMSMS. The samples were prepared using simple protein precipitation and cleanup using Phree phospholipid removal plates (Phenomenex, Torrance, CA). The chromatographic separation was performed using a Luna, C8, 2.0 × 150 mm 5 μm column (Phenomenex) with 75:25 Acetonitrile:0.1% Formic Acid in Water mobile phase at a flow rate of 0.3 mL/min at a temperature of 40°C with an Agilent 1100/1200 HPLC and an Agilent 6410B QQQ Mass Spectrometer. AVL-3288 was quantified using DMRM with 388>261. The mass spectrometer parameters were: FV 100, CE 8, with positive ion monitoring.
Data Analysis
The pharmacokinetic data were analyzed by standard methods using Agilent Mass Hunter Software Version 4.0, SAS JMP Version 7 and Microsoft Excel. The NIMH (DSMB-C) determined that there were no safety concerns. The primary efficacy outcome was the RBANS Total Scale Score. The P50 ratio was a secondary outcome, intended to assess whether AVL-3288 had its intended effect of increasing alpha7-nicotinic receptor activation. Both outcomes were measured as change during AVL-3288 compared to baseline. Both efficacy outcomes were considered exploratory because the cohorts lacked sufficient power.
Results
Conduct of the Trial
Forty subjects were screened, 38 were found eligible, and 32 entered the trial. After enrollment, all subjects completed the trial (Table 1).
Table 1.
Demographics of enrolled subjects
| Sex | Male 50% |
|---|---|
| Age | 3.2 ± 9.9 years |
| Caucasian non-Hispanic | 67% |
| Asia | 16% |
| Hispanic | 16% |
| Education | 15.4 ± 2.5 years |
Pharmacokinetics of AVL-3288
AVL-3288 was well absorbed, with peak blood levels reached in 1 to 2 hours (Figure 1). The half-life of the primary phase of elimination was 3 hours (Table 2). The plasma level appeared to be approaching a plateau value at the 30 mg dose (Figure 2).
Table 2.
Pharmacokinetic parameters of AVL-3288
| Kel1 (Hr1) | Kel1 (Hr1) | Kabs (Hr1) | AUC (ng/mL•Hr) | Calculated Half-Life Kel1 (Hr) | Calculated Half-Life Kel2 (Hr) | Observed Cmax (ng/mL) | Calculated Tmax (Hr) | |
|---|---|---|---|---|---|---|---|---|
| 3 mg mean | 0.26 | 2.63 | 385 | 2.87 | 79.3 | 1 | ||
| SD | 0.08 | 0.70 | 108 | 0.78 | 21.3 | 0.2 | ||
| 10 mg mean | 0.45 | 0.07 | 1.86 | 1488 | 2.45 | 24.8* | 239.0 | 1.1 |
| SD | 0.32 | 0.07 | 0.53 | 764 | 2.16 | 26.2 | 55.8 | 0.3 |
| 30 mg mean | 0.25 | .005 | 2.09 | 4231 | 2.83 | 222.3* | 310.73 | 1.3 |
| SD | 0.10 | .003 | 0.92 | 1763 | 1.02 | 201.8 | 80.6 | 0.5 |
The difference in half-life between 10 and 30 mg is a result of the limitation of the assay for the LCMS method with the levels at the lower dose being closer to the lower limit of quantitation.
Figure 2.
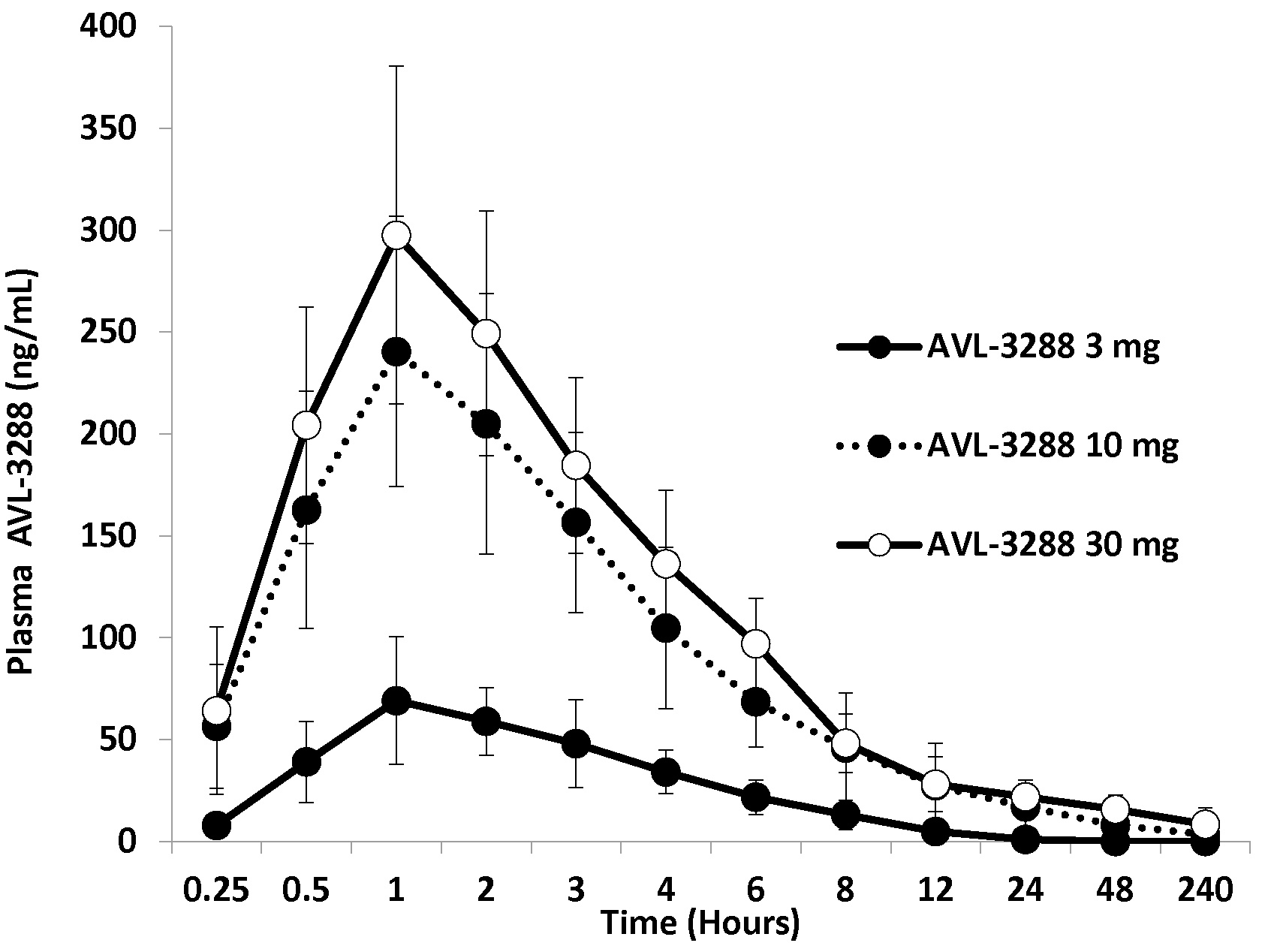
AVL-3288 plasma levels for cohorts of 6 non-smokers at each dose. Means and standard deviations.
Preliminary evidence for cognitive effect
The principal outcome for establishing preliminary evidence for efficacy was the RBANS Total Scale Score, assessed two hours after the administration of AVL-3288 or placebo. There was limited power for detecting effect in any individual cohort. Therefore results from all 6 non-smoking subjects who received placebo were combined, and the groups who received AVL-3288 10 or 30 mg were combined, because their plasma AVL-3288 levels were overlapping. The RBANS Total Scale Score of the 12 subjects who received AVL-3288 increased from their baseline by 5.33 ± 5.18 T-score units (t = 3.57, df 11, P = 0.004). The increase was greater than that experienced by the subjects who received placebo 2.51 ± 6.32, but the difference between the two groups was not significant. Table 3 shows the values obtained for each group.
Table 3.
Effects of AVL-3288 on the Repeatable Battery for the Assessment of Neuropsychological Status as change from baseline
| Non-smokers: | Total Scale Score | Immediate Memory | Visuospatial/Construction | Language | Attention | Delayed Memory | |
|---|---|---|---|---|---|---|---|
| 3 mg | mean | −1.5 | −1.5 | −4.0 | −1.0 | 5.5 | 0.7 |
| sd | 5.7 | 12.8 | 6.2 | 21.2 | 11.0 | 6.9 | |
| 10 mg | mean | 5.2 | 3.5 | −10.0 | 11.3 | 8.3 | 4.5 |
| sd | 5.3 | 5.8 | 12.3 | 12.9 | 13.7 | 6.9 | |
| 30 mg | mean | 5.5 | 2.0 | 9.7 | 2.5 | 1.2 | 3.7 |
| sd | 5.6 | 14.8 | 12.4 | 7.3 | 11.4 | 5.3 | |
| Placebo | mean | 2.5 | 4.2 | 7.7 | −7.5 | 1.7 | 3.0 |
| sd | 6.3 | 12.1 | 13.7 | 12.8 | 5.4 | 8.7 | |
| Smokers: | |||||||
| 3 mg | mean | −1.0 | −2.0 | −1.5 | −2.5 | 9.0 | −4.5 |
| sd | 4.3 | 13.6 | 17.4 | 11.5 | 11.9 | 11.0 | |
| Placebo | mean | −1.0 | 1.0 | 13.5 | 7.0 | −16.5 | −10.0 |
| sd | 9.9 | 25.5 | 4.9 | 2.8 | 6.4 | 14.1 |
As a biomarker of effect, the inhibition of the P50 auditory evoked potential in response to paired stimuli was measured, based on its previous response to alpha7-nicotinic agonists in schizophrenia. The agonist decreased the ratio of the amplitude of the second response to the first, indicating increased cerebral inhibition (Olincy et al. 2006). The 12 subjects who received 10 or 30 mg of AVL-3288 decreased the P50 ratio from 0.53 ± 0.38 to 0.32 ± 0.35, a non-significant reduction. The amplitude of the response to the first stimulus increased slightly from 2.35 ± 1.17 to 2.42 ± 1.14 microv, and the response to the second stimulus decreased from 1.03 ± 0.93 to 0.87 ± 1.10 microv. The 6 subjects who received placebo increased their P50 ratio from 0.24 ± 0.23 to 0.34 ± 0.30. There was no significant difference between the two groups. All the P50 ratios are within the range previously established for normal healthy subjects 0.37 ± 0.02 standard error (Olincy et al. 2010). Table 4 shows the values obtained for each group.
Table 4.
Effects of AVL-3288 on the P50 auditory evoked response to repeated stimuli as change from baseline
| Non-smokers | S1 response ms | S1 response mmv | S2 response mmv | P50 Ratio S2/S1 | |
|---|---|---|---|---|---|
| 3 mg | mean | −8.83 | 0.30 | 0.41 | 0.02 |
| sd | 10.30 | 2.55 | 1.02 | 0.32 | |
| 10 mg | mean | 0.50 | 0.30 | 0.00 | −0.25 |
| sd | 2.26 | 1.07 | 1.92 | 0.79 | |
| 30 mg | mean | 0.83 | −0.17 | −0.32 | −0.18 |
| sd | 6.62 | 1.01 | 0.60 | 0.20 | |
| Placebo | mean | 1.67 | 0.01 | 0.42 | 0.10 |
| sd | 2.34 | 1.06 | 1.33 | 0.39 | |
| Smokers | |||||
| 3 mg | mean | −3.17 | −0.50 | −0.19 | 0.02 |
| sd | 8.28 | 0.54 | 0.68 | 0.42 | |
| Placebo | mean | 1.50 | −0.40 | −0.79 | −0.23 |
| sd | 0.71 | 0.76 | 0.65 | 0.11 |
Safety and adverse effects
There were no serious adverse events, and no subject required medical attention. The QTc interval was modestly correlated with the plasma level of AVL-3288 for the 18 non-smoking subjects (r = 0.25, p > 0.3; Figure 3) but not significant. No subject had a QTc interval that exceed normal limits for his or her age, sex, and ethnicity, and QTc intervals at any dose were not significantly elevated above the level in 6 placebo controls. Clinical laboratory values were generally within the laboratory norms. Those that exceeded this level are listed in Table 5. None were considered medically significant, and all resolved spontaneously. None required treatment. Subject reports of adverse effects were minimal and all resolved spontaneously (Table 6).
Figure 3.
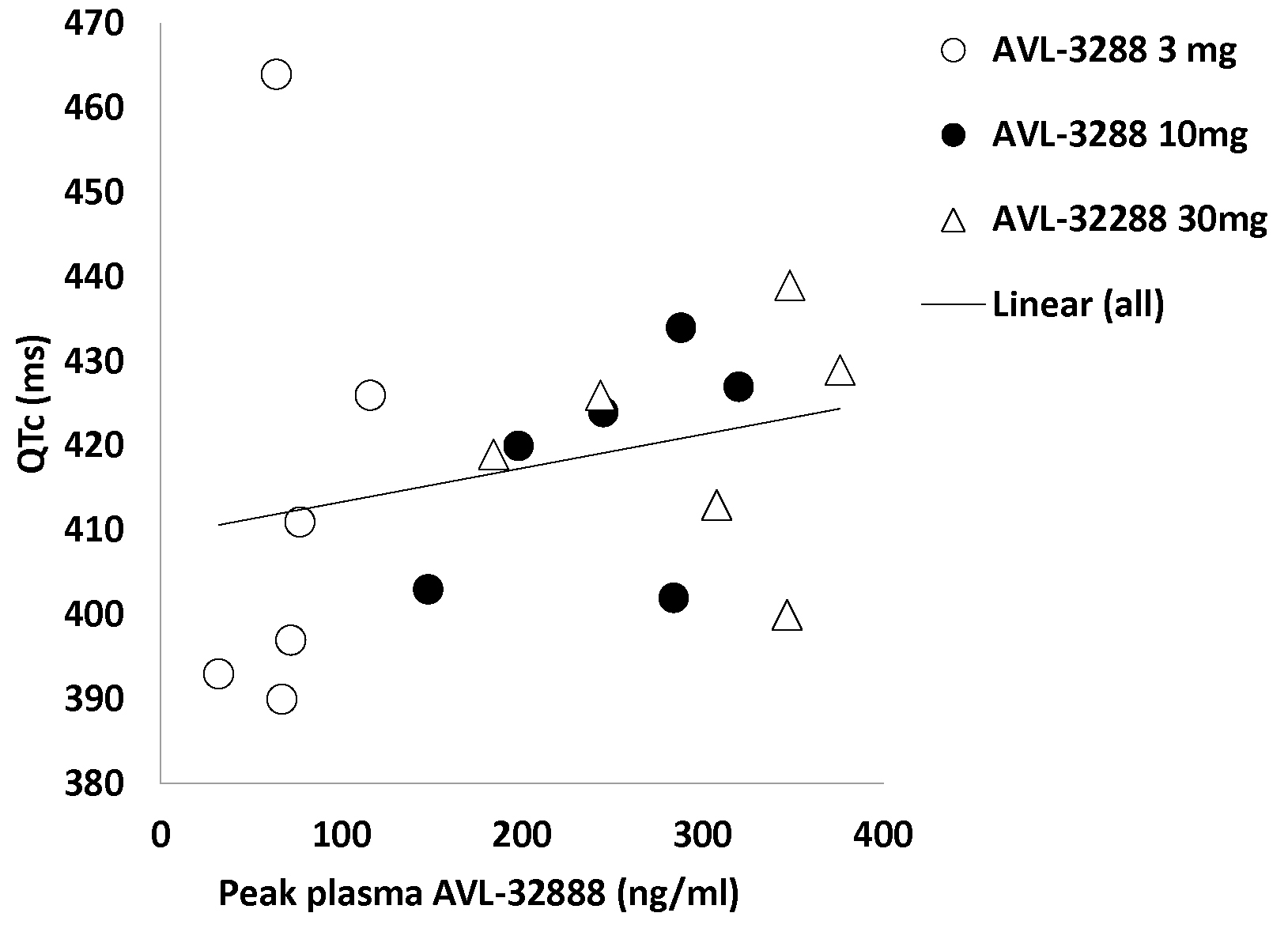
AVL-3288 peak plasma levels and QTc intervals for individual subjects. Linear regression line r = 0.25, P > 0.3.
Table 5.
Effects of AVL-3822 on clinical laboratory measures: numbers of subjects who developed abnormalities during treatment
| Placebo | 3 mg | 10 mg | 30 mg | Smokers: | Placebo | 3 mg | |
|---|---|---|---|---|---|---|---|
| RBC | 1 | 1 | 1 | ||||
| HGB | 1 | ||||||
| PT | 1 | ||||||
| APTT | 1 | 1 | 2 | 1 | 1 | ||
| K | 1 | ||||||
| Tot Prot | 1 | ||||||
| T bili | 1 | ||||||
| ALT | 1 | 1 | |||||
| GGT | 1 | ||||||
| Cholest | 1 | 1 | 1 | 1 | |||
| Glucose | 1 | 1 | 2 | 2 | |||
| Uric Acid | 1 |
Six subjects in all groups, except for 2 in the smoker placebo group.
Table 6.
Subject reports of adverse effects to AVL-3288: numbers of subjects
| Non-smokers: Placebo | 3 mg | 10 mg | 30 mg | Smokers: Placebo | 3 mg | |
|---|---|---|---|---|---|---|
| Insomnia | 2 | 0 | 1 | 0 | 0 | 0 |
| Sedation | 3 | 3 | 2 | 3 | 1 | 2 |
| Malaise | 0 | 0 | 0 | 0 | 0 | |
| Headache | 1 | 1 | 1 | 1 | 1 | 0 |
| Smell Disturbance | 1 | 0 | 0 | 0 | 0 | 0 |
| Dry Mouth | 1 | 1 | 1 | 0 | 0 | 0 |
| Chest Pain | 0 | 0 | 0 | 0 | 0 | 0 |
| Back Pain | 2 | 0 | 0 | 0 | 0 | 0 |
| Abdominal Pain | 2 | 0 | 0 | 0 | 0 | 0 |
| Enuresis | 2 | 0 | 0 | 0 | 0 | 0 |
| Joint Pain | 0 | 0 | 0 | 0 | 0 | 0 |
| Limb Pain | 0 | 0 | 0 | 0 | 0 | 0 |
| Paresthesia | 0 | 0 | 0 | 0 | 0 | 0 |
| Stiffness | 0 | 0 | 0 | 2 | 0 | 0 |
| Flatulence | 1 | 0 | 0 | 1 | 0 | 1 |
| Sore Throat | 1 | 0 | 0 | 0 | 0 | 0 |
| Nervousness | 1 | 0 | 0 | 0 | 0 | 0 |
| Rash | 0 | 0 | 0 | 0 | 0 | 1 |
| Hypersalivation | 0 | 0 | 0 | 0 | 0 | 1 |
Six subjects in all groups, except for 2 in the smoker placebo group.
Subjects who smoked
For the cigarette-smoking group who received AVL-3288 3 mg, the RBANS Total Scale T-score showed no significant change. Nor was there a significant change in P50 ratio (Tables 3, 4). Results were comparable to those seen in non-smokers at this AVL-3288 dose. Maximum AVL-3288 levels were modestly increased in smokers (71.9 ± 15.0 ng/ml) compared to non-smokers (58.5 ± 16.3 ng/ml). The QTc interval at maximal dose was not increased 415.5 ± 33.4 ms in smokers compared to 413.5 ± 28.1 ms in non-smokers. There were no unusual side effects or laboratory abnormalities. Plasma nicotine levels during AVL-3288 administration were 21.1 ± 16.3 ng/ml, and cotinine levels were 102.6 ± 134.3 ng/ml.
Discussion
AVL-3288 was safe and well-tolerated. The solubilized formulation yielded consistent plasma levels across individuals. In smoking subjects, there was a non-significant increase in plasma level, but otherwise no evidence of any differences. Neither the putative increased effect due to the presence of nicotine as an agonist nor the possible increased toxicity were observed.
The apparent plateau in peak mean blood level between 10 and 30 mg would appear to be protective against increases in plasma level to the in vitro IC50 hERG levels. The level obtained is well over an order of magnitude above those predicted for efficacy in preclinical measures of cognitive dysfunction (Ng et al., 2007; Thomsen et al., 2011; Nikiforuk et al., 2015, 2016). No significant effect on QTc interval was observed at any of the doses tested. The basis for this plateau is unknown, but it could indicate an increase in drug metabolism. The increase in AUC indicates that the additional dose was absorbed. The initial in vitro assessment of metabolism of AVL-3288 by Xytis did not reveal any strong P450 enzyme activity. The effects of nicotine to raise AVL-3288 plasma levels suggests that nicotine may be inhibiting a P450 enzyme involved in its metabolism. Thus, CYP 2E1 is a possible candidate (VanVleet et al. 2001).
A significant effect on the RBANS Total Scale Score was observed in the 12 subjects in the combined 10 and 30 mg cohorts, compared to baseline but not compared to the 6 placebo subjects. The high variance in the placebo group may have contributed to the lack of significant effect. Nonetheless, the effect size d’ = 0.49 appears sufficient to support further clinical trials. The effect size for the agonist 3-(2,4-dimethoxybenzylidene)-anabaseine was d’ = 0.51 after single dose administration in patients with schizophrenia (Olincy et al. 2006). The effect on the P50 evoked potential marker likewise appeared positive, but did not reach significance. However, the effect on P50 inhibition is consistent with the RBANS effect being caused by an increase in alpha7-nicotinic receptor activation. These observations on the type I PAM AVL-3288, in light of the failure of a type II PAM to show any effects in a similarly designed trial (Winterer et al. 2013), suggest that the type I PAM’s preservation of both the spatiotemporal integrity of receptor activation and normal kinetics of desensitization of the alpha-7 nicotinic receptor are necessary for its effect.
Funding:
Grants NIMH U01MH094247; NCATS UL1TR001082
Footnotes
Disclosure of conflicts of interest: KWG, DH and TJ are inventors on a patent covering AVL-3288 (U.S. Patent #7,820,663 B2) which is assigned to the University of California. AO, RK, LJ, JH, MT, SAE, RY and RF report no disclosures.
References
- Dempster EL, Toulopoulou T, McDonald C, Bramon E, Walshe M, Wickham H, Sham PC, Murray RM, Collier DA. 2006. Episodic memory performance predicted by the 2bp deletion in Exon 6 of the “Alpha 7-like” nicotinic receptor subunit gene. Am J Psychiatry 163:1832–1834 [DOI] [PubMed] [Google Scholar]
- Freedman R, Olincy A, Buchanan RW, Harris JG, Gold JM, Johnson L, Allensworth D, Guzman-Bonilla A, Clement B, Ball MP, Kutnick J, Pender V, Martin LF, Stevens KE, Wagner BD, Zerbe GO, Soti F. Kem WR. 2008. Initial phase 2 trial of a nicotinic agonist in schizophrenia. Am J Psychiatry 165:1040–1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freedman R 2014. Alpha7-nicotinic receptor agonists for cognitive enhancement in schizophrenia. Annu Rev Med 65:245–261. [DOI] [PubMed] [Google Scholar]
- Giniatullin R, Nistri A, Yakel JL. 2005. Desensitization of nicotinic ACh receptors: shaping cholinergic signaling. Trends Neurosci 28:371–378. [DOI] [PubMed] [Google Scholar]
- Gold JM, Queern C, Iannone VN, Buchanan RW. 1999. Repeatable battery for the assessment of neuropsychological status as a screening test in schizophrenia, I: sensitivity, reliability, and validity. Am J Psychiatry 156:1944–1950. [DOI] [PubMed] [Google Scholar]
- Grolien JH, Kakerud M, Ween H, Thorin-Hagene K, Briggs CA, Gopalkrishnan M. Maludz J. 2007. Distinct profiles of a7-nAChR positive allosteric modulators revealed by structurally diverse chemotypes. Mol Pharmacol 72:715–724. [DOI] [PubMed] [Google Scholar]
- Haig G, Bain E, Robieson W, Baker J, Othman A. 2016. A Randomized, Double-blind Trial to Assess the Efficacy and Safety of ABT-126, A selective α7 nicotinic acetylcholine receptor agonist in the treatment of cognitive Impairment in subjects with schizophrenia. Am J Psychiatry 173: 827–835. [DOI] [PubMed] [Google Scholar]
- Harris JG, Kongs S, Allensworth D, Martin L, Tregellas J, Sullivan B, Zerbe G, Freedman R. 2004. Effects of nicotine on cognitive deficits in schizophrenia. Neuropsychopharmacol 29:1378–1385. [DOI] [PubMed] [Google Scholar]
- Hu M, Gopalakrishnan M, Li J. 2009. Positive allosteric modulation of alpha7 neuronal nicotinic acetylcholine receptors: lack of cytotoxicity in PC12 cells and rat primary cortical neurons. Br J Pharmacol 158:1857–1864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keefe RSE, Meltzer HA Dgetluck N, Gawryl M, Koenig G, Moebius HJ, Lombardo I, Hilt DC. 2015. Randomized, double-blind, placebo-controlled study of encenicline, an α7 nicotinic acetylcholine receptor agonist, as a treatment for cognitive impairment in schizophrenia. Neuropsychopharmacol 40: 3053–3060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa H, Takenouchi T, Azuma R, Wesnes KA, Kramer WG, Clody DE, Burnett AL. 2003. Safety, pharmacokinetics, and effects on cognitive function of multiple doses of GTS-21 in healthy, male volunteers. Neuropsychopharmacol 28:542–551. [DOI] [PubMed] [Google Scholar]
- Lieberman JA; Dunbar G; Segreti AC; Girgis RR; Seoane F; Beaver JS; Duan N; Hosford DA. 2013. A randomized exploratory trial of an alpha-7 nicotinic receptor agonist (TC-5619) for cognitive enhancement in schizophrenia. Neuropsychopharmacol 38:968–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikiforuk A, Kos T, Holuj M, Potasiewica, Popik P. 2016. Positive allosteric modulators of alpha 7 nicotinic acetylcholine receptors reverse ketamine-induced schizophrenia-like deficits in rats. Neuropharmacol 101:389–400. [DOI] [PubMed] [Google Scholar]
- Nikiforuk A, Kos T, Potasiewica A and Popik P. 2015. Positive allosteric modulation of alpha 7 nicotinic acetylcholine receptors enhances recognition memory and cognitive flexibility in rats. Eur Neuropsychopharmacol 25:1300–1313. [DOI] [PubMed] [Google Scholar]
- Ng HJ, Whittemore ER, Tran MB, Hogenkamp DJ, Broide RS, Johnstone TB, Zheng L, Stevens KE, Gee KW. 2007. Nootropic alpha7 nicotinic receptor allosteric modulator derived from GABAA receptor modulators. Proc. Natl. Acad. Sci. USA. 104(19):8059–8064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olincy A, Harris JG, Johnson LL, Pender V, Kongs S, Allensworth D, Ellis J, Zerbe GO, Leonard S, Stevens KE, Stevens JO, Martin L, Adler LE, Soti F, Kem WR, Freedman R. 2006. Proof-of-concept trial of an α7 nicotinic agonist in schizophrenia. Arch Gen Psychiatry 63:630–638. [DOI] [PubMed] [Google Scholar]
- Olincy A, Braff DL, Adler LE, Cadenhead KS, Greenwood TA, Gur RE, Gur RC, Light GA, Mintz J, Nuechterlein KH, Radant AD, Schork NJ, Seidman LJ, Swerdlow NR, Twuand DW, Tsuang MT, Turetsky BI, Wagner BD, Freedman R. 2010. Inhibition of the P50 cerebral evoked response to repeated auditory stimuli: results from the Consortium on Genetics of Schizophrenia. Schizophr Res 119(1–3):175–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Katz M, Gerzanich V, Anand R, Lindstrom J. 1994. Human alpha 7 acetylcholine receptor: cloning of the alpha 7 subunits from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional alpha 7 homomers expressed in Xenopus oocytes. Mol Pharmacol 45: 546–54. [PubMed] [Google Scholar]
- Quick MW, Lester RA. 2002. Desensitization of neuronal nicotinic receptors. J Neurobiol 53:457–78. [DOI] [PubMed] [Google Scholar]
- Thomsen MS, El-Sayed M, Mikkelsen JD. 2011. Differential immediate and sustained memory enhancing effects of alpha7 nicotinic receptor agonists and allosteric modulators in rats. PLoS One 11:e27014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winterer G, Gallinat J, Brinkmeyer J, Musso F, Kornhuber J, Thuerauf N, Rujescu D, Favis R, Sun Y, Franc MA, Ouwerkerk-Mahadevan S, Janssens L, Timmers M and Streffer JR. 2013. Allosteric alpha-7 nicotinic receptor modulation and P50 sensory gating in schizophrenia: a proof-of-mechanism study. Neuropharmacol 64:197–204. [DOI] [PubMed] [Google Scholar]
- Van Vleet TR, Bombick DW, Coulombe RA. Inhibition of Human Cytochrome P450 2E1 by Nicotine, Cotinine, and Aqueous Cigarette Tar Extract in Vitro. 2001.J Toxico Sci 64, 185–191. [DOI] [PubMed] [Google Scholar]