

Abstract
Histone variants are key epigenetic players, but their functional and physiological roles remain poorly understood. Here, we show that depletion of the histone variant H2A.Z in mouse skeletal muscle causes oxidative stress, oxidation of proteins, accumulation of DNA damages, and both neuromuscular junction and mitochondria lesions that consequently lead to premature muscle aging and reduced life span. Investigation of the molecular mechanisms involved shows that H2A.Z is required to initiate DNA double strand break repair by recruiting Ku80 at DNA lesions. This is achieved via specific interactions of Ku80 vWA domain with H2A.Z. Taken as a whole, our data reveal that H2A.Z containing nucleosomes act as a molecular platform to bring together the proteins required to initiate and process DNA double strand break repair.
Graphical Abstract
Graphical Abstract.
Introduction
In all eukaryotic cells, the DNA is wrapped around an octamer of histones composed of two copies of H2A, H2B, H3 and H4 to form the fundamental unit of the chromatin i.e. the nucleosome core particule (1). Replacement of histones by histone variants, the non-allelic isoforms of conventional histones, confers novel structural properties to nucleosomes and changes the functional chromatin landscape (2–6). Conversely to conventional histones, the expression of histone variants is not restricted to the S phase and a growing number of observations indicate that in post-mitotic cells, histone variants gradually replace conventional histones (7–10). However, the functional and physiological requirement of the replacement of conventional histones by histone variants in post-mitotic cells remains poorly described in vivo.
H2A.Z belongs to the H2A family members, the largest histone variant family (11–13). In mammals, H2A.Z is encoded by two different genes (H2AFZ and H2AFV) to give H2A.Z-1 and H2A.Z-2 proteins which differ by only three amino-acids (14). H2A.Z is an essential protein in mammalian development (15,16) and is implicated in multiple nuclear processes (17–24). With age, H2A.Z accumulates in chromatin and H2A.Z accumulation has been linked to aged related cognitive decline (10). However, available data on the physiological role of H2A.Z are still scarce.
To characterize the physiological functions of H2A.Z during post-natal life, we have recently used the Cre-loxP system to develop the H2A.Z mdKO (muscle specific double knock-out) mouse model in which both H2A.Z isoforms were inactivated in post-mitotic muscle cells. This model allowed to show that in post-mitotic muscle cells, H2A.Z is not required for transcription nor for transcriptional regulation and that the enrichment of H2A.Z on active promoters is a marker but not an active driver of transcription (25).
This former study was conducted in 7 weeks old animals and here we have further pursued our investigations of the role of H2A.Z in older animals. We have observed that one-year-old H2A.Z mdKO muscles presented all the features of premature aging. Skeletal muscle aging causes sarcopenia which is mainly characterized at the organ level by muscle atrophy and neuromuscular junction dysfunction. At the cellular level, skeletal muscle aging is associated with increased ROS production, accumulation of DNA damage, and alterations in mitochondria and protein homeostasis (26,27).
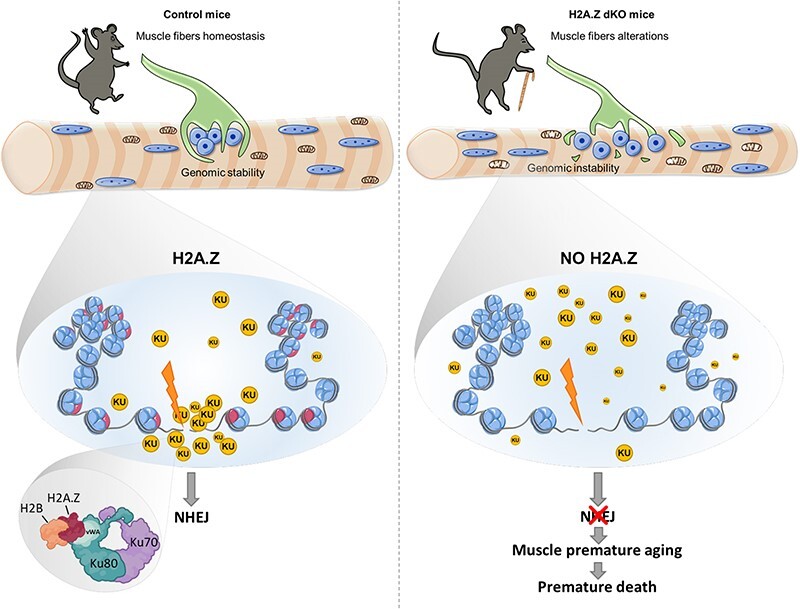
Investigation of the molecular mechanisms at play in one-year-old H2A.Z mKO muscles revealed accumulation of DNA damage. Ku70/80 proteins are essential to initiate DNA repair by non-homologous end joining (NHEJ). Here, we show that H2A.Z is required to recruit Ku80 to DNA lesions via direct protein-protein interaction and is therefore essential to initiate NHEJ. Altogether, our results demonstrate that in muscle fibers H2A.Z is required to prevent the accumulation of DNA damage, thus preventing mitochondrial dysfunction and premature aging.
Materials and methods
Mice care
The double cKO H2A.Z-1fl/fl:H2A.Z-2fl/fl HSA-Cre mouse line was previously described in (25). Animals were provided with mouse chow and water ad libitum in a restricted-access, specific pathogen–free animal care facility at the ‘Ecole Normale Supérieure of Lyon’ (Plateau de Biologie Expérimentale de la Souris). All procedures were performed in accordance with national and European legislation on animal experimentation (APAFIS#9964-2017032917131564; APAFIS#I0445-20170630 l3523347, Ceccap-ENS-2014–019 and APAFIS#27408-2020081914304420).
Body composition
Body composition was assessed in 12-month-old mice using Time Domain Nuclear Magnetic Resonance (Minispec LF90II - Bruker). Fat and lean mass were adjusted to body weight.
Treadmill exercise
Mice was progressively acclimated and trained to run-to-exhaustion during 10 days on the rodent treadmill (Ugo Basile 47 303) using different speed. Manual in the first intention then electric (0.2 Hz and 0.5 mA) stimuli were used to compel the mice to run. Exhaustion time is recorded when the mice are no longer able to run, even after several stimuli.
Electroporation of hindlimb muscles
Twelve-month-old CTL and H2A.Z mdKO mice were anesthetized with an intraperitoneal injection of ketamine (100 mg kg−1) and xylazine (10 mg kg−1). 20 μg of plasmid pCAG-Mito-DsRED in 30 μl of PBS were injected transcutaneously into the tibialis anterior (TA) muscle using a sterile U-100 insulin needle. Caliper electrode plates of 1 cm2 (Q-biogen, France) were immediately applied on each side of the muscle, and a series of 8 electrical pulses (200 V cm−1, 2 Hz, 20 ms each) was delivered with a standard square-wave electroporator (BTX ECM 830, Q-biogen). Electrical contact was ensured by shaving the leg of the animal and by conductive gel application. TA was collected 3 days after electroporation for tissue analysis.
Histology analysis
Hind limb muscles surrounding the tibial bone: Gastrocnemius, Plantaris, Soleus (GPS), Tibialis Anterior (TA) and Extensor Digitorum Longus (EDL) were collected from 12-month-old CTL and H2A.Z mdKO mice, frozen in isopentane cooled on dry ice, and cross-sectioned at 10 μm thickness in a cryostat. Transverse sections were stained with hematoxylin-phloxine-saffron (HPS), modified Gomori's trichrome, SDH-Cox, NADH-TR for immunohistochemistry as previously described in (25) and according to standard procedures for acid phosphatase and Sirius Red staining. TUNEL assays were performed according to the manufacturer's instruction (Invitrogen, C10617).
Immunofluorescence analysis
Cross sections were rehydrated in PBS, fixed with 4% paraformaldehyde for 10 min, permeabilized in 1X PBS, 0.1% Triton for 30 min at room temperature then saturated in 1× PBS 1% BSA 1% normal goat serum for 1 h. Staining were performed overnight at 4°C in the saturation buffer followed by 3 × 1 h wash in PBS and revealed with secondary antibody coupled with the indicated fluorophore mixed with DAPI. Slides were then mounted with Fluoromount (Invitrogen) and sealed with nail polish.
For myofibers staining, EDL muscles were collected and fixed 15 min in 4% paraformaldehyde. Muscles were slowly teased with fine forceps to isolate individual fibers and small fiber bundles. Fibers were permeabilized in 1× PBS, 0.1% Triton for 30 min at room temperature, saturated in 1× PBS 1% BSA for 1 h before overnight incubation with the primary antibody diluted in blocking buffer. After washing, fibers were incubated with a secondary antibody conjugated with indicated fluorophore (Molecular probes). Nuclei and neuromuscular junctions were respectively stained with DAPI and α-bungarotoxin Alexa555 (Molecular probes) for 2 h at room temperature. After washing, fibers were mounted on glass slide with Fluoromount (Invitrogen).
Immunofluorescence where analysed using a slide scanner (ZEISS Axio Scan.Z1) or confocal laser scanning microscopes (Leica SP5, Zeiss LSM880, Zeiss Elyra) as indicated in legends. Images were processed using ImageJ software. Morphometrics analysis were performed with a macro adapted from Desgeorge et al. (28).
Myoblasts derivation and culture
Myoblast cells were isolated from hindlimb muscle of 6- to 8-week-old of the double H2A.Z-1fl/fl:H2A.Z-2fl/fl mice. Collected muscles were washed 3 times in PBS. They are then mechanically dissociated and digested with 0.15% of Pronase type XIV solution (Sigma P-5147) for 1 h at 37 °C under gentle shaking. Pronase was inactivated with HAM F10 culture media and the resulted digested tissue was filtered through 40-μm Nylon Cell Strainers (Falcon – 352 340). Pronase was removed by three successive centrifugations at 450 g for 20 min at RT. The cellular pellet containing muscle-derived cells was resuspended and cultured in HAM F10 20% HS for 2 days before being supplemented with basic fibroblast growth factor (FGF2 – Sigma F0291) at 5 ng/ml. Every 2 days, the medium was replaced with a fresh medium.
To establish immortalized myoblast cell line, primary cells are infected with a retrovirus coding for mCDK4.
Myoblasts electroporation and differentiation
Immortalized myoblasts are electroporated with pEGFP-C1-hKu80. Briefly, cells were trypsinized, washed with OptiMEM, and 1 × 106 cells were resuspended in a volume of 100 μl containing 10 μg of DNA. The suspension is then placed in an electroporation cuvette of 2 mm gap (MBP 5520) and cells are electroporated with NEPA 21 electroporator (NEPAGENE) with the following poring pulse set up (voltage: 175 V; pulse interval: 50 ms; pulse length: 5 ms; number of pulses: 2). After 12h incubation myoblasts are infected with Adeno-Cre particles to induce H2A.Z gene defloxing. Cells are then plated on ibidi μ-dish and medium is changed to HAM F10 2% HS to induced myotube differentiation.
Laser irradiation
In order to locally generate DNA damage in living cells, we used a tuneable near-infrared pulsed laser (Cameleon Vision II; Coherent, Inc.) directly coupled to an inverted confocal microscope equipped with a 40×/1.3-NA oil objective in a thermostatic chamber maintained at 37°C, 5% CO2 (LSM 880; Zeiss). A small circular area (3 μm in diameter) within the nucleus of a living myotube was targeted three times to 10% capacity of the laser (800 nm). Subsequent time-lapse imaging of targeted cells was performed every 20 s until 60 s and every 60 s for 1140 s to evaluate the accumulation kinetics of EGFP-hKu80 on laser-induced DNA damage.
Metabolite analysis
Approximately 50 mg of tibialis anterior muscle sample was collected, snap frozen in nitrogen liquid, and conserved at −80°C. Metabolome extraction and measurements were carried out by Human Metabolome Technologies (HMT, Yamagata, Japan).
DHE staining for ROS measurement
Muscle cryosections were incubated for 30 min at 37 °C with 2.5 μM DHE in PBS. DHE staining revealed anion superoxide production at nuclear level. After staining, sections were rinsed and mounted in Vectashield (Vector Laboratories, Burlingame, CA). Slides were examined under a fluorescence microscope. The emission signal was recorded using a Zeiss 573–637 nm filter and high fluorescence intensity nuclei were quantified.
Mitochondrial DNA copy number
Tibialis anterior muscle was digested overnight with 100 μl of extraction buffer (1 mg/ml proteinase K, 1% SDS, Tris/HCl 10 mM pH 8.0) and DNA was extracted by phenol-chloroform extraction followed by ethanol precipitation. Mitochondrial DNA content was determined by quantitative PCR analysis. The quantities of cytochrome oxidase 2 (Cox2; mitochondrial DNA) were normalized to the levels of fatty-acid synthase (Fas) and 2 other genomic DNA (on Chr4 and Chr7). Primers are listed in Supplementary Table 1.
Quantitative RT-PCR
RNA was extracted from frozen tibialis anterior using 500 μl of TRI Reagent (Sigma) in tubes containing ceramics beads (Lysing Matrix D – MP biomedicals) for homogenization in a PreCellys (Bertin Technologies) (6500 RPM, 3 × 10 s). After centrifugation, TRI Reagent was removed and beads were washed once with 500 μl of TRI reagent. Total RNA was then extracted following provider's instruction. To generate cDNA, total RNA was treated with DNase (Ambion) and reverse transcribed with RevertAid H Minus Reverse Transcriptase (ThermoFisher) primed with random hexamers. cDNA was analysed with SYBR Green Mastermix (Qiagen) using the CFX-connect system (Bio-Rad) and the primer listed in Supplementary Table S1. Relative expression levels were normalized to GusB and Rpl41 housekeeping genes using the ΔΔCt method.
RNA-seq and computational analyses
RNA-seq experiments and analysis has been done as previously described (25). For analysis, the molecular signature of up-regulated genes with a log2 (H2A.Z dKO/CTL) >1 and P-value adjusted <0.05 was analyzed using Enrichr server and represented with a Bonferroni adjusted P-value: *P < 0.1; **P < 0.05; ***P < 0.001.
To address the concordance between H2A.Z mdKO and aging effects, we used the RRHO package (29) in two-sided mode and after Benjamini/Yekutieli correction, which quantifies the agreement between the order of two gene lists, ranked here according to their adjusted P-values, having taken into account the sign of the fold change. We alternatively employed the GeneOverlap R package (30), to measure how significant is the overlap between restricted gene lists, as compared to a background list.
Protein oxidation analysis
Oxidative protein carbonylation on tibialis anterior muscle was tested using an OxyBlot Protein Detection Kit (Millipore) according to the manufacturer's instructions. The carbonyl groups in protein side chains were derivatized to DNP-hydrazone with DNPH. The modified extract is then loaded on a 12% SDS-PAGE, transferred on a PVDF membrane and blotted with an anti-DNP antibody. Band intensities were quantified with the ImageJ software and normalized to the total protein content.
Immunoblot
Tissue preparations were separated by 18% SDS-PAGE electrophoresis and transferred onto polyvinylidene fluoride (PVDF) Immobilon-P membranes (Millipore). Immunoblots were performed with enhanced chemiluminescence (ECL) PLUS reagent (GE Healthcare) according to the manufacturer's instructions. Antibodies are listed in Supplemental Table S1.
Ku70/80 heterodimer purification
Ku70 and His-tagged Ku80 subunits were co-expressed in E. coli strain BL21-CodonPlus-RIL-pLysS (Stratagene) using a bicistronic pET70H80 vector as previously described (31). Bacteria were grown in 4.5 l of LB media at 37°C until the culture reached OD600 1.0. Cells were induced at 37°C with 1 mM IPTG and cultured for 3 h. Cells were harvested by centrifugation and resuspended in 120 ml of lysis buffer (50 mM Tris pH 8.0, 10% glycerol, 1 M NaCl, 0.4 M (NH)4OAc, 0.01% NP40, 2 mM dTT, 10 mM imidazole and 0.2 mM PMSF) on ice in the presence of lysozyme at 1 mg/ml. Cells were lysed by 3 cycles of through freezing/thawing followed by sonication for 3 × 1 min. Insoluble material was removed by centrifugation (30 min, 4°C, 12 000 g). The clarified supernatant was applied to 500 μl of Ni-NTA-agarose (Qiagen) equilibrated was batch washed with lysis buffer, then washed with 20 mM imidazole and eluted with 300 mM imidazole using a buffer containing 50 mM Tris pH 8.0, 10% glycerol, 500 mM NaCl, 0.01% NP40 and 1 mM dTT. The eluate fraction was desalted with PD-10 Sephadex G-25 columns (GE Healthcare) equilibrated with buffer containing 20 mM Tris–HCl pH 7.65, 150 mM NaCl, 10% glycerol, 3 mM MgCl2, 0.1 mM EDTA and 0.01% NP40.
H2A/H2B, H2A.X/H2B and H2A.Z/H2B histone dimers purification
FLAG-H2B and His-H2A, His-H2AX or His-H2A.Z human histones were co-expressed in E. coli strain BL21-CodonPlus-RIL-pLysS (Stratagene) using a homemade bicistronic pET28b (32). Bacteria were grown in 1.5 l of LB media at 37°C until the culture reached OD600 0.5. Cells were induced at 37°C with 1 mM IPTG and cultured for 3 h. Cells were harvested by centrifugation and resuspended in 40 ml of lysis buffer (50 mM Tris pH 8.0, 10% glycerol, 1 M NaCl, 0.4 M (NH)4OAc, 0.01% NP40, 2 mM dTT, 10 mM imidazole and 0.2 mM PMSF) on ice in the presence of lysozyme at 1 mg/mL. Cells were lysed by 3 cycles of through freezing/thawing followed by sonication for 3 × 1 min. Insoluble material was removed by centrifugation (30 min, 4 °C, 12 000 g). The clarified supernatant was applied to 450 μl of Ni-NTA-agarose (Qiagen) equilibrated with lysis buffer, then washed with 20 mM imidazole and eluted with 300 mM imidazole using a buffer containing 50 mM Tris pH 8.0, 10% glycerol, 500 mM NaCl, 0.01% NP40 and 1 mM dTT. The eluate fraction was desalted with PD-10 Sephadex G-25 columns (GE Healthcare) equilibrated with buffer containing 20 mM Tris–HCl pH 7.65, 150 mM NaCl, 10% glycerol, 3 mM MgCl2, 0.1 mM EDTA and 0.01% NP40.
Purification of GST-fusion Ku subdomains complexes expressed in bacteria
GST-fusion deletion mutants of Ku70 and Ku80 (cloned into a pGEX-5X-1 vector) were co-expressed with H2A/H2B, H2A.X/H2B or H2A.Z/H2B (cloned into a homemade bicistronic pET28b) in E. coli strain BL21-CodonPlus-RIL-pLysS (Stratagene). Cells were grown at 37°C until an OD600 of 0.5–0.7 was achieved. Cells were then induced with 1 mM IPTG 3 hours at 37°C. After induction, cells were harvested and resuspended in lysis buffer containing 20 mM Tris pH 7.65, 10% glycerol, 1 M NaCl, 3 mM MgCl2, 0.1% triton X-100, 0.01% NP40 and 0.2 mM PMSF. After adding lysozyme at 1 mg/mL, cells were lysed by 3 cycles of through freezing/thawing followed by sonication for 3 × 1 min. Insoluble material was removed by high speed centrifugation, and the clarified supernatant was applied to glutathione Sepharose 4B beads (GE Healthcare). The resin-bound proteins were washed with buffer containing 20 mM Tris–HCl pH 7.65, 1 M NaCl, 10% glycerol, 3 mM MgCl2, 0.1 mM EDTA and 0.01% NP40, and eluted with 20 mM l-glutathione. The eluted proteins were then fractionated on SDS-PAGE and stained with colloidal blue. Results were subsequently validated by Western blotting using anti-Flag and anti-His antibodies.
Results
H2A.Z mdKO mice progressively develop structural and functional muscle defects and reduced lifespan
The muscle specific double KO H2A.Z-1fl/fl:H2A.Z-2fl/fl HSA Cre+ (H2A.Z mdKO) mouse line, previously characterized at young age (25), was analyzed throughout adulthood, and compared with control H2A.Z-1fl/fl:H2A.Z-2fl/fl HSA Cre- mice (CTL).
The expression of both H2A.Z isoforms in various tissues was evaluated by RT-PCR. Both isoforms were expressed in heart, kidney, brain, liver and spleen and no difference could be observed between samples from 12-month-old H2A.Z mdKO and control mice (Supplementary Figure S1A and S1B). These results indicate that H2A.Z isoforms depletion is specific to skeletal muscle cells and does not affect other tissues, consistently with the initial characterization of the HSA-Cre line we used (33). In muscle, the specificity was confirmed by immunofluorescence on H2A.Z mdKO tibialis anterior (TA) muscles, showing the absence of H2A.Z proteins in myofibers while it remained present in interstitial cells (Supplementary Figure S1C) and by Western blot with a remaining signal (∼15–20%) in mutant muscles reflecting the presence of non-muscle cells (Figure 1A). Lifespan was monitored over 24 months on cohorts of CTL (n = 24) and H2A.Z mdKO (n = 23) mice. H2A.Z mdKO mice presented a median survival rate of 19 months whereas that of controls exceeded 24 months. Pairwise comparison revealed a significant difference in survival between the two groups (P < 0.0001 by log-rank test). At 24 months, only 10% of H2A.Z mdKO mice had survived, compared to >60% in CTL mice (Figure 1B). In addition to their reduced lifespan, H2A.Z mdKO mice gradually underwent characteristic physical changes beginning at 12 months of age, including systematic thinning of the body, often associated with a reduction in hair quality and color intensity, a slight kyphosis of the spine and a marked reduction in muscle size compared to control littermates (Figure 1C). These observations were consistent with body weight measures that showed a significant 19% decrease in 12-month-old H2A.Z mdKO compared to control littermates (Figure 1D). The reduction of lean mass correlated with a significant increase in fat mass (Figure 1E, F). Similar measures were obtained in males and females (data not shown).
Figure 1.

H2A.Z loss in muscle fibers leads to reduced lifespan and display physical changes. (A) TA muscle extracts from 12-month-old mice were immunoblotted with the indicated antibodies. (B) Kaplan–Meier survival curves of males and females CTL (n = 24) and H2A.Z mdKO (n = 23) mice. The cumulative survival rate was plotted against age in months. The Log-rank test was performed to compare the two mouse groups (***P < 0.0001). (C) Dorsal (upper panel), lateral (middle panel) and dissected tibialis anterior (TA) muscles (lower panel) photographs of 12-month-old mice reveal a thinner body, loss of hair quality, a kyphosis and muscle loss of H2A.Z mdKO mice compare to their CTL littermate. (D) Body weight in 12-old-month males (CTL n = 9; H2A.Z mdKO n = 10). Whole body fat mass (E) and lean mass (F) evaluated by TD-NMR (n = 12, *P < 0.05, Mann–Whitney test).
To evaluate the overall composition and structure of muscles, tibialis anterior (TA) cryosections were stained with hematoxylin-phloxine-saffron (HPS) and immunostained for laminin and DAPI (Figure 2A). Compared to CTL mice, H2A.Z mdKO mice presented smaller muscle fibers and increased inter-fiber space indicative of fibrosis (Figure 2A). Saffron staining indicated the presence of interstitial fibrotic tissue in H2A.Z mdKO muscles compared to control muscles. The presence of fibrotic tissue in H2A.Z mice muscle was confirmed by Sirius Red staining (Figure 2A). Fibers cross-sectional area evaluation confirmed the high proportion of small fibers in mutant mice (Figure 2B). Overall, the total surface of H2A.Z mdKO TA muscles was decreased by 38% compared to CTL muscles. The total number of fibers per TA muscle was unchanged but the number of fibers/mm2 was increased, consistent with the strong reduction in fiber size (Figure 2C–E). Consistently, western blot analysis of protein lysates showed a reduction in PKB/Akt phosphorylation in H2A.Z mdKO muscles compared to control muscles (Figure 2F, G), as observed in sarcopenia (34–36).
Figure 2.

H2A.Z loss in muscle fibers leads to muscle alterations at 12-month-old. (A) Representative image of hematoxylin phloxine saffron (upper panel), laminin/Dapi staining (middle panel) and Sirius Red (lower panel) in transversal sections of TA muscle (scale bar = 50 μm). (B) Quantification of cross-sectional area (CSA) of the whole TA with the measure of tissue surface in μm2 (C). The density (D), and the fiber number per tissue. (E) The data are shown as box-and-whisker plot with tukey style. Statistical analysis was performed using Mann–Whitney test. (*P < 0.05, **P < 0.01, ns: not significant, n = 5 per genotype). (F) Representative immunoblots for phosphorylated and total proteins of PKB/Akt in H2A.Z mdKO and CTL TA muscles. Tubulin was shown as loading control. (G) Densitometry quantification of phosphorylated bands relative to the total bands. Error bars are the SEM calculated from four independent CTL and 5 H2A.Z mdKO. Statistical analysis was performed using Mann–Whitney test. (*P < 0.05; ns: not significant). (H) Percentage of centronucleated fibers. (I) Relative mRNA expression of the regenerative marker MYH8. The data are shown as box-and-whisker plot with tukey style. Statistical analysis was performed using Mann–Whitney test (*P < 0.05, **P < 0.01, ns: not significant, n = 5 per genotype). (J) Quantification of F4/80+ cells/mm2 in the CTL and H2A.Z mdKO TA muscles. Error bars are the SEM calculated from 3 mice per group. Statistical analysis was performed using unpaired t test (*P < 0.05). (K) Representative images of IgG immunostaining in CTL and H2A.Z mdKO TA muscles. (L) Protocol of exercise to exhaustion test on the treadmill. (M) Percentage of running population on the treadmill by the distance of CTL (n = 13) and H2A.Z mdKO (n = 20) mice. The Log-rank test was performed to compare the two mouse groups (***P < 0.0001).
In addition to severe muscle atrophy, at 12-month of age H2A.Z mdKO mice contained 37% of fibers with central nuclei compared to 3% in CTL mice (Figure 2H). The presence of numerous centronucleated fibers suggested ongoing muscle fiber degeneration/regeneration in H2A.Z mdKO muscle. This was confirmed by the evaluation of the level of neonatal myosin MYH8 mRNA. In adult, this gene is re-expressed during muscle regeneration and MYH8 mRNA level was significantly increased in H2A.Z mdKO muscles compared to CTL (Figure 2I). The presence of regenerating fibres and fibrosis suggested that infiltration of immune cells may be present. As expected, immunostaining with the F4/80 antibody revealed the presence of infiltrating macrophages in mutant muscles compared to controls (15.73 cells/mm2 in CTL muscles compared to 98.77 cell/mm2 in H2A.Z mdKO muscles) (Figure 2J and Supplementary Figure S2A). Western blot confirmed the presence of elevated levels of immunoglobulins in H2A.Z mdKO extracts compared to controls (Supplementary Figure S2B). Mouse immunoglobulins were also visualized on H2A.Z mdKO TA cross-sections by immunostaining. In addition to the interstitial areas, some immunoglobulins were also detected inside few muscle fibers, indicating compromised membrane integrity (Figure 2K).
To confirm the phenotype of H2A.Z mdKO muscles, gastrocnemius, plantaris and soleus (GPS) muscles were also examined. As shown in TA muscle, GPS muscles displayed muscle atrophy (Supplementary Figure S3A). The histology of GPS and TA muscles were similar, both in the fast twitch EDL muscles (data not shown), and in the slow-twitch soleus muscles, with reduced muscle fiber cross-sectional area, increased inter-fiber space indicative of fibrosis and the presence of centronucleated fibers (17.2% in H2A.Z mdKO compared to 1.3% in the CTL soleus muscles) indicative of regenerative process (Supplementary Figure S3B-D).
To evaluate muscle function, H2A.Z mdKO and CTL mice were subjected to forced exercise running on a treadmill with progressive increase of speed in order to determine their maximum exercise capacity (Figure 2L). As shown in Figure 2M, the running performance of H2A.Z mdKO mice was significantly reduced compared to CTL mice.
Altogether, H2A.Z mdKO mice have reduced lifespan associated with decreased lean mass, increased fat mass, decreased exercise capacity, muscle atrophy, fibrosis, muscle fibers degeneration/regeneration and muscle infiltration by immune cells.
H2A.Z mdKO exhibits NMJ defects
In vertebrates, the neuromuscular junction (NMJ) is a specialized cholinergic synapse in charge of transmitting the nerve influx to muscle fibers. NMJ defects cause muscle weakness and fatigability (37) and have been identified as central actors in muscle aging (38). Staining of post-synaptic acetylcholine receptors (AChR) with α-bungarotoxin showed classical ‘bretzel shaped’ NMJs in control muscles, contrasting with the presence of highly fragmented and scattered NMJs in H2A.Z mdKO muscle (Figure 3A, B). To observe the presynaptic compartment of the NMJ, H2A.Z mdKO mice were crossed with Thy1-YFP transgenic mice expressing the YFP in axons and nerve terminals (39). In addition, synaptic vesicles were immunostained with an anti-SV2 antibody. Compared to the normal presynaptic branching pattern and SV2 punctuated staining colocalized with AChRs observed in control muscles, H2A.Z mdKO muscles presented denervated AChR clusters as well as axonal and terminal sprouting indicative of presynaptic remodelling consecutive to neurotransmission alterations (Supplementary Figure S4A–D). Acetylcholinesterase (AChE) staining confirmed these observations by showing a high proportion of fragmented NMJs (Figure 3E, E’, E’’) compared to CTL NMJ (Figure 3D). AChE staining of H2A.Z mdKO muscles also revealed the presence of ‘en passant’ synapses (Figure 3E’’’) on some fibers, and even the presence of muscle fibers with double NMJs (Figure 3E’’’’). Altogether, these results are indicative of an ongoing denervation/reinnervation process in many muscle fibers. This was confirmed by the strong upregulation of denervation markers such as the expression of AChR α subunit and of the embryonic γ subunit (Figure 3F).
Figure 3.

H2A.Z loss in muscle fibers leads to NMJ alterations. α-BTX staining of post-synaptic nAChRs (red) with nuclei (blue) in CTL (A) and H2A.Z mdKO (B) mice of 12-month-old. (C) Percentage of NMJ distribution in function of fragment number. Acethylcholinesterase activity by Koelle staining of CTL (D) and H2A.Z mdKO (E–E’’’’) mice. Neocluster of AChRs (E’’), «en passante» junction (E’’’) and double-innervated fiber (E’’’’) in H2A.Z mdKO mice (n = 5 CTL and 6 H2A.Z mdKO). (F) Relative mRNA expression of nAChRs subunits (n = 3; unpaired t-test, **P < 0.01, ***P < 0.001).
Such changes in NMJ morphology associated with increased expression of denervation markers, are indicative of pathological muscles or of muscles at old age.
H2A.Z mdKO mice exhibit mitochondrial defects
Muscle atrophy is often associated with mitochondrial defects (40). In addition, modified Gomori's trichrome staining of TA muscle cross sections from 12-month-old mice showed that H2A.Z mdKO muscles contained many fibers with no central staining, indicative of abnormal mitochondria distribution (Figure 4A, black arrows).
Figure 4.

H2A.Z loss exhibits enhanced mitochondrial alteration. Transversal sections stained with Modified Gomori's Trichrome (A), NADH (D), SDH-Cox (E) and acid phosphatase (K) (scale bar 20μm). Black arrows indicate fibers with abnormal mitochondria distribution. (B) Fibers electroporated with MitoDsRed (false color: yellow) in CTL and H2A.Z mdKO mice, and with higher magnification (C). (F) Mitochondrial DNA content determined on DNA from CTL and H2A.Z mdKO mice by quantitative PCR analysis using primers for COX2 (mitochondrial gene) and 3 nuclear control genes (n = 3, unpaired t-test, **P< 0.01). (G) Immunoblot analysis of steady-state levels of respiratory chain subunits, autophagic and lysosomal components in TA lysates of CTL and H2A.Z mdKO mice. (H) Representative electron microscopy of CTL and H2A.Z mdKO mice muscle structure of longitudinal sections. M: mitochondria, sM: subsarcollema mitochondria. (I) Representative tissue abnormalities observed in H2A.Z mdKO mice with a higher proportion: of minicore (left) and autophagic vacuoles with debris and multilamellar material (middle and right). (J) Representative immunostaining of p62 on EDL myofibers of CTL and H2A.Z mdKO mice. Each fiber is delimited by a dash line (scale bar: 10 μm).
In muscle fibers, mitochondria are regularly spaced and form a characteristic pattern. To evaluate this, TA muscles of 12-month-old H2A.Z mdKO and CTL mice were electroporated with an expression vector for a mitochondria-targeted red fluorescent protein. Muscles were collected 5 days after electroporation and individual fibers were isolated to visualize fluorescence. H2A.Z mdKO fibers showed a profound disruption of the striated mitochondrial pattern (Figure 4B, C and Supplementary Figure S5A). To evaluate mitochondrial activity, nicotinamide adenine dinucleotide (NADH, mitochondrial respiratory chain complex I enzyme) and combined succinate dehydrogenase and cytochrome c oxidase (SDH-Cox, mitochondrial respiratory chain complex II and IV enzymes) staining was performed on TA cross sections. The results confirmed altered mitochondria distribution (black arrows) in H2A.Z mdKO muscles compared to CTL muscles (Figure 4D, E). These alterations were present in both oxidative and glycolytic fibers (Supplementary Figure S5B, C). Similar results were also obtained in the more oxidative soleus muscle, although to a lesser extent (Supplementary Figure S6). To determine if the deregulation of the mitochondrial pattern could be associated with perturbations in the content of the mitochondrial genome, qPCR was used to compare the proportion of a mitochondrial gene, COX2, relative to nuclear DNA. The results showed that the mitochondrial DNA content was decreased by 2.75 times in H2A.Z mdKO muscles (Figure 4F). To determine if this decrease was associated with a loss of proteins in the respiratory chain, Western blots were performed on skeletal muscle lysates. Unexpectedly, the levels of the mitochondria encoded TMCO1 and ATP5A proteins, respectively belonging to complex IV and V and, were significantly higher in H2A.Z mdKO muscles than in controls. Levels of the mitochondrial encoded UQCRC2 subunit of complex III were also increased in H2A.Z mdKO muscles, although the difference with control muscles was less pronounced than with TMCO1 and ATP5A. The levels of the NDUFB8 and SDHB proteins, respectively involved in complex I and II were not changed (Figure 4G). These results indicated an imbalance between the different complexes of the respiratory chain in H2A.Z mdKO muscles, which could lead to electron leakage and ROS production.
To confirm altered mitochondrial distribution, transmission electron microscopy (TEM) was performed on H2A.Z mdKO and CTL TA muscles. In the absence of H2A.Z, mitochondria in some fibers were strongly swollen and displayed aberrant cristae evocative of mitochondrial dysfunction. In addition, a higher proportion of minicores (small zones of sarcomeric disorganization free of mitochondria) and autophagic vacuoles were observed (Figure 4H, I). The ultrastructural abnormalities observed in H2A.Z-deficient skeletal muscle were reminiscent of age-related changes in mitochondria (41).
To confirm autophagy/mitophagy features observed in electron microscopy, Western blot analysis revealed elevated levels of the autophagic markers p62 and LC3b in H2A.Z mdKO muscle extracts compared to controls (Figure 4G). Immunostaining confirmed this by showing that while p62 levels were quite low in control muscle fibers, they were more elevated in most H2A.Z mdKO muscle fibers, with some fibers even showing a striking accumulation of p62 (Figure 4J). In addition, a high proportion of muscle fibers showed a p62/LC3 colocalization suggesting the presence of autophagosomes (Supplementary Figure S7). This was confirmed by Western blot showing increased protein levels of the autophagosome marker Atg12 and the lysosomal marker LAMP2A in H2A.Z mdKO muscle extracts compared to controls (Figure 4G). This was further corroborated by the elevated intensity of acid phosphatase staining indicating high lysosomal activity in H2A.Z mdKO compared to controls (Figure 4K).
Altogether, in 12-month-old H2A.Z mdKO muscles, mitochondria activity, number, structure and distribution were severely perturbed. Such mitochondrial perturbations suggests that the metabolism of H2A.Z mdKO muscles may be perturbed. A metabolomic analysis was therefore performed on TA muscle from 12-month-old CTL and H2A.Z mdKO mice. 69 metabolites were quantified by CE-TOFMS and significant metabolic changes were observed (Table 1). Interestingly, putrescine was not detected in CTL muscles and was among the most upregulated metabolites in H2A.Z mdKO muscles. Accumulation of putrescine is known to be associated with diseases and aging. At the molecular level, putrescine, spermidine and spermine are polyamines derived from arginine. These polyamines have been described as negative regulators of muscle atrophy, activators of autophagy and of response to oxidative stress. Putrescine can also be metabolized into succinate to enter the Krebs cycle in mitochondria (42–46). Although their molecular targets remain poorly characterized, polyamines and putrescine have also been involved in DNA metabolism at various levels, such as for example genome stability or in DNA damage where they play a role in DNA double-strand breaks (47,48).
Table 1.
List of significant metabolites detected in CE-TOFMS in 12 month-old CTL and H2A.Z mdKO TA muscle. Data are expressed as mean of the relative area (peak value of each metabolite normalized by the sample volume) to give a relative concentration of each metabolite. The ratio is the comparative value of the relative areas of CTL versus H2A.Z mdKO. The P-value was calculated using Welch's t-test (n = 3 CTL and 3 H2A.Z mdKO)
| CTL | H2A.Z dKO | P-value | Ratio | ||
|---|---|---|---|---|---|
| Polyamines | Putrescine | 0.0E+00 | 8.5E-04 | 0.000 | |
| Arg | 2.1E-02 | 3.5E-02 | 0.015 | 1.6 | |
| methylation | SAM | 1.1E-03 | 8.8E-04 | 0.0497 | 0.8 |
| Met | 9.4E-03 | 6.4E-03 | 0.0323 | 0.7 | |
| Parp-1 regulator DNA repair | 3′,5′-ADP | 0.0E+00 | 6.2E-05 | 0.00 | |
| GSSG synthesis | Gly | 7.0E-02 | 1.1E-01 | 0.019 | 1.5 |
| Redox | GSSG | 1.2E-02 | 2.6E-02 | 0.0316 | 2.2 |
| non-protein nitrogenous (NPN) waste product | Urea | 3.4E-01 | 2.8E-01 | 0.034 | 0.8 |
| non-protein nitrogenous (NPN) waste product | Creatinine | 4.9E-03 | 3.6E-03 | 0.001 | 0.7 |
| energy shuttle (ATP ↔ ADP)) | Creatine | 2.9E+00 | 2.5E+00 | 0.030 | 0.8 |
| ATP | 2.3E-01 | 1.9E-01 | 0.0457 | 0.8 | |
| Carnosine Pathway | Anserine | 6.7E-01 | 8.6E-01 | 0.004 | 1.3 |
| Homocarnosine | 1.1E-01 | 1.4E-01 | 0.004 | 1.3 | |
| Carnosine | 6.5E-02 | 1.0E-01 | 0.011 | 1.6 | |
| Histamine | 2.8E-03 | 6.5E-03 | 0.000 | 2.3 | |
| b-Ala | 1.1E-03 | 2.3E-03 | 0.044 | 2.2 | |
| Uptake Arg and His (polyamines, Carnosines) | Asn | 3.7E-03 | 6.3E-03 | 0.010 | 1.7 |
| Carnitine Synthesis | Actinine | 3.3E-03 | 6.3E-03 | 0.01 | 1.9 |
| Trimethyllysine | 1.1E-03 | 2.1E-03 | 0.03 | 2.0 | |
| Betaine | 1.0E-02 | 1.4E-02 | 0.01 | 1.4 | |
| Lys | 4.9E-02 | 7.7E-02 | 0.01 | 1.6 | |
| Ketone Body | 3-HBA | 7.6E-04 | 1.4E-03 | 0.03 | 1.9 |
| Glycogenesis | GAP | 2.1E-05 | 1.2E-04 | 0.0180 | 5.7 |
| DPG | 1.3E-04 | 2.3E-04 | 0.0053 | 1.7 | |
| 3-PG | 5.8E-04 | 9.8E-04 | 0.0248 | 1.7 | |
| 2-PG | 1.0E-04 | 1.4E-04 | 0.0416 | 1.4 | |
| PEP | 2.5E-04 | 5.4E-04 | 0.0103 | 2.1 | |
| Glycolysis | G1P | 5.1E-03 | 4.0E-03 | 0.0266 | 0.8 |
| F6P | 2.0E-02 | 1.6E-02 | 0.0259 | 0.8 | |
| G6P | 7.1E-02 | 5.5E-02 | 0.0159 | 0.8 | |
| G3P | 1.9E-02 | 1.4E-02 | 0.0096 | 0.7 | |
| Lactic acid | 1.7E-01 | 1.2E-01 | 0.0045 | 0.7 | |
| Insulin Resistance | UDP-N-acetylgalactosamine UDP-GlcNAc | 5.3E-04 | 8.0E-04 | 0.0217 | 1.5 |
| Insulin Signaling | Myoinositol 1/3-phosphate | 7.7E-04 | 1.5E-03 | 0.0003 | 1.9 |
| Onco metabolite | 2-HG | 3.2E-04 | 4.2E-04 | 0.0125 | 1.3 |
| GMP, Urate, b-Ala synthesis | Ru5P | 1.7E-04 | 2.3E-04 | 0.0461 | 1.3 |
| NOS, smooth muscle relaxation | GMP | 2.8E-05 | 7.6E-05 | 0.0153 | 2.7 |
| anti-oxidant, Vit C substitute | Uric acid | 8.0E-05 | 1.2E-04 | 0.0036 | 1.5 |
| polysaccharide Biosynthesis | UDP-GlcA | 9.4E-05 | 1.4E-04 | 0.0090 | 1.5 |
| cofactor | NAD+ | 1.3E-02 | 1.1E-02 | 0.0503 | 0.8 |
| purine synthesis | Succinyl AMP | 9.5E-05 | 2.6E-05 | 0.0049 | 0.3 |
| muscle purine metabolite | IMP | 3.8E-03 | 1.9E-03 | 0.0209 | 0.5 |
| NAA donor | CMP-NeuNAc | 3.5E-04 | 2.2E-04 | 0.0553 | 0.6 |
| Fatty acid synthesis | Citric acid | 7.9E-03 | 9.9E-03 | 0.0518 | 1.3 |
| cis-Aconitic acid | 2.9E-04 | 4.4E-04 | 0.0311 | 1.5 |
Loss of H2A.Z causes accelerated muscle aging
All the results presented above suggested that H2A.Z mdKO muscles may be undergoing accelerated aging. Aging is associated with characteristic transcriptional changes. Comparative transcriptome analysis of H2A.Z mdKO and CTL muscle revealed strong transcriptional perturbation with respectively 1073 up-regulated genes and 462 downregulated genes (P < 0.05 and |log2 fold change| > 1) in mutant muscle tissue (Figure 5A). Gene Set Enrichment Analysis revealed that several molecular pathways were activated upon H2A.Z depletion (Figure 5B). The most affected ones were related to immune response, TNF-alpha signaling, complement activated genes, and interferon gamma-response genes and IL6/JAK/STAT3 signaling (Figure 5B). Such features are commonly considered as markers of aging in most tissues including skeletal muscle (49–52). Consistently, apoptotic and p53 pathways that are known to be activated during aging (51) were upregulated in H2A.Z mdKO muscles (Figure 5B and Supplementary Figure S8A, B).
Figure 5.

H2A.Z loss leads to the transcriptional signature of premature aging and oxidative damages. (A) Scatter plots comparing global gene expression levels between CTL and H2A.Z mdKO in TA muscle of 12-month-old mice. (B) Bar plots of significant MSigDB hallmark signatures of up-regulated genes with a log2FC (H2A.Z mdKO/CTL) > 1 and P-value adjusted < 0.05. MSigDB Hallmark pathway enrichment was analyzed using Enrichr server (Bonferroni adjusted P-value: *P < 0.1; **P < 0.05; ***P < 0.001). Data are from three biologically independent replicates. (C) DHE staining of tibialis anterior muscle section of CTL and H2A.Z mdKO mice and quantification of nuclei with high DHE intensity (D) (n = 5; scale bar = 50 μm). (E) Protein carbonylation was detected by Oxyblot assay. TA muscle extracts were derivatized with DNP and oxidized proteins were detected by western blot analysis using an anti-DNP antibody. The histogram represents the quantification and normalization of the anti-DNP on the coomassie staining (n = 3 CTL and 4 H2A.Z mdKO). (F) Representative western blot of Catalase expression in CTL and H2A.Z mdKO TA muscle. Tubulin was used as loading control.
Aging is also associated with progressive mitochondrial dysfunction (53,54). Our RNA-Seq data indicated that the expression of mitochondrial fission factor (Mff), mitochondrial fission 1 (Fis1) and peroxisome markers (Pex11a/b) was downregulated in the absence of H2A.Z (Supplementary Figure S8C, D), suggesting defects in mitochondria fission that could explain the mitochondria swelling observed in H2A.Z mdKO muscles. In contrast, mitofusin (Mfn1/2) expression was not affected (Supplementary Figure S8C). The expression of optic atrophy-1 (Opa1) that was shown to regulate cristae remodelling and maintenance (55,56) was also downregulated in H2A.Z mdKO muscles, which could explain the presence of aberrant cristae in these muscles. Genes involved in mitochondria biogenesis such as Tfam, mitochondrial ribosomal proteins Mrpl11, Mrpl16, Mrps28 and mitochondrial Ribosome Associated GTPase 1 (Mtg1) were also strongly downregulated in H2A.Z mdKO muscles compared to controls (Supplementary Figure S8E). Finally, genes involved in the redox balance were also dysregulated in H2A.Z mdKO muscles, with increased expression of the uncoupling protein isotype 2 (Ucp2) and Glutathion synthase (Gss) genes, and decreased expression of Gstm5, Mgst1, Gpx7 and Txnl4b genes (Supplementary Figure S8F). Altogether, this transcriptional profile pointed towards altered mitochondrial dynamics and dysregulated redox balance.
Since the transcriptome of 12-month-old H2A.Z mdKO muscles seemed to possess all the signatures of aging, it was compared to the transcriptome of aged muscle recently performed in aged rats. From the 21 898 mouse genes with an orthologue in the rat genome, we focused on the 10 475 that were found to be expressed in both data sets. We first ranked both lists of expressed genes by their adjusted P-values of differential expression, with the sign of their fold-change. Both data sets provided the same order and the calculated probability to obtain a similar order by chance was null. In addition, we specifically aligned the differentially expressed genes (P < 0.001) from both data sets (Supplementary Figure S9). The calculated probability to obtain such an overlap by chance was of 1.3e-12, and 2.4e-19 for up-regulated and down-regulated genes, respectively.
Altogether, this analysis indicated that 12-month-old H2A.Z mdKO muscles were undergoing premature aging.
Loss of H2A.Z causes oxidative stress
Oxidative stress and DNA damage are central features of aging and both metabolomic and transcriptomic analysis suggested that they were present in H2A.Z mdKO muscles. Glutathione is a major intracellular antioxidant and detoxifying agent. Severe oxidative stress can overwhelm the ability of the cell to reduce GSSG to GSH leading to accumulation of GSSG. In H2A.Z mdKO muscles the concentration of GSSG was significantly increased and the GSH/GSSG ratio was decreased by 62% compared to controls (Table 1). The levels of glycine and betaine that are involved in glutathione metabolism were also significantly increased in H2A.Z mdKO muscles (Table 1, Supplementary Figure S10).
To further evaluate oxidative stress in H2A.Z mdKO mice, intracellular ROS were quantified by dihydroethidium (DHE) staining. DHE reacts with superoxide anions, then is oxidized to Etd + to form a red fluorescent product (ethidium) which intercalates into DNA (57). H2A.Z mdKO TA muscles showed a 20-fold increase in the number of highly DHE positive nuclei compared to control muscles (Figure 5C,D), suggesting an important production of ROS by H2A.Z mdKO muscles. Once ROS are produced, they react with lipids, proteins and nucleic acids causing oxidative damage to these molecules. Protein carbonylation was thus evaluated and found to be 50% higher in H2A.Z mdKO muscles compared to controls (Figure 5E). Consistent with the presence of oxidative stress, the expression of Catalase, a ROS scavenger, was strongly increased in H2A.Z mdKO compared to controls (Figure 5F).
Thus, these results show that skeletal muscle in absence of H2A.Z present more oxidative stress.
H2A.Z loss causes accumulation of DNA damage in muscle fibers
ROS generate DNA lesions (58,59), such as oxidized DNA bases or DNA strand breaks in nuclei and in mitochondria, ultimately leading to genomic instability (60,61). We thus investigated for the presence of DNA lesions in H2A.Z mdKO muscles. A TUNEL assay was performed on muscle sections to detect DNA breaks. In parallel, immunofluorescence was performed to detect 8-OH-dG, the main oxidative DNA damage occurring in non-dividing cells (60,62). H2A.Z mdKO muscles presented a much higher number of TUNEL and 8-OH-dG foci compared to CTL muscles (Figure 6A and Supplementary Figure S11).
Figure 6.

H2A.Z loss leads to increase DNA damages. (A) Representative image of TUNEL (A488) and 8-OHdG (A647) staining in transversal sections of TA muscle. (B) Representative image of 53BP1 and γH2A.X staining (scale bar 10 μm). Each myonucleus is spotted with an arrow. (C, D) Quantification of the fluorescence intensity of each myonucleus on 500 μm2 of the 53BP1 (C) and γH2A.X staining (D). (Error bars are the SEM calculated from 4 mice per group, Mann–Whitney test ***P< 0.0001). (E) Representative Western blot analysis of total protein extract of 12-month-old TA muscle from CTL and H2A.Z mdKO mice immunoblotted with H2AX and γH2AX. H4 was used as loading control.
γ-H2A.X and 53BP1 foci indicate the presence of DNA damage (63,64). Western blot experiments showed a two-fold increase in the levels of γ-H2A.X in H2A.Z mdKO muscles (Figure 6E). Consistently, immunofluorescence to detect 53BP1 and γH2A.X on muscle cross sections generated stronger signals and identified more nuclei presenting a higher number of foci in H2A.Z mdKO muscles compared to the control muscles (Figure 6B–D).
Altogether, we show that one-year-old H2A.Z mdKO muscles accumulate DNA damage.
H2A.Z is required for the early steps of DNA repair
Although the precise molecular mechanism involved is not known yet, it was shown that H2A.Z might be involved in DNA repair via the Ku proteins (65–68). Ku proteins are recruited on double strand breaks (DSB) to initiate DNA repair by Non-Homologous End Joining (NHEJ), a major pathway involved in the repair of DNA lesions caused by oxidative stress. It was previously shown that H2A.Z is likely to participate in the recruitment of Ku70/80 to DNA lesions (66,32).
To evaluate the requirement of H2A.Z in the early steps of DNA repair of DSBs by NHEJ, immortalized primary muscle cells were isolated from H2A.Z double floxed mice and electroporated with an expression vector for a GFP-Ku80 protein fusion (Figure 7A). The cells were next infected with a Cre-adenovirus to inactivate both H2A.Z genes. The absence of H2A.Z in infected myotubes was confirmed by immunofluorescence 8 days after infection (Supplementary Figure S12A). Note that Ku70 and Ku80 protein expression was the same in CTL and H2A.Z KO myotubes (Supplementary Figure S12B). DNA lesions, including DSBs, were then induced by laser irradiation of selected nuclear regions (Figure 7B). In control cells, laser irradiation leads to a rapid (∼40 s post-laser burst) accumulation of GFP-Ku80, the ‘sensor’ of DSBs, which is further progressively increasing over a 10 min window. In contrast, in H2A.Z dKO myotubes, GFP-Ku80 was not recruited on the irradiated areas (Figure 7B), which corroborates with the published data in 293T cells (66).
Figure 7.

H2A.Z is necessary for GFP-Ku80 recruitment at DNA lesions sites. (A) Scheme of the sequential steps to obtain immortalized myoblasts (MB) and differentiated myotubes (MT) expressing the GFP-Ku80 in presence or absence of both H2A.Z for Frap analysis. (B) Accumulation of GFP-Ku80 after laser irradiation in CTL or H2A.Z dKO nuclei of seven-day differentiated myotubes. The damaged area has been delimited by a circle area and was spotted with a white arrow for each time point (scale bar 5 μm). (C) Chromatin extract fraction of isolated muscle nuclei immunoblotted with the indicated antibodies.
To determine if the absence of H2A.Z affects NHEJ in vivo, the chromatin fraction from 12-months-old H2A.Z mdKO and control muscles was analyzed by Western blot to evaluate the presence of the NHEJ proteins XRCC4 and Lig4 that act downstream of H2A.Z and Ku70/Ku80. As expected, the chromatin fraction of H2A.Z mdKO muscles contained significantly less XRCC4 and Lig4 compared to controls (Figure 7C).
Therefore, in post-mitotic muscle cells, H2A.Z is required to initiate NHEJ, via the recruitment of Ku80 at damaged sites. The open question was then how this is achieved? Since we had previously shown that Ku70/Ku80 associated with the H2A.Z deposition complex (32), we investigated a possible direct interaction between H2A.Z and Ku70/Ku80 dimers.
As in cells, histone H2A or its variants are exclusively present as a dimer with H2B, we performed pull-down experiments using purified H2A/H2B (Supplementary Figure S13A), H2A.X/H2B (Supplementary Figure S13B) or H2A.Z/H2B (Supplementary Figure S13C) histone dimers as bait, where H2B was Flag-tagged. Briefly, histone dimers were immobilized on anti-Flag resin and incubated or not with Ku dimers. After centrifugation, the resin-bound proteins were washed with increasing salt concentration (0.15, 0.5 and 1 M NaCl) and eluted with Flag peptide. SDS PAGE and western blotting of the eluted fractions show that Ku dimer binds specifically to the H2A.Z/H2B dimer at high salt concentration indicating a strong interaction affinity and specificity in-between these complexes. We included negative control to exclude any non-specific interaction of Ku heterodimer with anti-Flag resin (Supplementary Figure S13D). We conclude that Ku70/80 proteins interact directly and specifically with H2A.Z/H2B histone dimers.
To further dissect the specific molecular features of this interaction, the H2A.Z/H2B histone dimers were co-expressed in bacteria with various deletion mutants of Ku70 and Ku80 fused to GST (Figure 8A) and assayed for interaction using GST pull-down experiments. While GST pull-down failed to detect H2A.Z/H2B when the different Ku70 deletion mutants were used as bait, we detected a specific interaction between H2A.Z/H2B and the vWA domain of Ku80 (Figure 8B). We further identified amino-acids (40-101) as being sufficient for binding (Figure 8C, E). Interestingly, and in agreement with the data shown in Figure 8, the vWA domain of Ku80 was found to bind specifically to H2A.Z/H2B dimer, since no interaction was detected with H2A/H2B nor H2A.X/H2B dimers (Figure 8D). The importance of the Ku80-vWA domain in the H2A.Z context is confirmed in our post-mitotic cell model. CTL myotubes are electroporated with GFP-Ku80 devoid of its vWA domain (GFP-Ku80ΔvWA). After laser irradiation of selected nuclear region, we clearly observed that Ku80 is not able to be recruited without its vWA domain (Figure 8F), in contrast to GFP-Ku80 wild-type as observed in Figure 7B. These results unambiguously demonstrate that Ku80 interacts directly and specifically with H2A.Z/H2B dimers through its vWA domain. Consistently, the vWA domain of Ku80 was previously shown to be required for the recruitment of Ku80 to the chromatin surrounding DSBs (69), thus revealing its crucial importance for the in vivo function of Ku heterodimer in DSBs repair.
Figure 8.

The von Willebrand factor type A domain of Ku80 recognizes and binds specifically to H2A.Z/H2B histone dimer. (A) Functional domains of Ku70 and Ku80 proteins. Both proteins consist of an N-terminal von Willebrand factor type A (vWA) domain, a central DNA binding core domain and a nuclear localization sequence (NLS). C-terminal domain of Ku70 comprises an SAP (SAF-A/B, Acinus and PIAS) domain while C-terminal domain Ku80 comprises of sequence responsible for binding with DNA-PKcs. (B) The various deletion mutants of Ku70 and Ku80 fused to GST were assayed for interaction with H2A.Z/H2B histone dimer using GST pull-down assays. The eluates were analyzed by both SDS PAGE (top panel) and Western blotting (bottom panels). (C) Schematic representation of the different deletion mutants of the vWA domain of Ku80 analyzed in GST pull-down assays. (D) GST pull-down assays using GST-tagged vWA domain (09–244) of Ku80 as bait. Upper panel, the vWA domain of Ku80 specifically interacts with H2A.Z/H2B but not with H2A/B or H2A.X/H2B dimers. Lower panels, immunoblotting using anti-Flag (H2A, H2A.X or H2A.Z) and antis-His (H2B) antibodies. (E) Same as in (B) using different the GST tagged subdomains of vWA domain presented in C. (F) Accumulation of GFP-Ku80ΔvWA domain after laser irradiation in CTL nuclei of seven-day differentiated myotubes. The damaged area has been delimited by a circle area and was spotted with a white arrow for each time point (scale bar 5 = μm). (G) Schematic representation of the role of the H2A.Z nucleosome in the repair of DSBs in muscle fibres.
Discussion
The histone variant H2A.Z is involved in several functions of DNA metabolism (70,12,71), however its functions have mainly been investigated in cycling cells in which canonical histones are actively expressed. In post-mitotic neurons H2A.Z was shown to accumulate with age, possibly to compensate for the loss of H2A expression (10). However, the role of H2A.Z in post-mitotic tissues during aging remains unknown. Skeletal muscle fibers constitute an ideal system to conduct functional studies in long lived post-mitotic cells. We have previously shown that the HSA-Cre driver mouse line could be used to efficiently inactivate both H2A.Z isoforms in post-mitotic muscle cells using the Cre-loxP system (25). In the present study, muscle specific H2A.Z-1/H2A.Z-2 double KO mice (H2A.Z-1fl/fl:H2A.Z-2fl/fl: HSA-Cre) were characterized one year after birth. At this age, skeletal muscles had the appearance of muscles of a 28 months old geriatric mice and exhibited the hallmarks of severe muscle aging (72,73): muscle atrophy, loss of muscle strength, neuromuscular junction fragmentation, accumulation of DNA damage, mitochondrial alterations, oxidative stress and metabolic alterations, together with transcriptomic signatures of muscle aging.
Our results indicate that the primary cause of aging in H2A.Z mdKO muscles is the accumulation of DNA damage. Genomic instability and accumulation of DNA damage in cells lead to chronic stress and impaired organelle functions. It is well known that DNA repair deficiency causes premature aging and progeroid syndromes, and the current view is that it is responsible for mitochondrial alterations and impairment of nucleus-to-mitochondria signaling (54,74). DNA lesions accumulate with age and in highly metabolic post-mitotic cells like neurons and muscles, oxidative lesions are the main source of DNA damage. DNA lesions have been proposed to be the primary source of the degenerative processes associated with aging (75,63,76,77). Our mouse model pleads in favour of this claim. Indeed, by affecting the recruitment of Ku proteins on DNA lesions, H2A.Z depletion is very likely associated with the accumulation of lesions in muscle fibers. In other words, H2A.Z mdKO muscles provide an in vivo model in which the rate of DNA damage accumulation during aging is increased, and this appears to be sufficient to induce premature aging. This is consistent with previous studies that demonstrated that Ku80 inactivation in mouse induced early aging (78–80), and that Ku80 was required to prevent premature muscle aging (81).
The role of H2A.Z in genome stability and DNA damage repair was initially demonstrated in yeast where H2A.Z loss leads to increased sensitivity to DNA damage, defective DSB repair, and increased genomic instability (65,82). In mammals, H2A.Z was shown to be present at sites of DNA damage and was proposed to reduce chromatin compactness to facilitate DNA repair (66,83,67,84). However, the extent of compaction of H2A.Z nucleosomes is very likely to be similar to ‘conventional’ H2A chromatin (85–87). Actually, recent data reveal that the initial decondensation of chromatin upon DNA damage induction is associated with the removal of linker histone H1 (88), which COOH-terminus is responsible for the compaction of the chromatin fiber (for a recent review see (89)). It was also proposed that the presence of H2A.Z at DSBs was required for the loading of repair proteins such as Ku70/Ku80 (32,90). Our results confirm and provide a molecular basis to this hypothesis by showing that H2A.Z recruits Ku80 at DSBs via a direct interaction. Multiple DNA repair mechanisms have been characterized since the early 80′s, involving different proteins and pathways. In the future it will be interesting to evaluate a possible role of H2A.Z isoforms in DNA repair mechanisms beyond NHEJ. For DSB repair in muscle fibers, the homologous recombination pathway can be ruled out since it was recently shown that it was not involved in DSB repair in myotubes (91). Conversely, it was recently shown that H2A.Z could facilitate the repair of oxidative damages by the base excision repair pathway (92). It will therefore be of interest to investigate if H2A.Z is able to modulate this pathway in skeletal muscle fibers. DNA repair has been poorly described in post-mitotic cells compared to cycling cells, it is therefore possible that H2A.Z could be involved in other DNA repair pathways.
Taken as a whole, our data reveal a crucial role of H2A.Z as a gatekeeper against aging. They shed light on its mechanism of action to prevent the accumulation of DSBs by allowing the recruitment of Ku proteins around the DSB lesion. We propose that by directly interacting with Ku80, H2A.Z nucleosomes initiate the formation of a molecular platform dedicated to DSB repair.
Human epidemiologic studies and animal models indicate that skeletal muscle is a key determinant of lifespan (93). Most studies on the systemic role of skeletal muscle were performed in an aging or pathological context in which it is not easy to isolate the respective contribution of the various tissues in lifespan expectancy. By specifically inducing premature aging in muscle fibers, our model provides unbiased evidence that provoking accelerated aging in post-mitotic muscle fibers is sufficient to reduce lifespan. It also provides a unique in vivo model that will allow future studies to understand the impact of muscle aging on other organs and whole body homeostasis.
Supplementary Material
Acknowledgements
We thank the animal facility (PBES) of the research federation SFR Biosciences (UMS3444) and particularly C. Angleraux and P. Contard for their help in maintaining the mouse colony; the physiological explorations platform (ANIPHY, UMS 3453); and the imaging facility on SFR Santé Lyon Est (CIQLE, UMS3453), particularly B. Chapuis and D. Ressnikoff for their help with the imageJ software and image acquisition; and SFR Bioscience (PLATIM, UMS3444). Illustration in Figure 7A was created with BioRender.com.
Author contributions: L.S., S.D., A.H., E.B. conceived the project and L.S., S.D., A.H. secured funding. E.B. performed the mice breeding. N.L. and E.B. performed RT-QPCR, immunofluorescences and biochemistry analysis on mice model. E.B., N.L., A.O. and N.S. performed histology analysis. E.B., E.G. performed NMJ analysis. E.B. performed the metabolomics analysis. E.B. generated the H2A.Z myoblast cell model. E.B., P.-O.M. and G.G-M performed DNA repair analysis. C.P., T.S. performed bioinformatics analysis. A.I., C.P. performed the recombinant protein expression and biochemistry analysis. L.S., S.D. and A.H. supervised the work, and E.B., L.S., S.D. and A.H. wrote the paper.
Contributor Information
Edwige Belotti, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Nicolas Lacoste, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Arslan Iftikhar, For Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), CNRS/INSERM/ULP, Parc d’innovation, 1 rue Laurent Fries, 67404 Ilkirch Cedex, France.
Thomas Simonet, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Christophe Papin, For Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), CNRS/INSERM/ULP, Parc d’innovation, 1 rue Laurent Fries, 67404 Ilkirch Cedex, France.
Alexis Osseni, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Nathalie Streichenberger, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Pierre-Olivier Mari, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Emmanuelle Girard, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Mohamed Graies, Institute for Advanced Biosciences (IAB), Université Grenoble Alpes, CNRS UMR 5309, INSERM U1209, Site Santé - Allée des Alpes, 38700 La Tronche, France.
Giuseppina Giglia-Mari, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France.
Stefan Dimitrov, Institute for Advanced Biosciences (IAB), Université Grenoble Alpes, CNRS UMR 5309, INSERM U1209, Site Santé - Allée des Alpes, 38700 La Tronche, France.
Ali Hamiche, For Institut de Génétique et de Biologie Moléculaire et Cellulaire (IGBMC), CNRS/INSERM/ULP, Parc d’innovation, 1 rue Laurent Fries, 67404 Ilkirch Cedex, France.
Laurent Schaeffer, Laboratoire Physiopathologie et Génétique du Neurone et du Muscle (PGNM), Institut NeuroMyoGène, Université Claude Bernard Lyon 1, INSERM U1315, CNRS UMR 5261, 8 avenue Rockefeller, 69008 Lyon, France; Centre de Biotechnologie Cellulaire, Hospices Civils de Lyon, Lyon, France.
Data availability
RNA-seq datasets have been deposited in the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE216935.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
E.B. was funded by AFM through the MyoNeurAlp strategic plan; ANR through the « ZFUN » project [ANR-18-CE12-0010]; F.R.M. through an équipe FRM grant (to L.S.); A.F.M. through the MyoNeurAlp strategic plan and by the project 101 086 923 – AEGIS-IMB.
Conflict of interest statement. None declared.
References
- 1. Luger K. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 1997; 389:251–260. [DOI] [PubMed] [Google Scholar]
- 2. Ausió J. Histone variants–the structure behind the function. Brief. Funct. Genomic. Proteomic. 2006; 5:228–243. [DOI] [PubMed] [Google Scholar]
- 3. Doyen C.-M., An W., Angelov D., Bondarenko V., Mietton F., Studitsky V.M., Hamiche A., Roeder R.G., Bouvet P., Dimitrov S.. Mechanism of polymerase II transcription repression by the histone variant macroH2A. Mol. Cell Biol. 2006; 26:1156–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ouararhni K., Hadj-Slimane R., Ait-Si-Ali S., Robin P., Mietton F., Harel-Bellan A., Dimitrov S., Hamiche A.. The histone variant mH2A1.1 interferes with transcription by down-regulating PARP-1 enzymatic activity. Genes Dev. 2006; 20:3324–3336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cheema M.S., Ausió J.. The structural determinants behind the epigenetic role of histone variants. Genes (Basel). 2015; 6:685–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Roulland Y., Ouararhni K., Naidenov M., Ramos L., Shuaib M., Syed S.H., Lone I.N., Boopathi R., Fontaine E., Papai G.et al.. The flexible ends of CENP-A nucleosome are required for mitotic fidelity. Mol. Cell. 2016; 63:674–685. [DOI] [PubMed] [Google Scholar]
- 7. Maze I., Wenderski W., Noh K.-M., Bagot R.C., Tzavaras N., Purushothaman I., Elsässer S.J., Guo Y., Ionete C., Hurd Y.L.et al.. Critical role of histone turnover in neuronal transcription and plasticity. Neuron. 2015; 87:77–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Piazzesi A., Papić D., Bertan F., Salomoni P., Nicotera P., Bano D.. Replication-independent histone variant H3.3 controls animal lifespan through the regulation of pro-longevity transcriptional programs. Cell Rep. 2016; 17:987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tvardovskiy A., Schwämmle V., Kempf S.J., Rogowska-Wrzesinska A., Jensen O.N.. Accumulation of histone variant H3.3 with age is associated with profound changes in the histone methylation landscape. Nucleic Acids Res. 2017; 45:9272–9289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Stefanelli G., Azam A.B., Walters B.J., Brimble M.A., Gettens C.P., Bouchard-Cannon P., Cheng H.-Y.M., Davidoff A.M., Narkaj K., Day J.J.et al.. Learning and age-related changes in genome-wide H2A.Z binding in the mouse hippocampus. Cell Rep. 2018; 22:1124–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bönisch C., Schneider K., Pünzeler S., Wiedemann S.M., Bielmeier C., Bocola M., Eberl H.C., Kuegel W., Neumann J., Kremmer E.et al.. H2A.Z.2.2 is an alternatively spliced histone H2A.Z variant that causes severe nucleosome destabilization. Nucleic Acids Res. 2012; 13:5951–5964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Giaimo B.D., Ferrante F., Herchenröther A., Hake S.B., Borggrefe T.. The histone variant H2A.Z in gene regulation. Epigenetics Chromatin. 2019; 12:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sarma K., Reinberg D.. Histone variants meet their match. Nat. Rev. Mol. Cell Biol. 2005; 6:139–149. [DOI] [PubMed] [Google Scholar]
- 14. Eirín-López J.M., González-Romero R., Dryhurst D., Ishibashi T., Ausió J.. The evolutionary differentiation of two histone H2A.Z variants in chordates (H2A.Z-1 and H2A.Z-2) is mediated by a stepwise mutation process that affects three amino acid residues. BMC Evol. Biol. 2009; 9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Faast R., Thonglairoam V., Schulz T.C., Beall J., Wells J.R.E., Taylor H., Matthaei K., Rathjen P.D., Tremethick D.J., Lyons I.. Histone variant H2A.Z is required for early mammalian development. Curr. Biol. 2001; 11:1183–1187. [DOI] [PubMed] [Google Scholar]
- 16. Dijkwel Y., Tremethick D.J.. The role of the histone variant H2A.Z in Metazoan development. JDB. 2022; 10:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Fan J.Y., Rangasamy D., Luger K., Tremethick D.J.. H2A.Z alters the nucleosome surface to promote HP1α-mediated chromatin fiber folding. Mol. Cell. 2004; 16:655–661. [DOI] [PubMed] [Google Scholar]
- 18. Greaves I.K., Rangasamy D., Ridgway P., Tremethick D.J.. H2A.Z contributes to the unique 3D structure of the centromere. Proc. Natl Acad. Sci. U.S.A. 2007; 104:525–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ishibashi T., Dryhurst D., Rose K.L., Shabanowitz J., Ausió J.. Acetylation of vertebrate H2A.Z and its effect on the structure of the nucleosome. Biochemistry. 2010; 48:5007–5017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Nekrasov M., Soboleva T.A., Jack C., Tremethick D.J.. Histone variant selectivity at the transcription start site: H2A.Z or H2A.Lap1. Nucleus. 2013; 4:431–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Domaschenz R., Kurscheid S., Nekrasov M., Han S., Tremethick D.J.. The histone variant H2A.Z is a master regulator of the epithelial-mesenchymal transition. Cell Rep. 2017; 21:943–952. [DOI] [PubMed] [Google Scholar]
- 22. Tyagi M., Cheema M.S., Dryhurst D., Eskiw C.H., Ausió J.. Metformin alters H2A.Z dynamics and regulates androgen dependent prostate cancer progression. Oncotarget. 2018; 9:37054–37068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Cheema M.S., Good K.V., Kim B., Soufari H., O’Sullivan C., Freeman M.E., Stefanelli G., Casas C.R., Zengeler K.E., Kennedy A.J.et al.. Deciphering the enigma of the histone H2A.Z-1/H2A.Z-2 Isoforms: novel insights and remaining questions. Cells. 2020; 9:1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cole L., Kurscheid S., Nekrasov M., Domaschenz R., Vera D.L., Dennis J.H., Tremethick D.J.. Multiple roles of H2A.Z in regulating promoter chromatin architecture in human cells. Nat. Commun. 2021; 12:2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Belotti E., Lacoste N., Simonet T., Papin C., Padmanabhan K., Scionti I., Gangloff Y.-G., Ramos L., Dalkara D., Hamiche A.et al.. H2A.Z is dispensable for both basal and activated transcription in post-mitotic mouse muscles. Nucleic Acids Res. 2020; 48:4601–4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Börsch A., Ham D.J., Mittal N., Tintignac L.A., Migliavacca E., Feige J.N., Rüegg M.A., Zavolan M.. Molecular and phenotypic analysis of rodent models reveals conserved and species-specific modulators of human sarcopenia. Commun. Biol. 2021; 4:194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Szczesny B., Tann A.W., Mitra S.. Age- and tissue-specific changes in mitochondrial and nuclear DNA base excision repair activity in mice: susceptibility of skeletal muscles to oxidative injury. Mech. Ageing Dev. 2010; 131:330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Desgeorges T. Open-CSAM, a new tool for semi-automated analysis of myofiber cross-sectional area in regenerating adult skeletal muscle. 2019; [DOI] [PMC free article] [PubMed]
- 29. Rosenblatt J., Stein J.. RRHO: Test overlap using the Rank-Rank Hypergeometric test. 2014; R package version 1.42.0.
- 30. Shen L. GeneOverlap: an R package to test and visualize gene overlaps. 2021;
- 31. Hanakahi L.A. 2-Step purification of the Ku DNA repair protein expressed in Escherichia coli. Protein Expression Purif. 2007; 52:139–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Obri A., Ouararhni K., Papin C., Diebold M.-L., Padmanabhan K., Marek M., Stoll I., Roy L., Reilly P.T., Mak T.W.et al.. ANP32E is a histone chaperone that removes H2A.Z from chromatin. Nature. 2014; 505:648–653. [DOI] [PubMed] [Google Scholar]
- 33. Miniou P., Tiziano D., Frugier T., Roblot N., Meur M.L., Melki J.. Gene targeting restricted to mouse striated muscle lineage. Nucleic Acids Res. 1999; 27:e27–e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Joseph G.A., Wang S.X., Jacobs C.E., Zhou W., Kimble G.C., Tse H.W., Eash J.K., Shavlakadze T., Glass D.J.. Partial Inhibition of mTORC1 in aged rats counteracts the decline in muscle mass and reverses molecular signaling associated with sarcopenia. Mol. Cell. Biol. 2019; 39:e00141-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sasako T., Umehara T., Soeda K., Kaneko K., Suzuki M., Kobayashi N., Okazaki Y., Tamura-Nakano M., Chiba T., Accili D.et al.. Deletion of skeletal muscle Akt1/2 causes osteosarcopenia and reduces lifespan in mice. Nat. Commun. 2022; 13:5655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Bodine S.C. The role of mTORC1 in the regulation of skeletal muscle mass. Fac. Rev. 2022; 11:32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Gilhus N.E., Tzartos S., Evoli A., Palace J., Burns T.M., Verschuuren J.J.G.M.. Myasthenia gravis. Nat. Rev. Dis. Primers. 2019; 5:30. [DOI] [PubMed] [Google Scholar]
- 38. Ibebunjo C., Chick J.M., Kendall T., Eash J.K., Li C., Zhang Y., Vickers C., Wu Z., Clarke B.A., Shi J.et al.. Genomic and proteomic profiling reveals reduced mitochondrial function and disruption of the neuromuscular junction driving rat sarcopenia. Mol. Cell. Biol. 2013; 33:194–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Feng G., Mellor R.H., Bernstein M., Keller-Peck C., Nguyen Q.T., Wallace M., Nerbonne J.M., Lichtman J.W., Sanes J.R.. Imaging neuronal subsets in transgenic mice expressing multiple spectral variants of GFP. Neuron. 2000; 28:41–51. [DOI] [PubMed] [Google Scholar]
- 40. Romanello V., Sandri M.. Mitochondrial quality control and muscle mass maintenance. Front. Physiol. 2016; 6:422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Palla A.R., Ravichandran M., Wang Y.X., Alexandrova L., Yang A.V., Kraft P., Holbrook C.A., Schürch C.M., Ho A.T.V., Blau H.M.. Inhibition of prostaglandin-degrading enzyme 15-PGDH rejuvenates aged muscle mass and strength. Science. 2021; 371:eabc8059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Suppola S., Pietila M., Parkkinen J.J., Korhonen V.-P., Alhonen L., Halmekyto M., Porter C.W., Jänne J.. Overexpression of spermidine/spermine N1-acetyltransferase under the control of mouse metallothionein I promoter in transgenic mice: evidence for a striking post-transcriptional regulation of transgene expression by a polyamine analogue. Biochem. J. 1999; 338:311–316. [PMC free article] [PubMed] [Google Scholar]
- 43. Minois N., Carmona-Gutierrez D., Madeo F.. Polyamines in aging and disease. Aging (Albany NY). 2011; 3:716–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Manna S.K., Krausz K.W., Bonzo J.A., Idle J.R., Gonzalez F.J.. Metabolomics reveals aging-associated attenuation of noninvasive radiation biomarkers in mice: potential role of polyamine catabolism and incoherent DNA damage-repair. J. Proteome Res. 2013; 12:2269–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Uchitomi R., Hatazawa Y., Senoo N., Yoshioka K., Fujita M., Shimizu T., Miura S., Ono Y., Kamei Y.. Metabolomic analysis of skeletal muscle in aged mice. Sci. Rep. 2019; 9:10425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sagar N.A., Tarafdar S., Agarwal S., Tarafdar A., Sharma S.. Polyamines: functions, metabolism, and role in human disease management. Med. Sci. 2021; 9:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hofer S.J., Simon A.K., Bergmann M., Eisenberg T., Kroemer G., Madeo F.. Mechanisms of spermidine-induced autophagy and geroprotection. Nat. Aging. 2022; 2:1112–1129. [DOI] [PubMed] [Google Scholar]
- 48. Lee C.-Y., Su G.-C., Huang W.-Y., Ko M.-Y., Yeh H.-Y., Chang G.-D., Lin S.-J., Chi P.. Promotion of homology-directed DNA repair by polyamines. Nat. Commun. 2019; 10:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lin I.-H., Chang J.-L., Hua K., Huang W.-C., Hsu M.-T., Chen Y.-F.. Skeletal muscle in aged mice reveals extensive transformation of muscle gene expression. BMC Genet. 2018; 19:55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. de Magalhães J.P., Curado J., Church G.M.. Meta-analysis of age-related gene expression profiles identifies common signatures of aging. Bioinformatics. 2009; 25:875–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Shavlakadze T., Morris M., Fang J., Wang S.X., Zhu J., Zhou W., Tse H.W., Mondragon-Gonzalez R., Roma G., Glass D.J.. Age-related gene expression signature in rats demonstrate early, late, and linear transcriptional changes from multiple tissues. Cell Rep. 2019; 28:3263–3273. [DOI] [PubMed] [Google Scholar]
- 52. Zhu X., Chen Z., Shen W., Huang G., Sedivy J.M., Wang H., Ju Z.. Inflammation, epigenetics, and metabolism converge to cell senescence and ageing: the regulation and intervention. Sig. Transduct. Target Ther. 2021; 6:245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Akbari M., Kirkwood T.B.L., Bohr V.A.. Mitochondria in the signaling pathways that control longevity and health span. Ageing Res. Rev. 2019; 54:100940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Fang E.F., Scheibye-Knudsen M., Chua K.F., Mattson M.P., Croteau D.L., Bohr V.A.. Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 2016; 17:308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Frezza C., Cipolat S., Brito O.M.d., Micaroni M., Beznoussenko G.V., Rudka T., Bartoli D., Polishuck R.S., Danial N.N., Strooper B.D.et al.. OPA1 controls apoptotic cristae remodeling independently from mitochondrial fusion. Cell. 2006; 126:177–189. [DOI] [PubMed] [Google Scholar]
- 56. Tezze C., Romanello V., Desbats M.A., Fadini G.P., Albiero M., Favaro G., Ciciliot S., Soriano M.E., Morbidoni V., Cerqua C.et al.. Age-associated loss of OPA1 in muscle impacts muscle mass, metabolic homeostasis, systemic inflammation, and epithelial senescence. Cell Metab. 2017; 25:1374–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Cho S., Hwang E.S.. Fluorescence-based detection and quantification of features of cellular senescence. Methods in Cell Biology. 2011; 103:Elsevier; 149–188. [DOI] [PubMed] [Google Scholar]
- 58. Cadet J., Wagner J.R.. DNA base damage by reactive oxygen species, oxidizing agents, and UV radiation. Cold Spring Harb. Perspect. Biol. 2013; 5:a012559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. De Bont R. Endogenous DNA damage in humans: a review of quantitative data. Mutagenesis. 2004; 19:169–185. [DOI] [PubMed] [Google Scholar]
- 60. Markkanen E. Not breathing is not an option: how to deal with oxidative DNA damage. DNA Repair (Amst.). 2017; 59:82–105. [DOI] [PubMed] [Google Scholar]
- 61. da Silva P.F.L., Schumacher B.. DNA damage responses in ageing. Open Biol. 2019; 9:190168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Iyama T., Wilson D.M.. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst.). 2013; 12:620–636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Schumacher B., Pothof J., Vijg J., Hoeijmakers J.H.J.. The central role of DNA damage in the ageing process. Nature. 2021; 592:695–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Petr M.A., Tulika T., Carmona-Marin L.M., Scheibye-Knudsen M.. Protecting the aging genome. Trends Cell Biol. 2020; 30:117–132. [DOI] [PubMed] [Google Scholar]
- 65. Morillo-Huesca M., Clemente-Ruiz M., Andújar E., Prado F.. The SWR1 histone replacement complex causes genetic instability and genome-wide transcription misregulation in the absence of H2A.Z. PLoS One. 2010; 5:e12143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Xu Y., Ayrapetov M.K., Xu C., Gursoy-Yuzugullu O., Hu Y., Price B.D.. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell. 2012; 48:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Gursoy-Yuzugullu O., Ayrapetov M.K., Price B.D.. Histone chaperone Anp32e removes H2A.Z from DNA double-strand breaks and promotes nucleosome reorganization and DNA repair. Proc. Natl. Acad. Sci. U.S.A. 2015; 112:7507–7512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Begum N.A., Haque F., Stanlie A., Husain A., Mondal S., Nakata M., Taniguchi T., Taniguchi H., Honjo T.. Phf5a regulates DNA repair in class switch recombination via p400 and histone H2A variant deposition. EMBO J. 2021; 40:e106393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Koike M., Koike A.. Accumulation of Ku80 proteins at DNA double-strand breaks in living cells. Exp. Cell. Res. 2008; 314:1061–1070. [DOI] [PubMed] [Google Scholar]
- 70. Billon P., Côté J.. Precise deposition of histone H2A.Z in chromatin for genome expression and maintenance. Biochim. Biophys. Acta. 2013; 1819:290–302. [DOI] [PubMed] [Google Scholar]
- 71. Ferrand J., Rondinelli B., Polo S.E.. Histone variants: guardians of genome integrity. Cells. 2020; 9:2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G.. The Hallmarks of Aging. Cell. 2013; 153:1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. McCormick R., Vasilaki A.. Age-related changes in skeletal muscle: changes to life-style as a therapy. Biogerontology. 2018; 19:519–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Patel J., Baptiste B.A., Kim E., Hussain M., Croteau D.L., Bohr V.A.. DNA damage and mitochondria in cancer and aging. Carcinogenesis. 2020; 41:1625–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Schumacher B., Vijg J.. Age is in the nucleus. Nat. Metab. 2019; 1:931–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Vijg J. From DNA damage to mutations: all roads lead to aging. Ageing Res. Rev. 2021; 68:101316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Yousefzadeh M., Henpita C., Vyas R., Soto-Palma C., Robbins P., Niedernhofer L.. DNA damage—how and why we age?. eLife. 2021; 10:e62852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Vogel H., Lim D.-S., Karsenty G., Finegold M., Hasty P.. Deletion of Ku86 causes early onset of senescence in mice. Proc. Natl. Acad. Sci. U.S.A. 1999; 96:10770–10775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Hasty P., Vijg J.. Accelerating aging by mouse reverse genetics: a rational approach to understanding longevity. Aging Cell. 2004; 3:55–65. [DOI] [PubMed] [Google Scholar]
- 80. Li H., Vogel H., Holcomb V.B., Gu Y., Hasty P.. Deletion of Ku70, Ku80, or both causes early aging without substantially increased cancer. Mol. Cell. Biol. 2007; 27:8205–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Didier N., Hourdé C., Amthor H., Marazzi G., Sassoon D.. Loss of a single allele for Ku80 leads to progenitor dysfunction and accelerated aging in skeletal muscle: ku80 insufficiency causes muscle stem cell aging. EMBO Mol. Med. 2012; 4:910–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Papamichos-Chronakis M., Watanabe S., Rando O.J., Peterson C.L.. Global regulation of H2A.Z localization by the INO80 chromatin-remodeling enzyme is essential for genome integrity. Cell. 2011; 144:200–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Rona G., Roberti D., Yin Y., Pagan J.K., Homer H., Sassani E., Zeke A., Busino L., Rothenberg E., Pagano M.. PARP1-dependent recruitment of the FBXL10-RNF68-RNF2 ubiquitin ligase to sites of DNA damage controls H2A.Z loading. eLife. 2018; 7:e38771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Alatwi H.E., Downs J.A.. Removal of H2A.Z by INO80 promotes homologous recombination. EMBO Rep. 2015; 16:986–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Abbott D.W., Ivanova V.S., Wang X., Bonner W.M., Ausió J.. Characterization of the Stability and Folding of H2A.Z Chromatin Particles. J. Biol. Chem. 2001; 276:41945–41949. [DOI] [PubMed] [Google Scholar]
- 86. Thakar A., Gupta P., Ishibashi T., Finn R., Silva-Moreno B., Uchiyama S., Fukui K., Tomschik M., Ausio J., Zlatanova J.. H2A.Z and H3.3 histone variants affect nucleosome structure: biochemical and biophysical studies. Biochemistry. 2009; 48:10852–10857. [DOI] [PubMed] [Google Scholar]
- 87. Zhou M., Dai L., Li C., Shi L., Huang Y., Guo Z., Wu F., Zhu P., Zhou Z.. Structural basis of nucleosome dynamics modulation by histone variants H2A.B and H2A.Z.2.2. EMBO J. 2021; 40:e105907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Fortuny A., Chansard A., Caron P., Chevallier O., Leroy O., Renaud O., Polo S.E.. Imaging the response to DNA damage in heterochromatin domains reveals core principles of heterochromatin maintenance. Nat. Commun. 2021; 12:2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Hao F., Kale S., Dimitrov S., Hayes J.J.. Unraveling linker histone interactions in nucleosomes. Curr. Opin. Struct. Biol. 2021; 71:87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xu Y., Ayrapetov M., Xu C., Gursoy-Yuzugullu O., Hu Y., Price B.. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol. Cell. 2012; 48:723–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Sutcu H.H., Rassinoux P., Donnio L.-M., Neuillet D., Vianna F., Gabillot O., Mari P.-O., Baldeyron C., Giglia-Mari G.. Decline of DNA damage response along with myogenic differentiation. Life Sci. Allian. 2024; 7:e202302279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Li C., Rioux K.L., Delaney S.. Histone variants H3.3 and H2A.Z/H3.3 facilitate excision of uracil from nucleosome core particles. DNA Repair (Amst.). 2022; 116:103355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Demontis F., Piccirillo R., Goldberg A.L., Perrimon N.. Mechanisms of skeletal muscle aging: insights from Drosophila and mammalian models. Dis. Models Mech. 2013; 6:1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq datasets have been deposited in the Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under the accession number GSE216935.