


Abstract
RNA helicases function as versatile enzymes primarily responsible for remodeling RNA secondary structures and organizing ribonucleoprotein complexes. In our study, we conducted a systematic analysis of the helicase-related activities of Escherichia coli HrpA and presented the structures of both its apo form and its complex bound with both conventional and non-canonical DNAs. Our findings reveal that HrpA exhibits NTP hydrolysis activity and binds to ssDNA and ssRNA in distinct sequence-dependent manners. While the helicase core plays an essential role in unwinding RNA/RNA and RNA/DNA duplexes, the N-terminal extension in HrpA, consisting of three helices referred to as the APHB domain, is crucial for ssDNA binding and RNA/DNA duplex unwinding. Importantly, the APHB domain is implicated in binding to non-canonical DNA structures such as G-quadruplex and i-motif, and this report presents the first solved i-motif-helicase complex. This research not only provides comprehensive insights into the multifaceted roles of HrpA as an RNA helicase but also establishes a foundation for further investigations into the recognition and functional implications of i-motif DNA structures in various biological processes.
Graphical Abstract
Graphical Abstract.

Introduction
RNA helicases are pivotal enzymes responsible for unwinding double-stranded RNA, restructuring RNA secondary structures, and facilitating ribonucleoprotein (RNP) complex assembly through ATP-dependent mechanisms (1). These enzymes belong to the SF1 and SF2 superfamilies, encompassing six RNA helicase families (2). The DEAH/RHA family stands out as the second largest, with representatives found in both eukaryotes and bacteria (3). Eukaryotic DEAH/RHA proteins predominantly function in pre-mRNA splicing, RNA processing, RNA decay, ribosome biogenesis, ribosome biogenesis, translation initiation, with yeast helicases such as Prp2p, Prp16p, Prp22p, Prp43p, Dhr1p, Dhr2p, exemplifying these roles (4–10). In mammals, DEAH/RHA proteins have been implicated in various diseases, underscoring their significance (11–13).
While DEAH/RHA-type RNA helicases primarily act on RNA substrates as mentioned above, their activities can directly or indirectly influence interactions between RNA and DNA in various cellular processes (14,15). DEAH/RHA helicases may influence transcription termination events where RNA polymerase encounters DNA-RNA hybrid regions (16). While these helicases don’t directly unwind RNA-DNA duplexes, their actions in the vicinity of these structures could impact the dynamics of transcription termination. During pre-mRNA splicing, DEAH/RHA helicases participate in resolving RNA structures. While their main role is in the RNA domain, the dynamics of RNA-DNA interactions in the spliceosome machinery might be influenced by the helicase activity (17). In the context of certain RNA viruses, DEAH/RHA helicases can be involved in unwinding viral RNA structures. This might indirectly impact the formation of RNA–DNA hybrids if the viral replication process involves interactions with host genomic DNA. In processes such as DNA repair, where RNA-DNA hybrids can form transiently (18), DEAH/RHA helicases might indirectly influence the dynamics of these structures by interacting with the RNA component involved in the repair process. The intricate interplay between RNA and DNA transactions in the cell involves a network of proteins, including helicases with specific substrate preferences.
Structurally, DEAH/RHA helicases share common features, including two RecA-like domains (RecA1/RecA2) connected by a flexible linker, with RecA2 featuring a unique β-hairpin insertion. Additional domains like winged-helix (WH), ratchet and oligonucleotide/oligosaccharide-binding (OB) domains play crucial roles in binding, translocation, and duplex-unwinding activities (19–21). Notably, N- and C-terminal extensions beyond these conserved domains confer unique functions to DEAH/RHA helicases, including substrate recognition and protein-protein interactions.
Eukaryotic DEAH/RHA proteins have been extensively studied, while bacterial counterparts remain comparatively less explored (22). Yet, bacterial DEAH/RHA helicases like HrpA, HrpB and Z5898 are associated with critical roles in bacterial virulence and host infection (23). HrpA, in particular, is involved in endonucleolytic cleavage of specific transcripts in Escherichia coli and has been implicated in the virulence of Borrelia burgdorferi, the Lyme disease agent (24,25). Additionally, HrpA exhibits RNA helicase and RNA-dependent ATPase activities, suggesting involvement in RNA stability and degradation (26).
While the structure of the bacterial DEAH/RHA proteins HrpB and HrpA were previously elucidated by Grass et al, knowledge gaps persist regarding the functional significance of the N- and C-terminal extensions in HrpA (27–29). Our study delves into these extensions, uncovering novel insights into E. coli HrpA’s helicase-associated activities. Specifically, we reveal a previously uncharacterized nucleoside-binding domain within the N-terminal extension and demonstrate HrpA’s unique ability to recognize single-stranded DNA and quadruplex nucleic acids. Furthermore, our study unveils the first structural complex involving HrpA, ADP, and the i-motif, marking a significant milestone in our understanding of i-motif-protein interactions.
Materials and methods
Protein expression and purification
The gene encoding wild-type Escherichia coli HrpA (residues 1–1300) was generated via PCR and cloned into a modified pET-15b vector (Invitrogen) as an EcoRI–XhoI fragment. This vector was equipped with an N-terminal sumo tag, resulting in the creation of His-sumo-HrpA. The recombinant plasmid was subsequently transformed into the C2566H E. coli strain (New England Biolabs). Cultures were grown in Luria broth (LB) supplemented with 100 mg/ml ampicillin at 37°C until the OD600 reached 0.6–0.7. Then, cells were induced with 0.3 mM IPTG and further incubated at 18°C for an additional 16 h. After harvest, cells were resuspended in lysis buffer (20 mM Tris–HCl, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol and 5 mM imidazole) and lysed by passing the suspension through a French press (1200 bar) three times, followed by 2–3 rounds of sonication to disrupt DNA. The resulting cell lysate was centrifuged at 12 000 rpm for 40 min, and the supernatant containing the target protein was loaded onto a pre-equilibrated nickel affinity column (GE Healthcare). Elution was carried out using a high-imidazole buffer (20 mM Tris–HCl, pH 7.5, 500 mM NaCl, 10% (v/v) glycerol and 500 mM imidazole). The eluted protein was subjected to Sumo protease treatment overnight at 18°C and then diluted to a concentration of 100 mM NaCl. Subsequently, it was loaded onto a HiTrap SP column (GE Healthcare) pre-equilibrated with 20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 5% (v/v) glycerol and 2 mM EDTA. The target protein was eluted using an 80-ml gradient of 100–500 mM NaCl. Finally, the protein underwent purification using a Superdex-200 gel filtration column (S200, GE Healthcare) with size-exclusion buffer (300 mM NaCl, 20 mM Tris–HCl, pH 7.5 and 1 mM MgCl2). Fractions containing the target protein were concentrated through centrifugation, frozen in liquid nitrogen, and stored at -80°C.
Recombinant plasmids and protein expression for all non-wild-type HrpA variants followed the same procedure as the wild type, ensuring equal activity.
For HrpA1-696 (residues 1–696), initial purification was performed using a nickel affinity column, as described earlier. The Sumo-treated protein was then diluted to 170 mM NaCl and loaded onto an SP column. The eluate from the SP column was immediately loaded onto a Resource Q column (GE Healthcare) and eluted with an 80-ml gradient ranging from 170 to 500 mM NaCl. The identified fractions were subsequently diluted to 150 mM NaCl and loaded onto a HiTrap Heparin column (GE Healthcare). Elution was achieved using an 80-ml gradient ranging from 150 to 500 mM NaCl. During this process, two proteins with different migration distances were observed on 8% SDS-PAGE analysis. The smaller protein was purified using the S200 column, as previously described, and identified as HrpA1–630 based on the final crystal structure.
Oligonucleotides preparation
The DNA and RNA substrates used in this study were commercially obtained and chemically synthesized from Sangon Biotech (Shanghai) and Eurogentec (Belgium). Duplexes and G4 structures were generated by mixing complementary oligonucleotides in annealing buffer A (100 mM KCl and 20 mM Tris–HCl, pH 8.0). The mixture was heated to 98°C for 5 min using a PCR instrument and then slowly cooled to room temperature at a rate of 2 min/°C. For the Bcl2 i-motif (imBcl2) and Bcl2T14 i-motif (imBcl2T14), they were manufactured in annealing buffer B (100 mM KCl and 50 mM PBS, pH 6.2) using the same heating and annealing process as the duplexes. The sequences and labeling positions of all the oligonucleotides used in this study can be found in Supplementary Table S1.
Crystallization screening and structure determination
To generate the HrpA1–630•ADP•DNA complex (with a concentration of HrpA1–630 at 106 μM), HrpA1–630 in size-exclusion buffer was mixed with 11 nt polyC DNA (D-C11) in a molar ratio of 1:1.2. Subsequently, ADP was added to a final concentration of 1 mM. The HrpA1–630•ADP•DNA complex was then combined with the reservoir solution in a 1:1 ratio (0.25 μl:0.25 μl) using the sitting-drop vapor diffusion method at 20°C. Crystals of the HrpA1–630•ADP•C2 complex were obtained in a solution containing 0.03 M NaNO3, 0.03 M Na2HPO4, 0.03 M (NH4)2SO4, 0.1 M MES-imidazole (pH 6.5), 20% (v/v) ethylene glycol, and 10% (w/v) PEG 8000. Crystals of the HrpA1–630•ADP•i-motif complex were obtained in a solution containing 10% (w/v) PEG 3350 and 4% (v/v) Tacsimate (pH 7.0). For HrpA1-758(residues 1–758), with a concentration of 8 mg/ml, a similar sitting-drop method was used for crystallization screening. The optimized crystallization conditions consisted of 0.1 M Hepes (pH 7.0) and 7% (w/v) PEG 20K.
Data collection and processing
The crystals of the HrpA1–630•ADP•i-motif complex were cryoprotected using a solution containing 20% (w/v) PEG MME 2000 and 20% (v/v) ethylene glycol. In contrast, the HrpA1-758 apo crystals were cryoprotected by supplementing their reservoir solution with 30% (w/v) PEG 400. The crystals of the HrpA1–630•ADP•C2 complex were directly frozen in liquid nitrogen from the crystallization drop. All X-ray diffraction data were collected at beamline BL17U of the Shanghai Synchrotron Radiation Facility. The crystallographic data were processed using XDS and scaled with Aimless through the Autoproc pipeline (30). Detailed statistics regarding the X-ray diffraction data are presented in Table 1.
Table 1.
Data collection and refinement statistics
| HrpA1–630•ADP•C2 | HrpA1-758 apo | HrpA1–630•ADP•i-motif | |
|---|---|---|---|
| Wavelength (Å) | 0.9792 | 0.9785 | 0.9778 |
| Resolution range (Å) | 73.34–2.25 (2.33–2.25) | 29.73–2.66 (2.75–2.66) | 39.48 - 2.52 (2.61 - 2.52) |
| Space group | P212121 | P21 | P212121 |
| Unit cell parameters a, b, c (Å) | 80.47, 106.12, 178.27 | 39.60, 114.89, 94.01 | 80.27, 105.99, 177.57 |
| Total reflections | 912 871 (97908) | 149 540 (9907) | 299 445 (35223) |
| Unique reflections | 70 047 (7102) | 23 533 (2187) | 45 125 (5086) |
| Multiplicity | 13.0 (13.8) | 6.4 (4.5) | 6.6 (6.9) |
| Completeness (%) | 96.55 (99.76) | 98.88 (92.30) | 86.94 (100.00) |
| Mean I/sigma(I) | 20.26 (3.17) | 17.21 (2.16) | 15.18 (2.29) |
| Wilson B-factor (Å2) | 28.46 | 55.74 | 50.01 |
| R-merge | 0.1638 (1.296) | 0.08523 (0.5895) | 0.09691 (0.8385) |
| CC1/2 | 0.999 (0.88) | 0.999 (0.904) | 0.998 (0.784) |
| Refinement | |||
| Reflections used in refinement | 70 036 (7101) | 23 510 (2183) | 45 107 (5086) |
| Reflections used for R-free | 3464 (379) | 1118 (100) | 2230 (225) |
| R work/Rfree (%) | 18.72/23.06 | 20.59/25.73 | 19.45/23.88 |
| Number of non-hydrogen atoms | 10 066 | 6072 | 10 335 |
| macromolecules | 9422 | 6031 | 10 132 |
| ligands | 61 | 5 | 72 |
| solvent | 583 | 36 | 131 |
| Protein residues | 1173 | 754 | 1222 |
| RMS (bonds) (Å) | 0.008 | 0.003 | 0.005 |
| RMS (angles) (°) | 1.18 | 0.61 | 0.84 |
| Ramachandran favored (%) | 99.14 | 97.34 | 98.18 |
| Ramachandran outliers (%) | 0.00 | 0.00 | 0.00 |
| Average B-factor (Å2) | 46.71 | 72.75 | 62.01 |
| PDB code | 8PO7 | 8PO6 | 8PO8 |
Statistics for the highest-resolution shell are shown in parentheses.
Structure solution and refinement
Initial attempts to solve the structure of the HrpA1–630•ADP•C2 complex in space group P212121 using molecular replacement (MR) with Phaser (31) utilizing various models such as the HrpB structure (PDB code: 6HEG), different DEAH/RHA helicase core structures, or separated RecA domains did not yield satisfactory results, with a final translation Z-score (TFZ) below 6.5. Eventually, the structure of the HrpA1–630•ADP•C2 complex was successfully solved using molecular replacement with the automated pipeline MRage (32). The best model for the molecular replacement was the DHX15 structure (PDB code: 5XDR), which provided a TFZ of 12.8, compared to the Prp43 structure (PDB code: 2XAU) with a TFZ of only 6.5. The initial model was then automatically built in Phenix using an auto-build protocol without rebuilding in place. The data up to 2.0 Å resolution were used, and non-crystallographic symmetry (NCS) was applied to the two molecules in the asymmetric unit. The final model was manually adjusted in Coot during refinement using Phenix. The solved structure of the HrpA1–630•ADP•C2 complex served as the model for solving the other HrpA structures, including the structure with an extended C-terminus (HrpA1–758) that crystallized in different space groups (P21).
In the electron density map of the HrpA1–630•ADP•C2 complex, two cytosine nucleotides were successfully built, while the electron density map of the HrpA1–630•ADP•i-motif complex allowed the complete building of the i-motif. All structure representations were generated using PyMOL (http://www.pymol.org).
Fluorescence anisotropy assays
The nucleic acid binding affinities of HrpA were assessed using fluorescence anisotropy measurements performed on an Infinite F200 instrument (TECAN). To determine the binding affinity, 5 nM of 3′-FAM-labeled single-strand and double-strand nucleic acids in binding buffer A (50 mM NaCl, 20 mM Tris–HCl, pH 7.5, 1 mM MgCl2 and 1 mM DTT) and 5 nM of 3′-FAM-labeled imBcl2 in binding buffer B (50 mM KCl and 50 mM PBS, pH 6.2) were titrated with increasing concentrations of HrpA protein. Each sample was incubated at room temperature for 5 min to allow the system to reach equilibrium, and the fluorescence anisotropy signal was monitored and recorded. An additional reading was taken after 10 min to ensure the mixture had fully equilibrated and the signal was stable.
The equilibrium dissociation constant (Kd) was determined by fitting the binding curves using Equation (1):
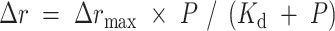 |
(1) |
where Δrmax represents the maximal amplitude of anisotropy (equal to rmax, complex – rfree DNA), P is the protein concentration, and Kd corresponds to the midpoint of the curve, reflecting the apparent dissociation constant. The Kd values were obtained from at least three independent experiments, ensuring reliable measurements of binding affinity.
Stopped-flow unwinding assay
Fluorescence-based stopped-flow FRET assays, following the methodology described by Liu et al. (33), were employed to conduct duplex unwinding experiments. Unwinding kinetics were measured using a two-syringe mode setup. In this configuration, 100 nM HrpA and 4 nM fluorescently labeled duplex substrates were pre-incubated in the unwinding buffer (30 mM NaCl, 20 mM Tris–HCl, pH 8.0, 2 mM MgCl2 and 2 mM DTT) at 37 °C for 5 min (in syringe 3). The unwinding reaction was initiated by rapid mixing with 1 mM ATP (in syringe 4).
To convert the output data from volts to percentage unwinding, calibration experiments were performed in a two-syringe mode. In these calibration experiments, HrpA and hexachlorofluorescein (HF)-labeled single-stranded oligonucleotide were placed in syringe 3, while FAM-labeled single-stranded oligonucleotide was placed in syringe 4. Both syringes were incubated in the unwinding buffer. The fluorescence signal of the mixed solution from the two syringes represented 100% unwinding.
All stopped-flow kinetic traces were averaged from over 10 individual traces. The fraction of duplex unwound (η) at time t was calculated using Equation (2):
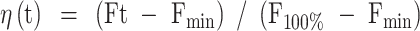 |
(2) |
Here, Ft represents the measured fluorescence signal at time t, Fmin is the minimum fluorescence signal observed after unwinding initiation, and F100% corresponds to the signal obtained from the calibration measurement.
NTPase activity assays
NTPase activity measurements were conducted using the ATPase/GTPase Activity Assay Kit (Sigma-Aldrich), which detects the release of free Pi resulting from NTP hydrolysis. A 20-μl reaction mixture containing 100 nM HrpA in the binding buffer was incubated for 10 min at 20°C in the presence or absence of 1 μM RNA (DNA). The reactions were initiated by adding NTP and stopped by adding 100 μl of the malachite green reagent solution to a white 96-well microplate. The NTP concentration was varied from 0 to 500 μM. After incubation at room temperature for 10 min, the absorbance at 620 nm was measured using a VICTOR Nivo Multimode Microplate Reader (PerkinElmer). The concentration of Pi in each sample was calculated using a standard curve provided in the manufacturer's instructions. The kcat value was determined by fitting the experimental data to the Michaelis-Menten equation using Origin 9.0 software.
Circular dichroism (CD) spectropolarimetry
The conformation of imBcl2 was analyzed using circular dichroism (CD) experiments performed with a Bio-Logic MOS450/AF-CD optical system (Bio-Logic Science Instruments, Seyssinet-Pariset, France). The experiments were conducted in a temperature-controlled cell holder using a quartz cell with a 1 mm path length. The imBcl2 sample, prepared as described previously, was diluted to a concentration of 3 μM in a buffer with pH ranging from 5.8 to 8.0. CD scans were recorded in the UV region (220–320 nm) with 0.75-nm increments, averaging for 2 s at 25°C. To obtain the CD profile of imBcl2, the spectra of samples containing only the buffer were subtracted. The data were further processed and analyzed using Origin 9.0 software.
Single-molecule fluorescence data acquisition and analysis
The single-molecule FRET (smFRET) assay was conducted following the protocol described in our previous publication (34). Briefly, a chamber was prepared and 50 pM of fluorescently labeled DNA (Cy3 and Cy5) was introduced and allowed to immobilize for 10 min. The chamber was then washed with the imaging buffer (50 mM PBS, pH 6.2, 100 mM KCl, 0.8% D-glucose, 1 mg/ml glucose oxidase, 0.4 mg/ml catalase, and 4 mM Trolox) to remove any free DNA. All single-molecule measurements were performed at a constant temperature of 22°C with an exposure time of 100 ms. The FRET efficiency was calculated using the equation E = IA/(ID+ IA), where ID represents the donor intensity and IA represents the acceptor intensity. Basic data analysis, including the generation of transition density plots (TDP), was carried out using custom MATLAB scripts. Data fitting was performed using Origin 9.0 software. Histograms were fitted using Gaussian distributions, with the peak positions left unrestrained.
Molecular modeling and molecular dynamics
Structures of APHB-ssDNA (7-mer polyC) and APHB-ssRNA (7-mer polyU) were generated with RosettaFoldNA (35) installed on local computer. From the HrpA-15polyU structure (PDB 6ZWW), the ssRNA bound to APHB was manually merged to 15polyU and geometry regularized in Coot. HrpA-ssRNA was then subjected to molecular dynamics simulation. The inputs were prepared using CHARMM-GUI with the Solution Builder module. The CHARMM36m force field was used to describe the full system consisting of a box with explicit water and KCl (reaching 150 mM). The CHARMM-GUI protocol was followed using GROMACS (36). The NVT equilibration phase was performed during 50 ps. Trajectory of 50 ns was obtained and further analyzed with the GROMACS tools. RMSD-based clustering was performed by gromos clustering algorithm. Snapshot structures were visualized in PyMOL which served to create all the illustrations.
Results
HrpA’s NTP usage promiscuity and efficient 3′-5′ RNA/RNA and RNA/DNA duplex unwinding
HrpA exhibits substantial similarity (58.87%) and identity (27.62%) with HrpB within the helicase core, encompassing two RecA domains, WH, OB, and CON modules (Figure 1A). Nonetheless, HrpA features additional structural elements—a 74-residue N-terminal extension (APHB) and a 542-residue C-terminal extension. To explore the functional relevance of these structural components, we initially assessed HrpA’s nucleic acid binding capability using fluorescently labeled DNA and RNA oligonucleotides. Equilibrium binding experiments involved titrating labeled DNA/RNA with escalating concentrations of HrpA, with the binding fraction determined through fluorescence anisotropy changes. Our results (Figure 1B, C, and D) unveiled two distinct binding characteristics of HrpA: (i) HrpA binds both ssDNA and ssRNA with a pronounced preference for binding ssRNA (KdU12 = 21.3 nM) over ssDNA (KdT12 = 307 nM), with reduced affinity for double-stranded blunt-ended RNA(R-Blunt); (ii) HrpA displays selectivity for guanine and cytosine (G/C) bases in ssDNA but exhibits a preference for guanine and adenine (G/A) bases in ssRNA.
Figure 1.

HrpA’s NTP usage promiscuity and efficient 3′-5′ RNA/RNA and RNA/DNA duplex unwinding (A) HrpA structure diagram and domain comparison with HrpB and Prp43. (B–D) The binding curves of HrpA1–1300 binds to DNA and RNA. Substrates D-G-rich, D-C12, D-T12, and D-A12 are 12 nt ssDNA with G-rich, polyC, polyT and polyA sequences, respectively. R-G-rich, R-C12, R-U12 and R-A12 are 12 nt ssRNA with G-rich, polyC, polyU and polyA sequences, respectively. D-Blunt and R-Blunt are 12 bp dsDNA and dsRNA, respectively. All assays were performed in 50 mM NaCl, 20 mM Tris–HCl, pH 7.5, 1 mM MgCl2, and 1 mM DTT. Kd values of each binding curve were obtained by fitting Equation (1). (E) Nucleotide and nucleic acid specificity of NTPase activity of HrpA1–1300 in the case with and without R-SS33, respectively. R-SS33 are 33 nt ssRNA. The kcat was calculated by fitting with the Michaelis–Menten equation. (F) Stopped-flow helicase activities of HrpA1–1300 for partial duplexes. All helicase activities were assayed with 100 nM HrpA1–1300, 4 nM fluorescently labeled partial duplex, and 1 mM ATP in the unwinding buffer. Structural diagrams of all five partial duplexes are indicated inside the graph.
The DEAD proteins family of RNA helicases harbors a central region featuring eight consensus motifs, including the DEAD box (Asp-Glu-Ala-Asp) ATPase site, which facilitates ATP hydrolysis and couples it to helicase activity. As HrpA (DEAH) and HrpB (DEFH) differ in their DExH-box sequences, we investigated HrpA’s NTPase activity with various nucleotides (A, U, C and G), both in the presence and absence of single-stranded RNA (R-SS33) (Figure 1E). We determined NTP hydrolysis activity using a colorimetric ATPase/GTPase Activity Assay Kit. Our findings unveiled HrpA’s versatile NTP usage, as it displayed heightened NTPase activity with UTP and CTP. Notably, in the absence of nucleic acids, HrpA exhibited significant UTPase and CTPase activities, with the addition of single-stranded RNA enhancing NTPase activities to 2–2.5-fold (Supplementary Table S2).
To ascertain whether HrpA possesses duplex unwinding activity for DNA or RNA substrates, we conducted unwinding experiments using fluorescently labeled 12-bp duplex substrates with a 3′- or 5′-10 nt tail. Real-time monitoring of changes in fluorescence resonance energy transfer (FRET) between the donor fluorophore-FAM and the acceptor fluorophore-Hexachlorofluorescein was achieved via a stopped-flow experiment. Loss of FRET corresponded to duplex unwinding and strand separation, resulting in increased FAM fluorescence. As illustrated in Figure 1F, HrpA efficiently unwound duplex RNA with a 10 nt overhang at the 3′-end, while the DNA/DNA duplex remained intact. This indicates that HrpA possesses intrinsic RNA unwinding activity but lacks the ability to unwind DNA, distinguishing it from HrpB (28,37). Furthermore, when assessing the unwinding polarity, we observed that HrpA exhibits a 3′–5′ polarity for RNA unwinding, as it could not unwind an RNA substrate with a 5′-end protruding overhang.
In cellular processes, the formation of RNA-DNA duplexes plays a crucial role in various nucleic acid metabolism pathways, such as transcription and RNA primer synthesis in DNA replication (38,39). These processes underscore the dynamic nature of nucleic acid metabolism within cells, where RNA and DNA interact in diverse ways to execute essential cellular functions like gene expression, DNA replication and repair. To delve into the potential mechanism behind HrpA-mediated RNA duplex unwinding specificity, we utilized RNA/DNA hybrid molecules with different loading strands (RNA or DNA) and 3′-overhangs for unwinding assays. Intriguingly, HrpA efficiently unwound the RNA/DNA hybrids with 3′-overhang single-stranded RNA, while those with 3′-overhang single-stranded DNA remained intact. These results suggest that HrpA selectively binds to single-stranded RNA and translocates along the RNA strand from the 3′ to 5′ direction. Notably, these helicases are not typically associated with the direct unwinding of RNA-DNA duplexes. In summary, these findings showcase HrpA’s versatility as an RNA helicase, encompassing functions in DNA and RNA binding, NTP hydrolysis, and duplex unwinding.
Structural and functional analysis of N-terminal domain APHB
In Escherichia coli, HrpA is encoded by one of the largest open reading frames (ORFs) and features an extensive C-terminal extension that extends beyond the CON domain (Figure 1A and Supplementary Figure S1). Unfortunately, our efforts to obtain the full-length crystal structure of HrpA, similar to HrpB, were unsuccessful. Instead, we successfully determined the crystal structure of HrpA1-758 (Figure 2A). The overall structure and folding of Apo-HrpA1-758 are identical to the previously reported HrpA1-783 structure by Grass et al. (29). It appears that the N- and C-terminal extensions of HrpA, which are distinct from HrpB, may present challenges in crystal packing. Additionally, HrpA harbors an extended C-terminal region encompassing the CON domain (residues 696–758) and a subsequent extended region (residues 759–1300), which was truncated for structural analysis. The CON domain features a β-sheet composed of three antiparallel strands, followed by two short helices, and shares both structural and sequence similarity with the CON domain found in the HrpB structure (Supplementary Figures S1 and S2A). HrpA1-758 exhibits 37% identity with Saccharomyces cerevisiae Prp43 and maintains the characteristic organization observed in DEAH/RHA helicases, exemplified by Prp43. Upon superimposing HrpA domains (RecA1, RecA2, WH, Ratchet, and OB) onto their Prp43 counterparts, root-mean-square deviations (r.m.s.d.) of 0.74 Å (154 Cα), 1.67 Å (168 Cα), 1.03 Å (49 Cα), 1.2 Å (102 Cα) and 1.07 Å (58 Cα), respectively, were observed (Supplementary Figure S2B-D). Of particular note, the ratchet domain in HrpA exhibits the same topology as Prp43, while in HrpB, the ratchet domain contains fewer helices, as revealed previously by Grass et al. (29).
Figure 2.

Structural and Functional Analysis of N-terminal domain APHB (A) Overall Structures of HrpA1-758 apo. All domains are colored and labeled according to Figure 1A. (B) Interactions between APHB and RecA1 domains in the structure of Apo-HrpA1-758 complex structure. Detailed contacts and their schematic representations are shown in the down panel. The dashed line indicates all hydrogen bonds. (C) Dissociation equilibrium constants of HrpA1–1300 and its variants to ssRNA. (D) ATPase assays show that APHB reduces the ATPase activity both in the presence and absence of RNA. (E) Kinetic unwinding curves of HrpA1–1300 and its variants were determined by stopped-flow assay with 4 nM partial dsRNA (with 3′-tail), 100 nM proteins and 1 mM ATP.
The N-terminal region of HrpA comprises three closely packed α-helices (α1, α2, α3), forming an antiparallel helix-bundle domain known as APHB. Additionally, spatial conformation is reinforced by hydrogen bonds between residues on the three alpha-helices and those from RecA1 (Figure 2A and B). APHB represents a novel extension within DEAH/RHA helicases, to investigate the nucleic acid binding capability of APHB, we conducted binding studies using full-length HrpA1–1300 and various truncations with diverse RNA substrates, including R-G-rich, R-C12, R-U12 and R-A12 (Figure 2C). In the absence of APHB, both HrpA75–1300 and HrpA75–758 displayed over a 3-fold reduction in affinity for all four single-stranded RNA substrates, confirming the crucial role of APHB in efficiently binding single-stranded RNA. Compared to HrpA1–1300, HrpA75–1300 exhibited more than a 9-fold reduction in affinity for both R-C12 and R-G-rich substrates, along with a more than 2-fold reduction for R-U12. Notably, APHB truncated protein (HrpA75–1300) lost its ability to bind D-C12 and D-G-rich substrates, underscoring the significance of the APHB domain in single-stranded DNA binding and nucleotide selectivity (Supplementary Figure S3A).
APHB is situated adjacent to the RecA1 domain and forms hydrogen bonds with residues D17, L21, R64, T72 and Y73 from APHB, as well as residues K122, G120, I121, T168, K112 and E116 from RecA1 (Figure 2B and Supplementary Figure S3B). Mutational analysis on amino acids involved in at least two hydrogen bond interactions (R64K/K122A) did not result in a significant difference in binding activity (Figure 2C). However, unlike single-stranded DNA, both HrpA75–1300 and HrpA75-758 maintained nucleotide selectivity for single-stranded RNA even after mutation, suggesting that HrpA’s sequence-dependent binding to single-stranded RNA is primarily governed by the helicase core (RNA binding tunnel) rather than APHB. These results confirm the role of the N-terminal APHB domain of HrpA as a nucleic acid-binding domain.
Moreover, APHB contributes to ATPase and helicase activities. The apparent Kobs values of ATPase activities are determined as increasing concentration of ATP using the full-length wild type and these bearing mutations in APHB. The APHB truncated HrpA with or without mutations were analyzed in parallel. The apparent ATP binding affinities determined from the ATPase activity curves demonstrate that truncation or mutations in APHB lead to a significant decrease in ATP binding affinities (Figure 2D). Since kcat values may reflect how efficiently the enzyme is converting ATP into products, these results indicated that APHB plays an essential role in ATP hydrolysis, showing that the APHB truncation or mutations lead to a significant decrease in ATP binding affinities and consequently affect the function of HrpA. Consistently, while the C-terminal truncated HrpA1–758 displays comparable unwinding activity with the full-length protein, the N-terminal truncated HrpA75–758 is completely compromised in its unwinding activity, the presence of the C-terminal domain can restore or compensate the APHB truncated results in unwinding activity (Figure 2E). The above results demonstrated that N- and C-terminal domains cooperatively regulate HrpA unwinding activity.
APHB dominates nucleic acid recognition
In order to reveal the structural details of APHB domain-mediated nucleic acid binding and unwinding, HrpA truncated prior to the CON and almost OB domain (residues 1–630) was complexed with a ssDNA in the presence of ADP (Figure 3A and Table 1). In the structure of the HrpA1–630•ADP•C2 complex, a single asymmetric unit contained two molecules, with only one molecule binding to DNA (Supplementary Figure S4A). The molecule lacking DNA binding was designated as the HrpA•ADP complex, whereas the one binding to C2-DNA was labeled the HrpA1–630•ADP•C2 complex.
Figure 3.

The APHB domain shows functions in binding, ATPase, and helicase activities. (A) Structure of HrpA1–630• ADP•C2 complex. Detail interactions between APHB and DNA are shown in zoomed-in panels. The cartoon shows DNA with strong electron density (blue lines). (B) Interactions between helices of APHB domain in the structure of HrpA1–630• ADP•C2 complex. Hydrogen bonds are represented by the blue dashed line. (C) Schematic diagram of APHB’s contacts with two cytosine nucleotides. (D) Dissociation equilibrium constants of HrpA1–1300 and its variants to D-C12 and D-G-rich. (E) The electrostatic surface potential of HrpA1–630• ADP•C2 complex. Blue and red indicate positive and negative charges, respectively.
Within the HrpA1–630•ADP•C2 complex structure, residues Q13 (α1), D17 (α1), D23 (α2), R30 (α2) and V34 (α2) formed hydrogen bonds with S28 (α2), R24 (α2), K60 (α3), E53 (α3) and Q43 (α3), respectively (Figure 3B). Most of these bonds were conserved in the apo structure as well (Supplementary Figure S4B). Compared to the structure of apo-HrpA in which a stable alpha-helix bundle is maintained through three pairs of hydrogen bonds among α1, α2, and α3, the electron density map within the HrpA1–630•ADP•C2 complex clearly revealed the presence of C2-DNA (Figure 3A). These structural features suggest that the N-terminal domain of HrpA may enhance DNA binding, given its relatively lower affinity for ssDNA compared to ssRNA. Notably, R30 and E49 formed distinct hydrogen bonds with the C2-DNA, providing additional evidence of the N-terminal domain's involvement in DNA binding (Figure 3C). To delve deeper into the mechanism of APHB domain binding to ssDNA/ssRNA, three arginine residues from α2 of APHB were independently mutated to alanine, in addition to R30 and E49 mutations. The results demonstrated that the binding affinities of the R22A, R26A, R29A and R30A mutants for D-C12, as well as the R26A, R29A and R30A mutants for D-G-rich, were significantly reduced compared to the wild type, underscoring the involvement of these arginine residues in interactions with guanine and cytosine bases in ssDNA (Figure 3D). Notably, despite the presence of a hydrogen bond between E49 and the C1 base, the Kd values of the E49A mutant for D-G-rich and D-C12 did not exhibit a substantial increase. Similar experiments were conducted with ssRNA, but no mutations were found to severely compromise the affinity of HrpA for the four tested RNA substrates (Supplementary Figure S4C). In light of the combined structural, truncated, and mutational data, it is evident that the α2 helix of the APHB domain plays a pivotal role in nucleic acid recognition and binding.
It is notable that the C2-DNA observed was not situated within the conserved RNA-binding tunnel formed by the RecA1, RecA2, WH, Ratchet and OB domains. Instead, it resided on the positively charged surface of the APHB domain, which is rich in arginine residues (Figure 3E). Additionally, the C2-DNA exhibited a defined polarity of 3′-5′, and its alignment was not continuous with the ssRNA bound in the binding tunnel, as observed in PDB 6ZWW by Grass et al.
To further explore the function of nucleic acids binding to the APHB domain, we conducted additional modeling experiments using RoseTTAFoldNA (35). In both ab initio complexes, the predicted structures accurately represent the APHB structure, with nucleic acids bound to the α2 helix in a reverse orientation compared to our crystallographic structures (Supplementary Figure S5A). This alignment suggests that the predicted orientation supports the potential connection between nucleic acids bound to APHB and those bound in the ssRNA tunnel. We generated a model from HrpA-15polyU structure (PDB 6ZWW) with a longer ssRNA exiting from the tunnel and binding to APHB according to the model generated by RoseTTAFoldNA. This model was then subjected to 50ns molecular dynamics. After 15ns equilibration the molecular dynamics trajectory shows that APHB with ssRNA adopts a stable mean conformation corresponding to the major 2 Å rmsd-cluster comprising 65% of the conformations (Supplementary Figure S5B). ssRNA occupies the positively charged surface of APHB, with phosphates of ssRNA establishing stable electrostatic interactions with Arg residues of α2 helix of APHB (Supplementary Figure S5C).
Structure of HrpA1–630•ADP•i-motif complex
To explore the binding of HrpA to various nucleic acid structures, attempts were made to co-crystallize HrpA with different substrates, including G-quadruplex (G4) and intercalated motif (i-motif) structures. Ultimately, successful co-crystallizations of the HrpA1–630•ADP•i-motif complex were achieved (Figure 4A). The structure of this complex was determined in the P212121 space group and refined to a resolution of 2.52 Å, with Rfree and Rwork values of 23.88% and 19.45%, respectively (Table 1). In the asymmetric unit, there were two HrpA1–630 molecules, two ADP molecules and two polyC DNA molecules. The nucleic acid structure was positioned between two APHB domains. Each molecule interacted with a single polyC chain primarily through its APHB domain, and these two chains formed an i-motif structure with typical cytosine-cytosine interactions. The i-motif structure was precisely nestled within a positively charged pocket formed by the RecA1, WH and APHB domains of the two HrpA molecules (Figure 4B).
Figure 4.

Structure analysis of HrpA1–630•ADP•i-motif complex. (A) Structural overview of HrpA1–630•ADP•i-motif complex. One of the two HrpA1–630 molecules is presented as a semi-transparent model. All domains are colored as in Figure 1A. The phosphate backbones of the two polyC chains forming i-motif are colored in yellow and red, respectively. (B) The electrostatic surface potential of HrpA1–630•ADP•i-motif complex. Blue and red indicate positive and negative charges, respectively. (C) Detailed structural information of this intermolecular i-motif. Cartoon structure (left panel) in strong electron density (green lines) and schematic (right panel) showing that two poly(C) DNA formed an i-motif with 3′-E topology. In the schematic structure, nucleotides involved in forming C:C+ base pairs in the two molecules are colored cyan and green, respectively, and the rest are in gray. (D) Detailed view of i-motif-HrpA interactions. (E) Interactions between HrpA and i-motif are shown schematically. Black dotted lines indicate H-bonds between i-motif and HrpA.
In the i-motif structure, the initial three cytosine bases (C1, C2, C3) of both polyC chains did not participate in i-motif formation (Figure 4C–E). Each polyC chain folded back to form an antiparallel hairpin, with the 3′ end closely associated with the 5′ end. Every base, except for C7 and C8, engaged in symmetric pairings with cytosine bases from the other DNA chain through cytosine-hemiprotonated cytosine interactions. Specifically, C4, C5, C6, C9, C10 and C11 paired with C9, C10, C11, C4, C5 and C6, respectively, from the other DNA chain (Figure 4C). Together, these four antiparallel chains constituted an intermolecular i-motif structure featuring a 3′-E topology (40). Apart from Q184, which interacted with the sugar-phosphate backbone of the i-motif, other residues of HrpA were engaged with the bases of the i-motif (Figures 4D and E). Among the 21 nucleotides that were resolved (excluding C1 in the 5′ end of one polyC chain), 12 were involved in the formation of C:C+ base pairs, resulting in limited interactions between the i-motif and HrpA1–630, with only nine hydrogen bonds identified (Figure 4E). Besides Q184 (RecA1), T476 (WH) and D478 (WH), the remaining six hydrogen bonds were contributed by the APHB domains of the two HrpA molecules. This indicates that the APHB domain is a pivotal domain for HrpA binding to the i-motif. To experimentally validate this hypothesis, wild-type HrpA, three truncations, and four point-mutations of HrpA were independently subjected to binding assays with the Bcl2-i-motif (imBcl2), a well-studied substrate known for its ability to form an i-motif structure.
Based on the results illustrating the capability of the annealed Bcl2 oligonucleotide to adopt an i-motif conformation in a PBS pH 6.2 buffer (Figure 5A), i-motif binding assays were subsequently carried out in the same buffer supplemented with 50 mM KCl. The presence of the APHB domain demonstrated a notable affinity for the i-motif, while its absence significantly diminished the binding affinity, as illustrated in Figure 5B. Mutations in the residues R22, R26, R29, and R30, responsible for binding to the 5′ end of polyC ssDNA, markedly impaired i-motif binding abilities. Notably, mutations of R26 and R30 resulted in 3- and 10-fold increases in the Kd values, respectively (Figure 5C and Supplementary Table S3). To confirm the specificity of HrpA in binding to i-motifs, we conducted assays using two additional i-motif structures (imMyc and imVEGF, sequences were collected in Supplementary Table S4) along with hairpins structured DNAs to evaluate their respective affinities for i-motif binding. In a consistent pattern, HrpA demonstrated effective binding to all i-motif structures, while showing no affinity for the hairpin structures (Supplementary Table S4). Additionally, HrpA1–1300 effectively unwound the i-motif structure, as evidenced by smFRET measurements of the interaction between HrpA1–1300 and the i-motif (Figures 5D and E). In the absence of APHB, there was no significant alteration in the smFRET signal (Figure 5F). smFRET assays were also conducted with mutations affecting the binding to the 5′ end of polyC ssDNA to compare the impact of APHB on the measured FRET values. The results revealed that the smFRET values decreased less than the full range after R22, R26, and R29 mutations (Figures 5G, H, and Supplementary Figure S6A–C). smFRET values of full-length proteins did not decrease after R30 mutation (Figure 5I). These findings indicate that APHB plays a pivotal role in the structural remodeling of the i-motif, thereby influencing the efficiency of unwinding through its impact on binding activity with the i-motif.
Figure 5.

Functional analysis of APHB in i-motif remodeling. (A) CD spectra of imBcl2 (3 μM) in a 50 mM sodium phosphate solution with 50 mM KCl at the required pH. (B, C) Dissociation equilibrium of HrpA1–1300, three truncations, and four point-mutations to Bcl2 i-motif (imBcl2). All assays were performed in 50 mM KCl and 50 mM PBS, pH 6.2. (D) FRET histograms were constructed from about 300 individual records of imBcl2 alone (imBcl2 only), imBcl2 with HrpA1–1300 and HrpA75–1300, respectively. Gaussian fittings yield two populations peaked at FRET = 0.39 and 0.89. (E, F) Individual FRET traces were recorded with 1 μM partial imBcl2 in the presence of HrpA1–1300 and HrpA75–1300. (G) FRET histograms were constructed from about 300 individual records of imBcl2 with HrpA1–1300, HrpA1–1300, R22A, and HrpA1–1300, R30A. Gaussian fittings yield two populations peaked at FRET = 0.39 and 0.89. (H, I) Individual FRET traces recorded with 1 μM partial
Discussion
HrpA and HrpB are members of the DEAH-family RNA helicase-like proteins, showing substantial sequence similarity with other DEAH/RHA proteins (41). These proteins play crucial roles in bacterial virulence, contributing significantly to essential processes in most bacteria (22). Previous research, including our own, has shed light on the crystal structure of full-length HrpB from E. coli in conjunction with ADP•AlF4. This unveiling showcased the classical helicase core along with a sizable C-terminal extension (28). The present study contributes additional insights into the properties of these proteins, further expanding our understanding of their functions.
Interactions between ratchet and RecA2 domains in RNA-independent NTPase activity
HrpA encompasses all conserved domains characteristic of the DEAH/RHA helicase family. The binding of ADP in the crevice between the two RecA-like domains, where the nucleobase is positioned between Arg141 from RecA1 and Phe336 from RecA2 (Supplementary Figure S7), follows a mode akin to that seen in other helicases like Prp43/DHX15, known as the F-motif (20). Upon ADP release, RecA2 undergoes an opening movement toward RecA1, transitioning from an open state to a closed state, a conformational change typical in DEAH/RHA helicases' translocation mechanism (Supplementary Figure S8).
Notably, our crystal structures reveal a significant rotation of the C-terminal Ratchet domain, which plays a crucial role in obstructing the 5′ end of the RNA-binding tunnel (Supplementary Figure S9). This closure of the tunnel is more pronounced in HrpA compared to other helicases, and it is stabilized by an extensive interaction surface between the Ratchet domain and RecA2 (Supplementary Figure S10). This inactive translocation form of HrpA impedes RNA-binding tunnel accessibility. Unlike other DEAH/RHA helicases where the RecA2 domain and C-terminal accessory domains exhibit flexibility in the apo state, the specific conformation of the Ratchet domain in HrpA limits RecA2’s flexibility. This restraint in the absence of nucleic acid binding contributes to HrpA’s elevated basal NTPase activity, rendering it less sensitive to nucleic acid binding.
The unique APHB domain
One distinctive feature of HrpA is its N-terminal APHB domain, responsible for binding ssDNA and ssRNA. Unlike other DEAH/RHA helicases, such as Prp43 and Prp2, where N-terminal domains involved in nucleic acid binding are positioned near the 5′ end of the RNA-binding tunnel, APHB in HrpA occupies a unique position adjacent to RecA1 at the 3′ end of the RNA-binding tunnel. In our structures, we observed that the DNA bound to the APHB domain, whether C2 or i-motif, is not aligned in a continuous manner with the ssRNA bound in the binding tunnel, as seen in PDB 6ZWW by Grass et al. Specifically, since the 3′ end of the polyU ssRNA points toward the APHB domain, the ssDNA extending from the APHB domain towards the ssRNA tunnel also terminates with a 3′ end. Consequently, the ssDNA from our structure and the ssRNA from Grass et al. cannot be seamlessly connected. However ab initio modeling of APHB-ssDNA or APHB-ssRNA complexes predict the single-stranded nucleic acid to bind in the reverse orientation. The modeling results are thus consistent with the structural feature of APHB in which the positively charged surface of APHB will confer this domain to bind single-stranded nucleic acids in both orientations. The above results and interpretation are further confirmed by a series of mutants bearing key residues in nucleic acid binding.
While E. coli HrpA helicase shares close sequence and helicase core structural similarities with other DEAH/RHA helicases like Prp43 and HrpB (both from Pseudomonas aeruginosa and E. coli), its distinction lies in the presence of the N-terminal domain. Notably, HrpA exhibits robust RNA duplex unwinding activity, a feature absent in the entirely inactive HrpB. In line with these observations, we present the following novel and consistent findings: (i) HrpA demonstrates sequence-dependent ssDNA binding affinity, as depicted in Figure 1, with the N-terminal domain playing a pivotal role; (ii) in addition to its RNA/RNA unwinding activity, HrpA displays intrinsic RNA/DNA duplex unwinding, a unique characteristic among Hrp family helicases. This activity may be linked to other cellular functions of HrpA; (iii) intriguingly, both N-terminal domain truncation in HrpA1-758 and full-length HrpA lead to complete and partial compromises in unwinding activity, underscoring the significant contribution of the N-terminal domain to RNA/RNA and RNA/DNA duplex unwinding; (iv) furthermore, our study reveals that full-length HrpA exhibits strong binding activity toward non-canonical DNA structures, including DNA/RNA G-quadruplex and i-motif; (v) we not only determined the crystal structures of the N-terminal domain in the apo-form and in complex with ssDNA but also solved the first crystal structure of a protein binding with i-motif, wherein the N-terminal domain plays an essential role in i-motif binding (further discussion in the subsequent question). This wealth of functional and structural information offers valuable insights into the mechanisms by which DEAH/RHA proteins, particularly those in the Hrp family, carry out their functions. Understanding these processes is crucial for comprehending bacterial tolerance towards certain antibiotics and potential functional roles in the cell.
In E. coli, HrpA is involved in mRNA cleavage, similar to several DEAH/RHA proteins in yeast (23,25), such as Prp2p, Prp16p, Prp22p and Prp43p30. However, the N-terminal extension of Prp22p, which includes an RNA-binding domain known as the S1 motif, is dispensable for pre-mRNA splicing in vivo (42). In contrast, HrpA from B. burgdorferi, which lacks the APHB domain, still regulates gene expression and mammalian infection (25). These discrepancies imply that the requirement of APHB in vivo may depend on the distinct mRNA processing methods used by various organisms and the various roles HrpA performs in different circumstances. The capacity of APHB to enhance ssRNA-specific protein affinity, identify C- and G-rich ssDNA, and bind to quadruplex structures suggests that it helps HrpA target specific substrates in vivo, possibly analogous to the roles of dsRBDs in MLE and the DSM element in DHX36 (43,44).
The distinctive structural characterizations of the interaction between a protein and the i-motif
In this study, we elucidated the interaction between HrpA and the unique DNA secondary structure known as the i-motif. Our structural analysis revealed the formation of a stable HrpA1–630•ADP•i-motif complex, shedding light on the intriguing relationship between HrpA and this non-canonical DNA structure.
The i-motif structure is characterized by the association of cytosine-rich DNA sequences into a knotted or intercalated conformation under acidic conditions. Our complex structure displayed two HrpA1–630 molecules embracing an i-motif structure, primarily through their APHB domains. This binding arrangement positioned the i-motif within a positively charged pocket created by the RecA1, WH, and APHB domains of the HrpA molecules, providing an optimal environment for stable interaction. The i-motif structure itself exhibited unique characteristics, with the initial three cytosine bases on each polyC chain not participating in i-motif formation. Instead, the polyC chains folded back to form an antiparallel hairpin structure, with cytosine-hemiprotonated cytosine interactions between bases, resulting in a 3′-E topology i-motif. While the interactions between HrpA and the i-motif were not extensive, a few critical residues in HrpA were involved in hydrogen bonding with the i-motif bases, with the APHB domains contributing significantly to these interactions.
To further substantiate the importance of the APHB domain in i-motif binding, we conducted binding assays with various HrpA mutants and truncations. Our results revealed that the presence of the APHB domain conferred substantial affinity for the i-motif, while its absence significantly reduced binding. Additionally, mutations in residues R22, R26, R29 and R30, which are crucial for binding to the 5′ end of polyC ssDNA, were found to impact i-motif binding, with mutations in R26 and R30 leading to notable increases in Kd values. Importantly, HrpA1–1300 demonstrated the ability to efficiently unwind the i-motif structure, and this unwinding was influenced by the presence of the APHB domain, as demonstrated by smFRET measurements.
Collectively, our results highlight the importance of the APHB domain in the interaction between HrpA and the i-motif. This domain plays a pivotal role in the structural remodeling of the i-motif, ultimately affecting the efficiency of unwinding and providing insights into the dynamic interplay between HrpA and non-canonical DNA structures. The specific recognition of i-motifs by HrpA, facilitated by the APHB domain, expands our understanding of HrpA’s diverse nucleic acid binding abilities and highlights its potential role in modulating gene expression and other cellular processes involving i-motif-containing DNA.
The potential physiological significance of i-motif in HrpA
To date, direct genetic evidence establishing a functional link between the i-motif and the HrpA helicase is lacking. However, with continuous advancements in genetic studies and biotechnologies, we acknowledge that the landscape may evolve. While the identification of i-motif structures in bacterial cells, particularly E. coli, remains elusive at present, it is not beyond the realm of possibility. The following intriguing points could shed new light on developing a functional connection between the i-motif and HrpA in cells:
First, Grass et al. demonstrated that HrpA mutants exhibit significant impairment in survival in the presence of chloramphenicol, tetracycline, and ampicillin. However, in the presence of rifampicin, HrpA mutant cells showed enhanced survival compared to WT cells. This suggests a correlation between HrpA and transcription, indicating that HrpA may play a role not only in post-transcriptional RNA regulation.
Second, i-motifs have been identified as regulators of transcription. Transcription and replication induce DNA negative supercoiling in prokaryotes (45) and also in eukaryotes (46). Hurley's group revealed that negative superhelicity induced by transcription in c-Myc promoter can favor formation of G-quadruplex on G-rich strand and i-motif on C-rich strand (47). Under these conditions, i-motif can even form at neutral pH. Hurley's group showed also that hnRNP LL can bind to the Bcl2 i-motif and regulate the transcription of the Bcl2 promoter (48). Although they did not obtain the atomic structure of the hnRNP LL with Bcl2 i-motif, they described the structural determinants of the complex (49). This finding was independently confirmed by another study (50). Notably, in their search for proteins binding to the i-motif, the Hurley group identified the helicase DDX21 (48). This suggests a potential relationship between transcription, i-motif, and helicase, although it has not been conclusively demonstrated.
Finally, the expression of virulence factors in pathogenic bacteria involves intricate regulatory mechanisms (29). While HrpA is hypothesized to primarily operate at the post-transcriptional level, it cannot be dismissed that HrpA may also exert effects at the transcriptional level. The established role of i-motifs in transcription regulation is noteworthy: i-motif formation can be induced by transcription itself as it is a source of DNA negative supercoiling and i-motif structures can also act as regulatory elements, either repressing or activating transcription. They achieve this by creating structural impediments for transcription factors and RNA polymerase, thereby influencing the transcriptional activity of the associated gene. Therefore, in prokaryotes where DNA supercoiling is precisely regulated in the dynamic topological setting of coupled replication and transcription, i-motif could have potentially an important regulatory function.
One hypothesis posits that HrpA may repress the transcription of housekeeping survival genes while enhancing the transcription of virulence genes under favorable conditions. When transcription is inhibited by rifampicin, survival genes would be less repressed in HrpA mutants compared to wild-type cells. This could elucidate why HrpA mutants exhibit better growth than WT cells when exposed to this antibiotic, aligning with the concept of i-motif regulation by HrpA.
Supplementary Material
Acknowledgements
We thank Dr Yang-Xue Dai and Liu-Tao Zhai for assisting with collecting the initial data set at Shanghai Synchrotron Radiation Facility and National Center for Protein Science Shanghai. We thank Dr. ML Hao (Jilin University) for his assistance on i-motif binding assay. We are grateful for access to the SOLEIL (Proxima) synchrotron radiation facility for crystal data collection. We thank the beamline scientists at BL17U1 of the Shanghai Synchrotron Radiation Facility (China) and at BL18U1 and BL19U1 of the National Center for Protein Sciences, Shanghai, for assistance with data collections.
Contributor Information
Ben-Ge Xin, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Ling-Yun Huang, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Ling-Gang Yuan, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Na-Nv Liu, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Hai-Hong Li, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Xia Ai, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Dong-Sheng Lei, School of Physical Science and Technology, Electron Microscopy Centre of Lanzhou University, Lanzhou University, Lanzhou 730000, People's Republic of China; Key Laboratory of Magnetism and Magnetic Materials of Ministry of Education, Lanzhou University, Lanzhou 730000, People's Republic of China.
Xi-Miao Hou, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China.
Stephane Rety, LBMC, ENS de Lyon, CNRS, UMR 5239, Inserm, U1293, Universite Claude Bernard Lyon 1, 46 allee d'Italie F-69364 Lyon, France.
Xu-Guang Xi, College of Life Sciences, Northwest A&F University, Yangling, Shaanxi 712100, China; Laboratoirede de Biologie et Pharmacologie Appliquée(LBPA), CNRS UMR8113, ENS Paris-Saclay, Université Paris-Saclay, F-91190 Gif-sur-Yvette, France.
Data availability
The atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 8PO7, 8PO6, and 8PO8.
Supplementary data
Supplementary Data are available at NAR Online.
Funding
National Natural Science Foundation of China [32071291, 32201042, 32071225]; Northwest A&F University Startup Funding for Xu-Guang Xi [Z101021903]. Funding for open access charge: NIH.
Conflict of interest statement. None declared.
References
- 1. Jankowsky E., Fairman M.E.. Duplex unwinding and RNP remodeling with RNA helicases. Methods Mol. Biol. 2008; 488:343–355. [DOI] [PubMed] [Google Scholar]
- 2. Chen C.Y., Liu X., Boris-Lawrie K., Sharma A., Jeang K.T.. Cellular RNA helicases and HIV-1: insights from genome-wide, proteomic, and molecular studies. Virus Res. 2013; 171:357–365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fairman-Williams M.E., Guenther U.P., Jankowsky E.. SF1 and SF2 helicases: family matters. Curr. Opin. Struct. Biol. 2010; 20:313–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Company M., Arenas J., Abelson J.. Requirement of the RNA helicase-like protein PRP22 for release of messenger RNA from spliceosomes. Nature. 1991; 349:487–493. [DOI] [PubMed] [Google Scholar]
- 5. Schwer B., Guthrie C.. PRP16 is an RNA-dependent ATPase that interacts transiently with the spliceosome. Nature. 1991; 349:494–499. [DOI] [PubMed] [Google Scholar]
- 6. Kim S.H., Smith J., Claude A., Lin R.J.. The purified yeast pre-mRNA splicing factor PRP2 is an RNA-dependent NTPase. EMBO J. 1992; 11:2319–2326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Colley A., Beggs J.D., Tollervey D., Lafontaine D.L.. Dhr1p, a putative DEAH-box RNA helicase, is associated with the box C+D snoRNP U3. Mol. Cell. Biol. 2000; 20:7238–7246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Arenas J.E., Abelson J.N.. Prp43: an RNA helicase-like factor involved in spliceosome disassembly. Proc. Natl. Acad. Sci. U.S.A. 1997; 94:11798–11802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Combs D.J., Nagel R.J., Ares M. Jr, Stevens S.W. Prp43p is a DEAH-box spliceosome disassembly factor essential for ribosome biogenesis. Mol. Cell. Biol. 2006; 26:523–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Granneman S., Bernstein K.A., Bleichert F., Baserga S.J.. Comprehensive mutational analysis of yeast DEXD/H box RNA helicases required for small ribosomal subunit synthesis. Mol. Cell. Biol. 2006; 26:1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhou Y., Ma J., Bushan Roy B., Wu J.Y., Pan Q., Rong L., Liang C.. The packaging of human immunodeficiency virus type 1 RNA is restricted by overexpression of an RNA helicase DHX30. Virology. 2008; 372:97–106. [DOI] [PubMed] [Google Scholar]
- 12. Wang H., Yu J., Wang X., Zhang Y.. The RNA helicase DHX33 is required for cancer cell proliferation in human glioblastoma and confers resistance to PI3K/mTOR inhibition. Cell Signal. 2019; 54:170–178. [DOI] [PubMed] [Google Scholar]
- 13. Parsyan A., Shahbazian D., Martineau Y., Petroulakis E., Alain T., Larsson O., Mathonnet G., Tettweiler G., Hellen C.U., Pestova T.V.et al.. The helicase protein DHX29 promotes translation initiation, cell proliferation, and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A. 2009; 106:22217–22222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tanner N.K., Linder P.. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Mol. Cell. 2001; 8:251–262. [DOI] [PubMed] [Google Scholar]
- 15. Fuller-Pace F.V. DExD/H box RNA helicases: multifunctional proteins with important roles in transcriptional regulation. Nucleic Acids Res. 2006; 34:4206–4215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fullam A., Schroder M.. DExD/H-box RNA helicases as mediators of anti-viral innate immunity and essential host factors for viral replication. Biochim. Biophys. Acta. 2013; 1829:854–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. De Bortoli F., Espinosa S., Zhao R.. DEAH-box RNA helicases in pre-mRNA splicing. Trends Biochem. Sci. 2021; 46:225–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T.. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016; 167:1001–1013. [DOI] [PubMed] [Google Scholar]
- 19. Yang Q., Del Campo M., Lambowitz A.M., Jankowsky E.. DEAD-box proteins unwind duplexes by local strand separation. Mol. Cell. 2007; 28:253–263. [DOI] [PubMed] [Google Scholar]
- 20. Tauchert M.J., Fourmann J.B., Luhrmann R., Ficner R.. Structural insights into the mechanism of the DEAH-box RNA helicase Prp43. eLife. 2017; 6:e21510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Pyle A.M. Translocation and unwinding mechanisms of RNA and DNA helicases. Annu. Rev. Biophys. 2008; 37:317–336. [DOI] [PubMed] [Google Scholar]
- 22. Redder P., Hausmann S., Khemici V., Yasrebi H., Linder P.. Bacterial versatility requires DEAD-box RNA helicases. FEMS Microbiol. Rev. 2015; 39:392–412. [DOI] [PubMed] [Google Scholar]
- 23. Koo J.T., Choe J., Moseley S.L.. HrpA, a DEAH-box RNA helicase, is involved in mRNA processing of a fimbrial operon in Escherichia coli. Mol. Microbiol. 2004; 52:1813–1826. [DOI] [PubMed] [Google Scholar]
- 24. Xu Y., Xu X., Lan R., Xiong Y., Ye C., Ren Z., Liu L., Zhao A., Wu L.F., Xu J.. An O island 172 encoded RNA helicase regulates the motility of Escherichia coli O157:H7. PLoS One. 2013; 8:e64211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Salman-Dilgimen A., Hardy P.O., Dresser A.R., Chaconas G.. HrpA, a DEAH-box RNA helicase, is involved in global gene regulation in the Lyme disease spirochete. PLoS One. 2011; 6:e22168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Salman-Dilgimen A., Hardy P.O., Radolf J.D., Caimano M.J., Chaconas G.. HrpA, an RNA helicase involved in RNA processing, is required for mouse infectivity and tick transmission of the Lyme disease spirochete. PLoS Pathog. 2013; 9:e1003841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hausmann S., Geiser J., Vadas O., Ducret V., Perron K., Valentini M.. Auxiliary domains of the HrpB bacterial DExH-box helicase shape its RNA preferences. RNA Biol. 2020; 17:637–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Xin B.G., Chen W.F., Rety S., Dai Y.X., Xi X.G.. Crystal structure of Escherichia coli DEAH/RHA helicase HrpB. Biochem. Biophys. Res. Commun. 2018; 504:334–339. [DOI] [PubMed] [Google Scholar]
- 29. Grass L.M., Wollenhaupt J., Barthel T., Parfentev I., Urlaub H., Loll B., Klauck E., Antelmann H., Wahl M.C.. Large-scale ratcheting in a bacterial DEAH/RHA-type RNA helicase that modulates antibiotics susceptibility. Proc. Natl. Acad. Sci. U.S.A. 2021; 118:e2100370118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Evans P.R., Murshudov G.N.. How good are my data and what is the resolution. Acta. Crystallogr. D Biol. Crystallogr. 2013; 69:1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McCoy A.J., Grosse-Kunstleve R.W., Adams P.D., Winn M.D., Storoni L.C., Read R.J.. Phaser crystallographic software. J. Appl. Crystallogr. 2007; 40:658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bunkoczi G., Echols N., McCoy A.J., Oeffner R.D., Adams P.D., Read R.J.. Phaser.MRage: automated molecular replacement. Acta. Crystallogr. D Biol. Crystallogr. 2013; 69:2276–2286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liu N.N., Duan X.L., Ai X., Yang Y.T., Li M., Dou S.X., Rety S., Deprez E., Xi X.G.. The Bacteroides sp. 3_1_23 Pif1 protein is a multifunctional helicase. Nucleic Acids Res. 2015; 43:8942–8954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang Y.R., Guo T.T., Zheng Y.T., Lai C.W., Sun B., Xi X.G., Hou X.M.. Replication protein A plays multifaceted roles complementary to specialized helicases in processing G-quadruplex DNA. iScience. 2021; 24:102493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Baek M., McHugh R., Anishchenko I., Jiang H., Baker D., DiMaio F.. Accurate prediction of protein-nucleic acid complexes using RoseTTAFoldNA. Nat. Methods. 2024; 21:117–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Yekeen A.A., Durojaye O.A., Idris M.O., Muritala H.F., Arise R.O.. CHAPERONg: A tool for automated GROMACS-based molecular dynamics simulations and trajectory analyses. Comput. Struct. Biotechnol. J. 2023; 21:4849–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pietrzyk-Brzezinska A.J., Absmeier E., Klauck E., Wen Y., Antelmann H., Wahl M.C.. Crystal Structure of the Escherichia coli DExH-Box NTPase HrpB. Structure. 2018; 26:1462–1473. [DOI] [PubMed] [Google Scholar]
- 38. Xu B., Clayton D.A.. RNA-DNA hybrid formation at the human mitochondrial heavy-strand origin ceases at replication start sites: an implication for RNA-DNA hybrids serving as primers. EMBO J. 1996; 15:3135–3143. [PMC free article] [PubMed] [Google Scholar]
- 39. Hamperl S., Cimprich K.A.. The contribution of co-transcriptional RNA:DNA hybrid structures to DNA damage and genome instability. DNA Repair (Amst.). 2014; 19:84–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abou Assi H., Garavis M., Gonzalez C., Damha M.J.. i-Motif DNA: structural features and significance to cell biology. Nucleic Acids Res. 2018; 46:8038–8056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Moriya H., Kasai H., Isono K.. Cloning and characterization of the hrpA gene in the terC region of Escherichia coli that is highly similar to the DEAH family RNA helicase genes of Saccharomyces cerevisiae. Nucleic Acids Res. 1995; 23:595–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schneider S., Schwer B.. Functional domains of the yeast splicing factor Prp22p. J. Biol. Chem. 2001; 276:21184–21191. [DOI] [PubMed] [Google Scholar]
- 43. Fu Q., Yuan Y.A.. Structural insights into RISC assembly facilitated by dsRNA-binding domains of human RNA helicase A (DHX9). Nucleic Acids Res. 2013; 41:3457–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Booy E.P., Howard R., Marushchak O., Ariyo E.O., Meier M., Novakowski S.K., Deo S.R., Dzananovic E., Stetefeld J., McKenna S.A.. The RNA helicase RHAU (DHX36) suppresses expression of the transcription factor PITX1. Nucleic Acids Res. 2014; 42:3346–3361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Travers A., Muskhelishvili G.. DNA supercoiling - a global transcriptional regulator for enterobacterial growth?. Nat. Rev. Microbiol. 2005; 3:157–169. [DOI] [PubMed] [Google Scholar]
- 46. Kouzine F., Sanford S., Elisha-Feil Z., Levens D. The functional response of upstream DNA to dynamic supercoiling in vivo. Nat. Struct. Mol. Biol. 2008; 15:146–154. [DOI] [PubMed] [Google Scholar]
- 47. Sun D., Hurley L.H.. The importance of negative superhelicity in inducing the formation of G-quadruplex and i-motif structures in the c-Myc promoter: implications for drug targeting and control of gene expression. J. Med. Chem. 2009; 52:2863–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kang H.J., Kendrick S., Hecht S.M., Hurley L.H.. The transcriptional complex between the BCL2 i-motif and hnRNP LL is a molecular switch for control of gene expression that can be modulated by small molecules. J. Am. Chem. Soc. 2014; 136:4172–4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Roy B., Talukder P., Kang H.J., Tsuen S.S., Alam M.P., Hurley L.H., Hecht S.M.. Interaction of individual structural domains of hnRNP LL with the BCL2 promoter i-motif DNA. J. Am. Chem. Soc. 2016; 138:10950–10962. [DOI] [PubMed] [Google Scholar]
- 50. Lannes L., Young P., Richter C., Morgner N., Schwalbe H.. Interaction of the N-terminal tandem domains of hnRNP LL with the BCL2 promoter i-motif DNA sequence. ChemBioChem. 2017; 18:2033–2044. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The atomic coordinates and structure factors have been deposited in the Protein Data Bank under accession codes 8PO7, 8PO6, and 8PO8.