

Abstract
Down Syndrome (DS) is a common genetic condition caused by trisomy of chromosome 21. Among the complex clinical features including musculoskeletal, neurological and cardiovascular disabilities, individuals with DS develop progressive dementia and early onset Alzheimer’s Disease (AD). This is attributed to the increased gene dosage of amyloid precursor protein (APP), the formation of self-propagating Aβ and tau prion conformers, and the deposition of neurotoxic Aβ plaques and tau neurofibrillary tangles. Tau amyloid fibrils have previously been established to adopt many distinct conformations across different neurodegenerative conditions. Here we characterized 4 DS cases spanning 36 to 63 years in age by spectral confocal imaging with conformation-specific dyes and cryo-electron microscopy (cryo-EM) to determine structures of isolated tau fibrils. High-resolution structures reveal paired helical (PHF) and straight filament (SF) conformations of tau that are identical to those determined from AD. The PHFs and SFs are made of two C-shaped protofilaments with a cross-β/β-helix motif. Similar to AD, most filaments adopt the PHF form, while a minority (~20%) form SFs. For the youngest individual with no documented dementia samples exhibited sparse tau deposits. To isolate tau for cryo-EM from this challenging sample we employed a novel “affinity grid” method involving a graphene-oxide surface derivatized with anti-tau antibodies. This improved isolation and revealed primarily tau PHFs and a minor population of SSPE type II-like filaments are present at this early age. These findings expand the similarities between AD and DS to the molecular level providing insight into their related pathologies and the potential for targeting common tau filament folds by small molecule therapeutics and diagnostics.
Keywords: Down syndrome, Alzheimer’s disease, tauopathy, protein conformation, cryo-electron microscopy
INTRODUCTION
Down syndrome (DS) is the most common chromosomal disorder of intellectual disability, affecting ~400,000 people in the United States and ~5.5 million people worldwide[1]. DS results from the presence of a third copy of chromosome 21 (Chr21) and associated genetic dosage changes that cause varying and complex disabilities including heart defects, obesity, and diabetes throughout development and adulthood. With recent increases in lifespan of persons with DS, it is now realized that Alzheimer’s Disease (AD) is a substantial comorbidity in DS individuals but arises at a much earlier age than other populations[2]. Nearly all DS adults develop severe AD neuropathological changes by age 40, however there is evidence that the first appearance of plaques and tangles occurs as early as the teens or twenties[3, 4]. Further investigation is needed in order to understand the molecular pathology of early onset AD resulting from increased APP production in DS.
As with familial and sporadic forms of AD, extracellular amyloid-β (Aβ) plaques, and intraneuronal tau neurofibrillary tangles (NFTs) are hallmarks of AD that arises in DS, and thus strongly implicated in neurodegeneration and cognitive decline. Early onset Aβ plaque deposition occurs from increased expression of the amyloid precursor protein (APP) gene and resulting overabundance of its cleavage product, Aβ42, given APP is present on Chr21. Increases in hyperphosphorylated tau and NFTs are also likely linked to increased Aβ42 considering gene duplication of APP is sufficient to cause early-onset AD[5]. Conversely, a small fraction of DS cases bear only a partial trisomy of Chr21 and lack the extra copy of the APP gene, which does not lead to the neuropathologic changes of AD[6, 7]. Taken together, these findings bolster the notion that APP overexpression is the major catalyst for AD neuropathology in aged DS individuals. While the clinical and biomarker similarities between sporadic AD that arises in general populations and AD in DS individuals are now well established[8–10], the molecular pathology is less understood given the substantial gene dosage changes with trisomy 21. Previously, we have shown that the abundance of self-propagating prion conformers of Aβ and tau accumulate in DS brains with increasing age[11]; in contrast, sporadic AD samples showed the opposite trend with a decreasing abundance of Aβ and tau prions in long-lived patients despite an overall increase in total insoluble tau[12]. Thus, although there is strong evidence for the overall similarity in the histopathological and clinical biomarkers of tau NFT accumulation between DS and AD, it is not known if the molecular structure of the associated filaments is similar.
Recent breakthroughs using cryo-EM have revealed striking differences between tau filament structures isolated from sporadic AD (sAD), Pick’s disease (PiD), Corticobasal Degeneration (CBD), Progressive Supranuclear Palsy (PSP) and several other primary and secondary tauopathies[13–18]. Alternative splicing of tau results in isoforms differing in the number of N-terminal (N) regions and microtubule binding repeat (R) regions, which exhibit distinct interactions of tau β sheets resulting in different folds in each of these diseases. Both the 3R and 4R tau isoforms implicated in AD form paired helical filaments, whereas in PiD and PSP the predominant amyloid structures are single filaments and are enriched in 3R and 4R respectively. CBD is also a 4R tauopathy and presents both single as well as paired filaments. These findings indicate that tau filament conformations are disease specific. Thus, propagation of a particular tau filament fold is likely linked to specific cellular and molecular factors associated with different disease pathologies[18]. We postulate that over-expression of the 250 protein-coding genes on Chr21 in DS and the high prevalence early onset AD present unique cellular drivers of tau filament propagation. Additionally, given the importance of developing structure-guided molecular diagnostics and therapeutics that target tau filaments to more precisely define pathologies that arise in DS and in neurodegenerative diseases, we were motivated to characterize tau filament structures that develop in DS individuals.
Here we sought to characterize and determine cryo-EM structures of tau filaments isolated from several post-mortem tissue samples from DS individuals across different ages. Tau filaments were successfully purified from 4 individuals aged 63, 51, 46, and 36 years old (cases 1 to 4, respectively). Cases 1, 2 and 3 showed robust tau pathology similar to end-stage sporadic AD. Samples from case 4, the 36-year-old, however, exhibited sparse tau deposits in comparison. Tau PHF and SF structures from the 63-year-old case were determined to 2.7 and 2.9 Å resolution, respectively, establishing complementarity to AD tau filament conformations at high-resolution. Moreover, structures from all 4 individuals demonstrate tau adopts the canonical PHF and SF forms, and are present at similar percentages to what has been observed for AD. To overcome the low level of tau filaments obtained from the 36-year-old case we employed a custom-developed graphene-oxide (GO) affinity-grid method in which the cryo-EM sample grids were derivatized with anti-tau antibodies to capture and enrich for tau filaments prior to vitrification. Thus, we present an “on-grid” tau filament isolation method that may be broadly useful for structure determination of filaments from low-abundant sources. Overall, these results establish that, despite substantial differences in genetic background and age, tau filaments that arise in DS individuals with neurodegeneration are identical to AD. Our data provide the molecular framework required to identify conformation-specific diagnostic probes and design novel therapeutic compounds targeting tau polymorphs found in DS.
METHODS
Clinical history and neuropathology.
Deidentified human biospecimens from deceased individuals were obtained from academic biorepositories. This study is exempt from institutional review board approval (i.e., this study is not considered human subject research) in accordance with University of California, San Francisco (UCSF) IRB policy. We determined the cryo-EM structure of tau filaments from 0.5 grams of fresh frozen frontal cortex samples from four individuals with Down syndrome (Supplementary Table S1). Case 1 of Down syndrome patient was a 63-year-old man and has a documented dementia. Case 2 of Down syndrome was a 51-year-old woman with dementia documented. Case 3 was a 46-year-old Down syndrome individual with no documented dementia. Case 4 of Down syndrome was a 36-year-old female with no documented dementia. Formalin-fixed samples from additional cases of Down syndrome and sporadic AD were obtained for histological studies. See complete list of all cases and the demographic and clinico-neuropathological details in Supplementary Table S2.
Immunohistochemistry.
Deparaffinized fixed sections were pretreated in 98% formic acid for 6 min to enhance immunoreactivity. After blocking with 10% normal goat serum (ngs) in PBS with 0.2% Tween 20 (PBST), sections were incubated at room temperature in primary antibodies overnight followed by secondary antibodies for 2 h. Primaries were prepared in 10% normal goat serum and applied as combinations of either: 1) anti-Aβ1-40 rabbit polyclonal (Millipore Sigma #AB5074P, 1:200) and anti-Aβ1-42 12F4 mouse monoclonal (Biolegend #805502, 1:200); 2) anti-Aβ17-24 4G8 mouse monoclonal (Biolegend #800709 1:1000) and anti-tau (phospho-S262) rabbit polyclonal (Abcam #ab131354, 1:200); or 3) anti-tau Alzheimer’s Disease conformation specific GT-38 mouse monoclonal antibody (Abcam #ab109390, 1:150) and anti-tau (phospho-S396) rabbit monoclonal (Abcam #ab109390, 1:200). Polyclonal IgG H&L secondaries were Alexa Fluor 488- and 647-conjugates (Thermo Fisher #s A11029, A21235, A11008, and A21244) applied 1:500 in 10% ngs in PBST. Stained slides were scanned on a ZEISS Axio Scan Z1 digital slide scanner at 20x magnification. Excitation at 493, 553, and 653 nm was followed by detection at 517 nm (Aβ40 or pTau), 568 nm (autofluorescence), and 668 nm (Aβ42 or tau).
Dye staining brain sections for EMBER (Excitation Multiplexed Bright Emission Recordings) microscopy.
Formalin-fixed paraffin-embedded (FFPE) brains were sectioned (8-μm thickness) and glass mounted. To reduce the autofluorescence signals by greater than 90% intensity (e.g., lipofuscin or hemosiderin), FFPE slides were photobleached up to 48 h using a multispectral LED array in a cold room overnight to reduce the autofluorescence in the brain tissue[19]. The sections were deparaffinized, PBS washed, and stained with 5 μM of dye 60 for 30 min. The sections were washed with PBS buffer and coverslipped with PermaFluor (Thermo Scientific) as the self-curing mounting medium. See Yang et al. for additional details[20].
EMBER microscopy data collection.
Dye-stained fixed brain slices were imaged with Leica SP8 confocal microscope using a 40× water immersion lens (1.1 NA), a white light and 405 nm lasers, and a HyD detector at 512 × 512-pixel resolution at 2× zoom. For each field-of-view, the optical plane was manually focused. To reduce the background noise from the bottom of the slide, the LightGate was set to 0.5 to 18 ns. First, a total of 110 images were acquired using the Λ/λ-scan mode with excitations of 470, 490, 510, 530, 550, 570, 590, 610, 630, 650, and 670 nm wavelengths. The emission detection range started at 10 nm greater than the given excitation wavelength, and ended at 780 nM, with 20-nm window. For example, of 470 nm excitation, the images were collected at 480 to 500, 500 to 520, 520 to 540, 540 to 560, 560 to 580, 580 to 600, 600 to 620, 620 to 640, 640 to 660, 660 to 680, 680 to 700, 700 to 720, 720 to 740, 740 to 760, 760 to 780 nm. Then in the λ-scan mode, 18 additional images were collected at 405 nm excitation with emission detection intervals of 20-nm for 420 nm to 780 nm. See Yang et al. for additional details[20].
EMBER data postprocessing, particle segmentation, dimension reduction.
The EMBER analytical pipeline is described in detail in Yang et al. In brief, we developed a set of custom scripts in MATLAB to process the raw fluorescent images and segment the aggregated protein deposits. The signal-processing algorithm for the analysis of particle resolution EMBER spectra was executed in MATLAB with pca. Each identified EMBER particle from the particle segmentation was normalized to [0, 1] and then concatenated in an array for PCA. The principal component scores PC1 and PC2 were plotted. UMAP was performed in MATLAB with run_umap with default settings. See Yang et al. for additional details[20].
Filament purification.
For case 1, tau filaments were purified from the frontal cortex as previously described in Fitzpatrick et al[13]. For cases 2-4, we achieved improvements in the tau filament purification, including reduced background ferritin contamination, by following methods previously described for purification of amyloid-β filaments[21]. Here, 0.5g brain tissue was homogenized in 20 volumes (w/v) of extraction buffer and brought to 2% sarkosyl for incubation at room temperature for one hour. The homogenates were then centrifuged at 10,000 g for 10 minutes and the resulting supernatants were additionally spun at 100,000 g for 60 mins at 4°C. The pellet from the second spin was resuspended in 1 ml/g of extraction buffer and centrifuged at 3000 g for 5 minutes. The supernatants were diluted 3-fold in buffer containing 50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 10 % sucrose, and 0.2 % sarkosyl and centrifuged at 100,000 g for 30 mins. The resulting sarkosyl-insoluble pellets were resuspended in 100 ul/g of 20 mM Tris-HCl, pH 7.4 buffer containing 50 mM NaCl prior to vitrification for cryo-EM.
Affinity grid preparation, vitrification and data collection.
GO deposition and assembly of the affinity grids was performed essentially as described[22, 23]. Briefly, to functionalize the GO surface, grids (Au Quantifoil, R 1.2/1.3, 300 mesh) were submerged in dimethyl sulfoxide (DMSO) with 10 mM dibenzocyclooctyne-PEG-amine (DBCO-PEG4-amine; Click Chemistry Tools, A103P) overnight at room temperature, washed, and then incubated with 20 μl of a 1 mM solution of azide-PEG-maleimide and azide-PEG-methoxyl (5 and 2 kDa molecular weight, respectively, Nanocs) at a 1:9 ratio for 6 hours. Maleimide-functionalized grids were then washed with water and ethanol, dried, and stored at −20°C. Prior to vitrification, GO grids were first incubated with 3 μL of 250 nM recombinant protein G (Genscript Z02007) in resuspension buffer: 100mM KCl 40 mM HEPES pH7.4 for 15 minutes in a Vitrobot Mark IV (Thermo Fisher Scientific) at 100% humidity then washed and inactivated with 20 mM βME in buffer. Three μL of AT8 phospho-tau (S202 and T205) monoclonal antibody (Invitrogen MN1020) was then applied at a 1:100 dilution for 15 minutes and washed three times. Three μL of purified tau filaments from cases 2 or 4 were then applied and incubated for 15 minutes, washed, and mechanically blotted (3-4 seconds) and plunge frozen. For cases 1 and 3, conventional methods were used: 3 ul of purified tau filaments were applied to glow-discharged holey carbon grids (Quantifoil Au R 1.2/1.3, 300 mesh) and mechanically blotted (3-4 seconds) at 100 % relative humidity and plunge frozen. For all datasets, super-resolution movies were collected on Titan Krios microscope operating at 300 kV equipped with K3 direct electron detector and BioQuantum (Gatan Inc.) energy filter with a slit width of 20 eV. Super-resolution movies were recorded with a magnification of 105,000x magnification (pixel size: 0.417 A per pixel), defocus range of 0.8 to 1.8 um with a total exposure time of 2.024s fractionated into 0.025s subframes.
Helical reconstruction.
All data processing steps were done either in RELION 3.1 or 4. The collected movies were motion-corrected using MotionCor2[24] and Fourier cropped by a factor of 2 to give the final pixel size of 0.834 A/pixel. The dose-weighted summed micrographs were directly imported to RELION[25, 26] and were used for further processing. Contrast transfer function (CTF) was estimated using CTFFIND 4.1. The filaments were picked manually with a. box size of 1120 pixels down sampled to 280 pixels. 2D classification was used to separate homogeneous segments for further image processing. After the first round of 2D classification, image alignment was performed to correct for the variable in-plane rotation of the fibril projections, as previously described[27]. For each well-resolved 2D classes, the angle α between the x-axis of the image and fibril-growth direction and the displacement δ of the fibril along the y-axis from the center of the box were measured in ImageJ[28]. The non-zero values for α and δ were corrected using two MATLAB codes as follows:
The new values of these parameters were updated in the “particles.star” file. Following the manual alignment, further 2D classification were performed to make sure all the 2D classes were well-centered and aligned in the boxes. 3D initial models were then created de novo from the 2D class averages using an estimated helical rise of 4.75 Å and the cross-over distances of the filaments estimated from the 2D classes with 1120-pixel boxes using RELION’s relion_helix_inimodel2d feature. The 3D initial models were low pass filtered to 10 Å for further 3D classification. The particles corresponding to each initial models were re-extracted with 280-pixel boxes for further 3D classification. The best particles were selected from 3D classification for each dataset. The final rise and twist were optimized in 3D auto-refinement. The final 3D densities were sharpened using post-processing in RELION, and the final resolution was determined from Fourier Shell correlation at 0.143 from two independently refined half-maps using soft edged solvent mask resulting in the final resolution of 2.7 Å tau PHF from patient 1, 3.1 Å for patient 2, 2.9 Å for patient 3, and 5 Å for patient 4. The tau SF from patients 1, 2, and 3 are at 3.0 Å, 3.2 Å, and 3.1 Å respectively. For patient 4, the resolution of the minor population of CTE/SSPE-II-like filaments was 7.8 Å. Imaging and reconstruction statistics are summarized in table 3.
Model building and refinement.
For PHF and SF model building and refinement, previous models[29] were initially docked into final maps for cases 1 and 2. A single strand of previously solved PHF (PDB ID: 7NRQ) and SF (PDB ID: 7NRS) were used to rigid body fit with the final post-processed density map in ChimeraX[30]. An initial round of real space refinement was performed using PHENIX[31] after which the model was used against a single half-map in ChimeraX with the ISOLDE plugin[32] to check for clashes and rotamers. A final round of PHENIX real space refinement was performed and MOLPROBITY[33] statistics for each model are summarized in Supplementary Table S4.
RESULTS
Fluorescence characterization of DS brain samples from individuals ages 36 to 63 years
In prior work, we extensively characterized a large cohort of postmortem DS brains samples from several biorepositories in the United States and Europe using the following assays: 1) biochemical measurements of Aβ and tau species in soluble and insoluble fractions of bulk tissue homogenate, 2) immunohistochemical measurements of Aβ and tau species, and 3) cell-based prion bioassays for self-propagating Aβ and tau species[11, 34]. In addition to the demographic and clinico-neuropathological details provided by each biorepository, we used our rich phenotypic data set to prioritize a list of candidate samples from the medial frontal cortex for tau filament purification and structural characterization (see Supplementary Table S1).
Here our goal was to isolate and structurally characterize tau filaments from the frontal cortex of DS individuals who died at different ages to span neuropathological stages of AD that develops in DS. We present the histological evaluation of samples that led to sufficient tau filament purification and structure determination by cryo-EM. Using fixed samples adjacent to the frozen samples used for purification in a given DS case, we performed a series of immunofluorescent stains for Aβ and tau species.
In DS cases 1 (63 y), 2 (51 y), and 3 (46 y), we observed abundant and widespread 4G8-positive Aβ plaques and phospho-tau S262 positive NFTs (Figure 1). Many amyloid plaques stained positive for both the Aβ42-isotype and Aβ40-isotype specific antibodies (Supplementary Figure S1); the accumulation of Aβ40 rich parenchymal plaques and cerebral amyloid angiopathy is indicative of more mature, late-stage Aβ deposition in DS [35, 36]. In DS case 4 (36 y), we observed moderate 4G8-positive Aβ plaque load that were uniformly composed of Aβ42 but not Aβ40 isotypes; moreover, phospho-tau S262 positive tangles were sparse, indicating an early-stage of neuropathological burden (Figure 1 and Supplementary Figure S1).
Figure 1:
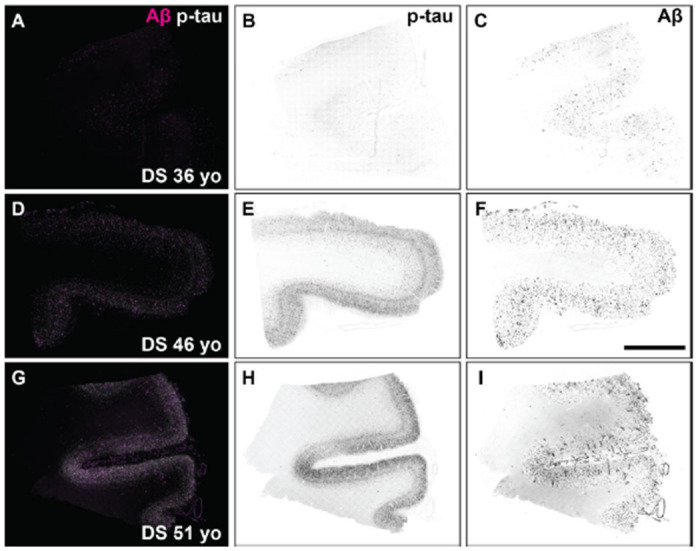
Fluorescence images of immunolabeled Aβ plaques and tau tangles in DS brain samples used for cryo-EM experiments. Formalin-fixed frontal cortex sections stained with antibodies against total Aβ (magenta) and the phosphorylated tau S262 epitope (white) in DS case 6, 36 year old (A-C), DS case 3, 46 year old (D-F), and DS case 2, 51 year old (G-I). Individual fluorescence channels were converted to greyscale and inverted for phospho-tau (B, E, H) and Aβ (C, F, I). Scale bar = 2 mm.
In situ assessment of tau tangle conformation in DS
Because aged DS brains feature AD neuropathological changes, we postulated that tau conformers should resemble those found in typical sporadic late-onset AD brain samples. Indeed, others have shown that neurofibrillary tangles are 3R and 4R tau isoform positive[37]. However, compared to sporadic AD, it has been shown that DS brains exhibit extensive tau pathology in the white matter[38] and produce greater amounts of 3R tau isoform likely due to other duplicated genes (e.g., DYRK1A) on Chr21 that affect the alternative splicing of tau exon 10[37, 39]. Thus, we performed two histological labeling methods in fixed slices to assess the conformational nature of tau tangles in DS brains compared to sporadic AD. First, we performed immunohistochemistry with an Alzheimer’s tau conformation specific antibody, known as GT-38[40, 41]. Like tau tangles in sporadic AD, we observed that phospho-tau S396 positive neurofibrillary tangles in samples from aged and young DS brains were also positive for GT-38 labeling (Figure 2A–D), suggesting that tau filaments in DS may adopt the AD fold.
Figure 2:
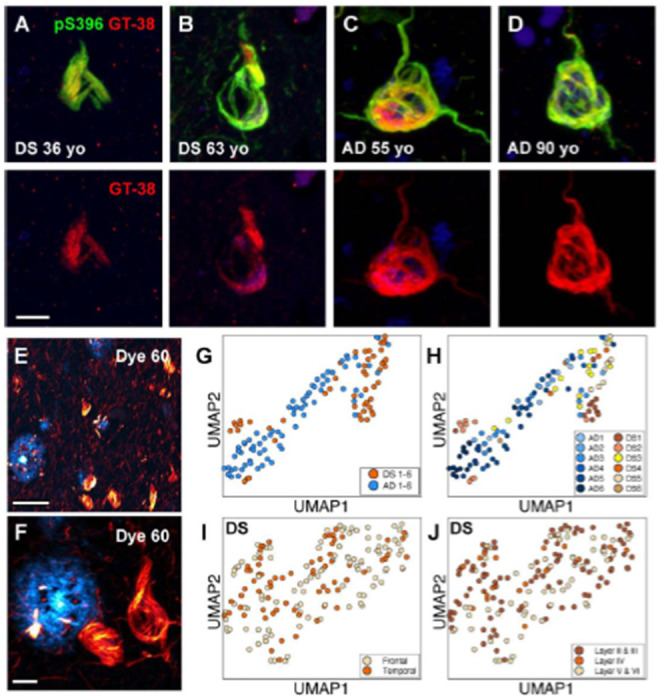
In situ assays show DS tau conformation is similar to sporadic AD. (A-D) Representative confocal images of tau tangles in DS brain fixed slices stained with antibodies targeting the phospho-tau S396 epitope (green) and the AD conformation-specific tau fold (red; GT-38). Scale bar = 10 βm. (E, F) Representative 40x confocal low (E) and high (F) zoom images of Aβ plaques (cyan) tau tangles (red) in DS brain fixed slices stained with “Dye 60”, a novel structure-sensitive dye used for EMBER amyloid strain discrimination. Scale bar =50 μm (E); Scale bar = 10 μm (F). (G, H) EMBER analysis of tau tangles in brain slices from six DS cases compared with tau tangles from brains slices of six AD cases. EMBER data plotted in UMAP as all DS cases versus all AD cases (G), or each individual DS case or AD case plotted separately (H). (I, J) EMBER analysis of tau tangles in brain slices from a different set of five DS cases. EMBER data plotted in UMAP as tau tangles in frontal cortex versus temporal cortex in the same brain (I), or by cortical layers in both frontal and temporal cortices combined (J).
In parallel experiments we used our novel spectral confocal microscopy method, called EMBER imaging paired with a new structurally-sensitive amyloid binding dye[20], to assess the conformational heterogeneity of tau tangles in DS and sporadic AD brain slices. Unlike traditional fluorescent amyloid dyes like thioflavin S, congo red, or their derivatives, our “dye 60” exhibits robust differences in both the excitation and emission spectra when bound to Aβ plaques versus tau tangles, thus making for facile spectral separation and morphological distinction between these two protein deposits (Figure 2E, F). We previously demonstrated EMBER could robustly discriminate tau deposits in sporadic AD and Pick’s disease[20]. Here, we stained six DS samples (including cases 1, 2 and 4) and six sporadic AD samples. Following our published protocol (see Methods), we collected and analyzed EMBER data for tau tangles only and presented the results in a UMAP plot. The EMBER signature of tau tangles in DS brain clustered tightly with tau tangles in sporadic AD brain (Figure 2G, H), suggesting that the DS tau tangle conformation may be like those in AD. Next, we examined the degree of conformational homogeneity between and within brain regions in the same DS cases. We obtained a new set of fixed postmortem tissues in which we had slices from both the frontal cortex and temporal cortex of five DS individuals with advanced AD neuropathology. We performed EMBER imaging and observed that the conformational signature of tau tangles was similar in the frontal and temporal cortices and across all cortical layers in those regions (Figure 2I, J). Taken together, these data corroborate the notion that tau filaments from DS are likely similar to the AD tau fold.
Cryo-EM structures reveal tau filaments adopt AD conformations in DS
We sought to determine structures of isolated tau filaments from DS cases 1-4 by cryo-EM. Sarkosyl-insoluble material was extracted from frontal cortex tissue essentially as described[13, 21] (see methods) and initially assessed by negative-stain EM (Supplementary Fig. S2). PHF-like filaments were readily identified in cases 1-3, however, very few fibrils were observed for case 4. We note this was expected based on the younger age of individual (36y) and the more minor presence of phospho-tau S262 positive tangles compared to other cases (Figure 1). For case 1, cryo-EM 2D and 3D analysis revealed the presence of both PHFs and SFs, totaling approximately 76% and 21% of the selected data, respectively (Figure 1A). Cryo-EM helical reconstructions using RELION[25, 26] resulted in well-resolved PHF and SF final maps at 2.7 and 2.9 Å-resolution, respectively. (Figure 1B, Supplementary Fig. S3 and Supplementary Table S3). The docked atomic models for tau residues G303 to E380 were in good agreement with the map and confirmed the tau conformations are identical to the AD PHF and SF structures (Figure 1C and Supplementary Fig. S3 and Supplementary Table S3). Indeed, alignment to the AD structures shows no substantial differences, with an overall α-carbon RMSD equal to 0.5 Å and 0.6 Å for the PHF and SF, respectively (Supplementary Fig. S3). As with AD, the filaments adopt a characteristic C-shaped fold comprised of a β-helix turn (residues 337-356) and an anti-parallel protofilament interaction interface across the 332PGGGQ336 motif for the PHF and residues 317–324 and 312–321 for the SF.
Considering the very low abundance of tau filaments and scarcity of tissue for case 4 we devised a scheme to enrich for tau filaments on cryo-EM grids prior to vitrification using an established affinity-grid methodology (Figure 4A)[23]. For this procedure, single-layer graphene oxide (GO) was applied to Quantifoil grids and then functionalized with a polyethylene glycol (PEG) maleimide and coupled to protein G for binding to the AT8 phospho-tau (S202 and T205) antibody (see Materials and Methods). This GO-AT8 affinity grid method was tested initially for capturing tau filaments purified from case 2, which exhibited robust NFTs. In control experiments, in which protein G was not added to the affinity grid assembly procedure, few to no filaments were observed in cryo-EM images (Figure 4B). We note this procedure involves wash steps after sample application to reduce background contaminants, such as ferritin[13], which can impact filament selection and refinement. Thus, the absence of filaments indicates successful removal of unbound material relative to typical vitrification procedures. Conversely, when AT8 is included in the grid preparation, we observe extensive tau filaments in cryo-EM images, although, some background contaminants remain present (Figure 4C). Together, these data indicate our GO-AT8 affinity grid method successfully enriches for tau-filaments in an AT8-dependent manner.
Figure 4:
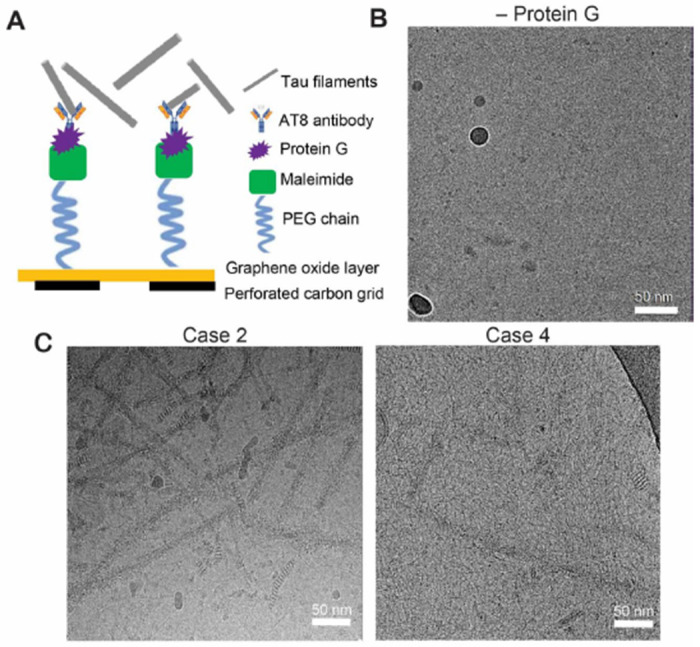
GO-AT8 antibody affinity grid assembly and isolation of tau filaments for cryo-EM. (A) Schematic showing affinity grid assembly procedure for the attachment of AT8 anti-tau antibodies to GO grid surface. (B) Control cryo-EM micrograph image where protein G is not included in the affinity grid assembly prior to incubation with tau filaments and vitrification. (C) Representative cryo-EM micrograph images following GO-AT8 affinity grid isolation of tau filaments from DS case 2 (left) and DS case 4 (right). Fewer filaments are observed in case 4 due to the low overall abundance of tau deposits.
Next, cryo-EM datasets were collected for tau filaments isolated form DS cases 2-4, with GO-AT8 affinity grids being used for cases 2 and 4. Resulting cryo-EM reconstructions show all cases contain predominantly AD PHF conformations, while cases 2 and 3 also exhibit substantial SFs, similar to case 1 (Figure 5). Notably, structure determination for case 4 remained limited by the low abundance of isolated filaments, resulting in a final resolution of 5 Å for the case 4 PHF. However, molecular models for both case 2 and 4 confirm PHF and SF conformations are resolved using the GO-AT8 grids (Supplementary Fig. S4). Interestingly, a small dataset (~17,000 segments) was classified and refined to a low-resolution (>7 Å) map that appears similar to the type II SSPE (subacute sclerosing panencephalitis)[42] or CTE (chronic traumatic encephalopathy) fold[16]. However, the map resolution and quality were not sufficient to build an atomic model and confirm the tau filament conformation. Overall, for the 4 cases we studied, these data identify that the tau AD PHF fold predominates across a wide age range of individuals with DS. The SF fold is also present at lower levels but is comparable to what has been previously observed by cryo-EM[13]. Therefore, both the structural conformation of tau filaments and relative presence of PHFs and SFs appears identical to AD for older DS individuals with a higher burden of tau deposits, supporting the general AD pathology that has been reported for DS[3].
Figure 5:
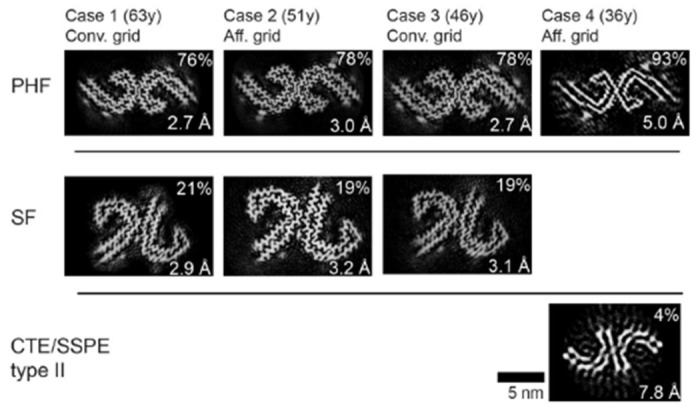
PHF and SF cross-section views of the tau filament cryoEM structures determined from four DS cases. Relative percentage of segments for the reconstructions is indicated with the final resolution and grid preparation method. In the 36y case a minor percentage refined to show a conformation similar to CTE or SSPE type II filaments (indicated). However, an atomic model was unable to be determined due to the low resolution and small data set size.
DISCUSSION
Cryo-EM structural studies have revealed that tau adopts a myriad of polymorphic folds that are associated with distinct etiological forms of primary and secondary tauopathy diseases[18]. In DS individuals, clinical biomarkers of tau pathology have been established to progress along a similar trajectory as compared to AD subjects—just decades sooner[8]. This early onset AD has long been attributed to the triplication of APP and exacerbated deposition of Aβ neuritic plaques[3, 4]. Moreover, isolated insoluble tau[43, 44] and NFTs [3, 45] that arise in Down syndrome individuals have been shown to share similar biochemical and morphological properties to the disease form of tau from AD. However, structures of tau filaments from DS individuals have not previously been determined. Thus, the specific tau fold that develops in DS has been unknown; a high-resolution structure of tau in DS would bear great significance for therapeutic development. Our novel workflow of in situ conformational assays and cryo-EM methods revealed that DS tau adopts the AD folds of paired helical filaments and straight filaments, in agreement with a recent study from Vidal and colleagues[46].
We previously developed EMBER imaging with structure-sensitive dyes to visualize conformational heterogeneity of intracellular tau deposits in situ and identified robust conformational discrimination between individual AD NFTs and Pick bodies at the cellular level[20]. We therefore employed EMBER imaging to screen and predict the conformational status of tau deposits in candidate brain samples and, indeed, demonstrated a robust overlap of conformational signatures for NFTs in AD and DS samples (Figure 2G, H). In AD, tau NFTs accumulate predominantly in deeper cortical layers V/VI[47]; in contrast, tau NFT accumulation is more widespread in DS involving all cortical layers II through VI[45, 48]. In a small cohort of well-matched DS cases, we found that the EMBER-based conformational signature of DS tau NFTs was homogenous across all cortical layers in frontal and temporal cortices (Figure 2I, J). These data suggest: 1) the same AD tau folds should be found in the temporal cortex of DS individuals, and 2) layer II/III cortical neurons in DS give rise to NFTs composed of tau filaments with the same AD tau fold as layer 5/6 neurons in DS or AD. Considering the overexpression of more than 250 protein-coding genes on Chr21, it is remarkable that the DS tau fold is identical to AD and the tau NFT conformations in DS appear homogenous within and between DS subjects.
It has been postulated that the distinct CTE tau fold[16], also found in SSPE[42] and ALS-PDC[49], derives from tau NFT deposition predominately in the upper cortical layers II/III of those diseases as a consequence of distinct disease mechanisms involving chronic neuroinflammation and/or environmental factors[50–53]. In contrast, we show that DS tau NFTs in layer II/III exhibit a similar EMBER signature as layer V/VI NFTs, suggesting that any putative microenvironmental differences specific to each cortical layer is not sufficient to drive CTE tau folds versus AD tau folds. Hence, it is reasonable to posit that a distinct neuroinflammatory state shared between CTE, SSPE, and ALS-PDC drives a common CTE tau fold that is unique from AD tau PHFs and SFs. Nevertheless, trisomy of Chr21 leads to a heightened state of neuroinflammation that is not typical of AD. In DS, there is duplication of four interferon receptor genes on Chr21 causing a lifelong, systemic type 1 interferonopathy phenotype (a chronic state of low-grade inflammation in all organs)[54, 55]. In the brain, proinflammatory cytokine profiles and aberrant microglial activation states are more pronounced in DS compared to AD[56]; in later stages of DS, a robust neuroinflammatory phenotype emerges including a prominent dystrophic and rod-shape microglia phenotype that aligns with neurons forming tau tangles[57, 58]. Therefore, it is somewhat surprising that the AD tau conformation predominates in the DS brain despite the overt differences in the biochemical microenvironment. Thus, we conclude that the Aβ pathology, and its associated cellular consequences, must be the major driver of the PHF and SF tau conformations no matter the etiological form of AD.
In DS, overt deposition of Aβ plaques and tau NFTs appear in the second and third decade of life, respectively; however, cognitive impairment and dementia do not emerge until the fifth or sixth decade of life[2]. The field now has a clear view of the spectrum of distinct tau folds present in advanced, late stages of disease, but the extent of conformational heterogeneity at disease onset remains unknown. To increase the potential for translational success, it is important to determine the structures of tau filaments from very early, asymptomatic stages of disease. Critical to the characterization of tau filaments from the younger (36y) individual with no documented dementia was our implementation of GO-AT8 affinity grids to isolate tau filaments from a very sparse sample (Figure 1). This procedure enabled small-scale affinity purification from an endogenous source, an advantageous step for future potential structural efforts targeting specific brain regions, early disease stages or other pathologies with reduced tau deposits. We additionally found this procedure to be beneficial in reducing background contaminants, such as ferritin, which can impact classification and refinement. Unfortunately, despite these advances, we were unable to complete high-resolution structures from the 36y case, thus data set size and filament abundance remained a challenge. Nonetheless, our structure determined to 5.0 Å resolution was sufficient to establish the AD PHF fold. Therefore, further optimizations were not pursued.
Strikingly, with the GO-AT8 affinity method we were able to identify that 4% of tau filaments from case 4 appear to have the Type II CTE fold, also found in SSPE and ALS-PDC[16, 42, 49]. However, due to the extreme rarity of these tau filaments, we only achieved a 7.8 angstrom map and thus cannot conclusively determine the fold to high resolution. Nevertheless, if we assume that more data would confirm there is a minor population of Type II CTE tau filaments in case 4, how do we reconcile that the other three cases with advanced AD pathology and abundant tau filaments do not have this morphology? One explanation could be that case 4 also had a well-documented history of epilepsy and the cause of death was respiratory failure. Notably, epilepsy (in non-DS young adults) is associated with the accumulation of tau histopathology[59–61], and neuroinflammatory pathways are activated by and exacerbate epileptic seizures[62]. Thus, we speculate that the neurodegenerative and neuroinflammatory processes in the epileptic brain may precipitate conditions that yield the Type II CTE fold. As DS individuals live longer into late middle age, the prevalence of comorbid epilepsy is estimated to reach upwards of 50% of the DS population with AD[63]. Future cryo-EM studies on tau filaments from greater numbers of DS individuals with comorbid epilepsy may identify cases with a heterogenous mix of AD tau and CTE tau folds, consistent with a prior study showing multiple tau polymorphs identified in a single primary tauopathy case[18].
Because modern clinical imaging and fluid biomarkers have revealed the trajectory of AD progression in DS individuals[8–10], there has been growing consideration to include DS subjects in clinical trials for therapeutics developed for AD. The urgency to do so has been tempered by the fact that the mechanism of action and/or safety profile of candidate drugs may be different in DS subjects despite the shared presence of Aβ plaques and tau tangles. Indeed, the first clinical trial (NCT05462106) testing an Aβ vaccine in DS participants is underway[64], and there may be future trials testing Aβ immunotherapies already showing promising results in sporadic AD subjects. However, compared to sporadic AD, the formation of tau tangles is much more rapid following the deposition of Aβ plaques in DS[65, 66], and thus effective therapeutics targeting pathological tau in DS will be required due to the condensed trajectory of AD pathogenesis. Given the intracellular localization of tau NFTs, immunotherapy approaches are unlikely to succeed, thus emphasizing the need to develop small molecule therapeutics. By solving the structure of tau filaments in DS, we now have a molecular framework to apply existing or design new conformation-specific small molecule drugs or diagnostics that target the AD tau fold. For example, we expect that the cleft of the PHF fold in DS bears an identical pocket for stacked small molecule binding as we demonstrated with PET ligand GTP-1[67] and Kunach et al. demonstrated with PET ligand MK-6240[68] in tau PHFs from sporadic AD. Based on this novel binding mode, new in silico docking methods[69] may predict small molecules with superior properties for development as next generation diagnostic ligands or therapeutic inhibitors targeting the tau fold in DS and AD.
Supplementary Material
Figure 3:
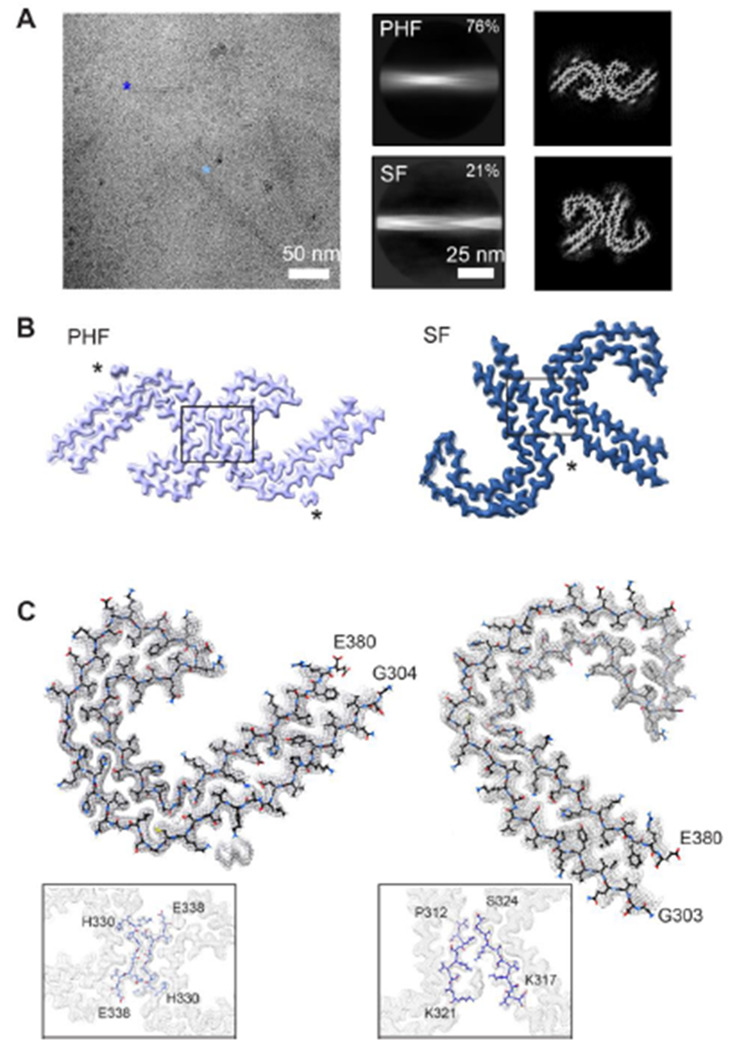
CryoEM structures of tau PHF and SF conformations from DS case 1. (A) Representative electron micrograph of amyloid filaments from the frontal cortex of DS case 1 showing PHF and SF segments (light and dark blue, respectively) and corresponding 2D class averages (middle) and cross section projection views of the conformations. (B) Final cryo-EM density maps of the PHF and SF at 2.7 and 2.9 Å resolution, respectively. Asterisks indicate extra densities previously observed in structures from AD, and boxes show protofilament interface, enlarged in (C). (C) Single protofilament cryo-EM map (mesh) with the fitted atomic model for the PHF (left) and SF (right), including tau residues G304 (or G303 the SF) to E380 and enlarged view of the protofilament interface.
ACKNOWLEDGEMENTS
This work was supported by NIH grants: P01AG002132 to S.B.P, C.C. and D.R.S, and RF1NS133651 to C.C. and D.R.S, the Henry M. Jackson Foundation (HU0001-21-2-065 subaward 5802 to S.B.P), Rainwater Charitable Foundation (A138221 to D.R.S). Tissue samples were supplied by the NIH NeuroBioBank (via the University of Maryland); the August Pi i Sunyer Biomedical Research Institute Biobank (Barcelona, Spain); the London Neurodegenerative Diseases Brain Bank (King’s College London, UK), which receives funding from the Medical Research Council UK and through the Brains for Dementia Research Project (jointly funded by the Alzheimer’s Society and Alzheimer’s Research UK); Autistica UK and the NIHR Oxford Biomedical Research Centre; and Professor Elizabeth Head and the University of California Alzheimer’s Disease Research Center (Irvine, CA), which is funded by the NIH National Institute on Aging (grant P30AG066519).
Footnotes
COMPETING INTERESTS
SBP is the founder of Prio-Pharma, which did not contribute financial or any other support to these studies. All other authors declare that they have no competing interests.
AVAILABILITY OF DATA & MATERIALS
Cryo-EM maps will be available in the Electron Microscopy Data Bank (EMDB) upon publication. Corresponding refined atomic models will be deposited in the Protein Data Bank (PDB) upon publication. Please address requests for materials to the corresponding authors.
REFERENCES CITED
- 1.Vos T, Allen C, Arora M (2016) Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet 388:1545–1602. 10.1016/S0140-6736(16)31678-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lott IT, Head E (2019) Dementia in Down syndrome: unique insights for Alzheimer disease research. Nat Rev Neurol 15:135–147. 10.1038/s41582-018-0132-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mann DM, Esiri MM (1989) The pattern of acquisition of plaques and tangles in the brains of patients under 50 years of age with Down’s syndrome. J Neurol Sci 89:169–179. 10.1016/0022-510x(89)90019-1 [DOI] [PubMed] [Google Scholar]
- 4.Davidson YS, Robinson A, Prasher VP, Mann DMA (2018) The age of onset and evolution of Braak tangle stage and Thal amyloid pathology of Alzheimer’s disease in individuals with Down syndrome. Acta Neuropathol Commun 6:56. 10.1186/s40478-018-0559-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sleegers K, Brouwers N, Gijselinck I, et al. (2006) APP duplication is sufficient to cause early onset Alzheimer’s dementia with cerebral amyloid angiopathy. Brain 129:2977–2983. 10.1093/brain/awl203 [DOI] [PubMed] [Google Scholar]
- 6.Prasher VP, Farrer MJ, Kessling AM, et al. (1998) Molecular mapping of Alzheimer-type dementia in Down’s syndrome. Ann Neurol 43:380–383. 10.1002/ana.410430316 [DOI] [PubMed] [Google Scholar]
- 7.Doran E, Keator D, Head E, et al. (2017) Down syndrome, partial trisomy 21, and absence of alzheimer’s disease: the role of APP. J Alzheimers Dis 56:459–470. 10.3233/JAD-160836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fortea J, Vilaplana E, Carmona-Iragui M, et al. (2020) Clinical and biomarker changes of Alzheimer’s disease in adults with Down syndrome: a cross-sectional study. Lancet 395:1988–1997. 10.1016/S0140-6736(20)30689-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bejanin A, Iulita MF, Vilaplana E, et al. (2021) Association of apolipoprotein E ε4 allele with clinical and multimodal biomarker changes of alzheimer disease in adults with down syndrome. JAMA Neurol 78:937–947. 10.1001/jamaneurol.2021.1893 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iulita MF, Bejanin A, Vilaplana E, et al. (2023) Association of biological sex with clinical outcomes and biomarkers of Alzheimer’s disease in adults with Down syndrome. Brain Commun 5:fcad074. 10.1093/braincomms/fcad074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Condello C, Maxwell AM, Castillo E, et al. (2022) Aβ and tau prions feature in the neuropathogenesis of Down syndrome. Proc Natl Acad Sci USA 119:e2212954119. 10.1073/pnas.2212954119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aoyagi A, Condello C, Stöhr J, et al. (2019) Aβ and tau prion-like activities decline with longevity in the Alzheimer’s disease human brain. Sci Transl Med 11:. 10.1126/scitranslmed.aat8462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fitzpatrick AWP, Falcon B, He S, et al. (2017) Cryo-EM structures of tau filaments from Alzheimer’s disease. Nature 547:185–190. 10.1038/nature23002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Falcon B, Zhang W, Schweighauser M, et al. (2018) Tau filaments from multiple cases of sporadic and inherited Alzheimer’s disease adopt a common fold. Acta Neuropathol 136:699–708. 10.1007/s00401-018-1914-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Falcon B, Zhang W, Murzin AG, et al. (2018) Structures of filaments from Pick’s disease reveal a novel tau protein fold. Nature 561:137–140. 10.1038/s41586-018-0454-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Falcon B, Zivanov J, Zhang W, et al. (2019) Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature 568:420–423. 10.1038/s41586-019-1026-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang W, Tarutani A, Newell KL, et al. (2020) Novel tau filament fold in corticobasal degeneration. Nature 580:283–287. 10.1038/s41586-020-2043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Shi Y, Zhang W, Yang Y, et al. (2021) Structure-based classification of tauopathies. Nature 598:359–363. 10.1038/s41586-021-03911-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Duong H, Han M (2013) A multispectral LED array for the reduction of background autofluorescence in brain tissue. J Neurosci Methods 220:46–54. 10.1016/jjneumeth.2013.08.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yang H, Yuan P, Wu Y, et al. (2023) EMBER multidimensional spectral microscopy enables quantitative determination of disease-and cell-specific amyloid strains. Proc Natl Acad Sci USA 120:e2300769120. 10.1073/pnas.2300769120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang Y, Arseni D, Zhang W, et al. (2022) Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 375:167–172. 10.1126/science.abm7285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Palovcak E, Wang F, Zheng SQ, et al. (2018) A simple and robust procedure for preparing graphene-oxide cryo-EM grids. J Struct Biol 204:80–84. 10.1016/jjsb.2018.07.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang F, Liu Y, Yu Z, et al. (2020) General and robust covalently linked graphene oxide affinity grids for high-resolution cryo-EM. Proc Natl Acad Sci USA 117:24269–24273. 10.1073/pnas.2009707117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zheng SQ, Palovcak E, Armache J-P, et al. (2017) MotionCor2: anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods 14:331–332. 10.1038/nmeth.4193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.He S, Scheres SHW (2017) Helical reconstruction in RELION. J Struct Biol 198:163–176. 10.1016/jJsb.2017.02.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zivanov J, Nakane T, Forsberg BO, et al. (2018) New tools for automated high-resolution cryo-EM structure determination in RELION-3. eLife 7:. 10.7554/eLife.42166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ghosh U, Thurber KR, Yau W-M, Tycko R (2021) Molecular structure of a prevalent amyloid-β fibril polymorph from Alzheimer’s disease brain tissue. Proc Natl Acad Sci USA 118:. 10.1073/pnas.2023089118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Collins TJ (2007) ImageJ for microscopy. BioTechniques 43:25–30. 10.2144/000112517 [DOI] [PubMed] [Google Scholar]
- 29.Shi Y, Murzin AG, Falcon B, et al. (2021) Cryo-EM structures of tau filaments from Alzheimer’s disease with PET ligand APN-1607. Acta Neuropathol 141:697–708. 10.1007/s00401-021-02294-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pettersen EF, Goddard TD, Huang CC, et al. (2021) UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci 30:70–82. 10.1002/pro.3943 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Afonine PV, Poon BK, Read RJ, et al. (2018) Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Crystallogr D Struct Biol 74:531–544. 10.1107/S2059798318006551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Croll TI (2018) ISOLDE: a physically realistic environment for model building into low-resolution electron-density maps. Acta Crystallogr D Struct Biol 74:519–530. 10.1107/S2059798318002425 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Williams CJ, Headd JJ, Moriarty NW, et al. (2018) MolProbity: more and better reference data for improved all-atom structure validation. Protein Sci 27:293–315. 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Maxwell AM, Yuan P, Rivera BM, et al. (2021) Emergence of distinct and heterogeneous strains of amyloid beta with advanced Alzheimer’s disease pathology in Down syndrome. Acta Neuropathol Commun 9:201. 10.1186/s40478-021-01298-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwatsubo T, Mann DM, Odaka A, et al. (1995) Amyloid beta protein (A beta) deposition: A beta 42(43) precedes A beta 40 in Down syndrome. Ann Neurol 37:294–299. 10.1002/ana.410370305 [DOI] [PubMed] [Google Scholar]
- 36.Lemere CA, Blusztajn JK, Yamaguchi H, et al. (1996) Sequence of deposition of heterogeneous amyloid beta-peptides and APO E in Down syndrome: implications for initial events in amyloid plaque formation. Neurobiol Dis 3:16–32. 10.1006/nbdi.1996.0003 [DOI] [PubMed] [Google Scholar]
- 37.Wegiel J, Kaczmarski W, Barua M, et al. (2011) Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J Neuropathol Exp Neurol 70:36–50. 10.1097/NEN.0b013e318202bfa1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ichimata S, Martinez-Valbuena I, Lee S, et al. (2023) Distinct Molecular Signatures of Amyloid-Beta and Tau in Alzheimer’s Disease Associated with Down Syndrome. Int J Mol Sci 24:. 10.3390/ijms241411596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shi J, Zhang T, Zhou C, et al. (2008) Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. J Biol Chem 283:28660–28669. 10.1074/jbc.M802645200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gibbons GS, Banks RA, Kim B, et al. (2018) Detection of Alzheimer Disease (AD)-Specific Tau Pathology in AD and NonAD Tauopathies by Immunohistochemistry With Novel Conformation-Selective Tau Antibodies. J Neuropathol Exp Neurol 77:216–228. 10.1093/jnen/nly010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gibbons GS, Kim S-J, Robinson JL, et al. (2019) Detection of Alzheimer’s disease (AD) specific tau pathology with conformation-selective anti-tau monoclonal antibody in comorbid frontotemporal lobar degeneration-tau (FTLD-tau). Acta Neuropathol Commun 7:34. 10.1186/s40478-019-0687-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Qi C, Hasegawa M, Takao M, et al. (2023) Identical tau filaments in subacute sclerosing panencephalitis and chronic traumatic encephalopathy. Acta Neuropathol Commun 11:74. 10.1186/s40478-023-01565-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanger DP, Brion JP, Gallo JM, et al. (1991) Tau in Alzheimer’s disease and Down’s syndrome is insoluble and abnormally phosphorylated. Biochem J 275 (Pt 1):99–104. 10.1042/bj2750099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Flament S, Delacourte A, Mann DM (1990) Phosphorylation of Tau proteins: a major event during the process of neurofibrillary degeneration. A comparative study between Alzheimer’s disease and Down’s syndrome. Brain Res 516:15–19. 10.1016/0006-8993(90)90891-e [DOI] [PubMed] [Google Scholar]
- 45.Hof PR, Bouras C, Perl DP, et al. (1995) Age-related distribution of neuropathologic changes in the cerebral cortex of patients with Down’s syndrome. Quantitative regional analysis and comparison with Alzheimer’s disease. Arch Neurol 52:379–391. 10.1001/archneur.1995.00540280065020 [DOI] [PubMed] [Google Scholar]
- 46.Fernandez A, Hoq MR, Hallinan GI, et al. (2024) Cryo-EM structures of amyloid-β and tau filaments in Down syndrome. Nat Struct Mol Biol. 10.1038/s41594-024-01252-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Pearson RC, Esiri MM, Hiorns RW, et al. (1985) Anatomical correlates of the distribution of the pathological changes in the neocortex in Alzheimer disease. Proc Natl Acad Sci USA 82:4531–4534. 10.1073/pnas.82.13.4531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mann DM, Yates PO, Marcyniuk B, Ravindra CR (1986) The topography of plaques and tangles in Down’s syndrome patients of different ages. Neuropathol Appl Neurobiol 12:447–457. 10.1111/j.1365-2990.1986.tb00053.x [DOI] [PubMed] [Google Scholar]
- 49.Qi C, Verheijen BM, Kokubo Y, et al. (2023) Tau filaments from amyotrophic lateral sclerosis/parkinsonism-dementia complex adopt the CTE fold. Proc Natl Acad Sci USA 120:e2306767120. 10.1073/pnas.2306767120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Tokuda T, Ikeda S, Yanagisawa N, et al. (1991) Re-examination of ex-boxers’ brains using immunohistochemistry with antibodies to amyloid beta-protein and tau protein. Acta Neuropathol 82:280–285. 10.1007/BF00308813 [DOI] [PubMed] [Google Scholar]
- 51.Miyahara H, Akagi A, Riku Y, et al. (2022) Independent distribution between tauopathy secondary to subacute sclerotic panencephalitis and measles virus: An immunohistochemical analysis in autopsy cases including cases treated with aggressive antiviral therapies. Brain Pathol 32:e13069. 10.1111/bpa.13069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hof PR, Nimchinsky EA, Buée-Scherrer V, et al. (1994) Amyotrophic lateral sclerosis/parkinsonism-dementia complex of Guam: quantitative neuropathology, immunohistochemical analysis of neuronal vulnerability, and comparison with related neurodegenerative disorders. Acta Neuropathol 88:397–404. 10.1007/BF00389490 [DOI] [PubMed] [Google Scholar]
- 53.Mimuro M, Yoshida M, Kuzuhara S, Kokubo Y (2018) Amyotrophic lateral sclerosis and parkinsonism-dementia complex of the Hohara focus of the Kii Peninsula: A multiple proteinopathy? Neuropathology 38:98–107. 10.1111/neup.12434 [DOI] [PubMed] [Google Scholar]
- 54.Galbraith MD, Rachubinski AL, Smith KP, et al. (2023) Multidimensional definition of the interferonopathy of Down syndrome and its response to JAK inhibition. Sci Adv 9:eadg6218. 10.1126/sciadv.adg6218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Waugh KA, Minter R, Baxter J, et al. (2023) Triplication of the interferon receptor locus contributes to hallmarks of Down syndrome in a mouse model. Nat Genet 55:1034–1047. 10.1038/s41588-023-01399-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wilcock DM, Hurban J, Helman AM, et al. (2015) Down syndrome individuals with Alzheimer’s disease have a distinct neuroinflammatory phenotype compared to sporadic Alzheimer’s disease. Neurobiol Aging 36:2468–2474. 10.1016/j.neurobiolaging.2015.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Flores-Aguilar L, lulita MF, Kovecses O, et al. (2020) Evolution of neuroinflammation across the lifespan of individuals with Down syndrome. Brain 143:3653–3671. 10.1093/brain/awaa326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Martini AC, Helman AM, McCarty KL, et al. (2020) Distribution of microglial phenotypes as a function of age and Alzheimer’s disease neuropathology in the brains of people with Down syndrome. Alzheimers Dement (Amst) 12:e12113. 10.1002/dad2.12113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tai XY, Koepp M, Duncan JS, et al. (2016) Hyperphosphorylated tau in patients with refractory epilepsy correlates with cognitive decline: a study of temporal lobe resections. Brain 139:2441–2455. 10.1093/brain/aww187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith KM, Blessing MM, Parisi JE, et al. (2019) Tau deposition in young adults with drug-resistant focal epilepsy. Epilepsia 60:2398–2403. 10.1111/epi.16375 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Toscano ECB, Vieira ÉLM, Grinberg LT, et al. (2023) Hyperphosphorylated tau in mesial temporal lobe epilepsy: a neuropathological and cognitive study. Mol Neurobiol 60:2174–2185. 10.1007/s12035-022-03190-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Vezzani A, Balosso S, Ravizza T (2019) Neuroinflammatory pathways as treatment targets and biomarkers in epilepsy. Nat Rev Neurol 15:459–472. 10.1038/s41582-019-0217-x [DOI] [PubMed] [Google Scholar]
- 63.Altuna M, Giménez S, Fortea J (2021) Epilepsy in down syndrome: A highly prevalent comorbidity. J Clin Med 10:. 10.3390/jcm10132776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rafii MS, Sol O, Mobley WC, et al. (2022) Safety, Tolerability, and Immunogenicity of the ACI-24 Vaccine in Adults With Down Syndrome: A Phase 1b Randomized Clinical Trial. JAMA Neurol 79:565–574. 10.1001/jamaneurol.2022.0983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zammit MD, Tudorascu DL, Laymon CM, et al. (2021) Neurofibrillary tau depositions emerge with subthreshold cerebral beta-amyloidosis in down syndrome. Neuroimage Clin 31: 102740. 10.1016/j.nicl.2021.102740 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zammit MD, Betthauser TJ, McVea AK, et al. (2024) Characterizing the emergence of amyloid and tau burden in Down syndrome. Alzheimers Dement 20:388–398. 10.1002/alz.13444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Merz GE, Chalkley MJ, Tan SK, et al. (2023) Stacked binding of a PET ligand to Alzheimer’s tau paired helical filaments. Nat Commun 14:3048. 10.1038/s41467-023-38537-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kunach P, Vaquer-Alicea J, Smith MS, et al. (2023) Cryo-EM structure of Alzheimer’s disease tau filaments with PET ligand MK-6240. BioRxiv. 10.1101/2023.09.22.558671 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Smith MS, Knight IS, Kormos RC, et al. (2024) Docking for Molecules That Bind in a Symmetric Stack with SymDOCK. J Chem Inf Model 64:425–434. 10.1021/acs.jcim.3c01749 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Cryo-EM maps will be available in the Electron Microscopy Data Bank (EMDB) upon publication. Corresponding refined atomic models will be deposited in the Protein Data Bank (PDB) upon publication. Please address requests for materials to the corresponding authors.