

Abstract
BACKGROUND
Cyclin-dependent kinase (CDK) 7 is aberrantly overexpressed in many types of cancer and is an attractive target for cancer therapy due to its dual role in transcription and cell cycle progression. Moreover, CDK7 can directly modulate the activities of estrogen receptor (ER), which is a major driver in breast cancer. Breast cancer cells have exhibited high sensitivity to CDK7 inhibition in pre-clinical studies.
Methods
In this review, we provide a comprehensive summary of the latest insights into CDK7 biology and recent advancements in CDK7 inhibitor development for breast cancer treatment. We also discuss the current application of CDK7 inhibitors in different molecular types of breast cancer to provide potential strategies for the treatment of breast cancer.
Results
Significant progress has been made in the development of selective CDK7 inhibitors, which show efficacy in both triple-negative breast cancer (TNBC) and hormone receptor-positive breast cancer (HR+). Moreover, combined with other agents, CDK7 inhibitors may provide synergistic effects for endocrine therapy and chemotherapy. Thus, high-quality studies for developing potent CDK7 inhibitors and investigating their applications in breast cancer therapy are rapidly emerging.
Conclusion
CDK7 inhibitors have emerged as a promising therapeutic strategy and have demonstrated significant anti-cancer activity in different subtypes of breast cancer, especially those that have been resistant to current therapies.
Subject terms: Breast cancer, Transcriptional regulatory elements
Introduction
Dysregulated cell division and gene expression are essential factors in the development of breast cancer [1]. Cyclin-dependent kinases (CDKs) play significant roles in these fundamental processes and are therapeutic targets in breast cancer. CDKs are serine/threonine protein kinases that cooperate with cyclin to regulate the progression of the cell cycle [2–4]. In the CDK family, CDK7 has garnered considerable attention because of its master regulatory features and biological functions. As a CDK-activating kinase (CAK), CDK7 binds to cyclin H and MAT1, resulting in the phosphorylation of specific cell cycle CDKs, thereby driving cell cycle progression, cell division, and cell proliferation [5, 6]. Additionally, CDK7, as part of the TFIIH complex, phosphorylates Ser5 residues in the C-terminal domain (CTD) of RNA polymerase II (PolII), facilitating transcription initiation [7–10]. Moreover, CDK7 regulates the activity of several transcription factors, including the estrogen receptor (ER) [11]. Hence, CDK7 is recognized as a crucial regulatory factor that drives both cell cycle progression and gene transcription, ensuring cell division and proliferation. Studies have confirmed that cancer cells are more vulnerable to CDK7 inhibition than normal cells due to the dysregulation of the cell cycle and high dependence on oncogenic transcription factors (TFs) needed for the growth and survival of cancer cells [12]. Preclinical studies revealed that CDK7 inhibitors caused cell cycle arrest, apoptosis and suppression of transcription, encouraging researchers to explore potent CDK7 inhibitors for breast cancer therapies [13–18]. However, research on the application of CDK7 inhibitors and the underlying mechanism of these inhibitors in breast cancer cells has rarely been summarized.
This review aims to provide a comprehensive summary of the latest insights into CDK7 biology and recent advancements in CDK7 inhibitor development for breast cancer treatment. Additionally, we discuss the current application of CDK7 inhibitors in different molecular types of breast cancer to provide potential strategies for the treatment of breast cancer.
Structure and function of CDK7
CDK7 structure
CDK7 is a kinase composed of 346 amino acids and has a typical kinase fold in the N-terminal lobe (residue 13–96), which is made up mostly of β-sheets and one α-helix, and a C-terminal lobe at residues 97–311 with mainly α-helices [19–21]. The activation segment in CDK7 is at residues 155–174 and appears to be a more open conformation than that in CDK2 [22]. A co-crystal structure has been reported for CDK7 bound to ATP [22, 23]. Notably, the adenine of ATP in the ATP–CDK7 structure established two hydrogen bonds with a hinge region between the two CDK7 lobes, thereby establishing a connection between the N and C lobes of CDK7. This binding model of ATP with CDK7 provides valuable insights for the development of selective CDK7 inhibitors.
The functional role of CDK7 is achieved through its polymerization with cyclin H and MAT1 to form CDK-activating kinase (CAK). CDK7 and cyclin H form a conventional CDK-cyclin pair, while MAT1 serves as a CAK assembly factor [24, 25] that enhances CAK activity toward target CDKs to regulate the cell cycle [24, 26] and anchors the CAK complex to the core section of the general transcription factor II H (TFIIH), which phosphorylates the CTD of RNA pol II and thus participates in transcription initiation [8, 20, 26, 27].
Recently, significant insights into the human CAK complex have emerged through cryo-EM-based structural studies,which completed the human TFIIH structure [21]. The combined structure illuminates the intricate interactions between MAT1 and both the TFIIH core complex and CDK7-cyclin H: the RING domain and helical regions near the MAT1 N-terminus, as well as the CAK anchor near the MAT1 C-terminus, engage with the TFIIH central complex and CDK-cyclin H, respectively [28]. This structural finding underscores the pivotal role of MAT1 as a regulator of TFIIH, as it can interact with elements on XPB, XPD, and CDK7. Furthermore, as a CAK assembly factor, MAT1 forms extensive and conserved interfaces with CDK7 and cyclin H. The contact between the CDK7 T-loop and MAT1 is a distinctive feature of the CAK complex [29]. This interaction contributes to the extended conformation of the T-loop, promoting CDK7 activation upon formation of the trimeric CAK. The trimeric CAK structure informs the functional mechanism and suggests a structural framework for a deeper understanding of the interactions between its regulatory elements.
The roles of CDK7 in the cell cycle
One of the fundamental biological activities is the cell cycle, which is the sequence of steps involved in cell division [30]. The cell cycle is separated into four phases: two intervening phases, G1 and G2, the DNA synthesis and mitotic segregation [31]. Three “checkpoints” regulate the cell cycle, the G1/S, G2/M transitions and mitotic spindle formation. An increasing number of studies has shown that CDKs, which facilitate DNA synthesis and chromosome segregation by phosphorylating their substrates, are part of a core mechanism that controls the cell cycle [32–34]. CDK1, CDK2, CDK4, CDK6, and CDK7 have been identified as key regulators of cell cycle progression and division in human cells [2]. Generally, CDKs are activated after binding with cyclins and phosphorylation on the T-loops by a CAK [35]. CDK7 is activated by two phosphorylation events (at Thr170 and Ser164), enhancing both its kinase activity and its ability to bind cyclin H [36]. More importantly, CDK7 plays a key role as the CAK responsible for the phosphorylation of CDK1, CDK2, CDK4 and CDK6, initiating their kinase activity [5, 6]. Thus, CDK7 indirectly influences all phases of the cell cycle and promotes downstream cell cycle progression (Fig. 1).
Fig. 1. CDK7 is a critical regulator of transcription.

Cell cycle progression is mediated through the phosphorylation of other CDKs (a). Phosphorylation of hormone receptors, Mediator and Pol II (b).
Mitogenic factors interact with cell cycle to produce intracellular signaling cascades that act on CDK4 and CDK6 to promote cell cycle progression from the G0 or G1 to the S phase [31, 37]. Previous research has established that CDK7 plays a role in phosphorylating CDK4/6 to facilitate the phosphorylation of the retinoblastoma (Rb) protein during G1 progression, phosphorylating CDK2 to promote progression from the G1 to the S phase, and phosphorylating CDK1 to regulate the G2/M transition [31, 37–39]. However, CDK4 and CDK6 activity rapidly diminishes after CDK7 inhibition, halting G1 progression [6, 40], implying the need for CDK7 activity to maintain CDK4/6 activation in the G1 phase.
The roles of CDK7 in transcription
In addition to the cell cycle, CDK7 plays a critical role in gene expression as a component of the general transcription factor II H (TFIIH), which mediates the phosphorylation of RNA polymerase II (Pol II) [20, 41]. The TFIIH complex consists of ten subunits, including helicase, ATPase, and protein kinase activities that are required for the initiation stage of transcription [42–44] (Fig. 2). Preinitiation complex formation facilitates promoter identification [45]. Transcription is initiated by RNA Pol II binding to multiple proteins, including the multi-subunit mediator complex and transcription factors. CAK is tethered to the core of TFIIH, ensuring the correct positioning of the active complex directly on the target site [26, 46], and phosphorylates the C-terminal domain (CTD) of RNA polymerase II (RNA Pol II) at Ser5 and Ser7 of the heptad repeat YSPTSPS [47–50]. CTD phosphorylation disrupts the interaction between the mediator and the RNA polymerase [20, 21], thereby triggering the release of RNA Pol II and enabling promoter clearance. Afterward, Pol II transcribes approximately 20–100 bases in a process known as promoter-proximal pausing. CDK7 is indispensable for recruiting two essential factors, DRB sensitivity-inducing factor (DSIF) and negative elongation factor (NELF), both of which are essential for promoter-proximal pausing. Additionally, CDK7 phosphorylates CDK9, a component of positive transcription elongation factor b (P-TEFb), which in turn phosphorylates the RNA Pol II CTD at Ser2 and Thr4, as well as DSIF and NELF, to facilitate transcriptional elongation [10, 51]. CDK12 and CDK13 are also involved in regulating transcription by phosphorylating the Pol II CTD at Ser2 and Ser5 during elongation [52, 53]. CDK7 mediates CDK12/13 activation through T-loop phosphorylation.
Fig. 2. CDK7 phosphorylates the CTD of RNA polymerase II (RNA Pol II) at serine 5 and serine 7 to regulate the initiation of transcription and promoter escape.

Yellow dots represent serine 2; red dots represent serine 5; and purple dots represent serine 7.
CDK7 plays a critical role in orchestrating various steps in transcription regulation. When CDK7 activity is inhibited, the phosphorylation levels of the CTD are reduced, hindering the transcription process. However, the specific role of CDK7 in transcription is still controversial. Inhibiting CDK7 activity specifically with YKL-5–124 was reported and did not greatly affect CTD phosphorylation or global gene expression but inhibited E2F-driven gene expression and promoted G1-S cell cycle arrest, indicating that certain CDK7 inhibitors may exert a greater impact on the cell cycle than on transcription [54].
The roles of CDK7 in the regulation of transcription factor activity
CDK7 has been shown to modulate the activity of various transcription factors, including nuclear hormone receptors such as androgen receptors (ARs) and estrogen receptors (ERs), thereby regulating their activation and target gene expression [11, 55]. As part of the TFIIH complex, CDK7 mediates the phosphorylation of ER at Ser118, modulating the activity of this transcription factor [11]. The recruitment of TFIIH to the hormone binding domain of ER mediates the phosphorylation of CDK7 target sites involved in the transcription activation of the ER [56] (Fig. 3). The importance of TFIIH/CDK7 in controlling the activities of certain transcription regulators, such as in breast cancer, for which ER is a major driver, emphasizes the potential application of CDK7 inhibitors in cancer therapy.
Fig. 3. As a component of the general transcription factor complex TFIIH, CDK7 regulates estrogen receptor (ER) activity.

Phosphorylation of Ser118 promotes ER activity.
CDK7-specific inhibitors
CDKs are essential for controlling transcription and the cell cycle. Numerous CDK inhibitors have been thoroughly investigated over the years, and some of them, including BS-181 [57], SY5609 [58], THZ1 [47] and ICEC0942 [59, 60], are currently in the clinical development stage for the treatment of breast cancer. Moreover, ATP-competitive inhibitors of CDK7 have been developed; these inhibitors include SY-1365 [61] and ICEC0942 [59]. The first selective CDK7 inhibitor, BS-181, demonstrated inhibition of CDK7 substrate phosphorylation, leading to cell cycle arrest, apoptosis, and inhibition of cancer cell growth, delivering the first proof of concept for the use of a CDK7 inhibitor as a therapeutic approach to breast cancer by targeting transcription [57, 62]. However, the in vivo activity of BS-181 was low as it showed poor bioavailability and cell permeability. Therefore, efforts should be made to improve the drug-like properties.
THZ1, a covalent inhibitor of CDK7, exhibited strong efficacy against TNBC [63]. THZ1 sensitivity depends on and is mediated by specific oncogenes regulated by super-enhancer elements and the associated transcriptional circuits. Importantly, THZ1 is usually well tolerated in mouse models, suggesting that this drug may have therapeutic value in combination therapy. However, THZ1 also prevents CDK12 and CDK13 from performing their normal functions. It is also related to off-target effects; that is, the inhibitor may disrupt catalysis mediated by other kinases in addition to the intended target [64]. Although findings to date demonstrate remarkable selectivity for cancer cells over normal cells, the aforementioned inhibition of other kinases may result in adverse effects.
To address the lack of selectivity of THZ1, a more potent and selective molecule, SY-1365, is undergoing clinical evaluation as a single agent and with other standard-of-care medications for breast cancer [61]. SY-1365 is the first selective CDK7 inhibitor authorized for use in hormone receptor (HR)+ breast cancer. In vitro, SY-1365 showed a higher susceptibility to induce apoptosis in cancer cell lines compared to non-cancer lines. Further investigation revealed that SY-1365 sensitivity was increased in cancer cells with low BCL2L1 (BCL-XL) expression. These results indicate SY-1365 use in combination with a BCL2 inhibitor in clinical settings. Furthermore, the same study found that SY-1365 exerted no significant effect on blood cell counts. These results imply that SY-1365 may show therapeutic efficacy in combination therapy and induce few side effects. Further research is being conducted to understand how SY-1365 affects the cell cycle, DNA repair, and apoptotic pathways. Interestingly, SY-1365 has been found to downregulate genes involved in DNA repair pathways, suggesting a potential synergistic effect when used with PARP inhibitors.
Researchers believe that greater or more frequent dosing is required for CDK inhibitor activity, which may result in an excessive dosing regimen for patients, as indicated by preclinical and clinical data. Thus, an oral non-covalent CDK7 inhibitor, SY-5609, is given greater priority for clinical development than SY-1365. In combination with fulvestrant, SY-5609 showed antitumor effects in preclinical TNBC and ER+ breast cancer models [65], and sustained tumor regression was associated with alterations in the RB pathway. Treatment with SY-5609 resulted in a dose-dependent reduction of cell cycle CAK targets and TFIIH targets of RNA polymerase II C-terminal domain in HCC70 cells. It led to cell cycle arrest in the G2/M phase and a decrease in the expression of crucial oncogenes such as c-Myc, ultimately inducing apoptosis in cancer cells. SY-5609 also induced regression in the HCC70 cell line-derived xenograft mouse model of TNBC. Due to these promising results, SY-5609 has been selected for clinical development and is currently being evaluated in a phase I trial with patients who have specific solid tumors.
ICEC0942 (samuraciclib) is a new orally bioavailable CDK7 inhibitor that showed substantial antitumor effects in studies of ER-positive breast cancer in vitro [59, 60]. It was also tested in TNBC model mice, where it resulted in significant tumor reduction with little effect on body weight. The favorable absorption and excellent safety profile has made ICEC0942 a promising clinical candidate in breast cancer. Additionally, combination therapy with tamoxifen and ICEC0942 showed complete growth arrest of ER-positive tumor xenografts. In clinical trials, ICEC0942 demonstrated acceptable safety and evidence of antitumor activity when administered in combination with fulvestrant to advanced HR+ breast cancer patients who showed disease progression with prior CDK4/6 inhibitors. Nevertheless, phase I clinical studies (CT7001) of samuraciclib (ICEC0942) showed that the clinical benefit rate (CBR) was 36% in patients with endocrine-resistant HR + BC after CDK4/6 inhibitor treatment and 20.0% in TNBC expansion [60]. Samuraciclib exhibited tolerable safety with clinical activity in both the patients with TNBC and those with HR+ after CDK4/6 inhibitor treatment. Since hematological adverse effects were rarely encountered, we expect that samuraciclib may be added to cytotoxic chemotherapy to improve treatment of TNBC.
Several more intriguing CDK7 inhibitors’ preclinical findings have recently been reported. Q901, a highly selective CDK7, induced G1 cell cycle arrest in cells and decreased tumor growth in ER + BC and CDK4/6 inhibitor-resistant models [66, 67]. Q901 was evaluated in a variety of solid tumor cell lines, and it revealed that TP53 wild-type cancer cells were more responsive to Q901 than TP53 mutant cells.
XL102 led to tumor regression and caused death in a variety of cancer cell lines [68]. In a phase I clinical trial, XL102 is being evaluated as a single agent and in combination in patients with HR + BC and TNBC,etc. At the data cutoff, no objective responses were observed, and XL102 is scheduled to be evaluated in a separate cohort of patients [69] (Table 1).
Table 1.
Summary of CDK7 inhibitors in clinical trials with breast cancer patients.
| Drug name | Administration | Condition/disease | Combination agent | Trial status | Clinical trial number |
|---|---|---|---|---|---|
| Samuraciclib (CT7001, ICEC0942) | Oral | Advanced solid malignancies, TNBC, HR + /HER2 − BC | Fulvestrant | Phase I/II | NCT03363893 |
| SY-1365 (Mevociclib) | Intravenous | Advanced solid tumor, ovarian and breast cancer | Carboplatin fulvestrant | Phase Iterminated | NCT03134638 |
| SY-5609 | Oral | ER+breast cancer; TNBC ovarian cancer;pancreatic cancer | Fulvestrant gemcitabine;gemcitabine+Nab-paclitaxel | Recruiting | NCT04247126 |
| Q901 | Orally | Advanced solid tumors, HR + /HER2 − BC | Phase I/II; recruiting | NCT05394103 | |
| XL102 (AUR102) | Solid tumors, ovarian cancer, TNBC, HR + BC, | Phase I; recruiting |
TNBC triple-negative breast cancer, HR + /HER2 − BC hormone receptor-positive/HER2-negative breast cancer.
The impact of CDK 7 inhibitors on different molecular subtypes of breast cancer
With advances in molecular biology, the molecular subtype of breast cancer has gradually become an important milestone in breast cancer treatment. Breast cancer is classified into four major sub-types: luminal A, luminal B, Her2-positive and basal-like [70]. Clinicians are committed to developing a suitable and effective treatment for each individual according to specific biological properties and different clinical prognoses. The development of CDK inhibitors gained momentum following the approval of CDK4/CDK6 inhibitors for breast cancer treatment [71–73]. CDK4/6 inhibitors primarily affect the cyclin D-CDK4/6-Rb pathway, leading to cell cycle arrest in the G1 phase and thereby inhibiting cell proliferation [74]. In contrast, CDK7 showed higher inhibitory effects on tumor cell proliferation due to its dual regulatory roles in cell cycle progression and transcription. Thus, various CDK7 inhibitors have been extensively studied, with some being entered in phase I/II clinical trials to evaluate their efficacy in different molecular sub-types of breast cancer [47, 57–59, 61, 75]. Although recent studies suggest that CDK7 inhibitors are effective therapeutic drugs for breast cancer, the mechanisms underlying CDK7 inhibitor effects in different molecular types of breast cancer are different.
CDK7 inhibitors in ER+ breast cancer
Research indicates that CDK7, cyclin H, and MAT1 are upregulated and coregulated at the mRNA level in breast cancer compared to normal breast tissue, suggesting that CDK7 expression may serve as a prognostic factor in ER+ breast cancer [76]. These findings raise the possibility that tumors, especially in ER+ breast cancer, with increased CDK7 expression may be particularly sensitive to CDK7 inhibition. Further studies revealed that the inhibition of ER+ breast cancer cells by CDK7 inhibitors depended mainly on the phosphorylation of ER. As one of the important complexes involved in CDK7 function, CAK activates ER through a ligand-dependent receptor activation process by inducing serine 118 (S118) phosphorylation of ER, which enhances ER-mediated transcriptional activity and thus promotes cell cycle progression [11]. Thus, CDK7 inhibitors exert significant inhibitory effects on ER+ breast cancer cells by reducing ER activity.
Several selective CDK7 inhibitors have been confirmed to block the CDK7-mediated oncogenic effects on transcription, prevent cell cycle release from the G1 phase and slow the release from the S phase in ER-positive xenograft and have been entered in phase I/II clinical trials [59, 61, 77]. The effects of CDK7 inhibitors on apoptosis and the cell cycle are associated with the inhibition of CDK7-mediated phosphorylation of CDK1/2/4/6 and RB. These inhibitors also blocked hormone receptor signaling and showed synergistic effects when administered with endocrine therapy in the HR+ breast cancer. They inhibited ER phosphorylation at Ser118, which was associated with ER-mediated transcriptional activity and response to endocrine therapies in clinical samples. The efficiency of combining CDK7 inhibitors and endocrine drugs may be explained by the importance of Ser118 phosphorylation for ER activity and the direct function of CDK7 in transcription. In addition, the CDK7 inhibitor ICEC0942 was well tolerated and did not exhibit adverse histological or functional effects on the liver or kidneys, in contrast to CDK4/6 inhibitors. The safety profile suggests strong cancer cell selectivity compared to normal cells. These findings reveal that CDK7 inhibitors offer a novel approach, particularly with potential utility as a single agent or in combination with hormone therapies, for ER+ breast cancer. However, the timing of CDK7 inhibitor therapy for patients with ER-positive breast cancer, including whether it should be used as adjuvant therapy or after the emergence of resistance to existing treatments, needs to be determined. We anticipate that more clinical trials will be designed to answer these issues.
Wang et al. indicated that upregulation of functional p53 has been shown to contribute to the increased sensitivity of breast cancer cells to CDK7 inhibitors in HR + BC. Inhibition of CDK7 increased p53 protein expression in wild-type (WT) p53-expressing breast cancer cells while decreasing p53 protein levels in mutant (MT) p53 breast cancer cells, suggesting that significant anticancer effects require the presence of wild-type p53 protein and its specific threshold level [78].
The E3 ligase murine double minute 2 (MDM2) regulates p53 [79]. Nutlin-3, a small-molecule MDM2 inhibitor, disrupts MDM2-p53 binding [80–82]. Doxorubicin, a classic chemotherapeutic drug for breast cancer, increases p53 expression by damaging DNA [83]. Breast cancer cells expressing WT p53 were more sensitive to THZ1 when nutlin-3 or doxorubicin was concurrently administered with THZ1. These findings indicate that combining a CDK7 inhibitor with nutlin-3 exerts an increased anticancer impact, and patients with wild-type p53 breast cancers may benefit from this combination treatment.
Moreover, treating MCF-7 cells with doxorubicin and THZ1 reduced survival compared to the effect of either treatment alone, implying that CDK7 inhibitors may enhance the anticancer effects of chemotherapy [78]. This information suggests new choices of treatments for patients with breast cancer resistant to standard chemotherapies.
Approximately 70% of patients with ER-positive breast cancer carry wild-type TP53 even after multiple treatments. If p53 status can be used as a predictive biomarker, the potential for significant benefit of CDK inhibitors combined with p53 inducers is anticipated, especially for patients with resistance to endocrine therapy plus CDK4/6 inhibitors. However, the safety of the combination of nutlin-3 and THZ1 needs to be confirmed. In addition to that of p53, the impact of genomic aberrations on the sensitivity to CDK7 inhibitors in breast cancer cell lines remains unclear. Therefore, further investigation into the interactions between CDK7 and proteins encoded by other mutant genes is needed.
Additionally, Kalan et al. reported that activating the p53 tumor suppressor protein in human colon cancer-derived cells induced transcriptional dependency on CDK7 [82]. They found that selective inhibition of CDK7 alone did not arrest division nor disrupt transcription to trigger apoptosis. Once p53 was activated by 5-fluorouracil or nutlin-3, transcription depended on CDK7. These works suggest that p53-activating agents may synergize with CDK7 inhibitors to induce cell death.
Combination therapies with CDK7 inhibitors in patients with HR+ breast cancer
ER activity drives most instances of breast cancers; thus, tamoxifen, aromatase inhibitors and fulvestrant, which target ERα, are used for ER+ breast cancer treatment [84, 85]. CDK7 inhibitors have been studied in combination with anti-estrogens due to their role in activating ER, which is the key transcriptional driver of ER+ breast cancer. Attia et al. revealed that CDK7 expression was linked to poor prognosis and reduced response to tamoxifen [86]. In MCF-7 cells, treatment with THZ1 increased tamoxifen-induced cytotoxicity and inhibited the expression of genes associated with tumor growth. In vivo, THZ1 increased the apoptotic death rate and enhanced the effect of tamoxifen. This research suggests that CDK7 is a potential therapeutic target for breast cancer, regardless of patient susceptibility to tamoxifen therapy. We found similar results showing that treatment with ICEC0942 in combination with either tamoxifen or fulvestrant led to a greater inhibitory effects in ER+ breast cancer cell lines than were realized with a single drug [59]. When given as single agents, selective estrogen receptor degraders (SERDs), such as fulvestrant, showed limited effects after disease progressed on CDK4/6 inhibitors [87, 88]. A phase I study has demonstrated acceptable safety and evidence of anti-tumor activity when combining CDK7 inhibitors with fulvestrant for advanced HR+ breast cancer disease progressed on an aromatase inhibitor and a CDK4/6 inhibitor [60]. In addition, ER-mutant breast cancer cells were particularly sensitive to combination therapy with fulvestrant [89]. The effectiveness of novel oral SERDs is now being evaluated in clinical trials, and the effectiveness of CDK7 inhibitors may be further increased when combined with a more effective companion [90].
Combination therapies frequently show greater efficacy than single-agent therapy. The possibility of combining CDK7 inhibitors with other medications has been explored in many studies [59, 61]. We look forward to more clinical trials to confirm the efficacy of combination therapies with CDK7 inhibitors in breast cancer.
CDK7 inhibitors to treat drug-resistant HR+ breast cancer
Although most early ER-positive (ER + ) breast cancer is highly sensitive to endocrine therapy, endocrine resistance introduces an obstacle to ER+ breast cancer treatment. Mutations in the ESR1 gene are common in advanced ER+ breast cancer, and they result in estrogen-independent receptor activation and resistance to hormone therapy [87, 91–93]. Harrod et al. found that induction of the tyrosine mutation Y537S at position 537 of the ESR1 gene reduced the sensitivity of MCF-7 breast cancer cells to ER inhibitors and was accompanied by TFIIH aggregation and CDK7-mediated S118 phosphorylation [89]. The CDK7 inhibitor THZ1 significantly inhibited the growth and proliferation of anti-estrogen resistant derivatives of MCF-7 breast cancer cells carrying the Y537S mutation and promoted the apoptosis of these cells. Furthermore, CDK7 inhibitors inhibited the phosphorylation of mutant ER at Ser118 [94]. These findings suggest that CDK7 inhibitors may be beneficial for patients with ESR1 mutation-positive advanced breast cancer who are partially resistant to anti-estrogen treatments.
CDK4/6 inhibitors have brought significant clinical benefits for the treatment of ER+ breast cancer; however, a substantial number of patients eventually develop resistance to these inhibitors, typically within 2–3 years. Loss of function of the retinoblastoma (RB) protein is a well-known mechanism of CDK4/6 inhibitor resistance. Increased cyclin E levels and CDK2 overexpression reduce the dependence of resistant cells on cyclin D1-CDK4/6, allowing them to re-enter the S phase [92]. Thus, it is possible to inhibit cyclin E-CDK2 to overcome CDK4/6 inhibitor resistance. In both CDK4/6 inhibitor-resistant and ER+ breast cancer xenograft models, CDK7 inhibitors successfully slowed the growth of tumors [60].
There is currently no consensus regarding the treatment of women with advanced disease progressed on CDK4/6 inhibition. Except to switching to chemotherapy, the mTOR inhibitor everolimus and the PI3K inhibitor alpelisib are two choices for continuing endocrine treatment [88, 95, 96]. However, everolimus showed limited progression-free survival (PFS) as well as raised toleration concerns because of stomatitis and pneumonitis [95]. Alpelisib is restricted to patients with a PIK3CA mutation and has obtained to a median PFS of 7.3 months in a single arm post-CDK4/6 inhibitor group [96]. Although the majority of patients with advanced disease have experienced many lines of regimen, CDK7 inhibitors are still effective against breast cancer cells with acquired resistance to palbociclib. The possible reason is that CDK7 is upstream of CDK4, and one mechanism of resistance to CDK inhibitors involves amplification of CDK4, indicating that CDK7 inhibitors may be useful after resistance to drugs that target CDK4/6.
It is anticipated to overcome endocrine resistance by using CDK7 inhibitors, but it is crucial to account for the information about efficacy predictive factors and acquired drug-resistance from preclinical studies when designing clinical trials.
CDK7 inhibitors in triple-negative breast cancer (TNBC)
The highly aggressive nature of TNBC and the poor prognosis makes this disease a high priority for effective therapeutics [97–99]. Due to the genetic complexity of tumors and the lack of targets, the development of potent therapies has been limited for a long time [100]. Studies have shown that deregulation of super-enhancers (SEs) is common in cancer, leading to significant changes in gene expression and high transcriptional outputs that maintain the oncogenic cellular state [101–103]. This transcriptional addiction suggests that cancer cells may be more responsive to transcriptional inhibition than normal cells, providing a strong rationale for targeting transcriptional kinases, including CDK7, in cancer therapy. Wang et al. found that TNBC cells specifically depend on CDK7 for transcriptional activity, and inhibiting CDK7 resulted in cell death [75] (Fig. 4). The study revealed that CDK7 mediated transcriptional addiction to a cluster of genes in TNBC cells, and many of these genes were oncogenic drivers (e.g., MYC, MYCN, and RUNX1). These genes exhibited strong dependency on continuous active transcription. TNBC cells were highly dependent on uninterrupted transcription of this specific set of genes, making them particularly sensitive to CDK7 inhibition, especially in association with SEs. The study concluded that CDK7 inhibitors can significantly diminish the transcription of specific genes that are overexpressed in TNBC but not in HR+ breast cancer cells, which indicated that CDK7 inhibition may be a potential therapy for patients with TNBC.
Fig. 4. CDK7 activity is required for the expression of numerous genes involved in TNBC.
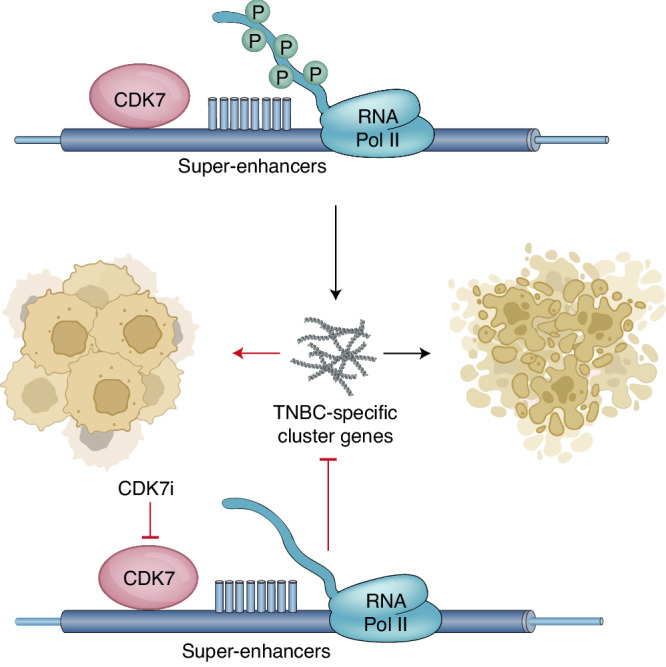
Tumour development is inhibited by a covalent CDK7 inhibitor.
The role of CDK7 in TNBC is related to super enhancers and closely related to specific oncogenic drivers, such as MYC in neuroblastoma [104, 105]. The MYC gene is a highly dysregulated oncogene in various cancer types and is considered a hallmark oncogene in many human cancers [106]. Abnormal expression of MYC family members often results in deregulated transcription and metabolism, leading to uncontrolled tumor growth and proliferation. Elevated expression of these oncogenes is typically indicative of a poor prognosis. The reported prognosis of breast cancer patients with high MYC expression is poor [107]. Studies indicating that CDK7 inhibitors induce tumor regression in mouse models of MYC-amplified neuroblastoma and MYC/MYCL-amplified small-cell lung cancer (SCLC) [63, 108] reinforced the idea that disruption of transcriptionally addicted malignancies can be realized via CDK7 inhibition. This effect was correlated with preferential downregulation of superenhancer-associated genes and mediated by downregulating MYC-driven transcription. These findings highlight the potential for targeting the general transcription machinery downstream of MYC through CDK7 inhibition as an attractive alternative approach to inhibiting difficult-to-target oncogenic transcription factors such as MYC.
Contrary to HR+ breast cancer, TNBC cells demonstrate high sensitivity to THZ1. Tang et al. identified a potential correlation between higher SOX9 expression and THZ1 sensitivity and poor prognosis in TNBC [109]. They found that higher SOX9 expression was substantially correlated with sensitivity to THZ1 and poor prognosis in TNBC. Additionally, THZ1 significantly reduced the ability of SOX9 to bind to an enhancer near the transcription factor FOXC1. The study also revealed an interaction between SOX9 and FOXC1, suggesting their cooperative regulation of the MYC signaling pathway in TNBC. These findings suggest that SOX9 may promote the sensitivity of TNBC cells to THZ1 in a FOXC1-related way, implying that SOX9 may function as a THZ1 predictive factor. In addition, TNBC patients with high expression of SOX9 show poor overall survival.
Li et al. analyzed publicly available transcriptomic data from TNBC patients and found a correlation between CDK7 mRNA levels and patient prognosis. High CDK7 protein expression was associated with a poor prognosis in a TNBC cohort [62]. This finding suggests that CDK7 may be a specific prognostic factor for TNBC patients. Mechanistic investigations revealed that the survival of MDA-MB-231 TNBC cells relied heavily on the BCL-2/BCL-XL signaling axis. Combining BCL-2/BCL-XL inhibitors, such as ABT-263/ABT199, with CDK7 inhibitors synergistically inhibited the growth of and induced apoptosis in human TNBC cells. These results provide mechanism-based proof of concept for combination treatment strategies to improve TNBC treatment effects especially in metastatic disease.
CDK7 inhibitors in Her2-positive breast cancer (Her2+ BC)
Human epidermal growth factor receptor 2 (Her2 + ) is frequently amplified and overexpressed in breast cancer [110, 111] and can be targeted with anti-Her2 therapies [112–115]. Her2-positive breast cancer (Her2+ BC) is more aggressive and has a higher recurrence and death rate than other breast cancer types [111]. Despite significant improvements achieved with anti-HER2 therapies, resistance can develop due to the overexpression of genes involved in cell cycle regulation in HER2 + BC cells [116]. Thus, blocking cell cycle progression with CDK4/6 inhibitors sensitizes these cells to Her2 inhibitors, suggesting the possibility that inhibiting CDK7, which activates CDK4, may be used to overcome resistance to Her2 inhibitors. Sun et al. demonstrated that combining HER2-targeted therapy with the CDK7 inhibitor THZ1 significantly suppressed Her2+ BC cell proliferation and promoted apoptosis in cancer cells that were resistant to Her2-targeted therapies [117]. Interestingly, the study revealed that CDK7 functioned largely as a TFIIH-associated kinase in Her2+ BC but did not appear to facilitate cell cycle progression in these cells via phosphorylation of CDKs. In contrast, the study identified SHP2 and PI3K/AKT as downstream signaling components that regulated the phosphorylation of the RNA pol II CTD via CDK7 activation. These findings suggest that targeting CDK7 may prevent the reactivation of multiple kinase pathways in HER2-resistant breast cancer, providing a potential therapeutic strategy.
Future prospects and key questions about CDK7 inhibitors
CDK7 inhibitors are in the early stage of development and clinical application, and therefore, there are many opportunities and related challenges for their clinical uses. First, CDK7 inhibitors are beneficial in HR+ breast cancer when administered alone or in combination with hormone therapy, and CDK7 inhibition might be effective even in advanced disease progressed on CDK4/6 inhibition. Second, the anti-tumor effects of CDK7 inhibitors have been confirmed in vitro as they inhibited the transcription of a cluster of cancer-specific genes in TNBC cells and showed survival benefits in clinical trials, suggesting their applicability to difficult-to-treat cancers. Finally, CDK7 inhibitors may enhance the anticancer effects of some standard chemotherapies, confirming that they can be used as potential combination agents for breast cancer, especially anthracycline-resistant disease. The most common adverse events were tolerable and reversible. Several clinical trials with CDK7 inhibitors are now being conducted to determine the maximum tolerated dose (MTD) and dose-limiting toxicities, as well as whether they can be safely given to patients.
However, before CDK7 inhibitors can be considered a novel treatment for breast cancer, several concerns remain. Although ER is particularly vulnerable to CDK7 inhibition, it is likely that other transcription factors play a role in the effects observed to date. We have an incomplete understanding of CDK7 function in controlling the actions of particular transcriptional regulators. Nevertheless, the transcription of certain genes may escape CDK7 inhibition, which may be compensated by other kinases. Compensation occurs quickly and involves the activation of other kinases (possibly including other CDKs). Since not all CDKs with compensatory activity are tumor-promoting factors and some may even play opposing roles, inhibiting certain CDKs may have an opposite effect of that intended [118, 119]. Additionally, the biological effects of different inhibitors are contradictory; for example, THZ1 typically caused cell cycle arrest and promoted apoptosis, whereas no substantial apoptosis was seen with YKL-5–124, which may be attributed to their different biochemical profiles. The mechanism underlying current CDK7 inhibitors appears to depend on the molecular genetics of the cancer cells. Even if CDK7 inhibitors prove to be innovative drugs, resistance may develop in certain patients. Analyses of tumor features associated with resistance to CDK7 inhibitors will contribute to understanding the mechanisms of CDK7 inhibitor resistance, which will assist in selecting the people who will most benefit from these drugs. A deeper comprehension of CDK7 biology and specific genes selectively dependent on CDK7 may lead to improve selection of particular breast cancer types and individualized therapy strategies. We anticipate that future research will reveal the value of this therapeutic approach.
Conclusions
CDK7 inhibitors have been developed as therapeutic strategy and have demonstrated promising anticancer activity in different subtypes of breast cancer, particularly those that elude current therapies. Further efforts to discover their clinical efficacy as a single agent or in rational therapeutic combinations will help in realizing CDK7 inhibitors as a brand new treatment for drug resistant breast cancer. We anticipate the availability of CDK7 inhibitors into clinical practice against different subtypes of breast cancer, as well as the potential impact on the treatment of breast cancer.
Acknowledgments
Funding
Zhaoyang project, Guangdong Provincial Hospital of Chinese Medicine
Author contributions
Xue Song: Conceptualization and writing. Chen Fang: Revision. Yang Sun, Xiaojie Lin and Yan Dai: Resources and literature search. Chang Qiu: Picture drawing. Rui Xu: Reviewing and Editing. All authors read and approved the manuscript.
Data availability
We declare that the materials described in the manuscript, including all relevant figures, are freely available to any scientist wishing to use them for noncommercial purposes, without breaching participant confidentiality.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–74. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 2.Malumbres M. Cyclin-dependent kinases. Genome Biol. 2014;15:122. doi: 10.1186/gb4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, et al. Cyclin-dependent kinases: a family portrait. Nat Cell Biol. 2009;11:1275–6. doi: 10.1038/ncb1109-1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Swaffer MP, Jones AW, Flynn HR, Snijders AP, Nurse P. CDK substrate phosphorylation and ordering the cell cycle. Cell. 2016;167:1750–61.e16. doi: 10.1016/j.cell.2016.11.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schachter MM, Fisher RP. The CDK-activating kinase Cdk7: taking yes for an answer. Cell cycle (Georget, Tex) 2013;12:3239–40. doi: 10.4161/cc.26355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schachter MM, Merrick KA, Larochelle S, Hirschi A, Zhang C, Shokat KM, et al. A Cdk7-Cdk4 T-loop phosphorylation cascade promotes G1 progression. Mol Cell. 2013;50:250–60. doi: 10.1016/j.molcel.2013.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Galbraith MD, Bender H, Espinosa JM. Therapeutic targeting of transcriptional cyclin-dependent kinases. Transcription. 2019;10:118–36. doi: 10.1080/21541264.2018.1539615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Serizawa H, Mäkelä TP, Conaway JW, Conaway RC, Weinberg RA, Young RA. Association of Cdk-activating kinase subunits with transcription factor TFIIH. Nature. 1995;374:280–2. doi: 10.1038/374280a0. [DOI] [PubMed] [Google Scholar]
- 9.Wong KH, Jin Y, Struhl K. TFIIH phosphorylation of the Pol II CTD stimulates mediator dissociation from the preinitiation complex and promoter escape. Mol Cell. 2014;54:601–12. doi: 10.1016/j.molcel.2014.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Larochelle S, Amat R, Glover-Cutter K, Sansó M, Zhang C, Allen JJ, et al. Cyclin-dependent kinase control of the initiation-to-elongation switch of RNA polymerase II. Nat Struct Mol Biol. 2012;19:1108–15. doi: 10.1038/nsmb.2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chen D, Riedl T, Washbrook E, Pace PE, Coombes RC, Egly JM, et al. Activation of estrogen receptor alpha by S118 phosphorylation involves a ligand-dependent interaction with TFIIH and participation of CDK7. Mol cell. 2000;6:127–37. doi: 10.1016/S1097-2765(05)00004-3. [DOI] [PubMed] [Google Scholar]
- 12.Bradner JE, Hnisz D, Young RA. Transcriptional addiction in cancer. Cell. 2017;168:629–43. doi: 10.1016/j.cell.2016.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Teng Y, Lu K, Zhang Q, Zhao L, Huang Y, Ingarra AM, et al. Recent advances in the development of cyclin-dependent kinase 7 inhibitors. Eur J Med Chem. 2019;183:111641. doi: 10.1016/j.ejmech.2019.111641. [DOI] [PubMed] [Google Scholar]
- 14.Fisher RP. Secrets of a double agent: CDK7 in cell-cycle control and transcription. J Cell Sci. 2005;118:5171–80. doi: 10.1242/jcs.02718. [DOI] [PubMed] [Google Scholar]
- 15.Wang M, Wang T, Zhang X, Wu X, Jiang S. Cyclin-dependent kinase 7 inhibitors in cancer therapy. Fut Med Chem. 2020;12:813–33. doi: 10.4155/fmc-2019-0334. [DOI] [PubMed] [Google Scholar]
- 16.Fisher RP. Cdk7: a kinase at the core of transcription and in the crosshairs of cancer drug discovery. Transcription. 2019;10:47–56. doi: 10.1080/21541264.2018.1553483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Diab S, Yu M, Wang S. CDK7 inhibitors in cancer therapy: The sweet smell of success? J Med Chem. 2020;63:7458–74. doi: 10.1021/acs.jmedchem.9b01985. [DOI] [PubMed] [Google Scholar]
- 18.Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–46. doi: 10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yankulov KY, Bentley DL. Regulation of CDK7 substrate specificity by MAT1 and TFIIH. EMBO J. 1997;16:1638–46. doi: 10.1093/emboj/16.7.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rimel JK, Taatjes DJ. The essential and multifunctional TFIIH complex. Protein Sci. 2018;27:1018–37. doi: 10.1002/pro.3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Greber BJ, Perez-Bertoldi JM, Lim K, Iavarone AT, Toso DB, Nogales E. The cryoelectron microscopy structure of the human CDK-activating kinase. Proc Natl Acad Sci. 2020;117:22849–57. doi: 10.1073/pnas.2009627117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lolli G. Structural dissection of cyclin dependent kinases regulation and protein recognition properties. Cell cycle (Georget, Tex) 2010;9:1551–61. doi: 10.4161/cc.9.8.11195. [DOI] [PubMed] [Google Scholar]
- 23.Lolli G, Lowe ED, Brown NR, Johnson LN. The crystal structure of human CDK7 and its protein recognition properties. Struct (Lond, Engl : 1993) 2004;12:2067–79. doi: 10.1016/j.str.2004.08.013. [DOI] [PubMed] [Google Scholar]
- 24.Fisher RP, Jin P, Chamberlin HM, Morgan DO. Alternative mechanisms of CAK assembly require an assembly factor or an activating kinase. Cell. 1995;83:47–57. doi: 10.1016/0092-8674(95)90233-3. [DOI] [PubMed] [Google Scholar]
- 25.Yee A, Nichols MA, Wu L, Hall FL, Kobayashi R, Xiong Y. Molecular cloning of CDK7-associated human MAT1, a cyclin-dependent kinase-activating kinase (CAK) assembly factor. Cancer Res. 1995;55:6058–62. [PubMed] [Google Scholar]
- 26.Busso D, Keriel A, Sandrock B, Poterszman A, Gileadi O, Egly JM. Distinct regions of MAT1 regulate cdk7 kinase and TFIIH transcription activities. J Biol Chem. 2000;275:22815–23. doi: 10.1074/jbc.M002578200. [DOI] [PubMed] [Google Scholar]
- 27.Shiekhattar R, Mermelstein F, Fisher RP, Drapkin R, Dynlacht B, Wessling HC, et al. Cdk-activating kinase complex is a component of human transcription factor TFIIH. Nature. 1995;374:283–7. doi: 10.1038/374283a0. [DOI] [PubMed] [Google Scholar]
- 28.Greber BJ, Toso DB, Fang J, Nogales E. The complete structure of the human TFIIH core complex. eLife. 2019;8:e44771. doi: 10.7554/eLife.44771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Peissert S, Schlosser A, Kendel R, Kuper J, Kisker C. Structural basis for CDK7 activation by MAT1 and Cyclin H. Proc Natl Acad Sci USA. 2020;117:26739–48. doi: 10.1073/pnas.2010885117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Norbury C, Nurse P. Animal cell cycles and their control. Annu Rev Biochem. 1992;61:441–70. doi: 10.1146/annurev.bi.61.070192.002301. [DOI] [PubMed] [Google Scholar]
- 31.Massague J. G1 cell-cycle control and cancer. Nature. 2004;432:298–306. doi: 10.1038/nature03094. [DOI] [PubMed] [Google Scholar]
- 32.Arellano M, Moreno S. Regulation of CDK/cyclin complexes during the cell cycle. Int J Biochem cell Biol. 1997;29:559–73. doi: 10.1016/S1357-2725(96)00178-1. [DOI] [PubMed] [Google Scholar]
- 33.Thu KL, Soria-Bretones I, Mak TW, Cescon DW. Targeting the cell cycle in breast cancer: towards the next phase. Cell cycle (Georget, Tex) 2018;17:1871–85. doi: 10.1080/15384101.2018.1502567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Canavese M, Santo L, Raje N. Cyclin dependent kinases in cancer: potential for therapeutic intervention. Cancer Biol Ther. 2012;13:451–7. doi: 10.4161/cbt.19589. [DOI] [PubMed] [Google Scholar]
- 35.Fisher RP, Morgan DO. A novel cyclin associates with MO15/CDK7 to form the CDK-activating kinase. Cell. 1994;78:713–24. doi: 10.1016/0092-8674(94)90535-5. [DOI] [PubMed] [Google Scholar]
- 36.Glover-Cutter K, Larochelle S, Erickson B, Zhang C, Shokat K, Fisher RP, et al. TFIIH-associated Cdk7 kinase functions in phosphorylation of C-terminal domain Ser7 residues, promoter-proximal pausing, and termination by RNA polymerase II. Mol Cell Biol. 2009;29:5455–64. doi: 10.1128/MCB.00637-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sherr CJ. G1 phase progression: cycling on cue. Cell. 1994;79:551–5. doi: 10.1016/0092-8674(94)90540-1. [DOI] [PubMed] [Google Scholar]
- 38.Giacinti C, Giordano A. RB and cell cycle progression. Oncogene. 2006;25:5220–7. doi: 10.1038/sj.onc.1209615. [DOI] [PubMed] [Google Scholar]
- 39.Larochelle S, Merrick KA, Terret ME, Wohlbold L, Barboza NM, Zhang C, et al. Requirements for Cdk7 in the assembly of Cdk1/cyclin B and activation of Cdk2 revealed by chemical genetics in human cells. Mol cell. 2007;25:839–50. doi: 10.1016/j.molcel.2007.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bisteau X, Paternot S, Colleoni B, Ecker K, Coulonval K, De Groote P, et al. CDK4 T172 phosphorylation is central in a CDK7-dependent bidirectional CDK4/CDK2 interplay mediated by p21 phosphorylation at the restriction point. PLoS Genet. 2013;9:e1003546. doi: 10.1371/journal.pgen.1003546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Egly JM, Coin F. A history of TFIIH: two decades of molecular biology on a pivotal transcription/repair factor. DNA Repair (Amst) 2011;10:714–21. doi: 10.1016/j.dnarep.2011.04.021. [DOI] [PubMed] [Google Scholar]
- 42.Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Dev (Camb, Engl) 2013;140:3079–93. doi: 10.1242/dev.091744. [DOI] [PubMed] [Google Scholar]
- 43.Compe E, Egly JM. TFIIH: when transcription met DNA repair. Nat Rev Mol cell Biol. 2012;13:343–54. doi: 10.1038/nrm3350. [DOI] [PubMed] [Google Scholar]
- 44.Kolesnikova O, Radu L, Poterszman A. TFIIH: A multi-subunit complex at the cross-roads of transcription and DNA repair. Adv protein Chem Struct Biol. 2019;115:21–67. doi: 10.1016/bs.apcsb.2019.01.003. [DOI] [PubMed] [Google Scholar]
- 45.Cramer P. Organization and regulation of gene transcription. Nature. 2019;573:45–54. doi: 10.1038/s41586-019-1517-4. [DOI] [PubMed] [Google Scholar]
- 46.Abdulrahman W, Iltis I, Radu L, Braun C, Maglott-Roth A, Giraudon C, et al. ARCH domain of XPD, an anchoring platform for CAK that conditions TFIIH DNA repair and transcription activities. Proc Natl Acad Sci USA. 2013;110:E633–42. doi: 10.1073/pnas.1213981110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Whittaker SR, Mallinger A, Workman P, Clarke PA. Inhibitors of cyclin-dependent kinases as cancer therapeutics. Pharmacol Ther. 2017;173:83–105. doi: 10.1016/j.pharmthera.2017.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schier AC, Taatjes DJ. Structure and mechanism of the RNA polymerase II transcription machinery. Genes Dev. 2020;34:465–88. doi: 10.1101/gad.335679.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Meinhart A, Cramer P. Recognition of RNA polymerase II carboxy-terminal domain by 3’-RNA-processing factors. Nature. 2004;430:223–6. doi: 10.1038/nature02679. [DOI] [PubMed] [Google Scholar]
- 50.Jeronimo C, Collin P, Robert F. The RNA polymerase II CTD: the increasing complexity of a low-complexity protein domain. J Mol Biol. 2016;428:2607–22. doi: 10.1016/j.jmb.2016.02.006. [DOI] [PubMed] [Google Scholar]
- 51.Akhtar MS, Heidemann M, Tietjen JR, Zhang DW, Chapman RD, Eick D, et al. TFIIH kinase places bivalent marks on the carboxy-terminal domain of RNA polymerase II. Mol cell. 2009;34:387–93. doi: 10.1016/j.molcel.2009.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bartkowiak B, Liu P, Phatnani HP, Fuda NJ, Cooper JJ, Price DH, et al. CDK12 is a transcription elongation-associated CTD kinase, the metazoan ortholog of yeast Ctk1. Genes Dev. 2010;24:2303–16. doi: 10.1101/gad.1968210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liang K, Gao X, Gilmore JM, Florens L, Washburn MP, Smith E, et al. Characterization of human cyclin-dependent kinase 12 (CDK12) and CDK13 complexes in C-terminal domain phosphorylation, gene transcription, and RNA processing. Mol Cell Biol. 2015;35:928–38. doi: 10.1128/MCB.01426-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Olson CM, Liang Y, Leggett A, Park WD, Li L, Mills CE, et al. Development of a Selective CDK7 Covalent Inhibitor Reveals Predominant Cell-Cycle Phenotype. Cell Chem Biol. 2019;26:792–803.e10. doi: 10.1016/j.chembiol.2019.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen D, Washbrook E, Sarwar N, Bates GJ, Pace PE, Thirunuvakkarasu V, et al. Phosphorylation of human estrogen receptor alpha at serine 118 by two distinct signal transduction pathways revealed by phosphorylation-specific antisera. Oncogene. 2002;21:4921–31. doi: 10.1038/sj.onc.1205420. [DOI] [PubMed] [Google Scholar]
- 56.Métivier R, Penot G, Hübner MR, Reid G, Brand H, Kos M, et al. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell. 2003;115:751–63. doi: 10.1016/S0092-8674(03)00934-6. [DOI] [PubMed] [Google Scholar]
- 57.Ali S, Heathcote DA, Kroll SH, Jogalekar AS, Scheiper B, Patel H, et al. The development of a selective cyclin-dependent kinase inhibitor that shows antitumor activity. Cancer Res. 2009;69:6208–15. doi: 10.1158/0008-5472.CAN-09-0301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marineau JJ, Hamman KB, Hu S, Alnemy S, Mihalich J, Kabro A, et al. Discovery of SY-5609: a selective, noncovalent inhibitor of CDK7. J Med Chem. 2022;65:1458–80. doi: 10.1021/acs.jmedchem.1c01171. [DOI] [PubMed] [Google Scholar]
- 59.Patel H, Periyasamy M, Sava GP, Bondke A, Slafer BW, Kroll SHB, et al. ICEC0942, an orally bioavailable selective inhibitor of CDK7 for cancer treatment. Mol Cancer Ther. 2018;17:1156–66. doi: 10.1158/1535-7163.MCT-16-0847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Coombes RC, Howell S, Lord SR, Kenny L, Mansi J, Mitri Z, et al. Dose escalation and expansion cohorts in patients with advanced breast cancer in a Phase I study of the CDK7-inhibitor samuraciclib. Nat Commun. 2023;14:4444. doi: 10.1038/s41467-023-40061-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hu S, Marineau JJ, Rajagopal N, Hamman KB, Choi YJ, Schmidt DR, et al. Discovery and characterization of SY-1365, a selective, covalent inhibitor of CDK7. Cancer Res. 2019;79:3479–91. doi: 10.1158/0008-5472.CAN-19-0119. [DOI] [PubMed] [Google Scholar]
- 62.Li B, Ni Chonghaile T, Fan Y, Madden SF, Klinger R, O’Connor AE, et al. Therapeutic rationale to target highly expressed CDK7 conferring poor outcomes in triple-negative breast cancer. Cancer Res. 2017;77:3834–45. doi: 10.1158/0008-5472.CAN-16-2546. [DOI] [PubMed] [Google Scholar]
- 63.Kwiatkowski N, Zhang T, Rahl PB, Abraham BJ, Reddy J, Ficarro SB, et al. Targeting transcription regulation in cancer with a covalent CDK7 inhibitor. Nature. 2014;511:616–20. doi: 10.1038/nature13393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Clopper KC, Taatjes DJ. Chemical inhibitors of transcription-associated kinases. Curr Opin Chem Biol. 2022;70:102186. doi: 10.1016/j.cbpa.2022.102186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sharma M, Bashir B, Hamilton E, Juric D, Papadopoulos K, Richardson D, et al. 518MO Tolerability and preliminary clinical activity of SY-5609, a highly potent and selective oral CDK7 inhibitor, in patients with advanced solid tumors. Ann Oncol. 2021;32:S587–S8. doi: 10.1016/j.annonc.2021.08.1040. [DOI] [Google Scholar]
- 66.Yu D, Jeon Y, Park D, Seo M, Ahn W, Kim J, et al. Abstract 4855: Development of highly selective CDK7 inhibitor Q901 for solid tumors. Cancer Res. 2020;80:4855. doi: 10.1158/1538-7445.AM2020-4855. [DOI] [Google Scholar]
- 67.DYYJDPMSWAJKK N. Abstract 1953: q901, a selective CDK7 inhibitor, a potential new strategy for primary and CDK4/6 inhibitor resistant ER-positive breast cancer. Cancer Res. 2021; 81:1953.
- 68.Satyam LK, Poddutoori R, Thiyagarajan S, Mukherjee S, Kaza LN, Charamanna K, et al. Potent anti-tumor activity of AUR102, a selective covalent inhibitor of CDK7. Eur J Cancer. 2020;138:S47. doi: 10.1016/S0959-8049(20)31201-6. [DOI] [Google Scholar]
- 69.Shapiro G, Barve MA, Bhave MA, Subbiah V, Uttamsingh S, Sharma K, Andrianova L, Patnaik A. A phase 1 dose-escalation and expansion-cohort study of the oral CDK7 inhibitor XL102 as a single-agent and in combination therapy in patients (pts) with advanced solid tumors. J Clin Oncol. 2022;40:TPS3176. doi: 10.1200/JCO.2022.40.16_suppl.TPS3176. [DOI] [Google Scholar]
- 70.Hammond ME, Hayes DF, Dowsett M, Allred DC, Hagerty KL, Badve S, et al. American Society of Clinical Oncology/College Of American Pathologists guideline recommendations for immunohistochemical testing of estrogen and progesterone receptors in breast cancer. J Clin Oncol. 2010;28:2784–95. doi: 10.1200/JCO.2009.25.6529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Slamon DJ, Neven P, Chia S, Fasching PA, De Laurentiis M, Im SA, et al. Phase III randomized study of ribociclib and fulvestrant in hormone receptor-positive, human epidermal growth factor receptor 2-negative advanced breast cancer: MONALEESA-3. J Clin Oncol. 2018;36:2465–72. doi: 10.1200/JCO.2018.78.9909. [DOI] [PubMed] [Google Scholar]
- 72.Sledge GW, Jr., Toi M, Neven P, Sohn J, Inoue K, Pivot X, et al. MONARCH 2: abemaciclib in combination with fulvestrant in women with HR+/HER2- advanced breast cancer who had progressed while receiving endocrine therapy. J Clin Oncol. 2017;35:2875–84. doi: 10.1200/JCO.2017.73.7585. [DOI] [PubMed] [Google Scholar]
- 73.Cristofanilli M, Turner NC, Bondarenko I, Ro J, Im SA, Masuda N, et al. Fulvestrant plus palbociclib versus fulvestrant plus placebo for treatment of hormone-receptor-positive, HER2-negative metastatic breast cancer that progressed on previous endocrine therapy (PALOMA-3): final analysis of the multicentre, double-blind, phase 3 randomised controlled trial. Lancet Oncol. 2016;17:425–39. doi: 10.1016/S1470-2045(15)00613-0. [DOI] [PubMed] [Google Scholar]
- 74.Klein ME, Kovatcheva M, Davis LE, Tap WD, Koff A. CDK4/6 inhibitors: the mechanism of action may not be as simple as once thought. Cancer cell. 2018;34:9–20. doi: 10.1016/j.ccell.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Y, Zhang T, Kwiatkowski N, Abraham BJ, Lee TI, Xie S, et al. CDK7-dependent transcriptional addiction in triple-negative breast cancer. Cell. 2015;163:174–86. doi: 10.1016/j.cell.2015.08.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Patel H, Abduljabbar R, Lai CF, Periyasamy M, Harrod A, Gemma C, et al. Expression of CDK7, Cyclin H, and MAT1 is elevated in breast cancer and is prognostic in estrogen receptor-positive breast cancer. Clin Cancer Res. 2016;22:5929–38. doi: 10.1158/1078-0432.CCR-15-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Papadopoulos KP, Sharma M, Hamilton EP, Richardson DL, Hodgson G, Zhou L, et al. First-in-human phase I study of SY-5609, an oral, potent, and selective noncovalent CDK7 inhibitor, in adult patients with select advanced solid tumors. J Clin Oncol. 2020;38:TPS3662. doi: 10.1200/JCO.2020.38.15_suppl.TPS3662. [DOI] [Google Scholar]
- 78.Wang Y, Zhang Z, Mi X, Li M, Huang D, Song T, et al. Elevation of effective p53 expression sensitizes wild-type p53 breast cancer cells to CDK7 inhibitor THZ1. Cell Commun Signal. 2022;20:96. doi: 10.1186/s12964-022-00837-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Thomasova D, Bruns HA, Kretschmer V, Ebrahim M, Romoli S, Liapis H, et al. Murine double minute-2 prevents p53-overactivation-related cell death (podoptosis) of podocytes. J Am Soc Nephrol. 2015;26:1513–23. doi: 10.1681/ASN.2014040345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Endo S, Yamato K, Hirai S, Moriwaki T, Fukuda K, Suzuki H, et al. Potent in vitro and in vivo antitumor effects of MDM2 inhibitor nutlin-3 in gastric cancer cells. Cancer Sci. 2011;102:605–13. doi: 10.1111/j.1349-7006.2010.01821.x. [DOI] [PubMed] [Google Scholar]
- 81.Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science. 2004;303:844–8. doi: 10.1126/science.1092472. [DOI] [PubMed] [Google Scholar]
- 82.Kalan S, Amat R, Schachter MM, Kwiatkowski N, Abraham BJ, Liang Y, et al. Activation of the p53 transcriptional program sensitizes cancer cells to Cdk7 inhibitors. Cell Rep. 2017;21:467–81. doi: 10.1016/j.celrep.2017.09.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aoubala M, Murray-Zmijewski F, Khoury MP, Fernandes K, Perrier S, Bernard H, et al. p53 directly transactivates Δ133p53α, regulating cell fate outcome in response to DNA damage. Cell Death Differ. 2011;18:248–58. doi: 10.1038/cdd.2010.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Clemons M, Danson S, Howell A. Tamoxifen (“Nolvadex”): a review. Cancer Treat Rev. 2002;28:165–80. doi: 10.1016/S0305-7372(02)00036-1. [DOI] [PubMed] [Google Scholar]
- 85.Nathan MR, Schmid P. A review of fulvestrant in breast cancer. Oncol Ther. 2017;5:17–29. doi: 10.1007/s40487-017-0046-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Attia YM, Shouman SA, Salama SA, Ivan C, Elsayed AM, Amero P, et al. Blockade of CDK7 reverses endocrine therapy resistance in breast cancer. Int J Mol Sci. 2020;21:2974. doi: 10.3390/ijms21082974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Jeselsohn R, Buchwalter G, De Angelis C, Brown M, Schiff R. ESR1 mutations—a mechanism for acquired endocrine resistance in breast cancer. Nat Rev Clin Oncol. 2015;12:573–83. doi: 10.1038/nrclinonc.2015.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.André F, Ciruelos EM, Juric D, Loibl S, Campone M, Mayer IA, et al. Alpelisib plus fulvestrant for PIK3CA-mutated, hormone receptor-positive, human epidermal growth factor receptor-2-negative advanced breast cancer: final overall survival results from SOLAR-1. Ann Oncol. 2021;32:208–17. doi: 10.1016/j.annonc.2020.11.011. [DOI] [PubMed] [Google Scholar]
- 89.Harrod A, Fulton J, Nguyen VTM, Periyasamy M, Ramos-Garcia L, Lai CF, et al. Genomic modelling of the ESR1 Y537S mutation for evaluating function and new therapeutic approaches for metastatic breast cancer. Oncogene. 2017;36:2286–96. doi: 10.1038/onc.2016.382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Patel HK, Tao N, Lee KM, Huerta M, Arlt H, Mullarkey T, et al. Elacestrant (RAD1901) exhibits anti-tumor activity in multiple ER+ breast cancer models resistant to CDK4/6 inhibitors. Breast Cancer Res. 2019;21:146. doi: 10.1186/s13058-019-1230-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Jeselsohn R, Yelensky R, Buchwalter G, Frampton G, Meric-Bernstam F, Gonzalez-Angulo AM, et al. Emergence of constitutively active estrogen receptor-α mutations in pretreated advanced estrogen receptor-positive breast cancer. Clin Cancer Res. 2014;20:1757–67. doi: 10.1158/1078-0432.CCR-13-2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Razavi P, Chang MT, Xu G, Bandlamudi C, Ross DS, Vasan N, et al. The genomic landscape of endocrine-resistant advanced breast cancers. Cancer Cell. 2018;34:427–38.e6. doi: 10.1016/j.ccell.2018.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Robinson DR, Wu YM, Vats P, Su F, Lonigro RJ, Cao X, et al. Activating ESR1 mutations in hormone-resistant metastatic breast cancer. Nat Genet. 2013;45:1446–51. doi: 10.1038/ng.2823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Jeselsohn R, Bergholz JS, Pun M, Cornwell M, Liu W, Nardone A, et al. Allele-specific chromatin recruitment and therapeutic vulnerabilities of ESR1 activating mutations. Cancer Cell. 2018;33:173–86.e5. doi: 10.1016/j.ccell.2018.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Cook MM, Al Rabadi L, Kaempf AJ, Saraceni MM, Savin MA, Mitri ZI. Everolimus plus exemestane treatment in patients with metastatic hormone receptor-positive breast cancer previously treated with CDK4/6 inhibitor therapy. ncologist. 2021;26:101–6. doi: 10.1002/onco.13609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rugo HS, Lerebours F, Ciruelos E, Drullinsky P, Ruiz-Borrego M, Neven P, et al. Alpelisib plus fulvestrant in PIK3CA-mutated, hormone receptor-positive advanced breast cancer after a CDK4/6 inhibitor (BYLieve): one cohort of a phase 2, multicentre, open-label, non-comparative study. Lancet Oncol. 2021;22:489–98. doi: 10.1016/S1470-2045(21)00034-6. [DOI] [PubMed] [Google Scholar]
- 97.Foulkes WD, Smith IE, Reis-Filho JS. Triple-negative breast cancer. N Engl J Med. 2010;363:1938–48. doi: 10.1056/NEJMra1001389. [DOI] [PubMed] [Google Scholar]
- 98.Zhang S, Huang S, Zhang H, Li D, Li X, Cheng Y, et al. Histo- and clinico-pathological analysis of a large series of triple-negative breast cancer in a single center in China: Evidences on necessity of histological subtyping and grading. Chin J Cancer Res. 2020;32:580–95. doi: 10.21147/j.issn.1000-9604.2020.05.03. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Abramson VG, Lehmann BD, Ballinger TJ, Pietenpol JA. Subtyping of triple-negative breast cancer: implications for therapy. Cancer. 2015;121:8–16. doi: 10.1002/cncr.28914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Asleh K, Riaz N, Nielsen TO. Heterogeneity of triple negative breast cancer: current advances in subtyping and treatment implications. J Exp Clin Cancer Res. 2022;41:265. doi: 10.1186/s13046-022-02476-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hnisz D, Abraham BJ, Lee TI, Lau A, Saint-André V, Sigova AA, et al. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–47. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lovén J, Hoke HA, Lin CY, Lau A, Orlando DA, Vakoc CR, et al. Selective inhibition of tumor oncogenes by disruption of super-enhancers. Cell. 2013;153:320–34. doi: 10.1016/j.cell.2013.03.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hnisz D, Schuijers J, Lin CY, Weintraub AS, Abraham BJ, Lee TI, et al. Convergence of developmental and oncogenic signaling pathways at transcriptional super-enhancers. Mol Cell. 2015;58:362–70. doi: 10.1016/j.molcel.2015.02.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chipumuro E, Marco E, Christensen CL, Kwiatkowski N, Zhang T, Hatheway CM, et al. CDK7 inhibition suppresses super-enhancer-linked oncogenic transcription in MYCN-driven cancer. Cell. 2014;159:1126–39. doi: 10.1016/j.cell.2014.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Balko JM, Giltnane JM, Wang K, Schwarz LJ, Young CD, Cook RS, et al. Molecular profiling of the residual disease of triple-negative breast cancers after neoadjuvant chemotherapy identifies actionable therapeutic targets. Cancer Discov. 2014;4:232–45. doi: 10.1158/2159-8290.CD-13-0286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kalkat M, De Melo J, Hickman KA, Lourenco C, Redel C, Resetca D, et al. MYC deregulation in primary human cancers. Genes (Basel) 2017;8:151. doi: 10.3390/genes8060151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Horiuchi D, Kusdra L, Huskey NE, Chandriani S, Lenburg ME, Gonzalez-Angulo AM, et al. MYC pathway activation in triple-negative breast cancer is synthetic lethal with CDK inhibition. J Exp Med. 2012;209:679–96. doi: 10.1084/jem.20111512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Christensen CL, Kwiatkowski N, Abraham BJ, Carretero J, Al-Shahrour F, Zhang T, et al. Targeting transcriptional addictions in small cell lung cancer with a covalent CDK7 inhibitor. Cancer Cell. 2014;26:909–22. doi: 10.1016/j.ccell.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tang L, Jin J, Xu K, Wang X, Tang J, Guan X. SOX9 interacts with FOXC1 to activate MYC and regulate CDK7 inhibitor sensitivity in triple-negative breast cancer. Oncogenesis. 2020;9:47. doi: 10.1038/s41389-020-0232-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hynes NE, Lane HA. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat Rev Cancer. 2005;5:341–54. doi: 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- 111.Loibl S, Gianni L. HER2-positive breast cancer. Lancet (Lond, Engl) 2017;389:2415–29. doi: 10.1016/S0140-6736(16)32417-5. [DOI] [PubMed] [Google Scholar]
- 112.Cameron D, Piccart-Gebhart MJ, Gelber RD, Procter M, Goldhirsch A, de Azambuja E, et al. 11 years’ follow-up of trastuzumab after adjuvant chemotherapy in HER2-positive early breast cancer: final analysis of the HERceptin Adjuvant (HERA) trial. Lancet (Lond, Engl) 2017;389:1195–205. doi: 10.1016/S0140-6736(16)32616-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.von Minckwitz G, Procter M, de Azambuja E, Zardavas D, Benyunes M, Viale G, et al. Adjuvant Pertuzumab and Trastuzumab in Early HER2-Positive Breast Cancer. N Engl J Med. 2017;377:122–31. doi: 10.1056/NEJMoa1703643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu B, Yan M, Ma F, Hu X, Feng J, Ouyang Q, et al. Pyrotinib plus capecitabine versus lapatinib plus capecitabine for the treatment of HER2-positive metastatic breast cancer (PHOEBE): a multicentre, open-label, randomised, controlled, phase 3 trial. Lancet Oncol. 2021;22:351–60. doi: 10.1016/S1470-2045(20)30702-6. [DOI] [PubMed] [Google Scholar]
- 115.Slamon D, Eiermann W, Robert N, Pienkowski T, Martin M, Press M, et al. Adjuvant trastuzumab in HER2-positive breast cancer. N. Engl J Med. 2011;365:1273–83. doi: 10.1056/NEJMoa0910383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Goel S, Wang Q, Watt AC, Tolaney SM, Dillon DA, Li W, et al. Overcoming therapeutic resistance in HER2-positive breast cancers with CDK4/6 Inhibitors. Cancer cell. 2016;29:255–69. doi: 10.1016/j.ccell.2016.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sun B, Mason S, Wilson RC, Hazard SE, Wang Y, Fang R, et al. Inhibition of the transcriptional kinase CDK7 overcomes therapeutic resistance in HER2-positive breast cancers. Oncogene. 2020;39:50–63. doi: 10.1038/s41388-019-0953-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Akoulitchev S, Chuikov S, Reinberg D. TFIIH is negatively regulated by cdk8-containing mediator complexes. Nature. 2000;407:102–6. doi: 10.1038/35024111. [DOI] [PubMed] [Google Scholar]
- 119.Fant CB, Taatjes DJ. Regulatory functions of the mediator kinases CDK8 and CDK19. Transcription. 2019;10:76–90. doi: 10.1080/21541264.2018.1556915. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
We declare that the materials described in the manuscript, including all relevant figures, are freely available to any scientist wishing to use them for noncommercial purposes, without breaching participant confidentiality.