

Abstract
Background
Perazine is an old phenothiazine derivative used for the treatment of people with schizophrenia and is reputed to have a low level of extrapyramidal adverse effects. As far as we are aware, its use is limited to Germany, Poland, the former Yugoslavia and the Netherlands.
Objectives
To examine the effects of perazine for those with schizophrenia or related psychoses in comparison with placebo, no treatment or other antipsychotic medications.
Search methods
We searched the Cochrane Schizophrenia Group Trials Register, which includes relevant randomised controlled trials from the bibliographic databases Biological Abstracts, CINAHL, The Cochrane Library, EMBASE, MEDLINE, PsycLIT, LILACS, PSYNDEX, Sociological Abstracts and Sociofile. We searched the references of all included studies for further trials. We contacted pharmaceutical companies and authors of trials. We updated this search on 16th July 2012.
Selection criteria
We selected all randomised controlled trials that compared perazine with other treatments for people with schizophrenia or schizophrenia‐like psychoses, or both.
Data collection and analysis
The review authors (SL, BH, BHe) independently inspected the citations and where possible abstracts and ordered papers for re‐inspection and quality assessment. We independently extracted data. We calculated the risk ratio (RR) and 95% confidence interval (CI) using a random‐effects model. For continuous data, we calculated mean differences (MD). We inspected all data for heterogeneity, assessed trials for risk of bias and created summary of findings tables using GRADE.
Main results
The review now includes seven trials with a total of 479 participants. In only one trial, with 95 participants, perazine appeared superior to 'active placebo' (trimipramine) at five weeks for the outcome of 'no important global improvement' (n = 95, RR 0.43 CI 0.2 to 0.8, low quality evidence), but there was no statistically significant difference in most measures of mental state. Perazine did not induce more general adverse events than placebo but more participants received at least one dose of antiparkinson medication (n = 95, RR 4.50 CI 1.0 to 19.5, very low quality evidence).
Six small trials comparing perazine with other antipsychotics, including 384 participants in total, were incompletely reported and the outcomes were presented in various ways so that meta‐analysis was not possible on most occasions. In the six studies, a similar number of participants receiving perazine or comparator antipsychotics (amisulpride, haloperidol, olanzapine, ziprasidone, zotepine) left the studies early (n = 384, RR 0.97 CI 0.68 to 1.38, low quality evidence). The results on efficacy could not be meta‐analysed because the authors presented their results in very different ways. No obvious differences in adverse events between perazine and other antipsychotics could be derived from the limited data. Two haloperidol comparisons did not present extrapyramidal side‐effects in a way that was suitable for use in meta‐analysis, but three small comparisons with the second‐generation antipsychotics zotepine and amisulpride showed no higher risk of akathisia (n = 111, RR 0.31 CI 0.1 to 1.1), dyskinesia (n = 111, RR 0.47 CI 0.1 to 3.5), parkinsonism (n = 81, RR 1.21 CI 0.5 2.8) or tremor (n = 40, RR 0.80 CI 0.3 to 2.6) with perazine.
Authors' conclusions
The number, size and reporting of randomised controlled perazine trials are insufficient to present firm conclusions about the properties of this antipsychotic. It is possible that perazine is associated with a similar risk of extrapyramidal side‐effects as some atypical antipsychotics but this is based on small comparisons. This should be clarified in larger, well‐designed trials.
Keywords: Humans, Antipsychotic Agents, Antipsychotic Agents/adverse effects, Antipsychotic Agents/therapeutic use, Perazine, Perazine/adverse effects, Perazine/therapeutic use, Randomized Controlled Trials as Topic, Schizophrenia, Schizophrenia/drug therapy
Plain language summary
Perazine for schizophrenia
Schizophrenia is often a severe and disabling illness that affects approximately one per cent of the worldwide population. Schizophrenia has 'positive' symptoms, such as strange and fixed beliefs (delusions), as well as hearing voices and seeing things (hallucinations). Schizophrenia also has 'negative' symptoms such as apathy, loss of emotion, lack of drive and disorganisation of behaviour and thought. The degree of disability is considerable with 80% ‐ 90% not working and up to 10% dying.
Antipsychotic drugs are the main treatment for schizophrenia, and are grouped into older drugs (first generation or ‘typical’) and newer drugs (second generation or ‘atypical’). However, antipsychotic drugs also have serious side effects, particularly movement disorders such as uncontrollable shaking, tremors, muscle stiffness, tiredness, weight gain and the inability to sit still.
Perazine is an older antipsychotic drug first introduced in the 1950s. It is suggested to have a low level of side effects (especially for movement disorders). Its use is regional and restricted to countries like Germany, Poland, the Netherlands and the former Yugoslavia.
A search for trials was carried out in July 2012. The review now includes seven studies with a total of 479 participants and assesses the effects of perazine for people with schizophrenia. Comparisons of perazine versus placebo (‘dummy’ treatment) and versus other antipsychotic drugs revealed no clear differences or superiority of perazine. However, only a handful of studies have been undertaken and the number of participants in each study was small. In addition the studies avialable were of limited quality with data for the main outcomes of interest rated as low or very low quality. As perazine is a cheap drug and there is some limited evidence that it may cause less side effects than other older antipsychotic drugs, further large scale, well designed and well‐reported studies are much needed.
This plain language summary has been written by a consumer Benjamin Gray from Rethink Mental Illness,
Email: ben.gray@rethink.org.
Summary of findings
Summary of findings for the main comparison. Perazine versus placebo for schizophrenia.
| Perazine versus placebo for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: hospital Intervention: perazine versus placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Perazine versus placebo | |||||
| Acceptability of treatment Leaving the study early due to any reason Follow‐up: mean 5 weeks | 422 per 1000 | 262 per 1000 (148 to 464) | RR 0.62 (0.35 to 1.1) | 95 (1 study) | ⊕⊝⊝⊝ very low1,2,3 | |
| Global state change over time (no better or deterioration) Follow‐up: mean 5 weeks | 467 per 1000 | 201 per 1000 (107 to 378) | RR 0.43 (0.23 to 0.81) | 95 (1 study) | ⊕⊕⊝⊝ low1,3 | |
| Overall mental state less than 30% BPRS reduction Follow‐up: mean 5 weeks | 733 per 1000 | 601 per 1000 (447 to 799) | RR 0.82 (0.61 to 1.09) | 95 (1 study) | ⊕⊕⊝⊝ low1,3 | |
| Movement disorders number of participants receiving antiparkinson medication Follow‐up: mean 5 weeks | 44 per 1000 | 200 per 1000 (46 to 864) | RR 4.5 (1.04 to 19.45) | 95 (1 study) | ⊕⊝⊝⊝ very low1,3,4 | |
| Overall tolerability Leaving the studies early due to adverse events | See comment | See comment | Not estimable | 0 (0) | See comment | No data available |
| Satisfaction with care ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No data available |
| Quality of life ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No data available |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: high risk of attrition bias. 2 Indirectness: acceptability was measured by the number of participants leaving the studies for any reason which is an indirect measure of acceptability. 3 Imprecision: very small number of events. 4 Indirectness: movement disorders were measured by the number of participants receiving antiparkinson medication which is an indirect measure of adverse events.
Summary of findings 2. Perazine versus other antipsychotics for schizophrenia.
| Perazine versus other antipsychotics for schizophrenia | ||||||
| Patient or population: patients with schizophrenia Settings: mainly hospital Intervention: perazine versus other antipsychotics | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | Perazine versus other antipsychotics | |||||
| Global state change over time (no better or deterioration) Follow‐up: mean 4 weeks | 200 per 1000 | 266 per 1000 (72 to 994) | RR 1.33 (0.36 to 4.97) | 30 (1 study) | ⊕⊕⊝⊝ low1,2 | |
| Overall mental state BPRS endpoint score. Scale from: 0 to 108. Follow‐up: mean 4 weeks | The mean overall mental state in the control groups was BPRS endpoint score | The mean overall mental state in the intervention groups was 0.4 lower (0.74 to 0.06 lower) | 40 (1 study) | ⊕⊕⊝⊝ low1,3 | ||
| Movement disorders number of participants receiving antiparkinson medication Follow‐up: mean 4 weeks | 200 per 1000 | 242 per 1000 (106 to 552) | RR 1.21 (0.53 to 2.76) | 81 (2 studies) | ⊕⊝⊝⊝ very low1,2,4 | |
| Overall tolerability Leaving the study early due to adverse events Follow‐up: mean 4 weeks | 126 per 1000 | 110 per 1000 (51 to 240) | RR 0.87 (0.4 to 1.9) | 193 (5 studies) | ⊕⊕⊝⊝ low1,2 | |
| Acceptability of treatment Leaving the studies early due to any reason Follow‐up: 1‐3 months | 296 per 1000 | 288 per 1000 (202 to 409) | RR 0.97 (0.68 to 1.38) | 384 (6 studies) | ⊕⊕⊝⊝ low1,2,5 | |
| Satisfaction with care ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No data available |
| Quality of life ‐ not reported | See comment | See comment | Not estimable | ‐ | See comment | No data available |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 Risk of bias: high risk of reporting bias. 2 Imprecision: Small number of events. 3 Imprecision: Very small population size. 4 Indirectness: adverse events were measured by the number of participants receiving antiparkinson medication which is an indirect measure of adverse events. 5 Indirectness: acceptability was measured by the number of participants leaving the studies for any reason which is an indirect measure of acceptability.
Background
Description of the condition
Schizophrenia is often a chronic and disabling psychiatric disorder. It afflicts approximately 1% of the population worldwide with few gender differences. Its typical manifestations are 'positive' symptoms such as fixed, false beliefs (delusions) and perceptions without cause (hallucinations); 'negative' symptoms such as apathy and lack of drive, disorganisation of behaviour and thought; and catatonic symptoms such as mannerisms and bizarre posturing (Carpenter 1994). The degree of suffering and disability is considerable with 80% to 90% of those affected not working (Marvaha 2004) and up to 10% dying (Tsuang 1978).
Description of the intervention
Perazine is a phenothiazine derivative which has been used as an antipsychotic in Germany since 1958. It is usually taken by mouth but an intramuscular formulation for the rapid treatment of very acutely disturbed psychotic patients is also available. It was developed and tested soon after chlorpromazine in the 1950s.
The customary dose range recommended for this antipsychotic under inpatient conditions is 75 to 600 mg/day (maximum 1000 mg/day). In acutely ill and severely disturbed patients, 50 mg perazine at time intervals of 30 minutes can be administered intramuscularly (Benkert 1996).
How the intervention might work
Perazine has a dopamine receptor binding action, which is similar to that of chlorpromazine and lower than that of haloperidol (Seeman 1981). This suggests that the risk of extrapyramidal adverse effects should be lower than high potency antipsychotics such as haloperidol. This low risk of extrapyramidal adverse effects might also be enhanced by a rather high binding affinity for cholinergic receptors that counteract dopamine receptor blockade (Menge 1988). Perazine is also a potent inhibitor of central histaminergic receptors, which explains its sedating effects. These can be of benefit in the case of psychotic agitation (Menge 1988). Antagonistic effects on alpha‐1 adrenergic receptors can be associated with cardiovascular adverse effects. Perazine has two metabolites, desmethyl‐perazin and perphenazine‐sulfoxide, which do not seem to have clinically significant effects (Menge 1988).
Why it is important to do this review
Early uncontrolled studies of perazine suggested good efficacy for the treatment of acutely ill people with schizophrenia (Enss 1958). Other open studies evaluated the use of perazine for antipsychotic relapse prevention and yielded promising results, especially as the rate of extrapyramidal adverse effects appeared to be low (Hippius 1962; Krüger 1963; Grohmann 1988). However, although perazine is frequently used in Germany, we understand its current use is limited to only a few other countries, Poland, the Netherlands and former Yugoslavia. We felt it important to evaluate the evidence derived from randomised controlled trials of an antipsychotic with regionally restricted use.
Objectives
To examine the effects of perazine for those with schizophrenia or related psychoses in comparison with placebo, no treatment or other antipsychotic medications.
Methods
Criteria for considering studies for this review
Types of studies
We included all relevant randomised controlled trials.
Types of participants
We included people with the diagnosis of schizophrenia, however diagnosed. We also included those with 'serious or chronic mental illness' or 'psychotic illness'. If possible, we excluded people with schizoaffective disorder, dementing illnesses, depression and problems primarily associated with substance misuse.
Types of interventions
1. Perazine monotherapy, any dose and pattern of administration. If different doses of perazine were randomised, we included these studies if there was also a comparison with a second drug, placebo or no treatment. 2. Placebo or no treatment. 3. Monotherapy with other antipsychotic drugs, any dose or pattern of administration.
Types of outcome measures
As schizophrenia is often a lifelong illness and perazine is used as an ongoing treatment, we grouped outcomes according to time periods: short term (up to three months), medium term (three months to one year) and long term (more than one year).
Primary outcomes
1. Clinical response
1.1 Clinically significant response in global state ‐ as defined by each of the studies (short term)
Secondary outcomes
1. Death ‐ suicide or natural causes
2. Leaving the study early
3. Service utilisation
3.1 Hospital admission 3.2 Days in hospital 3.3 Change in hospital status
4. Mental state
4.1 Clinically significant response in mental state ‐ as defined by each of the studies 4.2 Average score or change in mental state 4.3 Clinically significant response on positive symptoms ‐ as defined by each of the studies 4.4 Average score or change in positive symptoms 4.5 Clinically significant response on negative symptoms ‐ as defined by each of the studies 4.6 Average score or change in negative symptoms
5. Behaviour
5.1 Clinically significant response in behaviour (e.g. aggressive behaviour, behaviour on the ward etc.) ‐ as defined by each of the studies 5.2 Average score or change in behaviour
6. Extrapyramidal adverse effects
6.1 Incidence of use of antiparkinson drugs 6.2 Clinically significant extrapyramidal adverse effects ‐ as defined by each of the studies 6.3 Average score or change in extrapyramidal side‐effects
7. Other adverse effects, general and specific
7.1 Number of people dropping out due to adverse affects 7.2 Cardiac effects 7.3 Anticholinergic effects 7.4 Antihistamine effects 7.5 Prolactin related symptoms
8. Social functioning
8.1 Clinically significant response in social functioning ‐ as defined by each of the studies 8.2 Average score or change in social functioning
9. Economic outcomes
10. Quality of life or satisfaction with care for either recipients of care or carers 10.1 Significant change in quality of life or satisfaction ‐ as defined by each of the studies 10.2 Average score or change in quality of life or satisfaction 10.3 Employment status
11. Cognitive functioning.
12. Summary of findings table
We used the GRADEapproach to interpret findings (Schünemann 2008) and used the GRADE profiler to import data from Review Manager (RevMan) to create 'Summary of findings' tables. These tables provide outcome‐specific information concerning the overall quality of evidence from each included study in the comparison, the magnitude of effect of the interventions examined, and the sum of available data on all outcomes that we rated as important to patient care and decision making. We selected the following main outcomes for inclusion in the summary of findings table.
1. Global state.
2. Mental state (overall) ‐ clinically significant response in mental state, as defined by each of the studies.
3. Acceptability of treatment (leaving the study early due to any reason).
4. Overall tolerability (leaving the study early due to adverse events).
5. Movement disorders (number of participants receiving antiparkinson medication at least once).
6. Satisfaction with care.
7. Quality of life.
Search methods for identification of studies
Search methods for this 2012 update are below, for previous searches please see the Appendices.
No language restriction was applied within the limitations of the search tools.
Electronic searches
1. Cochrane Schizophrenia Group Trials Register
The Trials Search Co‐ordinator searched the Cochrane Schizophrenia Group Trials Register (16th July 2012).
1.1 Intervention search
The ‘Intervention’ field was searched using the phrase:
(*perazin* or *pernazin* or *taxilan* or *phenothiazine tran* or * piperazin*).
The Cochrane Schizophrenia Group Trials Register is compiled by systematic searches of major databases, handsearches of journals and conference proceedings (see Group Module). Incoming trials are screened by the Trial Search Co‐ordinator and assigned to the awaiting classification section of relevant existing or new review titles.
Searching other resources
1. Reference searching
We inspected the references of all identified studies for more citations.
2. Personal contact
We contacted the first author of each included study for more information regarding unpublished trials.
3. Drug company
We contacted the manufacturers of proprietary perazine (Lundbeck, Kopenhagen) for additional data (first version only, not done for updates).
Data collection and analysis
Methods used in data collection and analysis for this 2012 update are below, for previous methods please see Appendices.
Selection of studies
For this 2012 update the review authors SL and BHe independently inspected citations from the new electronic search and identified relevant abstracts. SL and BHe also inspected full articles of the abstracts which might have met the inclusion criteria.
Data extraction and management
1. Extraction
Review authors SL and BH extracted the data from the included studies (first version, there were no new included studies in the 2012 update). We extracted data presented only in graphs and figures whenever possible. When further information was necessary, we contacted the authors of studies in order to obtain missing data or for clarification.
2. Management
2.1 Forms
We extracted data onto standard, simple forms.
2.2 Scale‐derived data
We included continuous data from rating scales only if: a. the psychometric properties of the measuring instrument have been described in a peer‐reviewed journal (Marshall 2000); and b. the measuring instrument has not been written or modified by one of the trialists for that particular trial.
Ideally the measuring instrument should either be: i. a self‐report or ii. completed by an independent rater or relative (not the therapist). We realise that this is not often reported clearly; we have noted whether or not this is the case in the 'Description of studies'.
2.3 Endpoint versus change data
There are advantages of both endpoint and change data. Change data can remove a component of between‐person variability from the analysis. On the other hand, calculation of change needs two assessments (baseline and endpoint), which can be difficult in unstable and difficult to measure conditions such as schizophrenia. We decided to primarily use endpoint data, and to only use change data if the former were not available. We combined endpoint and change data in the analysis as we used mean differences (MD) rather than standardised mean differences throughout the review (section 9.4.5.2 of Higgins 2011).
2.4 Skewed data
Continuous data on clinical and social outcomes are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data, we aimed to apply the following standards to all data before inclusion:
a) standard deviations and means are reported in the paper or obtainable from the authors;
b) when a scale starts from the finite number zero the standard deviation, when multiplied by two, is less than the mean (as otherwise the mean is unlikely to be an appropriate measure of the centre of the distribution (Altman 1996));
c) if a scale started from a positive value (such as the Positive and Negative Syndrome Scale (PANSS), which can have values from 30 to 210), we modified the calculation described above to take the scale starting point into account. In these cases skew is present if 2SD > (S ‐ S min), where S is the mean score and S min is the minimum score.
Endpoint scores on scales often have a finite start and endpoint, and these rules can be applied. We entered skewed endpoint data from studies of fewer than 200 participants in 'Other tables' within the 'Data and analyses' section rather than into a statistical analysis. Skewed data pose less of a problem when looking at the mean if the sample size is large; we entered such endpoint data into the syntheses.
When continuous data are presented on a scale that includes a possibility of negative values (such as change data), it is difficult to tell whether data are skewed or not, we entered skewed change data into analyses regardless of the size of the study.
2.5 Common measure
To facilitate comparisons between trials, we intended to convert variables that can be reported in different metrics, such as days in hospital (mean days per year, per week or per month) to a common metric (for example mean days per month).
2.6 Conversion of continuous to binary
Where possible, we made efforts to convert outcome measures to dichotomous data. This can be done by identifying cut‐off points on rating scales and dividing participants accordingly into 'clinically improved' or 'not clinically improved'. It is generally assumed that if there is a 50% reduction in a scale‐derived score such as the Brief Psychiatric Rating Scale (BPRS) (Overall 1962) or the PANSS (Kay 1986) this could be considered as a clinically significant response (Leucht 2005; Leucht 2005a). If data based on these thresholds were not available, we used the primary cut‐off presented by the original authors.
2.7 Direction of graphs
Where possible, we entered data in such a way that the area to the left of the line of no effect indicated a favourable outcome for perazine.
Assessment of risk of bias in included studies
For this 2012 update, BHe worked independently by using the criteria described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011) to assess trial quality. SL supervised the process. This new set of criteria is based on evidence of associations between overestimate of effect and high risk of bias of the article, such as sequence generation, allocation concealment, blinding, incomplete outcome data and selective reporting.
Where adequate details of randomisation and other characteristics of the trials were not provided, we contacted the authors of the studies in order to obtain additional information.
We have noted the level of risk of bias in both the text of the review and in the Table 1.
Measures of treatment effect
1. Binary data
For binary outcomes we calculated a standard estimation of the risk ratio (RR) and its 95% confidence interval (CI). It has been shown that RR is more intuitive (Boissel 1999) than odds ratios and that odds ratios tend to be interpreted as RR by clinicians (Deeks 2000). The number needed to treat or harm (NNT or H) statistic with its confidence interval is intuitively attractive to clinicians but is problematic both in its accurate calculation in meta‐analyses and its interpretation (Hutton 2009). For binary data presented in the 'Summary of findings' tables, where possible, we calculated illustrative comparative risks.
2. Continuous data
For continuous outcomes we estimated the mean difference (MD) between groups. We preferred not to calculate effect size measures (standardised mean difference (SMD)). If scales of very considerable similarity were used, we presumed there was a small difference in measurement and we calculated effect size and transformed the effect back to the units of one or more of the specific instruments.
Unit of analysis issues
1. Cluster randomised trials
Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice), but analysis and pooling of clustered data pose problems. Authors often fail to account for intra‐class correlation in clustered studies, leading to a 'unit of analysis' error (Divine 1992) whereby P values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Bland 1997; Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra‐class correlation coefficients for their clustered data and to adjust for this using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we presented these data as if from a non‐cluster randomised study but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation coefficient (ICC) [Design effect = 1 + (m ‐ 1) * ICC] (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
If cluster studies have been appropriately analysed, taking into account intra‐class correlation coefficients and relevant data documented in the report, synthesis with other studies would have been possible using the generic inverse variance technique.
2. Cross‐over trials
A major concern of cross‐over trials is the carry‐over effect. It occurs if an effect (for example pharmacological, physiological or psychological) of the treatment in the first phase is carried over to the second phase. As a consequence, on entry to the second phase the participants can differ systematically from their initial state despite a wash‐out phase. For the same reason, cross‐over trials are not appropriate if the condition of interest is unstable (Elbourne 2002). As both effects are very likely in severe mental illness, we only used the data of the first phase of cross‐over studies.
3. Studies with multiple treatment groups
Where a study involved more than two treatment arms, if relevant, we presented the additional treatment arms in comparisons. If data were binary we simply added these and combined them within the two‐by‐two table. If data were continuous we combined the data following the formula in section 7.7.3.8 (Combining groups) of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). Where the additional treatment arms were not relevant, we did not reproduce these data.
Dealing with missing data
1. Overall loss of credibility
We agree that at some degree of loss of follow‐up, data must lose credibility (Xia 2009). It is, however, unclear from which degree of loss to follow‐up this becomes a problem. We therefore did not exclude studies on the basis of loss to follow‐up.
2. Binary
We presented data on a 'once‐randomised‐always‐analyse' basis (an intention‐to‐treat analysis). Those leaving the study early were assumed to have remained unchanged. This assumption is conservative for measures of response because those leaving the study early are assumed to have not responded. It is not conservative for side‐effects. We however felt that it would often be an overestimation of the risk if all participants who left the studies early were assumed to have developed a side‐effect.
3. Continuous
3.1 Attrition
We aimed to extract intention‐to‐treat data, but if these were not available we presented and used the data from the people who completed the study.
3.2 Standard deviations
If standard deviations were not reported, we first tried to obtain the missing values from the authors. If not available, where there are missing measures of variance for continuous data, but exact standard errors and confidence intervals available for group means, and either P values or T values available for differences in mean, we can calculate them according to the rules described in the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011). When only the standard error (SE) is reported, standard deviations (SDs) are calculated by the formula SD = SE * square root (n). Chapters 7.7.3 and 16.1.3 of the Cochrane Handbook for Systemic reviews of Interventions (Higgins 2011) present detailed formulae for estimating SDs from P values, T or F values, confidence intervals, ranges or other statistics. If these formulae did not apply, we would calculate the SDs according to a validated imputation method which is based on the SDs of the other included studies (Furukawa 2006). Although some of these imputation strategies can introduce error, the alternative would be to exclude a given study’s outcome and thus to lose information. We nevertheless examined the validity of the imputations in a sensitivity analysis excluding imputed values.
3.3 Last observation carried forward
We anticipated that in some studies the method of last observation carried forward (LOCF) would be employed within the study report. As with all methods of imputation to deal with missing data, LOCF introduces uncertainty about the reliability of the results (Leucht 2007). We reproduced these data but readers should be aware of the problem.
Assessment of heterogeneity
1. Clinical heterogeneity
We initially considered all included studies, without seeing the comparison data, to judge clinical heterogeneity. We simply inspected all studies for clearly outlying people or situations which we had not predicted would arise. When such situations or participant groups arose, we fully discussed them.
2. Methodological heterogeneity
We initially considered all included studies, without seeing comparison data, to judge methodological heterogeneity. We simply inspected all studies for clearly outlying methods which we had not predicted would arise. When such methodological outliers arose, we fully discussed them.
3. Statistical heterogeneity
3.1 Visual inspection
We visually inspected graphs to investigate the possibility of statistical heterogeneity.
3.2 Employing the I2 statistic
We investigated heterogeneity between studies by considering the I2 statistic alongside the Chi2 test P value. The I2 provides an estimate of the percentage of inconsistency thought to be due to chance (Higgins 2003). The importance of the observed value of I2 depends on: i. the magnitude and direction of the effects and ii. strength of evidence for heterogeneity (for example P value from Chi2 test, or a confidence interval for I2). An I2 estimate greater than or equal to around 50% accompanied by a statistically significant Chi2 statistic was interpreted as evidence of substantial levels of heterogeneity (Higgins 2011). When substantial levels of heterogeneity were found in the primary outcome, we explored reasons for heterogeneity (Subgroup analysis and investigation of heterogeneity).
Assessment of reporting biases
Reporting biases arise when the dissemination of research findings is influenced by the nature and direction of results (Egger 1997). These are described in section 10 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We are aware that funnel plots may be useful in investigating reporting biases but are of limited power to detect small‐study effects. We did not use funnel plots for outcomes where there were 10 or fewer studies, or where all studies were of similar sizes. In other cases where funnel plots were possible, we sought statistical advice in their interpretation.
Data synthesis
We understand that there is no closed argument for preference for use of fixed‐effect or random‐effects models. The random‐effects model method incorporates an assumption that the different studies are estimating different, yet related, intervention effects. To us this often seems to be true, and the random‐effects model takes into account differences between studies even if there is no statistically significant heterogeneity. There is, however, a disadvantage to the random‐effects model as it puts added weight onto small studies, which often are the most biased ones. Depending on the direction of effect these studies can either inflate or deflate the effect size. We chose the random‐effects model for all analyses. The reader is, however, able to choose to inspect the data using the fixed‐effect model.
Subgroup analysis and investigation of heterogeneity
1. Subgroup analyses
We proposed to undertake this review and provide an overview of the effects of perazine for people with schizophrenia in general. Information about subgroups (including first episode of the illness, elderly patients, and patients with subtypes of schizophrenia) was missing in the available set of data.
2. Investigation of heterogeneity
If inconsistency was high, this was reported. First we investigated whether the data had been entered correctly. Second, if the data were correct, we looked to see if there were methodological reasons in the studies that accounted for the heterogeneity. Finally, the graph was visually inspected and studies outside of the company of the rest were successively removed to see if heterogeneity was restored. When unanticipated clinical or methodological heterogeneity was obvious we simply stated hypotheses regarding the heterogeneity for future reviews or versions of this review. We did not anticipate undertaking analyses relating to these.
Sensitivity analysis
We applied all sensitivity analyses to the primary outcomes of this review.
1. Implication of randomisation
We aimed to include trials in a sensitivity analysis if they were described in some way so as to imply randomisation. For the primary outcomes we included these studies and if there was no substantive difference when the implied randomised studies were added to those with a better description of randomisation, then we entered all data from these studies.
2. Imputed values
We also undertook a sensitivity analysis to assess the effects of including data from trials where we used imputed values for ICC in calculating the design effect in cluster randomised trials.
If we noted substantial differences in the direction or precision of effect estimates in any of the sensitivity analyses listed above, we did not pool the data from the excluded trials with the other trials contributing to the outcome but presented them separately.
5. Fixed‐effect and random‐effects models
All data were synthesised using a random‐effects model, however we also synthesised the data for the primary outcome using a fixed‐effect model to evaluate whether the greater weights assigned to larger trials with greater event rates altered the significance of the results compared to the more evenly distributed weights in the random‐effects model.
Results
Description of studies
For a substantive description of studies please see the Characteristics of included studies and Characteristics of excluded studies tables.
Results of the search
Please compare with Figure 1.
1.

1. Original search in 2000
The original search of the Cochrane Schizophrenia Group (CSG) controlled Trials Register in the year 2000 yielded 40 studies ,which we ordered and inspected. After handsearching included trials and contacting the manufacturers of perazine, we did not identify any further trials. Sixteen publications of six studies met our inclusion criteria. Our contact with the first authors of the studies yielded no further studies or individual patient data.
1. Update search in 2005
The update search in the CSG controlled Trials Register in 2005 yielded 59 references. The vast majority of the references were clearly not related to perazine and hence we did not record them in the excluded studies table. We identified three references in two studies (Kemperdick 1967; Loza 2001) that were classified as excluded studies. We identified three further references in studies that had already been included in the first version. Most noteworthy was the final journal publication of Bender 1997. In contrast to the previously available abstracts and short versions of the trial, further outcome data were presented and some of the previously listed results had to be slightly changed because they were more clearly reported within the new reference.
1. Update search in 2012
The update search in 2012, made using the CSG controlled Trials Register, yielded four new references. Three studies did not meet the inclusion criteria. One was an additional publication of a study that had already been included (Dieterle 1991), one was not randomised and it was unclear whether the substance tested was perazine (Gerson 1964), and one used an inappropriate comparison (Ohlmeier 2007). The fourth study was a randomised, open trial comparing perazine with olanzapine and ziprasidone that met the inclusion criteria (Tybura 2012).
Included studies
We included seven studies. Six trial centres were in Germany, one was in Poland.
1. Methods
All included studies were described as randomised. Follow‐up periods ranged from 28 to 84 days. All seven were short term (up to three months).
2. Participants
All but one study (Tybura 2012) of the included studies had less than 100 participants; the range was 23 to 191. A total of 479 people participated in the seven trials, most of whom had a diagnosis of schizophrenia. Bender 1997 also included participants with schizophreniform disorder, and Rüther 1988 also included participants with schizophrenia‐like disorders (for example schizoaffective disorder). We included these studies because the majority of randomised participants had schizophrenia.
Five studies used International Classification of Diseases‐9 (ICD‐9) criteria, Bender 1997 used Diagnostic and Statistical Manual of Mental Disorders‐III‐R (DSM‐III‐R), and Tybura 2012 used ICD‐10. Klimke 1993, Rüther 1988 and Schmidt 1982 included only "acutely ill" participants, Wetzel 1991 restricted inclusion to participants with positive symptoms and Tybura 2012 to paranoid schizophrenia. Bender 1997 required a minimum score on the Brief Psychiatric Rating Scale (BPRS).
From those studies that included information on the gender of participants there were 169 males and 174 females. Ages ranged from 18 to 70 years; where the mean age was indicated it ranged between 31 and 37 years.
3. Setting
Five studies were from inpatient settings. In two studies (Dieterle 1991; Tybura 2012) the setting was not clear.
4. Interventions
The dose of perazine ranged between 75 and 1000 mg/day, with means between 300 and 650 mg/day where reported.
Only one study (Bender 1997) was considered to be placebo‐controlled. The comparator group received a tricyclic antidepressant, trimipramine. It turned out to be statistically significantly less effective than perazine at least according to the global impression of the raters. It was regarded as an active placebo.
Two studies compared perazine with oral haloperidol. Four studies compared perazine with the second‐generation antipsychotics zotepine (Dieterle 1991; Wetzel 1991), amisulpride (Rüther 1988), and ziprasidone and olanzapine in Tybura 2012, which was a three‐arm study. In these trials perazine was used as the gold standard for the evaluation of the second‐generation antipsychotics.
5. Outcomes
Many outcomes were presented as scale‐derived, continuous data. Often we could not use the data for meta‐analytic calculations because they were either skewed or the standard deviations were not indicated. Despite letters sent to the study authors (not done for the 2012 update), no original patient data that could have been dichotomised were obtained.
6. Scales
Details of the scales that provided usable data are shown below.
6.1. Global state
6.1.1 Clinical Global Impression (CGI)
CGI (Guy 1976) is a rating instrument commonly used in studies on schizophrenia that enables clinicians to quantify severity of illness and overall clinical improvement during therapy. A seven‐point scoring system is usually employed with low scores indicating decreased severity or greater recovery. Usable data were reported by Rüther 1988.
6.1.2 Global Assessment Scale (GAS)
In the GAS (Endicott 1976) a clinician rates the overall functioning of a patient on a scale of 1 to 100. Lower scores indicate poorer functioning. Usable data were reported by Wetzel 1991.
6.2. Mental state
6.2.1 Brief Psychiatric Rating Scale (BPRS)
BPRS (Overall 1962) is a brief rating scale used to assess the severity of a range of psychiatric symptoms, including psychotic symptoms. The original scale has 16 items, but a revised 18‐item scale is commonly used. Each item is defined on a seven‐point scale varying from 'not present' to 'extremely severe', scoring from 0 to 6 or 1 to 7. Scores can range from 0 to 126, with high scores indicating more severe symptoms. Usable data were reported by Bender 1997, Dieterle 1991, Schmidt 1982 and Wetzel 1991.
6.2.2 Positive and Negative Syndrome Scale (PANSS)
PANSS (Kay 1986) is a schizophrenia scale that has 30 items, each of which can be defined on a seven‐point scoring system varying from 1 ‐ absent to 7 ‐ extreme. It can be divided into three subscales for measuring the severity of general psychopathology, positive symptoms (PANSS‐P), and negative symptoms (PANSS‐N). A low score indicates lesser severity. Useable data (percentage PANSS total score from baseline) were obtained for Tybura 2012.
6.3. Missing outcomes
Relapse, issues of hospital admission, behaviour, cognitive functions, cost issues, quality of life and satisfaction with care were not addressed in the included studies.
Excluded studies
The majority of excluded studies were not stated to be randomised (n = 19). One study was excluded because it did not examine participants with schizophrenia, one used wrong interventions, and three did not provide any useable outcomes.
Risk of bias in included studies
For graphical representations of our judgements of risk of bias please refer to Figure 2 and Figure 3. Full details of judgements can be seen in the 'Risk of bias' tables.
2.
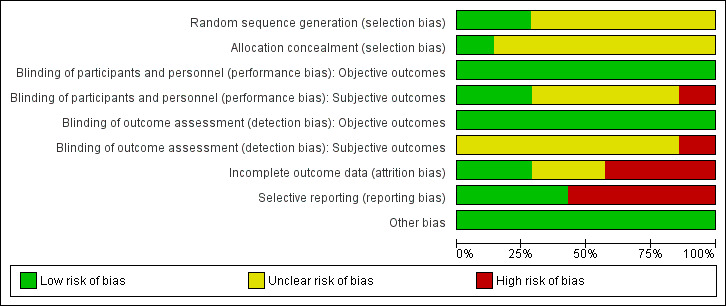
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
Allocation
In only two studies (Bender 1997: randomisation schedule generated by a validated computer program, Tybura 2012: randomisation programme written by statistician) was the method used to generate random allocation described. These two studies were classified as having a low risk of selection bias. None of the other studies provided a specific method used in the randomisation procedure and thus they were classified as having an unclear risk of selection bias.
Although no differences in baseline descriptions of the comparator groups were found on most occasions, the readers were given little assurance that bias was minimised during the allocation procedure in these other trials.
As for the allocation concealment, all studies were classified as unclear in this respect except for Tybura 2012 (see risk of bias table).
Blinding
Precautions taken to ensure blinding were described in three studies. Two used identical capsules (Dieterle 1991; Bender 1997), whereas in the third study blinding was attempted by treating one group with perazine tablets and placebo liquid and the other group with haloperidol liquid and placebo pills (Schmidt 1982). Tybura 2012 was an open trial; and the remaining trials reported that they were blind but did not describe the methods of blinding.
Incomplete outcome data
In all seven studies data were presented on losses to follow‐up.
Although all studies indicated the numbers of participants who left the study before its completion, the reasons for leaving the studies early were not consistently indicated.
Two studies were assessed as having a low risk of attrition bias, meaning that the problem of incomplete outcome data was addressed in an appropriate way (the number of dropouts was not very high and was evenly distributed between groups). Another two studies were judged as unclear, and another and the last three as having high risk of attrition bias.
Selective reporting
Four out of seven studies were judged as having a high risk of reporting bias due to the lack of information about some predefined outcomes. Standard deviations and means were often missing. The remaining three studies did not selectively report on any outcomes and thus were judged as having a low risk of bias in that respect.
Other potential sources of bias
The included studies were free of any other clear sources of bias.
Effects of interventions
1. Comparison 1: Perazine versus placebo
Ninety‐five participants from one study (Bender 1997) were randomised to this comparison.
1.1 Leaving the study early
No significant difference between perazine and trimipramine was found for the outcome 'leaving the study early for any reason' (n = 95, RR 0.62 CI 0.4to 1.1) and 'due to inefficacy of treatment' (n = 95, RR 0.30 CI 0.09 to 1.04, p = 0.06). The number of participants 'leaving the study early for adverse events' was not indicated.
1.2 Global state
For change in global state over time, one small five‐week study (Bender 1997) demonstrated a statistically significant difference between perazine and the tricyclic antidepressant trimipramine, which was considered to be an active placebo (n = 95, RR 0.43 CI 0.2 to 0.8). The definition of the response criterion was unimproved or worse according to the clinical global impression of the raters.
1.3 Mental state
Using a cut‐off of less than 20% BPRS reduction (n = 95, RR 0.70 CI 0.5 to 1.1) or less than 30% BPRS reduction (n = 95, RR 0.82 CI 0.6 to 1.1) no statistically signficant difference in the number of responders was found. The mean BPRS at the endpoint was not significantly different between both groups (n = 79, MD ‐6.20 CI ‐13.2 to 0.8). Results on the mean change of BPRS from baseline to the endpoint differed depending on whether the results were analysed in an intention‐to‐treat or a per protocol analysis. In the former a significant superiority of perazine compared to active placebo was found (n = 95, MD ‐7.10 CI ‐13.5 to ‐0.7), while the latter showed no significant difference (n = 79, MD ‐6.10 CI ‐13.1 to 0.9). Therefore, this result must be interpreted with caution. Further data from two studies on the BPRS at the endpoint (Rüther 1988) and the Association for Methodology and Documentation in Psychiatry (AMDP) system (Schmidt 1982) could only be presented in 'Other data' tables because they were skewed.
1.4 Adverse events
We found no significant differences in terms of 'at least one adverse event' (n = 95, RR 1.15 CI 0.9 to 1.5) and 'at least one extrapyramidal side‐effect' (n = 95, RR 13.53 CI 0.8 to 230.4). However, significantly more people in the perazine group received at least one dose of antiparkinson medication (n = 95, RR 4.50 CI 1.04 to 19.5).
1.5 Other outcomes
No data were presented on the other outcomes listed in our methods section.
2. Comparison 2: Perazine versus other antipsychotics
A total of 384 patients from six studies were randomised to this comparison. Two studies used haloperidol as a comparator (Schmidt 1982; Klimke 1993). The other three studies used atypical antipsychotics, one amisulpride (Rüther 1988), one olanzapine and ziprasidone (Tybura 2012), and two zotepine (Dieterle 1991; Wetzel 1991), with perazine being used as the gold standard for the evaluation of the atypical antipsychotics.
2.1 Leaving the study early
All six studies could be used for the outcome 'leaving the study early' and no difference was found between perazine and the other antipsychotics (n = 384, RR 0.97 CI 0.68 to 1.38).
2.2 Global state
According to limited data from one study (Wetzel 1991), participants allocated to perazine were as likely to be 'no better or worse' than those given amisulpride (n = 30, RR 1.33 CI 0.4 to 5.0) but had a higher mean severity of illness according to the Clinical Global Impression Scale (n = 30, MD 1.85 CI 1.01 to 2.7) and a higher severity of illness according to the Global Assessment Scale in the zotepine comparison (n = 41, MD ‐10.0 CI ‐17.1 to ‐2.9).
2.3 Mental state
As with the global outcomes, mental state data were reported in a variety of ways hampering comparison. Schmidt 1982 reported no significant difference between perazine and haloperidol on 'no better or worse' (BPRS, n = 32, RR 1.18 CI 0.5 to 2.6). Reporting endpoint BPRS data, Wetzel 1991 showed zotepine as significantly superior to perazine (n = 34, MD 7.92 CI 1.1 to 14.7); however, using a different method to calculate this outcome, Dieterle 1991 reported the superiority of perazine compared to zotepine (n = 40, MD ‐0.40 CI ‐0.7 to ‐0.1). If we understand Dieterle 1991 correctly, they calculated the mean score of all BPRS items first for each patient and then calculated the mean of all patients. Tybura 2012 presented the percentage PANSS total score change from baseline and found no difference between perazine and olanzapine or ziprasidone.
Dieterle 1991 also presented data on the positive symptoms subscore of the BPRS and found significant superiority of perazine compared to zotepine (n = 40, MD ‐0.40, CI ‐0.8 to ‐0.02).
The BPRS data from Rüther 1988 and the AMDP data from Schmidt 1982 appeared to be skewed so we were unable to analyse them (numbers are presented in the 'Other data' table).
2.4 Adverse events
The reporting of adverse events was incomplete in the included studies. Concerning extrapyramidal side‐effects, no statistically significant differences in terms of akathisia (n = 111, RR 0.31 CI 0.1 to 1.1), dyskinesia (n = 111, RR 0.47 CI 0.1 to 3.5), parkinsonism (n = 81, RR 1.21 CI 0.5 to 2.8) or tremor (n = 80, RR 0.80 CI 0.3 to 2.6) were found between perazine and the atypical antipsychotics zotepine and amisulpride. With the exception of one case of neuroleptic malignant syndrome in the study by Schmidt 1982 (equivocal data, n = 32, RR 0.30 CI 0.01 to 6.8) no data on extrapyramidal side‐effects could be used from the three haloperidol comparisons due to poor reporting.
A similar number of participants in five studies left the studies earlier due to adverse events (n = 193, RR 0.87 CI 0.4 to 1.9).
The incidence of anticholinergic side‐effects (two studies), arousal (one study), orthostatic hypotension (two studies), laboratory abnormalities (three studies), hypersalivation (one study), vegetative disorders (one study), electroencephalogram (EEG) changes (one study) and increase in transaminase (one study) were found to be similar for both groups. However, the equivalence of these two compounds cannot be assumed in this respect due to small numbers and insufficient power.
2.5 Other outcomes
No data were presented on all other outcomes listed in our method section.
3. Publication bias, subgroup and sensitivity analyses
Due to the paucity of studies we were unable to perform subgroup and sensitivity analyses or tests for publication bias as set out in the methods section. If more trials are published, these analyses will be completed in future updates of this review. The use of the fixed‐effect model did not lead to substantially different results compared to the random‐effects model in terms of the primary outcome.
Discussion
Six out of the seven randomised trials were undertaken in Germany, and one in Poland, which limits the generalisability of the results to other countries. Furthermore, most of the studies were undertaken in hospital settings. Although the studies involved people with a clear diagnosis of schizophrenia, it is difficult to make a definite statement on generalisability because the reports do not describe how many patients who would have met the inclusion criteria were not enrolled in the trials. The trials were all classified as short term, so the long‐term effects of perazine for an often chronic disorder were not assessed. Specific data for those presenting with a first episode of the illness, elderly patients, and patients with subtypes of schizophrenia are missing. The limitations in the available data are also reflected in the risk of bias table and the summary of findings table.
Summary of main results
1. Comparison 1: Perazine versus placebo
1.1 General
Surprisingly, the only trial which could be considered as a comparison of perazine with active placebo was the second most recently published one (Bender 1997); therefore perazine has been used for several decades without evidence derived from randomised controlled trials that it is more effective than placebo. This illustrates the dramatic shift of research paradigms and the recent increase in requirements for new drugs to be prescribed to patients. It is of course debatable whether the comparator in the trial, trimipramine, can be considered to be an 'active placebo'. Trimipramine is a tricyclic antidepressant and antidepressants have not been shown to have important antipsychotic effects. But the hypothesis of the trial was that trimipramine may have such properties because it affects the dopamine system. Given the paucity of data on perazine in general, and since overall perazine was more effective than trimipramine in the study, we decided that for the purpose of this review this classification was acceptable.
1.2 Leaving the study early
A similar number of the participants treated with perazine and with trimipramine left the study early. This is surprising as it would be expected that a proper antipsychotic treatment would be more acceptable for those with schizophrenia than an antidepressant.
1.3 Global state and mental state
There are very little data, derived from one small trial with 95 participants, to suggest that more people with schizophrenia who are treated with perazine globally improve in the short term than those treated with an antidepressant. However, looking at the mental state result this finding was only confirmed in the intention‐to‐treat analysis of the BPRS change from baseline to endpoint and hence cannot be considered as robust.
1.4 Adverse events
Compared to the original version, the update did provide some data on adverse events. While no differences in terms of the number of participants with 'any adverse event' and 'at least one extrapyramidal side‐effect' were found, more participants in the perazine group received antiparkinson medication indicating movement disorders as an adverse event. This result could call into question our hypothesis in the first version of the review that perazine may be an 'atypical antipsychotic'. However, only 20% of people in the perazine group received antiparkinson medication. This is a similar rate to that of atypical antipsychotics such as olanzapine (16%) (Duggan 2005). The fact that perazine induced significantly more movement disorders than 'active placebo' in this trial may be due to the anticholinergic effects of trimipramine. The studies on atypical antipsychotics used real placebo so that withdrawal extrapyramidal symptoms may have occurred in the placebo groups and obscured any differences between the groups. We would therefore like to maintain the hypothesis that perazine could be an atypical antipsychotic, but given the small amount of available data this is at present mere speculation.
2. Comparison 2: Perazine versus other antipsychotics
2.1 General
We were pleasantly surprised that in 2012 another perazine randomised controlled trial has been published, it is the largest included study (Tybura 2012). We welcome the research interest in old compounds such as perazine, which can make valuable contributions to treatment. The overall results of this comparison cannot be regarded as conclusive due to the small numbers randomised to the studies and outcomes that were either poorly presented or presented in different ways that made meta‐analysis impossible. On most occasions only one or two studies reported on an outcome in the same way.
2.2 Leaving the study early
In these short‐term studies 28% of those taking perazine and 30% in the control groups left before the trial was completed. This is a relatively low attrition rate compared to some trials dealing with newer antipsychotic drugs.
2.3 Global state
No usable data could be extracted from the trials comparing perazine with conventional antipsychotics such as haloperidol, thus it is not clear whether perazine is more or less effective in improving global state. According to two comparisons with the atypical antipsychotics zotepine and amisulpride, perazine faired worse regarding the mean global state of the participants at the end of the studies. Due to the very low number of participants that were randomised in these trials this result cannot be regarded as conclusive without further trials.
2.4 Mental state
On analysing a small database containing the results of two trials, perazine had a similar efficacy compared to haloperidol for the improvement of mental state. The data from two studies comparing perazine with the newer antipsychotic zotepine are equivocal; one trial found zotepine to be superior, whereas Dieterle 1991 favoured perazine in this regard. This might be the result of chance findings due to small sample sizes. The more reasonably sized study by Tybura 2012 (191 participants) found no difference between perazine and olanzapine or ziprasidone. Again, further larger trials are needed before definite conclusions can be drawn.
2.5 Adverse events
Perazine was not associated with significantly more or fewer adverse events than the drugs in the control groups, but this result might not be a true reflection of effect because the single outcomes were mostly only reported by one or two trials. However, looking at the more global parameter 'leaving the studies earlier because of adverse events', for which five studies could be included, similar rates were found indicating that perazine might generally be as well tolerated as other currently used antipsychotics. Although in each of the haloperidol comparisons perazine was associated with fewer extrapyramidal side‐effects, this could not be confirmed by a meta‐analysis due to poor reporting of data. However, according to three studies comparing perazine with the newer antipsychotics amisulpride and zotepine, perazine had a similar extrapyramidal side‐effect risk. It might therefore be possible that perazine has an atypical profile but, because of the low numbers of participants, more trials are necessary before we can reach a definite conclusion. Unfortunately the comparison of perazine with ziprasidone and olanzapine did not report side‐effect data, thus nothing can be said about the relative effects (Tybura 2012).
Overall completeness and applicability of evidence
The number of included studies is too small to conclusively judge the potential applicability of the obtained results. Trials with small sample sizes lack sufficient power to detect a small to moderate effect, and thus results from such trials are often inconclusive even when a real effect does exist. A review suggested that meta‐analyses based on the summation of small trials should be interpreted as inconclusive, regardless of whether the combined estimate was significant (Davey Smith 1998).
Quality of the evidence
There were major limitations in study quality according to the risk of bias tool. In addition to this tool, only a secondary publication of one study reported that the assessors rating the outcome were independent of the treatment (Schmidt 1982). As the scale‐rated outcomes were largely rated by a person who was unlikely to be disinterested in the final result, all results in this review need to be considered prone to bias. Continuous scale data were often poorly reported. Frequently they lacked explicit statements regarding the denominator or standard deviation, or were only presented in significance tests or within graphs. Three studies used the last observation carried forward (LOCF) approach for those who left the study (Klimke 1993; Bender 1997; Tybura 2012), which is prone to bias (Leucht 2007); other studies only presented completer analyses. This did not affect the results of the review because Bender 1997 is the only study in the first comparison and for Klimke 1993 usable data on efficacy outcomes were not available anyway. These limitations led to low or very low quality judgements in several important outcomes in the summary of findings table.
Potential biases in the review process
The evidence presented here is, to the best of our knowledge, complete. However, one can never be certain whether some additional (unpublished) material exists that was not pooled in the analysis.
Agreements and disagreements with other studies or reviews
We are not aware of any other systematic reviews of the effects of perazine in schizophrenia.
Authors' conclusions
Implications for practice.
1. For clinicians
Clinicians should be aware that perazine is an antipsychotic with a very limited dataset concerning the evidence from randomised controlled trials. The available data do not allow a clear judgement on whether it is more or less effective than other currently used antipsychotics. Although perazine is said to be associated with fewer extrapyramidal side‐effects than high‐potency conventional antipsychotics, the necessary information could not be extracted from the few randomised controlled trials that are available. However, in three small trials perazine was not associated with more extrapyramidal side‐effects than zotepine and amisulpride. Therefore perazine might have a similar risk for movement disorders as these second‐generation antipsychotics, but larger trials are needed to confirm this hypothesis.
2. For people with schizophrenia
People with schizophrenia should be aware that although German and Polish psychiatrists have more than 30 years experience of treating patients with perazine, there exists only very limited evidence from randomised controlled trials on its effectiveness and its tolerability.
3. For managers, founders, decision makers
They should be aware that perazine is a cheap antipsychotic which possibly has atypical properties (low extrapyramidal symptom risk) and that it is worth examining perazine in randomised trials. Indeed, the most recent randomised controlled trial found no major difference in efficacy and numbers leaving the study early between perazine and olanzapine or ziprasidone (Tybura 2012).
Implications for research.
The trials that were reviewed predated the CONSORT statement (Begg 1996). Had this been anticipated more data may have been available to inform practice. However, even with better reporting of the trials, the possibility of drawing firm conclusions would have been limited due to the low number of participants that were included (479). Since there seems to be an indication that perazine might have similar properties to some of the newer second‐generation antipsychotics, in terms of movement disorders, further well‐designed studies are called for. See also Table 3.
1. Suggested design of future study.
| Methods | Allocation: randomised ‐ clearly described generation of sequence and concealment of allocation. Blindness: double ‐ described and tested. Duration: 3 months, 12 months open follow‐up. |
| Participants | Diagnosis: schizophrenia with acute exacerbation. N = 900.* Age: any. Gender: both. History: any. |
| Interventions | 1. Perazine: dose 300‐800 mg/day. N = 300. 2. Haloperidol: dose 2‐15 mg/day. N = 300. 3. Olanzapine: dose 5‐20mg/day. N = 300. |
| Outcomes | Leaving study early (any reason, adverse events, inefficacy).
Service outcomes: hospitalised, time in hospital, attending out patient clinics.
Global impression: Number of participants much improved (CGI)**.
Mental state: PANSS. Quality of life, subjective well‐being. Adverse events. |
* Power calculation suggested 300/group would allow good chance of showing a 10% difference between groups for primary outcome. ** Primary outcome
What's new
| Date | Event | Description |
|---|---|---|
| 11 December 2013 | New citation required but conclusions have not changed | Review updated with new trials but no overall changes to conclusions. |
| 15 October 2012 | New search has been performed | Results of update search in July 2012 added to review; risk of bias tables and description of bias added; background information, results and discussion extended; structure of the review updated. |
History
Protocol first published: Issue 4, 2000 Review first published: Issue 1, 2002
| Date | Event | Description |
|---|---|---|
| 15 February 2010 | Amended | Contact details updated. |
| 11 November 2009 | Amended | Contact details updated. |
| 31 October 2008 | Amended | Converted to new review format. |
Acknowledgements
We would like to thank the team of the Cochrane Schizophrenia Group for all the help they gave us concerning the literature search and for their helpful comments on this review. We would also like to thank Jerzy Samochowiec and Promonta Lundbeck Hamburg for their information on perazine trials and for sending us a full report (Jerzy Samochowiec) and abstracts and congress posters which we would otherwise not have obtained.
The Cochrane Schizophrenia Group produces and maintains a template for the methods section, we have used this template and adapted it as required.
The search term and searches for the 2012 update were developed and carried out by the Trial Search Co‐ordinator of the Cochrane Schizophrenia Group, Samantha Robers.
Appendices
Appendix 1. Previous searches
1. June 2000 In the first version of the review we searched the Cochrane Schizophrenia Group Register (June 2000) with the phrase:
[perazin or perazine or pernazinum or taxilan or pernazine or #13 = "phenothiazine tranquilizers" or (#13 = piperazines) or perazin‐neuraxpharm or methylpiperazin or piperazinyl]
2. March 2005 In the 2005 update we searched the Cochrane Schizophrenia Group's Trials Register (March 2005) with a slightly different phrase:
[((* perazin* or *pernazin* or *taxilan* or *phenothiazine tran* or * piperazin*) in Ti, Ab and In fields in Reference) AND (perazin* in Study Intervention field)]
The Cochrane Schizophrenia Group’s Trials Register is compiled by systematic searches of major databases, handsearches of journals and conference proceedings (see Group Module).
Appendix 2. Previous data collection and analysis
1. Selection of trials We (SL and BH) independently inspected all citations of those studies identified by the search. Where disagreement occurred we resolved this by discussion,and where there was still doubt, we acquired the full article for further inspection. Once the full articles were obtained BH decided whether they met the review criteria and this was checked by SL (this sequence was reversed in the 2005 update). We resolved any disagreement by discussion but if doubt remained we put the study on the list of those awaiting assessment pending acquisition of more information.
2. Quality assessment We used criteria in the Cochrane Collaboration Handbook (Alderson 2004) to assess trial quality. This simple set of criteria is based on evidence of strong association between overestimate of effect and poor concealment of allocation, and is defined as follows.
a. Low risk of bias (adequate allocation concealment). b. Moderate risk of bias (some doubt about the results). c. High risk of bias (inadequate allocation concealment).
The Jadad scale (Jadad 1996) measures a wider range of factors that impact on the quality of a trial. The scale includes three items.
a. Was the study described as randomised? b. Was the study described as double blind? c. Was there a description of withdrawals and dropouts?
Each item scores one point if the answer is positive and an additional point for the first two items if the means of randomisation/blinding is described. In addition, a point can be deducted if either the randomisation or the blinding/masking described were inadequate.
For the purpose of analysis of this review we included studies if they met criteria A and B of the Handbook. We gave studies not described as randomised a C rating and we excluded these. In addition, we used a cut‐off of two points on the Jadad scale to check the assessment made by the handbook criteria. If a B rated study scored less than two on the Jadad scale we excluded it.
3. Data extraction BH and SL independently extracted data from selected trials . When disputes arose we attempted resolution by discussion. If doubt remained and further information was necessary to resolve the dilemma, we did not enter the data but added them to the list of those awaiting assessment, pending further information.
4. Data synthesis 4.1 Data types Outcomes are assessed using continuous (for example, average changes on a behaviour scale), categorical (for example, one of three categories on a behaviour scale, such as 'little change', 'moderate change' or 'much change') or dichotomous measures (for example, either 'no important changes' or 'important changes' in a person's behaviour). Currently RevMan software does not support categorical data so we only presented these in the text of the review.
4.2 Incomplete data With the exception of the outcome of leaving the study early, we did not include trial outcomes if more than 50% of people were not reported in the final analysis. We felt that such a degree of attrition would threaten the validity of the findings.
4.3 Dichotomous data Where the original authors of the studies gave outcomes such as 'clinically improved' or 'not clinically improved' based on their clinical judgement, predetermined criteria or any scale, we recorded this in RevMan. If data was from a rater not clearly stated to be independent then we included if it did not change the results, otherwise we presented it separately with a label 'prone to bias'. Where possible, we tried to convert relevant categorical or continuous outcome measures to dichotomous data by identifying cut‐off points on rating scales and dividing subjects accordingly into groups. This was with the cut‐off points 'moderate or severe impairment' for end of study data or 'no better or worse' for change data. For example, the Brief Psychiatric Rating Scale (BPRS) (Overall 1962) is frequently used as a measure of change of symptoms in studies. We defined a 50% change on this particular scale as clinically important, although, it was recognised that for many people, especially those with chronic or severe illnesses, a less rigorous definition of important improvement, for example, 20% on the BPRS, would be equally valid. If individual patient data were available we used the 50% cut‐off for the definition in the case of non‐chronically ill people and 20% for those with chronic illness.
We used an intention‐to‐treat analysis. As long as over 50% of people completed the study, everyone allocated to the intervention was counted whether or not they completed follow‐up. We assumed that those who left the study early had no change as regards outcome.
We used the relative risk (RR) and 95% confidence interval (CI) based on the random‐effects model, as this takes into account differences between studies even if heterogeneity is not statistically significant, as the preferred statistic for summation. We inspected data to see if analysis using a Mantel‐Haenszel odds ratio and fixed‐effect models made a substantive difference.
4.4 Continuous data 4.4.1 In the case of continuous data a post hoc application of intention to treat is not possible. Therefore, we could only use the data as presented by the original studies, i.e. either as intention to treat last observation carried forward data, or as data based on observed cases. If both analyses were presented we used the intention‐to‐treat results. 4.4.2 Rating scales: a wide range of instruments are available to measure mental health outcomes. These instruments vary in quality and many are not valid, or even ad hoc. For outcome instruments some minimum standards have to be set. They were that: (a) the psychometric properties of the instrument should have been described in a peer‐reviewed journal; (b) the instrument should either be: (i) a self‐report or (ii) completed by an independent rater or relative (not the therapist). As it was expected that therapists would frequently also be the rater, such data will be commented on with 'prone to bias'; or (c) the instrument should be a global assessment of an area of functioning.
4.5 Normal distribution of data Mental health continuous data are often not normally distributed. To avoid the pitfall of applying parametric tests to non‐parametric data we applied the following standards to all data before inclusion: 4.5.1 standard deviations and means were reported in the paper or were obtained from the authors; 4.5.2 if the data were finite measures from, for example 0‐100, when the standard deviation was multiplied by two, the result should be less than the mean. Otherwise the mean was unlikely to be an appropriate measure of the centre of the distribution (Altman 1996). Continuous data, if normally distributed, were summated using a calculation of the weighted mean difference (MD). We inspected data to see if analysis using a standardised mean difference made a substantive difference. Non‐normally distributed data were reported in the 'Other data types' tables. We did not consider continuous data presented without use of summary statistics (i.e. mean, SD, SE, median, interquartile range), although we noted the existence of these data in the table of included studies. Furthermore, continuous data may be presented from different scales, rating the same outcome. In this event, we presented all data separately without summation and inspected the general direction of effect.
Endpoint scale‐derived data is finite, ranging from one score to another. Change data is more problematic and you cannot apply the rule described above. Although most change scores are likely to be skewed, it cannot be proven so twe presented these in MetaView. Where both endpoint and change were available for the same outcome we presented endpoint in preference.
4.6 Cluster trials Studies increasingly employ 'cluster randomisation' (such as randomisation by clinician or practice) but analysis and pooling of clustered data poses problems. Firstly, authors often fail to account for intra class correlation in clustered studies, leading to a 'unit of analysis' error whereby p values are spuriously low, confidence intervals unduly narrow and statistical significance overestimated. This causes type I errors (Gulliford 1999).
Where clustering was not accounted for in primary studies, we presented the data in a table, with a (*) symbol to indicate the presence of a probable unit of analysis error. In subsequent versions of this review we will seek to contact first authors of studies to obtain intra class correlation co‐efficients of their clustered data and to adjust for this by using accepted methods (Gulliford 1999). Where clustering has been incorporated into the analysis of primary studies, we will also present these data as if from a non‐cluster randomised study, but adjusted for the clustering effect.
We have sought statistical advice and have been advised that the binary data as presented in a report should be divided by a 'design effect'. This is calculated using the mean number of participants per cluster (m) and the intra‐class correlation co‐efficient (ICC) Design effect = 1 + (m ‐ 1) * ICC (Donner 2002). If the ICC was not reported it was assumed to be 0.1 (Ukoumunne 1999).
4.7 Sensitivity analyses 4.7.1 We compared the outcome of the intention‐to‐treat analysis with a completer analysis.
5. Heterogeneity Firstly, we considered all the included studies within any comparison to judge clinical heterogeneity. Then we visually inspected graphs to investigate the possibility of statistical heterogeneity. This was supplemented using, primarily, the I2 statistic. This provides an estimate of the percentage of variability due to heterogeneity rather than chance alone. Where the I2 estimate was greater than or equal to 75%, we interpreted this as indicating the presence of high levels of heterogeneity (Higgins 2003). If inconsistency was high, we did not summate data, but presented them separately and investigated the reasons for heterogeneity.
6. Addressing publication bias We entered data from all identified and selected trials into a funnel graph (trial effect versus trial size) in an attempt to investigate overt publication bias (Egger 1997).
7. Tables and figures Where possible, we entered data into RevMan so the area to the left of the line of no effect indicated a favourable outcome for perazine.
Data and analyses
Comparison 1. Perazine versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Any reason | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.62 [0.35, 1.10] |
| 1.2 Inefficacy of treatment | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.09, 1.04] |
| 2 Global state: Change over time ‐ no better or deterioration | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.43 [0.23, 0.81] |
| 3 Mental state: 1a. General ‐ Less than 20% BPRS reduction | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.7 [0.47, 1.05] |
| 4 Mental state: 1b. General ‐ Less than 30% BPRS reduction | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.61, 1.09] |
| 5 Mental state: 1c. General ‐ BPRS endpoint score ‐ per protocol sample (high=poor) | 1 | 79 | Mean Difference (IV, Random, 95% CI) | ‐6.20 [‐13.20, 0.80] |
| 6 Mental state: 1d. General ‐ BPRS change from baseline to endpoint (high=poor) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 per protocol analysis | 1 | 79 | Mean Difference (IV, Random, 95% CI) | ‐6.1 [‐13.12, 0.92] |
| 6.2 intent‐to‐treat analysis | 1 | 95 | Mean Difference (IV, Random, 95% CI) | ‐7.10 [‐13.51, ‐0.69] |
| 7 Adverse events: 1. General ‐ at least one adverse event | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 1.15 [0.87, 1.51] |
| 8 Adverse events: 2. Movement disorder ‐ at least one extrapyramidal side‐effect | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 13.53 [0.79, 230.37] |
| 9 Adverse events: 3. Movement disorder ‐ use of antiparkinson medication | 1 | 95 | Risk Ratio (M‐H, Random, 95% CI) | 4.5 [1.04, 19.45] |
| 10 Sensitivity analysis: fixed effects model ‐ global state no better or worse | 1 | 95 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.43 [0.23, 0.81] |
1.1. Analysis.
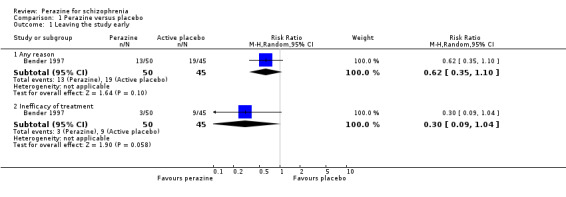
Comparison 1 Perazine versus placebo, Outcome 1 Leaving the study early.
1.2. Analysis.
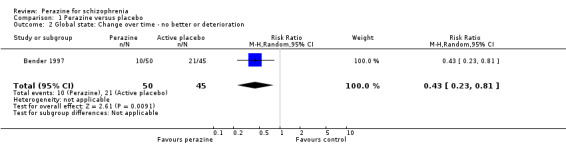
Comparison 1 Perazine versus placebo, Outcome 2 Global state: Change over time ‐ no better or deterioration.
1.3. Analysis.
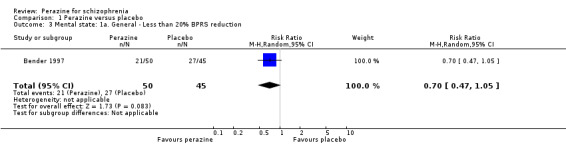
Comparison 1 Perazine versus placebo, Outcome 3 Mental state: 1a. General ‐ Less than 20% BPRS reduction.
1.4. Analysis.

Comparison 1 Perazine versus placebo, Outcome 4 Mental state: 1b. General ‐ Less than 30% BPRS reduction.
1.5. Analysis.

Comparison 1 Perazine versus placebo, Outcome 5 Mental state: 1c. General ‐ BPRS endpoint score ‐ per protocol sample (high=poor).
1.6. Analysis.
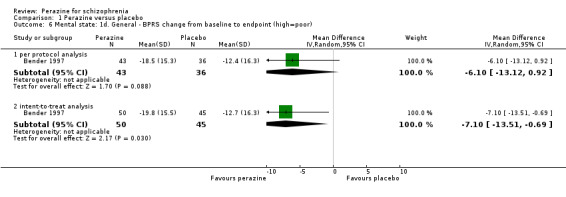
Comparison 1 Perazine versus placebo, Outcome 6 Mental state: 1d. General ‐ BPRS change from baseline to endpoint (high=poor).
1.7. Analysis.
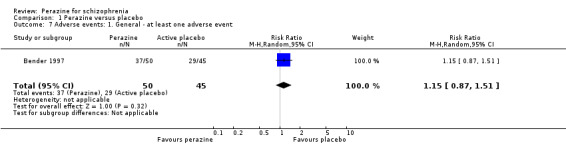
Comparison 1 Perazine versus placebo, Outcome 7 Adverse events: 1. General ‐ at least one adverse event.
1.8. Analysis.
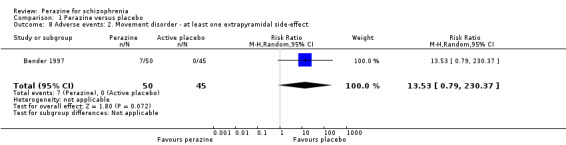
Comparison 1 Perazine versus placebo, Outcome 8 Adverse events: 2. Movement disorder ‐ at least one extrapyramidal side‐effect.
1.9. Analysis.
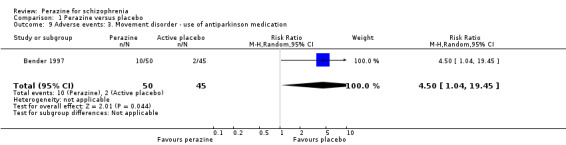
Comparison 1 Perazine versus placebo, Outcome 9 Adverse events: 3. Movement disorder ‐ use of antiparkinson medication.
1.10. Analysis.
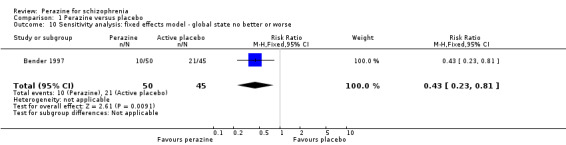
Comparison 1 Perazine versus placebo, Outcome 10 Sensitivity analysis: fixed effects model ‐ global state no better or worse.
Comparison 2. Perazine versus other antipsychotics.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Leaving the study early due to any reason | 6 | 384 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.68, 1.38] |
| 2 Global state: 1. No better or deterioration | 1 | 30 | Risk Ratio (M‐H, Random, 95% CI) | 1.33 [0.36, 4.97] |
| 3 Global state: 2. CGI severity score at endpoint (high = poor) | 1 | 30 | Mean Difference (IV, Random, 95% CI) | 1.85 [1.01, 2.69] |
| 4 Global state: 3. GAS endpoint score (low = poor) | 1 | 41 | Mean Difference (IV, Random, 95% CI) | ‐10.0 [‐17.12, ‐2.88] |
| 5 Mental state: 1a. General ‐ BPRS ‐ no better or deterioration | 1 | 32 | Risk Ratio (M‐H, Random, 95% CI) | 1.18 [0.53, 2.62] |
| 6 Mental state: 1b. General ‐ BPRS endpoint score (high = poor) | 1 | 34 | Mean Difference (IV, Random, 95% CI) | 7.92 [1.10, 14.74] |
| 7 Mental state: 1c. General ‐ BPRS endpoint score: separate items (high = poor) | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.74, ‐0.06] |
| 8 Mental state: 1d. General ‐ percentage PANSS change from baseline | 1 | 191 | Mean Difference (IV, Random, 95% CI) | 3.80 [‐2.80, 10.40] |
| 9 Mental state: 1e. Specific ‐ BPRS positive symptoms subscore at endpoint (high = poor) | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.78, ‐0.02] |
| 10 Mental state: 2. Unable to use (skewed data) | Other data | No numeric data | ||
| 11 Adverse events: 1. Movement disorder | 4 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 akathisia | 3 | 111 | Risk Ratio (M‐H, Random, 95% CI) | 0.31 [0.09, 1.05] |
| 11.2 dyskinesia | 3 | 111 | Risk Ratio (M‐H, Random, 95% CI) | 0.47 [0.06, 3.48] |
| 11.3 neuroleptic malignant syndrome | 1 | 32 | Risk Ratio (M‐H, Random, 95% CI) | 0.30 [0.01, 6.77] |
| 11.4 parkinsonism | 2 | 81 | Risk Ratio (M‐H, Random, 95% CI) | 1.21 [0.53, 2.76] |
| 11.5 tremor | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.8 [0.25, 2.55] |
| 12 Adverse events: 2. Leaving the study early due to adverse events | 5 | 193 | Risk Ratio (M‐H, Random, 95% CI) | 0.87 [0.40, 1.90] |
| 13 Adverse events: 3. Anticholinergic | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 13.1 dry mouth | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.16, 6.42] |
| 13.2 blurred vision | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.5 [0.05, 5.08] |
| 14 Adverse events: 4. Arousal | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 14.1 drowsiness / sedation | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.39, 1.95] |
| 15 Adverse events: 5. Abnormal laboratory results | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 15.1 leukopenia | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 15.2 leukocytosis | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 2.48] |
| 15.3 increase of transaminases | 3 | 103 | Risk Ratio (M‐H, Random, 95% CI) | 2.09 [0.67, 6.51] |
| 16 Adverse events: 6. Cardiovascular | 2 | 81 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.12, 5.72] |
| 16.1 hypotension ‐ orthostatic | 2 | 81 | Risk Ratio (M‐H, Random, 95% CI) | 0.83 [0.12, 5.72] |
| 17 Adverse events: 7. EEG‐changes | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 0.32 [0.04, 2.80] |
| 18 Adverse events: 8. Hypersalivation | 1 | 40 | Risk Ratio (M‐H, Random, 95% CI) | 0.33 [0.01, 7.72] |
| 19 Adverse events: 9. Vegetative | 1 | 41 | Risk Ratio (M‐H, Random, 95% CI) | 1.50 [0.73, 3.08] |
| 20 Sensitivity analysis: fixed effects model ‐ global state no better or worse | 1 | 30 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.33 [0.36, 4.97] |
2.1. Analysis.
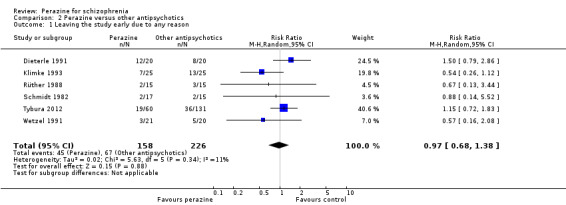
Comparison 2 Perazine versus other antipsychotics, Outcome 1 Leaving the study early due to any reason.
2.2. Analysis.

Comparison 2 Perazine versus other antipsychotics, Outcome 2 Global state: 1. No better or deterioration.
2.3. Analysis.
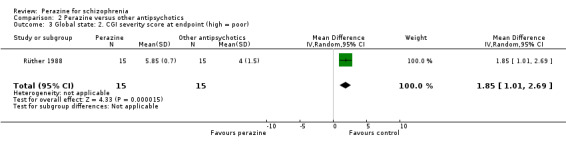
Comparison 2 Perazine versus other antipsychotics, Outcome 3 Global state: 2. CGI severity score at endpoint (high = poor).
2.4. Analysis.
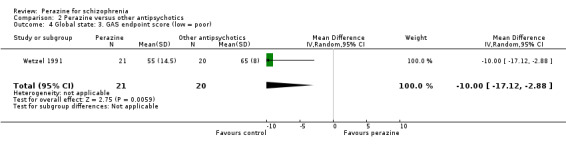
Comparison 2 Perazine versus other antipsychotics, Outcome 4 Global state: 3. GAS endpoint score (low = poor).
2.5. Analysis.

Comparison 2 Perazine versus other antipsychotics, Outcome 5 Mental state: 1a. General ‐ BPRS ‐ no better or deterioration.
2.6. Analysis.
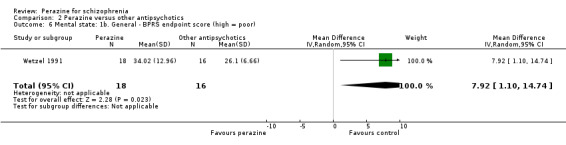
Comparison 2 Perazine versus other antipsychotics, Outcome 6 Mental state: 1b. General ‐ BPRS endpoint score (high = poor).
2.7. Analysis.
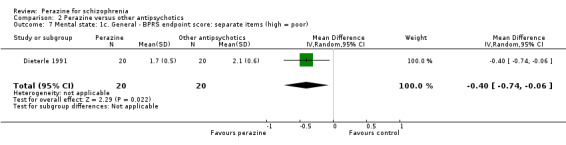
Comparison 2 Perazine versus other antipsychotics, Outcome 7 Mental state: 1c. General ‐ BPRS endpoint score: separate items (high = poor).
2.8. Analysis.
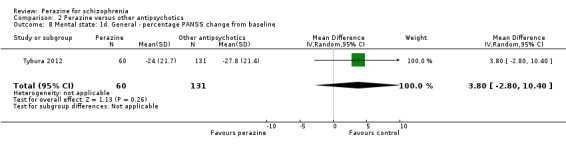
Comparison 2 Perazine versus other antipsychotics, Outcome 8 Mental state: 1d. General ‐ percentage PANSS change from baseline.
2.9. Analysis.

Comparison 2 Perazine versus other antipsychotics, Outcome 9 Mental state: 1e. Specific ‐ BPRS positive symptoms subscore at endpoint (high = poor).
2.10. Analysis.
Comparison 2 Perazine versus other antipsychotics, Outcome 10 Mental state: 2. Unable to use (skewed data).
| Mental state: 2. Unable to use (skewed data) | ||||
|---|---|---|---|---|
| Study | Intervention | Mean | SD | N |
| Rüther 1988 | Mean BPRS‐score at endpoint Perazine | 42.9 | 18.0 | 15 |
| Rüther 1988 | Amisulpride | 37.9 | 19.5 | 15 |
| Schmidt 1982 | Mean AMDP‐score at endpoint: Perazine | 10.2 | 11.8 | 17 |
| Schmidt 1982 | Haloperidol | 11.4 | 13.7 | 15 |
2.11. Analysis.
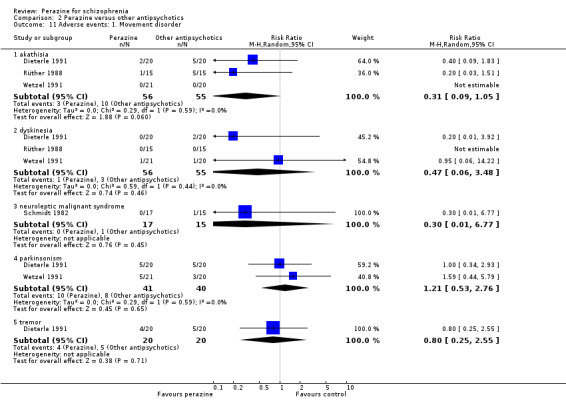
Comparison 2 Perazine versus other antipsychotics, Outcome 11 Adverse events: 1. Movement disorder.
2.12. Analysis.
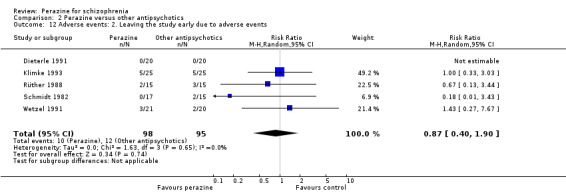
Comparison 2 Perazine versus other antipsychotics, Outcome 12 Adverse events: 2. Leaving the study early due to adverse events.
2.13. Analysis.
Comparison 2 Perazine versus other antipsychotics, Outcome 13 Adverse events: 3. Anticholinergic.
2.14. Analysis.
Comparison 2 Perazine versus other antipsychotics, Outcome 14 Adverse events: 4. Arousal.
2.15. Analysis.
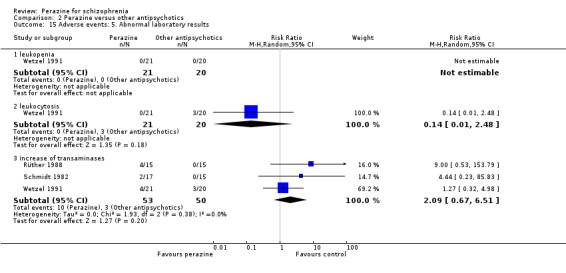
Comparison 2 Perazine versus other antipsychotics, Outcome 15 Adverse events: 5. Abnormal laboratory results.
2.16. Analysis.

Comparison 2 Perazine versus other antipsychotics, Outcome 16 Adverse events: 6. Cardiovascular.
2.17. Analysis.
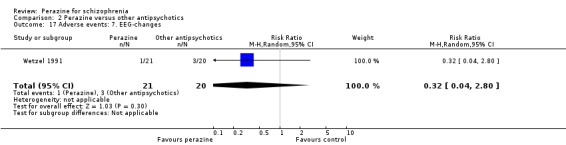
Comparison 2 Perazine versus other antipsychotics, Outcome 17 Adverse events: 7. EEG‐changes.
2.18. Analysis.
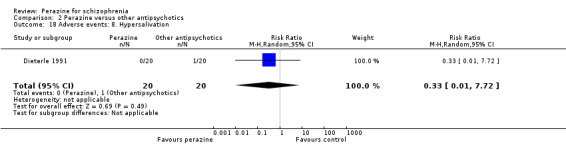
Comparison 2 Perazine versus other antipsychotics, Outcome 18 Adverse events: 8. Hypersalivation.
2.19. Analysis.
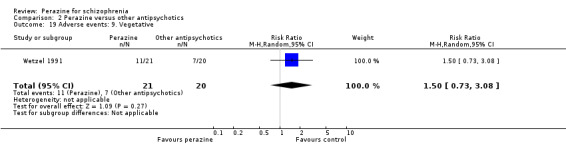
Comparison 2 Perazine versus other antipsychotics, Outcome 19 Adverse events: 9. Vegetative.
2.20. Analysis.
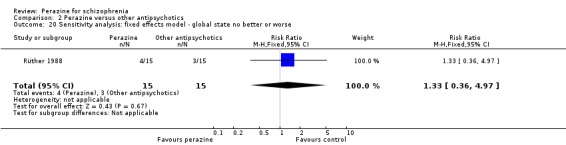
Comparison 2 Perazine versus other antipsychotics, Outcome 20 Sensitivity analysis: fixed effects model ‐ global state no better or worse.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Bender 1997.
| Methods | Allocation: randomised ‐ chronological randomisation using a validated computer program where eligible patients were assigned to the lowest yet unassigned treatment number available at the study centre. Blindness: double ‐ identical capsules. Duration: 35 days. Raters: not stated to be independent of treatment. Design: multi‐centre. | |
| Participants | Diagnosis: schizophrenia or schizophreniform disorder (DSM‐III‐R), BPRS > 39. N = 95. Sex: not indicated. Age: between 18 and 70 years. History: not indicated. Setting: hospital. | |
| Interventions | 1. Perazine: dose average 459 mg/day SD 62 mg/day. N = 50. 2. Trimipramine: dose average 309 mg/day SD 73 mg/day. N = 45. | |
| Outcomes | Improvement of global state.
Leaving the study early.
Mental state: BPRS.
Adverse events: EPS (participants with at least one movement disorder, at least one dose of antiparkinson medication).
Adverse events: number of patients with at least one adverse event. Unable to use ‐ Mental state: Depression scale (unclear N). |
|
| Notes | We considered trimipramine ‐ a tricyclic antidepressant ‐ to be an active placebo. Jadad = 5 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "randomized" (p.61); "A randomization schedule was produced by a validated computer program" (p.63) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double‐blind" (p.61); "Study medication was administered in capsules indistinguishable in colour, size, form, smell and consistency" (p.63) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "double‐blind" (p.61); "Study medication was administered in capsules indistinguishable in colour, size, form, smell and consistency" (p.63) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "double‐blind" (p.61) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "double‐blind" (p.61) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 37% dropout rate, not evenly distributed between groups (13/50 and 19/45) |
| Selective reporting (reporting bias) | Low risk | No selective reporting |
| Other bias | Low risk | No other bias |
Dieterle 1991.
| Methods | Allocation: randomised ‐ no further details. Blindness: double ‐ identical capsules. Duration: 28 days. Raters: not stated to be independent of treatment. Design: single‐centre. | |
| Participants | Diagnosis: schizophrenia (ICD‐9). Excluded: substance dependence, epilepsy, neurological diseases, hypotension, history of adverse events. N = 40. Sex: 27F, 13M. Age: mean ˜ 33 years (range: 20‐64). History: 16 participants were hospitalised for the first time. Setting: unclear. | |
| Interventions | 1. Perazine: dose mean 350 mg/frequency not reported (SD 100, range 75‐675). N = 20. 2. Zotepine: dose mean 240 mg/frequency not reported (SD 70, range 50‐450). N = 20. | |
| Outcomes | Leaving the study early.
Mental state: BPRS.
Adverse effects: vegetative side‐effects. Unable to use ‐ Global state: CGI (only P value). Mental state: SANS, AMDP (only P value). |
|
| Notes | Jadad = 3 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomised" (p18) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double blind"; "identical capsules" (p.18) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Low risk | "double blind"; "identical capsules" (p.18) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "double blind" (p.18) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "double blind" (p.18) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 50% dropout rate, not evenly distributed (12/20 and 8/20) |
| Selective reporting (reporting bias) | High risk | No SDs for CGI, SANS |
| Other bias | Low risk | No other bias |
Klimke 1993.
| Methods | Allocation: randomised ‐ no further details. Blindness: double ‐ no further details. Duration: 28 days. Raters: not stated to be independent from treatment. Design: single‐centre. | |
| Participants | Diagnosis: acute schizophrenia (ICD‐9) without antipsychotic treatment at admission. N = 50. Sex: 21M, 29F. Age: mean 36.6 years (SD 11.9, range 20‐62). History: duration since first episode: 80.4 months (SD 104.4, range 0‐400). Setting: hospital. | |
| Interventions | All patients were treated with 15 mg haloperidol intravenously for the first three days; then random assignment to: 1. Perazine: dose 300 mg/day. N = 25. 2. Haloperidol: dose 15 mg/day. N = 25. Flunitrazepam (2 mg) and biperiden (1‐2 times 5 mg) were allowed as concomitant medication. |
|
| Outcomes | Leaving the study early.
Mental state: BPRS. Unable to use ‐ Global state: CGI (only P value). Adverse effects: SAS (only P value). |
|
| Notes | Jadad = 3 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomly assigned" (p.25) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "double blind" (p.25); "Patients and treating psychiatrists were blind to the treatment conditions" (p.26) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "double blind" (p.25); "Patients and treating psychiatrists were blind to the treatment conditions" (p.26) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "double blind" (p.25) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "double blind" (p.25) |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 40% dropout rate, not evenly distributed (7/25 and 13/25) |
| Selective reporting (reporting bias) | Low risk | No selective reporting |
| Other bias | Low risk | No other bias |
Rüther 1988.
| Methods | Allocation: randomised ‐ no further details. Blindness: double ‐ no further details. Duration: 28 days. Raters: not stated to be independent of treatment. Design: single‐centre. | |
| Participants | Diagnosis: schizophrenia (‐like) disorders (ICD 9 295.1‐295.7); 'acutely ill', pretreatment with antipsychotics was allowed. N = 30. Sex: 14M, 16F. Age ˜ 35 years. History: 13 were first episode patients. Setting: hospital. | |
| Interventions | 1. Perazine: dose starting at 400 mg/day (max. 1000 mg/day). N = 15.
2. Aminosultoprid: dose starting at 400 mg/day (max. 1000 mg/day). N = 15. Allowed additional medication: chloraldurat (sleep medication), diazepam and biperiden (antiparkinson medication). |
|
| Outcomes | Leaving the study early.
Mental state: BPRS.
Global state: CGI.
Vital signs.
Laboratory. Unable to use ‐ Mental state: NGI, AMDP (no mean or no SD). Adverse effects: SAS, Webster Scale (no mean or no SD). Laboratory: most laboratory data (no data). |
|
| Notes | Jadad = 3 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "zufällige Zuteilung" (p.65) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "doppelblind" (p.70) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "doppelblind" (p.70) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "doppelblind" (p.70) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "doppelblind" (p.70) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low dropout rate, evenly distributed between groups (2/15 and 3/15) |
| Selective reporting (reporting bias) | High risk | Not all SDs provided |
| Other bias | Low risk | No other bias |
Schmidt 1982.
| Methods | Allocation: randomised ‐ no further details. Blindness: double ‐ HPL tablets + placebo liquid or perazine liquid + placebo tablets. Duration: 28 days. Raters: not stated to be independent of treatment. Design: single‐centre. | |
| Participants | Diagnosis: acute paranoid schizophrenia (ICD 9 295.3). Excluded: catatonic, schizoaffective and residual patients. N = 32. Sex: all male. Age: ˜ 31 years. History: duration ill ˜ 4 years, ˜ 3 previous admissions. Setting: hospital. | |
| Interventions | 1. Perazine: dose 300, 600 or 900 mg/day according to clinical requirements. N = 17. 2. Haloperidol: dose 15, 30 or 45 mg/day according to clinical requirements. N = 15. | |
| Outcomes | Leaving the study early.
Global state: CGI.
Adverse effects: number of EPS, dry mouth, tiredness, increase of transaminases, neuroleptic malignant syndrome. Unable to use ‐ Mental state: AMDP (no SD). Adverse effects: laboratory results (only P values), EWL, SAS (no SD or no mean). |
|
| Notes | 20 screened patients could not be randomised because they were too severely ill. Jadad = 3 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomisiert" (p.531) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Doppel‐Blind" (p.530) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Doppel‐Blind" (p.530) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Doppel‐Blind" (p.530) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Doppel‐Blind" (p.530) |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Low dropout rate, evenly distributed (2/17 and 2/15) |
| Selective reporting (reporting bias) | High risk | Not all SDs provided |
| Other bias | Low risk | No other bias |
Tybura 2012.
| Methods | Allocation: randomised, allocation concealed (see below). Blindness: open. Duration: 3 months. Raters: not stated to be independent of treatment. Design: two‐centre. | |
| Participants | Diagnosis: paranoid schizophrenia according to "Composite International Diagnostic Interview" and ICD‐10. Excluded: serious neurological and/or somatic disorders (e.g. stroke, hepatic insufficiency, diabetes). N = 191. Sex: 89 men 102 women. Age: ˜ 36 years. History: duration ill ˜ 10 years. Setting: unclear. | |
| Interventions | 1. Perazine 300‐600mg/day. N = 60. 2. Olanzapine 10‐20mg/day. N = 72. 3. Ziprasidone 120‐160mg/day. N = 59. |
|
| Outcomes | Leaving the study early.
General mental state: percentage PANSS change from baseline. No further outcomes were reported, the main focus of the study was genetics. |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Personal communication from Jerzy Samochowiec: "Both recruiting centers had a simple program generating random allocations. The program was writen by our statistician (P.M.) and allowed random allocation of each patient meeting the study criteria. The program was a simple randomization tool, i.e. it allowed random (with a probability of 33,3%) allocation of each qualified patient to one of three treatment groups." |
| Allocation concealment (selection bias) | Low risk | Personal communication from Jerzy Samochowiec: "The processes of initial qualification and randomization were separated. Senior researchers in each center were asked for a random allocation by physicians willing to qualify a patient to the study. The patient was registered in the study database and allocation to one of the study groups was performed as described above. Database records and randomization was a duty of a senior researcher. Hence, the physician deciding to qualify a patient to the study could not predict to which group the patient will be allocated. The allocation was done by another person and was final. The whole system worked like with two 'randomization centers'." |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Open study". Lack of blinding would not be so important for objective outcomes. |
| Blinding of participants and personnel (performance bias) Subjective outcomes | High risk | "Open study". |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Open study". Lack of blinding would not be so important for objective outcomes. |
| Blinding of outcome assessment (detection bias) Subjective outcomes | High risk | "Open study". |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropout rate 24%‐32%. Reasons for dropout not indicated. LOCF analysis. |
| Selective reporting (reporting bias) | Low risk | No indication of selective reporting. |
| Other bias | Low risk | No obvious other bias. |
Wetzel 1991.
| Methods | Allocation: randomised ‐ no further details. Blindness: double ‐ no further details. Raters: not stated to be independent of treatment. Design: single‐centre. | |
| Participants | Diagnosis: schizophrenia 'with positive symptoms' (ICD‐9). Excluded: people with substance dependence, epilepsy, pregnancy, organic psychoses, relevant other diseases or people currently treatment with depot antipsychotics. N = 41. Sex: 16M, 25F. Age: mean ˜ 40 years. History: not indicated. Setting: hospital. | |
| Interventions | 1. Perazine: dose starting at 50 mg/day then flexible dose (max. 900 mg/day). N = 21.
2. Zotepine: dose starting at 100 mg/day, then flexible dose (max. 600 mg/day). N = 20. Chloralhydrate (sleep medication), flurazepam, diazepam and biperiden (anticholinergic) were allowed as concomitant medication. |
|
| Outcomes | Leaving the study early.
Global state: GAS.
Mental state: BPRS.
Extrapyramidal side effects.
Vital signs: EEG, ECG, laboratory. Unable to use ‐ Mental state: AMDP, FSCL‐NL, FSUCL (no mean or no SD, or no usable data). Global state: CGI (no usable data). Adverse effects: AIMS, Gerlach scale (no mean or no SD). |
|
| Notes | Jadad = 3 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | randomized ("Zufallsprinzip") (p.24) |
| Allocation concealment (selection bias) | Unclear risk | Not indicated |
| Blinding of participants and personnel (performance bias) Objective outcomes | Low risk | "Double blind" (p.23) |
| Blinding of participants and personnel (performance bias) Subjective outcomes | Unclear risk | "Double blind" (p.23) |
| Blinding of outcome assessment (detection bias) Objective outcomes | Low risk | "Double blind" (p.23) |
| Blinding of outcome assessment (detection bias) Subjective outcomes | Unclear risk | "Double blind" (p.23) |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 20% dropout rate, not evenly distributed (3/21 and 5/20) |
| Selective reporting (reporting bias) | High risk | No SDs or mean values provided |
| Other bias | Low risk | No other bias |
General abbreviations: HPL = haloperidol EPS = extrapyramidal side‐effects. M = males F = females N = number mg = milligram SD = standard deviation ECG = electrocardiogram EEG = electroencephalogram
Diagnostic tools: DSM‐III‐R = Diagnostic and Statistical Manual of Mental disorders, third edition, revised ICD‐9 = International Classification of Diseases, ninth revision
Global effect scales: CGI = Clinical Global Impression (Guy 1976) GAS = Global Assessment of Symptoms Scale (Endicott 1976)
Mental state scales: AMDP = Arbeitsgemeinschaft für Methodik und Dokumentation in der Psychiatrie (AMDP 1981) BPRS = Brief Psychiatric Rating Scale (Overall and Gorham 1970) FSCL‐NL = Collegium Internationale Psychiatricae Scalarum, CIPS 1986 FSUCL = Collegium Internationale Psychiatricae Scalarum, CIPS 1986 SANS = Scale for Assessment of Negative Symptoms (Andreasen 1989)
Adverse effects scales: AIMS = Abnormal Involuntary Movement Scale (Guy 1976) EWL = Eigenschaftswörterliste (Janke 1977) Gerlach Scale (Gerlach 1983) NGI = (no reference) SAS = Simpson and Angus Scale (Simpson and Angus 1970) Webster Scale (no reference)
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bender 1999 | Allocation: not randomised. |
| Czekalla 1999 | Allocation: not randomised. |
| Fischer 1992 | Allocation: not randomised. |
| Gaebel 1981 | Allocation: not randomised. |
| Gaebel 1986 | Allocation: randomised. Participants: patients with neurotic instead of schizophrenic disorders. |
| Gaebel 1988 | Allocation: not randomised. |
| Gaebel 1992 | Allocation: randomised. Participants: patients with schizophrenia and depressive disorders. Interventions: perazine versus haloperidol. Outcomes: no data for the patients with schizophrenia only; patients with depression are compared with patients with schizophrenia. |
| Gerson 1964 | Allocation: not randomised. |
| Grabe 1999 | Allocation: not randomised. |
| Jarema 1999 | Allocation: randomised. Participants: patients with schizophrenic disorders. Interventions: conventional versus 'old' atypical neuroleptics, one of the latter being perazine. Outcomes: no data on the patients treated with perazine. |
| Jockers‐Scherübl1997 | Allocation: not randomised. |
| Kemperdick 1967 | Allocation: not randomised. |
| Kuhs 1988 | Allocation: not randomised. |
| Loza 2001 | Allocation: randomised. Participants: patients with schizophrenia. Interventions: various atypical antipsychotics versus various typical antipsychotics (among them perazine). Outcomes: data for the patients treated with perazine have not been presented separately. |
| Ohlmeier 2007 | Allocation: randomised. Participants: people with schizophrenia. Interventions: Perazine combined with carbamazepine versus olanzapine, i.e. no appropriate intervention group. |
| Pietzcker 1981 | Allocation: not randomised. |
| Pietzcker 1984 | Allocation: not randomised. |
| Rein 1983 | Allocation: not randomised. |
| Schied 1983 | Allocation: not randomised. |
| Schmidt 1988 | Allocation: not randomised. |
| Terminska 1989 | Allocation: not randomised. |
| Volz 1994 | Allocation: not randomised. |
| Wetterling 1996 | Allocation: not randomised. |
| Wetzel 1991 | Allocation: not randomised. |
Contributions of authors
Stefan Leucht: protocol development, literature search, data extraction, updating, writing of the review.
Benno Hartung: protocol development, literature search, data extraction.
Bartosz Helfer: updating, study extraction, writing of the review.
Sources of support
Internal sources
Freistaat Bayern, Germany.
External sources
No sources of support supplied
Declarations of interest
Stefan Leucht: has received honoraria for lectures from Abbvie, Astra Zeneca, Bristol‐Myers Squibb, ICON, Eli Lilly, Janssen, Johnson & Johnson, Roche, SanofiAventis, Lundbeck and Pfizer; honoraria for consulting and advisory boards from Roche, Eli Lilly, Medavante, Bristol‐Myers Squibb, Alkermes, Janssen, Johnson & Johnson and Lundbeck. Eli Lilly has provided medication for a study with SL as primary investigator.
Markus Dold: nothing to declare.
Bartosz Helfer: nothing to declare.
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Bender 1997 {published data only}
- Bender S, Olbrich H, Hornstein C, Schöne W, Falkai P. Antipsychotic efficacy of trimipramine: double‐blind comparison with a classical neuroleptic. Pharmacopsychiatry 1997;30(5):151. [DOI] [PubMed] [Google Scholar]
- Bender S, Olbrich H, Hornstein C, Schöne W, Falkai P, Trimipramine Study Group. Antipsychotic efficacy of the antidepressant trimipramine: double‐blind comparison with the phenothiazine perazine. 6th World Congress of Biological Psychiatry. Nice, France, June 22‐27 1997. [MEDLINE: ; PMID 4608707] 4608707
- Bender S, Olbrich HM, Fischer W, Hornstein C, Schoene W, Falkai P, et al. Antipsychotic efficacy of the antidepressant trimipramine: A randomized, double‐blind comparison with the phenothiazine perazine. Pharmacopsychiatry 2003;36(2):61‐9. [EMBASE: 2003210869] [DOI] [PubMed] [Google Scholar]
- Blaeser‐Kiel G. Schizoaffective disorders ‐ testing of antipsychotic components of trimipramine [Schizoaffektive Störungen. Antipsychotische Komponente von Trimipramin im Test]. TW Neurologie Psychiatrie 1996;10(10):780‐1. [Google Scholar]
Dieterle 1991 {published data only}
- Ackenheil M, Dieterle D, Kapfhammer HP, Müller‐Spahn F, Hippius H. not available [Therapievergleich von Perazin und Zotepin bei schizophrenen Patienten]. In: Helmchen H, Hippius H, Tölle R editor(s). Therapie mit Neuroleptika ‐ Perazin. Stuttgart, New York: Thieme Verlag, 1988:60‐4. [Google Scholar]
- Dieterle DM, Albus M, Blanke E, Eben E, Klein H, Mullerspahn F, et al. A double‐blind comparison of aminosultoprid (dan 2163) versus perazine in schizophrenic‐patients. International Journal of Neuroscience 1987;32(1‐2):438. [Google Scholar]
- Dieterle DM, Albus M, Blanke E, Eben E, Klein H, Müller‐Spahn, Rüther E. A double‐blind comparison of aminosultoprid (Dan 2163) versus perazine in schizophrenic patients. 4th World Congress of Biological Psychiatry; 1989 Sept 8‐13; Philadelphia, USA. 1989.
- Dieterle DM, Müller‐Spahn F, Ackenheil M. Comparison of zotepine and perazine in schizophrenia. Psychopharmacology 1988;96 Suppl 1:340. [Google Scholar]
- Dieterle DM, Müller‐Spahn F, Ackenheil M. Efficacy and tolerance of zotepine in a double‐blind comparison with perazine in schizophrenics [Wirksamkeit und Verträglichkeit von Zotepin im Doppelblindvergleich mit Perazin bei schizophrenen Patienten]. Fortschritte der Neurologie und Psychiatrie 1991;59 Suppl 1:18‐22. [DOI] [PubMed] [Google Scholar]
- Müller‐Spahn F, Dieterle D, Ackenheil M. Clinical efficacy of zotepine in the treatment of schizophrenic negative symptoms: results of an open and a double‐blind controlled study [Klinische Wirksamkeit von Zotepin in der Behandlung schizophrener Minussymptomatik: Ergebnisse einer offenen und einer doppelblind‐kontrollierten Studie]. Fortschritte der Neurologie und Psychiatrie 1991;59:30‐5. [DOI] [PubMed] [Google Scholar]
Klimke 1993 {published data only}
- Klimke A, Klieser E. Clinical relevance of the test dose model for drug choice and response in acute schizophrenia. Pharmacopsychiatry 1992;25:85. [DOI] [PubMed] [Google Scholar]
- Klimke A, Klieser E, Lehmann E, Miele L. Initial improvement as a criterion for drug choice in acute schizophrenia [Frühes Ansprechen als Kriterium der Medikamentenwahl bei akuter Schizophrenie]. Pharmacopsychiatry 1993;26:25‐9. [DOI] [PubMed] [Google Scholar]
Rüther 1988 {published data only}
- Rüther E, Blanke J. not available [Therapievergkeich von Aminosultoprid (DAN 2163) und Perazin bei schizophrenen Patienten]. In: Helmchen H, Hippius H, Tölle R editor(s). Therapie mit Neuroleptika ‐ Perazin. Stuttgart, New York: Thieme Verlag, 1988:65‐70. [Google Scholar]
Schmidt 1982 {published data only}
- Küstner U, Renfordt E, Müller‐Oerlinghausen B. Relationship between neuroleptic medication, handwriting area and therapeutic response. Proceedings of the 14. CINP‐Congress, Florence. 1984.
- Meya U, Renfordt E. Can changes in eye‐contacts predict therapeutic outcome in schizophrenic patients undergoing neuroleptic treatment?. Pharmacopsychiatry 1986;19:429‐33. [DOI] [PubMed] [Google Scholar]
- Renfordt E, Küstner U, Müller‐Oerlinghausen B. not available [Handschriftveränderungen unter Perazin und Haloperidol]. In: Helmchen H, Hippius H, Tölle R editor(s). Therapie mit Neuroleptika ‐ Perazin. Stuttgart, New York: Thieme‐Verlag, 1988:55‐60. [Google Scholar]
- Schmidt LG, Schüssler G, Kappes C‐V, Müller‐Oerlinghausen B. A double‐blind trial of 2 oral neuroleptics (perazine vs. haloperidol) in different dosages for acute schizophrenic patients [Vergleich einer höher dosierten Haloperidol‐Therapie mit einer Perazin‐Standard‐Therapie bei akut schizophrenen Patienten]. Nervenarzt 1982;53(9):530‐6. [PubMed] [Google Scholar]
- Schmidt LG, Schüssler G, Kappes V, Mühlbauer H, Müller‐Oerlinghausen B. A double‐blind comparison of the antipsychotic efficacy of perazine compared to haloperidol [Doppelblind‐Studie zur antipsychotischen Wirkung von Perazin verglichen mit Haloperidol]. Arzneimittel‐Forschung 1982;32(8):910. [Google Scholar]
- Schüssler G, Müller‐Oerlinghausen B, Schmidt LG. not available [Vergleich einer höher dosierten Haloperidoltherapie mit einer Perazinstandardtherapie bei akut schizophrenen Patienten]. Therapie mit Neuroleptika ‐ Perazin. Stuttgart, New York: Thieme‐Verlag, 1988:40‐50. [Google Scholar]
Tybura 2012 {published data only}
- Samochowiec J, Tybura P, Wysiecka JP. Pharmacogenetic studies of ziprasidone, olanzapine and perazine in paranoid schizophrenia. Proceedings of the 162nd Annual Meeting of the American Psychiatric Association; 2009 May 16‐21; San Francisco, CA 2009.
- Tybura P, Samochowiec A, Beszlej A, Grzywacz A, Mak M, Frydecka D, et al. Some dopaminergic genes polymorphisms are not associated with response to antipsychotic drugs in schizophrenic patients. Pharmacological Reports 2012;64:528‐35. [DOI] [PubMed] [Google Scholar]
Wetzel 1991 {published data only}
- Wetzel H, Bardeleben U, Holsboer F, Benkert O. Zotepine versus perazine in paranoid schizophrenia: a double‐blind controlled trial of antipsychotic efficacy [Zotepin versus Perazin bei Patienten mit paranoider Schizophrenie: eine doppeltblind‐kontrollierte Wirksamkeitsprüfung]. Fortschritte der Neurologie und Psychiatrie 1991;59 Suppl 1:23‐9. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bender 1999 {published data only}
- Bender S, Czekalla J, Hille S, Schlömer T, Schlebusch A, Gastpar M. Evaluation of dry mouth and its treatment in the course of antipsychotic pharmacotherapy. European Neuropsychopharmacology 1999;9 Suppl 5:293. [Google Scholar]
Czekalla 1999 {published data only}
- Czekalla J, Bender S, Dittmann RW, Gastpar M. Effect of olanzapine on salivation is less prominent than predicted by its in vitro muscarinic binding profile. European Neuropsychopharmacology 1999;9 Suppl 5:292‐3. [Google Scholar]
Fischer 1992 {published data only}
- Fischer S, Kissling W, Kuß H‐J. Schizophrenic patients treated with high dose phenothiazine or thioxanthene become deficient in polyunsaturated fatty acids in their thrombocytes. Biochemical Pharmacology 1992;44(2):317‐23. [DOI] [PubMed] [Google Scholar]
Gaebel 1981 {published data only}
- Gaebel W, Pietzcker A, Poppenberg A. Predictors of the course of schizophrenic diseases under neuroleptic long‐term medication [Prädiktoren des Verlaufs schizophrener Erkrankungen unter neuroleptischer Langzeitmedikation]. Pharmacopsychiatry 1981;14:180‐8. [DOI] [PubMed] [Google Scholar]
Gaebel 1986 {published data only}
- Gaebel W, Oschinsky AM. Low‐dose neuroleptic treatment in neurotic disorders. Abstract book of the the 15. CINP‐Congress. San Juan, Puerto Rico, 1986.
Gaebel 1988 {published data only}
- Gaebel W, Pietzcker A, Ulrich G, Schley J, Müller‐Oerlinghausen B. Predictors of neuroleptic treatment response in acute schizophrenia: results of a treatment study with perazine. Pharmacopsychiatry 1988;21:384‐6. [DOI] [PubMed] [Google Scholar]
- Gaebel W, Schley J, Müller‐Oerlinghausen B. Early pharmacokinetic and clinical data as predictors of response to acute antipsychotic treatment with perazine. Abstract book of the 15. CINP‐Congress at San Juan, Puerto Rico. 1986.
- Gaebel W, Schley J, Renfordt E, Müller‐Oerlinghausen B. Early treatment effects of perazine as predictor of acute antipsychotic treatment outcome in schizophrenics. Acta Pharmacolgica Toxicologica 1986;59 Suppl 5:43. [Google Scholar]
Gaebel 1992 {published data only}
- Gaebel W, Wölwer W. Facial expression and emotional face recognition in schizophrenia and depression. Psychiatry and Clinical Neuroscience 1992;242:46‐52. [DOI] [PubMed] [Google Scholar]
Gerson 1964 {published data only}
- Gerson IM, Chat E, Twigger NA. Clinical trial of a potentiated diketopiperazine derivative as a psychopharmacological agent for the treatment of psychotic patients. American Journal of Psychiatry 1964;121:179‐81. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Grabe 1999 {published data only}
- Grabe HJ, Wolf T, Grätz S, Laux G. The influence of clozapine and typical neuroleptics on information processing of the central nervous sstem under cinical conditions in schizophrenic disorders: implications for fitness to drive. Neuropsychobiology 1999;40:196‐201. [DOI] [PubMed] [Google Scholar]
Jarema 1999 {published data only}
- Jarema M, Konieczynska Z. Quality of life of schizophrenic patients treated with classic and "old" atypical neuroleptics. European Neuropsychopharmacology 1999;9 Suppl 5:269. [PubMed] [Google Scholar]
Jockers‐Scherübl1997 {published data only}
- Jockers‐Scherübl MC, Godemann F, Pietzcker A. Paroxetine augmentation of neuroleptic treatment in chronic schizophrenia: An open pilot study. Proceedings of the 20th Symposium of the AGNP, Nürnberg. 1997.
Kemperdick 1967 {published data only}
- Kemperdick KT, Nieling C, Steinig A. not available [Behandlung schizophrener Psychosen mit schwach potenten Neuroleptika]. Medizinische Klinik 1967;62:1512‐4. [PubMed] [Google Scholar]
Kuhs 1988 {published data only}
- Kuhs H, Eikelmann B. not available. In: Helmchen H, Hippius H, Tölle R editor(s). Therapie mit Neuroleptika‐Perazin. Stuttgart, New York: Thieme‐Verlag, 1988:51‐6. [Google Scholar]
Loza 2001 {published data only}
- Loza B, Kucharska‐Pietura K, Debowska G. Atypical versus typical antipsychotic treatment prognosis in first‐episode paranoid schizophrenia based on wcst and dichotic listening scores. European Neuropsychopharmacology 2001;11(3):285. [14th Congress of the European College of Neuropsychopharmacology [Congress Information System]: Conifer, Excerpta Medica Medical Communications BV, 2001 P2103#] [Google Scholar]
- Mazurek I, Loza B, Lecyk A. Atypical versus typical antipsychotic treatment prognosis based on veps and wcst scores in paranoid schizophrenia. Journal of the European College of Neuropsychopharmacology 2003;13(4):S347. [P.2.156] [Google Scholar]
Ohlmeier 2007 {published data only}
- Ohlmeier MD, Jahn K, Wilhelm‐Gossling C, Godecke‐Koch T, Hoffmann J, Seifert J, et al. Perazine and carbamazepine in comparison to olanzapine in schizophrenia. Neuropsychobiology 2007;55(2):81‐8. [MEDLINE: ] [DOI] [PubMed] [Google Scholar]
Pietzcker 1981 {published data only}
- Pietzcker A, Poppenberg A, Schley J, Müller‐Oerlinghausen B. Outcome and risks of ultra‐long‐term treatment with an oral neuroleptic drug. Archives of Psychiatry and Neurological Sciences 1981;229:315‐29. [DOI] [PubMed] [Google Scholar]
Pietzcker 1984 {published data only}
- Pietzcker A, Müller‐Oerlinghausen B. The outpatient clinic for patients under chronic lithium treatment as a phase‐IV research tool [Katamnesen für Patienten unter Lithium‐ und Neuroleptikabehandlung als Forschungshilfsinstrument in der Phase IV der Arzneimittelforschung]. Pharmacopsychiatry 1984;17:162‐7. [DOI] [PubMed] [Google Scholar]
Rein 1983 {published data only}
- Rein W, Schied HW, Breyer‐Pfaff U, Straube E. Prediction and evaluation criteria in perazine therapy of acute schizophrenics: Methodological considerations, sample population, and research variables [Prädiktion und Verlaufsbeurteilung bei Perazin‐Therapie akut Schizophrener: Methodologische Überlegungen, Patientengut und Untersuchungsvariable]. Pharmacopsychiatry 1983;16:147‐51. [DOI] [PubMed] [Google Scholar]
Schied 1983 {published data only}
- Schied HW, Rein W, Straube E, Jung H, Breyer‐Pfaff U. Prediction and evaluation criteria in perazine therapy of acute schizophrenics: psychopathological results [Prädiktion und Verlaufsbeurteilung bei Perazin‐Therapie akut Schizophrener: Psychopathologische Ergebnisse]. Pharmacopsychiatry 1983;16:152‐9. [DOI] [PubMed] [Google Scholar]
Schmidt 1988 {published data only}
- Schmidt LG, Siemetzki H. Assessment of neuroleptic therapy in schizophrenic patients admitted to a university psychiatric department [Differentielle Wirkprofile der neuroleptischen Therapie akut Schizophrener]. Nervenarzt 1988;59:721‐6. [PubMed] [Google Scholar]
Terminska 1989 {published data only}
- Terminska K, Mrowiec W. Comparison of influence of perazine, fluphenazine, trifluoroperazine, chloropromazine and haloperidol on primary and deficit schizophrenic symptoms in patients first hospitalized because of paranoid schizophrenia [Badanie Porownawcze Wplywu Perazyny, Flufenazyny, Trifluoroperazyny, Chloropramazyny I Haloperidolu Na Objawy Pierwotne I Deficytowe Pierwszego Zachorowania Na Schizofrenie Paranoidalna]. Psychatria Polska 1989;23(1):24‐30. [PubMed] [Google Scholar]
Volz 1994 {published data only}
- Volz HP, Mackert A, Diefenbacher A, Friedrich A, Gaebel W, Müller H, et al. Orthostatic challenge during neuroleptic test dose: A possible predictor of short‐term outcome. Pharmacopsychiatry 1994;30(2‐3):94‐100. [DOI] [PubMed] [Google Scholar]
Wetterling 1996 {published data only}
- Wetterling T, Müßigbrodt H. not available [Gewichtszunahme: eine Nebenwirkung von Zotepin (Nipolept)?]. Nervenarzt 1996;67:256‐61. [PubMed] [Google Scholar]
Wetzel 1991 {published data only}
- Wetzel H, Wiedemann K, Holsboer F, Benkert O. Savoxepine: Invalidation of an "atypical" neuroleptic response pattern predicted by animal models in an open clinical trial with acute schizophrenic patients. Psychopharmacology 1991;103(2):280‐3. [DOI] [PubMed] [Google Scholar]
Additional references
Alderson 2004
- Alderson P, Green S, Higgins JPT. Cochrane Reviewers' Handbook 4.2.2 [updated December 2003]. The Cochrane Library. Chichester, UK: John Wiley & Sons, Ltd, 2004, issue Issue 1. [In: The Cochrane Library, Issue 1, 2004. Chichester, UK: John Wiley & Sons, Ltd.]
Altman 1996
- Altman DG, Bland JM. Detecting skewness from summary information. BMJ 1996;313:1200. [PER170800] [DOI] [PMC free article] [PubMed] [Google Scholar]
Begg 1996
- Begg C, Cho M, Eastwood S, Horton R, Moher D, Olkin I, et al. Improving the quality of reporting of randomized controlled trials. The CONSORT Statement. JAMA 1996;276(8):637‐9. [DOI] [PubMed] [Google Scholar]
Benkert 1996
- Benkert O, Hippius H. Psychiatrische Pharmakotherapie. 6th Edition. Berlin: Springer Verlag, 1996. [Google Scholar]
Bland 1997
- Bland JM. Statistics notes. Trials randomised in clusters. BMJ 1997;315:600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Boissel 1999
- Boissel JP, Cucherat M, Li W, Chatellier G, Gueyffier F, Buyse M, et al. The problem of therapeutic efficacy indices. 3. Comparison of the indices and their use [Apercu sur la problematique des indices d'efficacite therapeutique, 3: comparaison des indices et utilisation. Groupe d'Etude des Indices D'efficacite]. Therapie 1999;54(4):405‐11. [PUBMED: 10667106] [PubMed] [Google Scholar]
Carpenter 1994
- Carpenter WT, Buchanan RW. Schizophrenia. New England Journal of Medicine 1994;330:681‐90. [DOI] [PubMed] [Google Scholar]
Davey Smith 1998
- Davey Smith G, Egger M. Meta‐analysis: Unresolved issues and future developments. BMJ 1998;16:221‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2000
- Deeks J. Issues in the selection for meta‐analyses of binary data. Proceedings of the 8th International Cochrane Colloquium; 2000 Oct 25‐28; Cape Town. Cape Town: The Cochrane Collaboration, 2000.
Divine 1992
- Divine GW, Brown JT, Frazier LM. The unit of analysis error in studies about physicians' patient care behavior. Journal of General Internal Medicine 1992;7(6):623‐9. [DOI] [PubMed] [Google Scholar]
Donner 2002
- Donner A, Klar N. Issues in the meta‐analysis of cluster randomized trials. Statistics in Medicine 2002;21:2971‐80. [DOI] [PubMed] [Google Scholar]
Duggan 2005
- Duggan L, Fenton M, Rathbone J, Dardennes R, El‐Dosoky A, Indran S. Olanzapine for schizophrenia. Cochrane Database of Systematic Reviews 2005, Issue 2. [DOI: 10.1002/14651858.CD001359.pub2] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey‐Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple graphical test. BMJ 1997;315:629‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Elbourne 2002
- Elbourne D, Altman DG, Higgins JPT, Curtina F, Worthingtond HV, Vaile A. Meta‐analyses involving cross‐over trials: methodological issues. International Journal of Epidemiology 2002;31(1):140‐9. [DOI] [PubMed] [Google Scholar]
Endicott 1976
- Endicott J, Spitzer RL, Fleiss JL, Cohen J. The Global Assessment Scale. Archives of General Psychiatry 1976;33:766‐71. [DOI] [PubMed] [Google Scholar]
Enss 1958
- Enss H, Hartmann K, Richter H‐E. not available [Klinische Erfahrungen mit einem neuen Piperazin‐Derivat des Phenothiazins in der Neuro‐Psychiatrie]. Archiv für Neurologie und Nervenkrankheiten 1958;197:534. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(7):7‐10. [DOI] [PubMed] [Google Scholar]
Grohmann 1988
- Grohmann R, Koch R, Rüther E, Schmidt LG. not available [Nebenwirkungen von Perazin im Vergleich zu anderen Neuroleptika]. In: Helmchen H, Hippius H, Tölle R editor(s). Therapie mit Neuroleptika ‐ Perazin. Stuttgart/New York: Georg Thieme Verlag, 1988:74‐83. [Google Scholar]
Gulliford 1999
- Gulliford MC, Ukoumunne OC, Chinn S. Components of variance and intraclass correlations for the design of community‐based surveys and intervention studies: data from the Health Survey for England 1994. American Journal of Epidemiology 1999;149:876‐83. [DOI] [PubMed] [Google Scholar]
Guy 1976
- Guy W, Bonato RR. Clinical Global Impressions. Manual for the ECDEU Assessment Battery, 2. Rev ed. National Institute of Mental Health, 1970. [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327:557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2011]. The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org..
Hippius 1962
- Hippius H. Long‐term pharmacotherapy of schizophrenia. Proceedings of the II International Conference of the Manfred Sakel Foundation. New York: CC Page and Co, 1962.
Hutton 2009
- Hutton JL. Number needed to treat and number needed to harm are not the best way to report and assess the results of randomised clinical trials. British journal of haematology 2009;146(1):27‐30. [DOI] [PubMed] [Google Scholar]
Jadad 1996
- Jadad A, Moore A, Carrol D, Jenkinson C, Reynolds DJM, Gavanagh DJ, McQuay HJ. Assessing the quality of reports of randomised controlled trials: is blinding necessary?. Controlled Clinical Trials 1996;17:1‐12. [DOI] [PubMed] [Google Scholar]
Kay 1986
- Kay SR, Opler LA, Fiszbein A. Positive and Negative Syndrome Scale (PANSS) Manual. North Tonawanda, NY: Multi‐Health Systems, 1986. [Google Scholar]
Krüger 1963
- Krüger HJ, Schwarz H. not available [Neue Aspekte der psychiatrischen Langzeittherapie in der psychiatrischen Ambulanz und Klinik]. Medizinische Welt 1963;9:463‐7. [Google Scholar]
Leucht 2005
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel RR. What does the PANSS mean?. Schizophrenia Research 2005;79(2‐3):231‐8. [PUBMED: 15982856] [DOI] [PubMed] [Google Scholar]
Leucht 2005a
- Leucht S, Kane JM, Kissling W, Hamann J, Etschel E, Engel R. Clinical implications of brief psychiatric rating scale scores. British Journal of Psychiatry 2005;187:366‐71. [PUBMED: 16199797] [DOI] [PubMed] [Google Scholar]
Leucht 2007
- Leucht S, Engel RR, Bauml J, Davis JM. Is the superior efficacy of new generation antipsychotics an artifact of LOCF?. Schizophrenia Bulletin 2007;33(1):183‐91. [PUBMED: 16905632] [DOI] [PMC free article] [PubMed] [Google Scholar]
Marshall 2000
- Marshall M, Lockwood A, Bradley C, Adams C, Joy C, Fenton M. Unpublished rating scales: a major source of bias in randomised controlled trials of treatments for schizophrenia. British Journal of Psychiatry 2000;176:249‐52. [DOI] [PubMed] [Google Scholar]
Marvaha 2004
- Marwaha S, Johnson S. Schizophrenia and employment? A review. Social Psychiatry and Psychiatric Epidemiology 224;39:337‐49. [DOI] [PubMed] [Google Scholar]
Menge 1988
- Menge HG, Brand U. not available [Pharmakologie von Perazin und seinen Hauptmetaboliten]. In: Helmchen H, Hippius H, Tölle R editor(s). Therapie mit Neuroleptika ‐ Perazin. Stuttgart/New York: Georg Thieme Verlag, 1988:9‐13. [Google Scholar]
Overall 1962
- Overall JE, Gorham DR. The Brief Psychiatric Rating Scale. Psychological Reports 1962;10:799‐812. [Google Scholar]
Schünemann 2008
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S editor(s). Cochrane Handbook for Systematic Reviews of Interventions. The Cochrane Collaboration, 2008:359‐83. [Google Scholar]
Seeman 1981
- Seeman P. Brain dopamine receptors. Pharmacological Review 1981;32:3. [PubMed] [Google Scholar]
Tsuang 1978
- Tsuang MT. Suicide in schizophrenics, manics, depressives, and surgical controls: a comparison with general population suicide mortality. Archives of General Psychiatry 1978;35:153‐55. [DOI] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JAC, Burney PGJ. Methods for evaluating area‐wide and organistation‐based intervention in health and health care: a systematic review. Health Technology Assessment 1999;3(5):1‐75. [PubMed] [Google Scholar]
Xia 2009
- Xia J, Adams CE, Bhagat N, Bhagat V, Bhoopathi P, El‐Sayeh H, et al. Loss to outcomes stakeholder survey: the LOSS study. Psychiatric Bulletin 2009;33(7):254‐7. [Google Scholar]