

Abstract
A rapidly expanding repertoire of neural antibody biomarkers exists for autoimmune central nervous system (CNS) disorders. Following clinical recognition of an autoimmune CNS disorder, the detection of a neural antibody facilitates diagnosis and informs prognosis and management. This review considers the phenotypes, diagnostic assay methodologies, and clinical utility of neural antibodies in autoimmune CNS disorders. Autoimmune CNS disorders may present with a diverse range of clinical features. Clinical phenotype should inform the neural antibodies selected for testing via the use of phenotype-specific panels. Both serum and cerebrospinal fluid (CSF) are preferred in the vast majority of cases but for some analytes either CSF (e.g. N-methyl-d-aspartate receptor [NMDA-R] IgG) or serum (e.g. aquaporin-4 [AQP4] IgG) specimens may be preferred. Screening using 2 methods is recommended for most analytes, particularly paraneoplastic antibodies. We utilize murine tissue-based indirect immunofluorescence assay (TIFA) with subsequent confirmatory protein-specific testing. The cellular location of the target antigen informs choice of confirmatory diagnostic assay (e.g. blot for intracellular antigens such as Hu; cell-based assay for cell surface targets such as leucine-rich glioma inactivated 1 [LGI1]). Titers of positive results have limited diagnostic utility with the exception of glutamic acid decarboxylase (GAD) 65 IgG autoimmunity, which is associated with neurological disease at higher values. While novel antibodies are typically discovered using established techniques such as TIFA and immunoprecipitation-mass spectrometry, more recent high-throughput molecular technologies (such as protein microarray and phage-display immuno-precipitation sequencing) may expedite the process of antibody discovery. Individual neural antibodies inform the clinician regarding the clinical associations, oncological risk stratification and tumor histology, the likely prognosis, and immunotherapy choice. In the era of neural antibody biomarkers for autoimmune CNS disorders, access to appropriate laboratory assays for neural antibodies is of critical importance in the diagnosis and management of these disorders.
Keywords: Autoimmune neurology, Neural antibody testing, Autoimmune central nervous system disorders, Antibody panels
1. Introduction
An expanding repertoire of disease-specific IgG biomarkers for autoimmune neurologic disorders has led to the emergence of autoimmune neurology as a clinical subspeciality [1]. Autoimmune neurological disorders may be paraneoplastic, where an immune response developed against an underlying cancer against the host nervous system, or non-paraneoplastic where the etiology is often unknown [2]. Sometimes a trigger for non-paraneoplastic neurologic autoimmunity is identified, including infections (particularly herpes viruses), immunization and, in some instances, immune-checkpoint inhibitor therapy [3–5]. Neurologic autoimmunity may affect any anatomic portion of the neuraxis. In this review we consider only autoimmune disorders affecting the central nervous system (CNS), though the principles are generalizable to autoimmune neurologic disorders which affect the peripheral nervous system. The number of clinically validated antibody biomarkers continues to expand at a rapid rate and will likely increase further in the future due to the availability of molecular diagnostic tools that can be deployed to unmask the antigen specificity of novel antibodies [6,7]. It is likely that increased availability of neural antibody biomarkers has led to improved recognition of autoimmune neurologic disorders over the past two decades [8]. Access to accurate and reliable diagnostic laboratory assays is therefore a critical component in the diagnosis and management of patients with autoimmune CNS disorders. In this review we will consider the clinical presentations, diagnostic assays, and clinical utility of neural antibodies for autoimmune CNS disorders.
2. Clinical phenotypes
Autoimmune central nervous system disorders may affect any anatomic region of the CNS from cerebrum to anterior horn cell. They may be unifocal, when symptoms localize to one anatomic region, or multifocal when more than one anatomic site is affected. The onset of autoimmune CNS disorders is usually subacute (<3 months by the time of clinical presentation), although chronic clinical courses have also been occasionally described which can mimic phenotypically atypical neurodegenerative disorders [9]. The clinical presentations of individual autoimmune neurologic disorders are considered below.
2.1. Encephalitis
2.1.1. Limbic
Autoimmune encephalitis presents typically with altered mental status and symptoms localizing to the cerebral cortex. Limbic encephalitis is an anatomically-defined subcategory of autoimmune encephalitis which presents with subacute-onset personality change, short-term memory deficits, seizures and psychosis[10]. Diagnostic criteria require the presence of characteristic MRI signal change affecting the mesial temporal lobes bilaterally, in addition to either elevation in cerebrospinal fluid (CSF) white cell count (pleocytosis) or electroencephalogram (EEG) abnormalities involving the temporal lobes[11]. Examples include antineuronal nuclear antibody (ANNA)-1 and gamma-aminobutyric acid-B receptor (GABAB-R) autoimmunity, both of which may present as a paraneoplastic limbic encephalitis often in association with small cell lung cancer (SCLC) [12,13]. Leucine-rich glioma inactivated 1 (LGI1) encephalitis may present with an autoimmune limbic encephalitis; a distinctive seizure semiology termed faciobrachial dystonic seizures (FBDS) frequently occurs and underlying tumors are rare (usually thymoma) [14,15]. Formes frustes of autoimmune encephalitis (limbic or extra-limbic) may also occur and are usually designated with phenotypically descriptive terms such as autoimmune dementia, autoimmune epilepsy or autoimmune movement disorders [16–18].
2.1.2. Extralimbic
In patients with clinical or paraclinical findings localizing to extra-temporal and non-limbic structures the term extra-limbic encephalitis is sometimes employed [19]. Examples of this phenotype include anti-GABAA-R extralimbic encephalitis which can present with seizures and multifocal cerebral cortical, subcortical or juxtacortical lesions on magnetic resonance imaging (MRI) [20]. Glial Fibrillary Acidic Protein (GFAP) astrocytopathy is a steroid-responsive encephalitis (often with co-existing myelopathy) that most frequently affects deep, periventricular white matter with characteristic perivascular radial MRI lesions. In addition to encephalopathy, patients may display diverse CNS findings including meningism, parkinsonism, ataxia and eye-movement abnormalities [21,22]. ANNA-1 encephalitis may rarely present with an extralimbic encephalitis and this subphenotype of ANNA-1 autoimmunity is most frequently associated with focal motor seizures localizing to the peri-Rolandic cortex [23]. Certain neural antibodies are associated with well-recognized biomarker-defined clinical syndromes and may be suspected on clinical grounds. Examples include N-methyl-d-aspartate receptor (NMDA-R) encephalitis, which presents with prodromal psychiatric symptoms, followed by psychosis, seizures, abnormal movements, sleep disorders and autonomic instability [24].
2.2. Rapidly progressive cerebellar ataxia
Autoimmune cerebellar ataxia usually consists of a pancerebellar syndrome (such as gait disorder, dysarthria and limb ataxia), though the initial presentation may be with a more isolated syndrome with diplopia, vertigo and unsteadiness. Onset is usually rapid (<3 months to clinical presentation) in contrast with genetic or degenerative ataxias. Gait ataxia may be more prominent than limb ataxia due to the preferential involvement of the vermis in autoimmune cerebellar ataxias [25]. The most frequent antibodies associated with autoimmune cerebellar ataxia are Purkinje cytoplasmic antibody type 1 (PCA-1, also known as anti-Yo) and glutamic acid decarboxylase (GAD) 65 IgG. Overall, stabilization or modest improvements may occur with immune therapies. Outcomes tend to be worse for patients who harbor antibodies to intracellular antigens with the exception of GAD65 cerebellar ataxia which sometimes demonstrates a robust immunotherapy response [26]. Certain clinical features may help differentiate between autoimmune ataxia syndromes such as eye-movement abnormalities in Septin-5 ataxia, co-existing stiff person spectrum disorder in GAD65 autoimmunity, or altered taste sensation in metatropic glutamate receptor (mGluR)-1 ataxia [26–28].
2.3. Brainstem encephalitis
Autoimmune brainstem encephalitis presents with oculomotor abnormalities, vestibulocochlear or bulbar dysfunction, often in addition to cerebellar findings [29]. Onset is subacute in the majority of cases. Notable exceptions exist such as IgLON5 autoimmunity which typically displays a more progressive course[30]. Antibodies frequently associated with brainstem encephalitis include kelch-like protein 11 (KLHL-11), antineuronal nuclear antibody (ANNA)-2 (anti-Ri) and IgLON5 IgG [31]. Bickerstaff’s Brainstem Encephalitis may present with multiple cranial nerve abnormalities in association with ganglioside Q1B (GQ1B) IgG autoimmunity [32]. Certain clinical features are evocative of particular antibody specificities such as jaw dystonia with ANNA-2 brainstem encephalitis or vestibulocochlear dysfunction with KLHL-11 [33,34] autoimmunity. Sleep disorders (insomnia, rapid-eye-movement [REM] sleep behavior disorder, sleep disordered breathing) are commonly encountered with IgLON5 autoimmunity but may occur in all forms of brainstem encephalitis presumably through involvement of brainstem arousal pathways [35]. In severe cases of brainstem encephalitis, life-threatening cardiorespiratory dysfunction may occur either directly as a result of medullary involvement or indirectly due to laryngospasm [36–38].
2.4. Myelopathy
Autoimmune myelopathy presents with ascending sensorimotor deficits in one or more limbs. Urinary retention/incontinence or sexual dysfunction may coexist. A sensory level in the trunk in conjunction with upper motor neuron signs (spasticity or hyperreflexia) is characteristic, although early in the disease course upper motor neuron signs may not yet be present [39]. L’hermitte phenomenon (sensory symptoms provoked by neck flexion) or Uhtoff phenomenon (worsening of neurologic symptoms due to heat) suggest a demyelinating etiology [40]. Autoimmune myelopathies frequently associate with myelin oligodendrocyte glycoprotein (MOG) or aquaporin-4 (AQP4) antibodies. Optic neuritis (unilateral or bilateral) may also occur with or without evidence of cerebral demyelination. Although both disorders have overlapping features (and were historically included under the term neuromyelitis optica spectrum disorder [NMOSD]), MOG antibody disease (MOGAD) is now considered a separate entity [41]. In both disorders, spinal cord lesions are typically longitudinally extensive (affecting 3 or more vertebral segments), usually affecting the central grey matter [42,43]. Autoimmune paraneoplastic myelopathies, such as those encountered with amphiphysin and collapsin-responsive mediator protein (CRMP)-5 IgG, may demonstrate tract-specific lesions (for example lesions selective for the dorsal columns) [44]. Certain clinical and radiographic features can point towards a particular diagnosis, such as conus involvement in MOG antibody disease (MOGAD) or longitudinally-extensive spinal cord lesions often with central canal enhancement in GFAP IgG astrocytopathy [21,45].
2.5. Encephalomyelitis
Autoimmune encephalomyelitis is a multifocal disorder where patients have symptoms localizing to cerebrum and spinal cord, which may also extend to the peripheral nervous system[46]. This phenotype is considered high-risk for an underlying paraneoplastic etiology (for example, in the case of ANNA-2 autoimmunity and SCLC)[12]. Encephalomyelitis may be autoimmune non-paraneoplastic as in autoimmune GFAP astrocytopathy or MOGAD. Both of these disorders are typically preceded by a flu-like prodrome, but meningeal symptoms (neck stiffness, photophobia and vomiting) are frequently encountered in GFAP astrocytopathy. MOGAD may present with large enhancing fluffy or tumefactive lesions in the brain or cerebellum along with co-existing myelitis or optic neuritis. Acute disseminated encephalomyelitis (ADEM) is a monophasic autoimmune disorder with encephalopathy and other multifocal neurologic deficits, usually affecting pediatric patients. Onset classically occurs following infection or immunization, and can occur in association with MOG or (less commonly) AQP4 antibodies [47,48].
2.6. Opsoclonus-myoclonus syndrome
Opsoclonus-myoclonus syndrome (OMS) is a disorder characterized by the onset of arrhythmic multidirectional conjugate saccadic eye movements (opsoclonus) and myoclonic jerks. Ataxia, cognitive and behavioral changes may also feature [49]. The disorder is more frequently seen in children where there is a strong association with neuroblastoma [50]. In adults, seropositive OMS occurs most commonly in association with ANNA-2 IgG [49]. Less commonly, NMDA-R, GABAB, GAD65 and dipeptidyl-peptidase-like protein (DPPX) antibodies have been described [51].
2.7. Stiff person syndrome
Stiff person syndrome (SPS) is a disorder characterized by progressive muscle rigidity (usually axial), spasms and abnormal posturing. Symptoms may be exacerbated with heightened emotion or startle [52]. It associates with GAD65 IgG in the majority of cases. SPS is rarely paraneoplastic but in such cases amphiphysin is the most frequently antibody encountered (usually in women with breast cancer) [53]. The term stiff person spectrum disorder (SPSD) has been defined to include variants of SPS. Variants include a limb restricted phenotype (‘stiff limb syndrome’) which usually affects a single lower extremity [52]. On the other end of the clinical spectrum, a severe phenotype termed Progressive Encephalomyelitis with Rigidity and Myoclonus (PERM) may occur usually in association with glycine receptor IgG [54]. This presents with features of both SPS and a disseminated encephalomyelitis [55].
3. Diagnostic testing
3.1. Clinical and paraclinical evaluation
Autoimmune CNS disorders are almost always subacute in onset. Clinical assessment typically localizes the disorder within the central nervous system and may identify pertinent individual risk factors for an autoimmune etiology (a strong family or personal history of autoimmunity, history of malignancy, smoking status or immune check-point inhibitor exposure). Where present, an inflammatory CSF profile (pleocytosis, elevated IgG index, CSF-exclusive oligoclonal bands [OCBs] or elevated CSF kappa free light chains) increases the level of suspicion for an autoimmune etiology. MRI findings vary depending on the clinical phenotype, but characteristic radiographic features have been identified in autoimmune limbic encephalitis, GFAP, neuromyelitis optica spectrum disorder (NMOSD), and MOGAD which may direct the clinician towards a specific diagnosis [21,39,45]. EEG may be pursued to document encephalopathy and seizures, including subclinical events.
3.2. Importance of antibody biomarkers
A syndrome-based diagnostic algorithm for autoimmune encephalitis was proposed in 2016 and includes clinical criteria for definite, probable and possible autoimmune encephalitis. The criteria for ’definite’ autoimmune encephalitis can also be satisfied by meeting ‘possible’ clinical criteria in the presence of neural antibody positivity[11]. Separate disease-specific clinical criteria also exist for individual autoimmune disorders such as NMDA-R encephalitis, Bickerstaff’s brainstem encephalitis and Stiff Person Syndrome [11,56]. Syndrome-based autoimmune encephalitis criteria (possible, probable and definite) are less sensitive for patients with limited or atypical presentations (such as isolated seizures, dementia, or brainstem presentations) who harbor antibodies that are highly specific for neurologic autoimmunity [31,57]. With the exception of OMS associated with neuroblastoma or SCLC, more recent diagnostic criteria for closely related paraneoplastic neurologic disorders from 2021 require antibody seropositivity to reach a definite diagnosis [58]. Therefore, comprehensive laboratory testing for neural antibody biomarkers of autoimmune CNS disorders remains of vital clinical importance in reaching a confident diagnosis.
3.3. Antibody profiles are preferred over single-antibody testing
Individual neurologic phenotypes are associated with a diverse range of neural antibodies and therefore isolated analyte testing is not recommended [59]. The use of comprehensive antibody evaluations is recommended in patients with a suspected autoimmune CNS disorder, especially considering the rapidly expanding repertoire of antibody biomarkers discovered in the past two decades [58]. Isolated testing may lead to the failure to detect a diagnostically informative antibody and sequential testing of individual antibodies can lead to diagnostic delay [60]. Phenotype specific panels serve the purpose of testing for all antibodies known to associate with a specific clinical presentation and therefore provide assurance to the requesting physician [59]. The number of analytes may vary extensively depending on the neurological phenotype: for example, optic neuritis may be evaluated by testing a limited number of antibodies (such as AQP4 or MOG IgG) whereas over 20 antibodies are included in the movement disorder evaluation (Table 3). In children, neural antibody profiles associated with neurologic phenotypes differs from that observed in adults and therefore we recommend a separate pediatric-specific evaluation in children with suspected autoimmune CNS disorders [61]. Antibody evaluations associated with specific autoimmune CNS disorder phenotypes are listed in Table 3.
Table 3.
Phenotype-specific neural antibody evaluations.
| CNS Phenotype | Pertinent neural antibodies |
|---|---|
| Encephalitis [1–23] | AMPA-R, AK-5, Amphiphysin, AGNA-1, ANNA-1-3, CASPR2, CRMP-5, DPPX, GABA-A, GABA-B, GAD65, GFAP, IgLON5, LGI1, mGluR1, Neurexin 3α, Neurochondrin, NF-L, NMDA-R, PCA1,2 and Tr, Septin-7 |
| Ataxia and other movement disorders [1,3–7,9–11,13–16,18–33] | AMPA-R, Amphiphysin, AGNA-1, ANNA-1-3, AP3B2, CASPR2, CRMP-5, DPPX, GABA-A, GABA-B, GAD65, GFAP, GRAF-1, IgLON5, ITPR-1, KLHL-11, LGI1, mGluR1, Neurochondrin, NF-L, NMDA-R, PCA1, 2 and Tr, PDE10A; RGS8, SEZ6L2, Septin-5, Septin-7 |
| Brainstem Encephalitis [3,4,7,13,15,18–20,26,28,34–37] | ANNA-1, ANNA-2, Amphiphysin, AQP4, AGNA-1, GAD65, GFAP, IgLON5, ITPR1, KLHL-11, Neurochondrin, NF-L, NMDA-R, MOG, PCA-1 |
| Myelopathy [6,16,18,20,21,23,25,26,38,39] | Amphiphysin, AQP4, AGNA-1, ANNA-1-3, AP3B2, CRMP-5, DPPX, GAD65, GFAP, mGluR1, MOG, Neurochondrin, NF-L, PCA1, PCA2, Septin 7 |
| Stiff-person syndrome-spectrum (including PERM) [40–43] | Amphiphysin, DPPX, GABA-A, GAD65, Glycine |
| CNS demyelinating disease evaluation (optic neuritis, transverse myelitis, ADEM, UCE) [44,45] | AQP4, MOG |
| Paraneoplastic Retinopathy [46,47] | Recoverin, CRMP5 |
| Pediatric CNS evaluation [8,10,48–56] | ANNA-1, AQP4, CASPR2, DPPX, GABA-B, GAD65, GFAP, LGI1, mGluR1, MOG, Neurochondrin, NMDA-R, PCA-1 |
AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; AK: adenylate kinase; AGNA: anti-glial nuclear antibody; ANNA: antineuronal nuclear antibody; CASPR2: contactin-associated protein-like 2;; CRMP: collapsin-responsive mediator protein; DPPX: dipeptidyl-peptidase-like protein; GABAA: gamma-aminobutyric acid-A receptor; GABABR: gamma-aminobutyric acid-B receptor; GFAP: glial fibrillary acidic protein; GAD: glutamic acid decarboxylase; LGI1: leucine-rich glioma inactivated protein 1; mGluR: metabotropic glutamate receptor;; NF-L: neuronal intermediate filament; NMDA-R: N-methyl-d-aspartate receptor; PCA: Purkinje cell antibody; GRAF1: GTPase Regulator Associated with Focal Adhesion Kinase 1;; ITPR1, inositol 1,4,5-triphosphate receptor 1; KLHL-11: Kelch-like protein 11; AP3B2: adaptor protein 3B2; RGS8: regulator of G-protein signaling 8; SEZ6L2: seizure-like related 6 homolog like 2 antibodies; AQP4: aquaporin 4; MOG: myelin oligodendrocyte glycoprotein; PERM: progressive encephalomyelitis with rigidity and myoclonus.
3.4. Send both Serum and CSF where possible
The testing of both serum and CSF is recommended in the evaluation of patients with autoimmune CNS disorders [58,59]. For patients with suspected autoimmune CNS disorders, lumbar puncture is indicated for the markers of inflammation (e.g white cell count) and a phenotype-specific IgG profile. Testing serum only can pose difficulties for the interpretation of TIFA due to an increased likelihood of interference from non-organ-specific antibodies [62]. Some antibodies detected in the CSF are of greater clinical importance than when they are found in serum only: notable examples include NMDA-R IgG and GFAP IgG where isolated serum positivity is less likely to be clinically significant [63]. NMDA-R IgG in the serum alone is of uncertain clinical significance as it has been found in healthy patients and patients with other neurological disorders [64,65]. GFAP IgG in the serum alone is accompanied by clinical features of GFAP astrocytopathy in only 10 % of cases whereas this increases to 95 % in the presence of GFAP CSF antibodies [66]. In contrast, for other analytes such as LGI1-IgG and AQP4-IgG sensitivity is higher in the serum than the CSF [15,67]. MOG-IgG, similarly, is more readily detected in serum than CSF but recent evidence has demonstrated that a subgroup of MOGAD patients may harbor CSF-exclusive antibodies [68]. The presence of CSF antibodies may have clinical implications in MOGAD and LGI1 encephalitis where it appears to be associated with more severe disease [69,70]. A list of appropriate specimens to test for each neural antibody is provided in Tables 1 and 2.
Table 1.
Antibodies targeting intracellular antigens: preferred specimen type, recommended diagnostic assay, clinical phenotype, cancer risk and associated malignancy.
| Antibody (alternative name) | Preferred Specimen Type | Recommended Diagnostic Assay | Predominant CNS Phenotype | Cancer Risk | Associated neoplasm type |
|---|---|---|---|---|---|
| Adaptor protein 3B2 [1] | Serum/CSF | TIFA, CBA | Ataxia, sensory ataxia | Low | Renal carcinoma, B cell lymphoma |
| AGNA-1 (SOX1) [2] | Serum/CSF | TIFA*, Line blot | Ataxia, encephalitis | High | SCLC |
| Adenylate Kinase-5 (AK-5) [3] | Serum/CSF | TIFA, CBA | Encephalitis | n/a | No cancer association |
| Amphiphysin [4] | Serum/CSF | TIFA, Line blot | Stiff person syndrome, encephalitis, myelopathy, cerebellar syndrome | High | SCLC, breast adenocarcinoma |
| ANNA-1 (Hu) [5] | Serum/CSF | TIFA, Line blot | Ataxia, limbic encephalitis, opsoclonus myoclonus | High | SCLC, thymoma, neuroendocrine, neuroblastoma |
| ANNA-2 (Ri) [6] | Serum/CSF | TIFA, Line blot | Ataxia, opsoclonus myoclonus, dystonia and parkinsonism | High | Breast adeno, SCLC |
| ANNA-3 (DACH1) [7] | Serum/CSF | TIFA, CBA | Ataxia | High | SCLC |
| ARHGAP26 (GRAF1) [8] | Serum/CSF | TIFA, CBA | Brainstem encephalitis, ataxia | Intermediate | Ovarian adenocarcinoma |
| CRMP5 (CV2) [9] | Serum/CSF | TIFA, Western Blot, ELISA | Encephalomyelitis, ataxia, | High | SCLC, thymoma |
| GFAP [10] | CSF | TIFA, CBA | Meningoencephalitis | Low | Ovarian teratoma, various adenocarcinomas |
| GAD-65 [11] | Serum/CSF | TIFA*, RIPA, ELISA | Limbic encephalitis, stiff-person syndrome, ataxia | Low | SCLC, other neuroendocrine, malignant thymoma |
| IgLON-5 [12] | Serum/CSF | TIFA, CBA | Brainstem encephalitis, sleep disorder, movement disorders | n/a | No cancer association |
| ITPR1 [13] | Serum/CSF | TIFA, CBA | Brainstem encephalitis, ataxia, | Intermediate | Breast, lung, haematologic |
| KLHL-11 [14] | Serum/CSF | TIFA, CBA | Ataxia, brainstem encephalitis, vestibulocochlear symptoms | High | Testicular (seminoma), teratoma |
| PCA-1 (Yo) [15] | Serum/CSF | TIFA, Line blot | Ataxia | High | Ovarian & breast adenocarcinoma |
| MAP1B (PCA-2) [16] | Serum/CSF | TIFA, Line blot | Ataxia, encephalomyelitis, | High | Lung (SCLC & NCSLC), breast adenocarcinoma |
| Ma2 [17] | Serum/CSF | TIFA*, ELISA, CBA | Limbic encephalitis, brainstem encephalitis, narcolepsy/cataplexy | High | Testicular germinoma |
| NF-L [18] | Serum/CSF | TIFA, CBA | Ataxia, encephalopathy, myelopathy | High | Neuroendocrine |
| PDE10A [19] | Serum/CSF | TIFA, CBA | Encephalopathy, chorea | High | Lung, renal, pancreatic |
| RGS8[20] | Serum/CSF | TIFA, Line blot | Ataxia | High | Lymphoma |
| TRIM 46 [21] | Serum/CSF | TIFA, CBA | Encephalomyelitis, ataxia | High | SCLC |
| TRIM 9/67 [22] | Serum/CSF | TIFA,CBA | Ataxia | High | NSCLC |
TIFA: Tissue-based indirect immunofluorescence assay; CBA: cell-based assay; Ataxia = cerebellar ataxia; AGNA: anti-glial nuclear antibody; SOX1: SRY-box transcription factor 1; SCLC: small cell lung cancer; ANNA: antineuronal nuclear antibody; DACH1: Dachshund-homolog 1; ARHGAP26: Rho GTPase-activating protein; GRAF1: GTPase Regulator Associated with Focal Adhesion Kinase 1; CRMP: collapsin-responsive mediator protein; ELISA: enzyme-linked immunosorbent assay; GFAP: glial fibrillary acidic protein; GAD: glutamic acid decarboxylase; ITPR1, inositol 1,4,5-triphosphate receptor 1; KLHL-11: kelch-like protein 11; PCA: Purkinje cell antibody; MAP1B: microtubule-associated protein 1B; NSCLC: non-small cell lung cancer; NF-L: neuronal intermediate filament; PDE10A: phosphodiesterase 10A TRIM: tripartite motif-containing protein. *Can be detected by TIFA screening but with reduced sensitivity.
Table 2.
Antibodies targeting extracellular antigens: preferred specimen type, recommended diagnostic assay, clinical phenotype, cancer risk and associated malignancy.
| Antibody (alternative name) | Preferred Specimen Type | Recommended Diagnostic Assays | Predominant CNS Phenotype | Cancer Risk | Associated malignancy |
|---|---|---|---|---|---|
| AMPA-R [1] | Serum/CSF | TIFA, CBA | Limbic encephalitis | Intermediate | SCLC, malignant thymoma, breast, ovarian adenocarcinoma |
| Aquaporin 4 [2,3] | Serum | Live cell FACS, CBA | NMOSD | Low | Adenocarcinoma |
| CASPR2 [4] | Serum | TIFA, CBA | Limbic encephalitis, Morvan Syndrome (cancer risk 50 %) | Low-intermediate | Malignant thymoma |
| DPPX [5] | Serum/CSF | TIFA, CBA | Encephalitis, CNS hyperexcitability, PERM | Low | B-cell neoplasms |
| GABAA [6] | CSF | TIFA, CBA | Encephalitis | Intermediate | Thymoma |
| GABAB [7] | Serum/CSF | TIFA, CBA | Limbic encephalitis | Intermediate | SCLC |
| Glycine-R [8] | Serum/CSF | Live CBA | Limbic encephalitis, PERM | Low | Hodgkin lymphoma, malignant thymoma |
| LGI1 [4] | Serum | TIFA, CBA | Limbic encephalitis | Low | Malignant thymoma, neuroendocrine |
| mGluR1 [9] | Serum/CSF | TIFA, CBA | Ataxia | Low | Hodgkin lymphoma, cutaneous T cell lymphoma |
| mGluR2 [10] | Serum/CSF | TIFA, CBA | Ataxia | High | SCLC, rhabdomyosarcoma |
| mGluR5 [11] | Serum/CSF | TIFA, CBA | Encephalitis, Ophelia Syndrome | Intermediate | Hodgkin lymphoma, SCLC |
| MOG [12,13] | Serum (CSF in rare instances) | Live cell FACS, CBA | MOGAD | Low | Ovarian teratoma |
| Neurexin3α [14] | Serum/CSF | TIFA, CBA | Encephalitis | n/a | No cancer association |
| Neurochondrin [15] | Serum/CSF | TIFA, CBA | Ataxia | Low | Uterine adenocarcinoma |
| NMDAR [16,17] | CSF | TIFA, CBA | NMDAR encephalitis | Intermediate | Teratoma (ovarian or extra-ovarian) |
| PCA-Tr (DNER) [18] | Serum/CSF | TIFA, CBA | Ataxia | High | Hodgkin lymphoma |
| P/Q VGCC [19] | Serum | RIPA | Ataxia | Intermediate | SCLC |
| SEZ6L2 [20–22] | Serum/CSF | TIFA, CBA | Ataxia | Low | Invasive lobular breast carcinoma, SCLC |
| Septin 5 [23] | Serum/CSF | TIFA, CBA | Ataxia | n/a | No cancer association |
| Septin 7 [24] | Serum/CSF | TIFA, CBA | Encephalopathy, myelopathy, psychiatric symptoms | Low | Breast adenocarcinoma, NHL |
AMPAR: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor; TIFA: Tissue indirect immunofluorescence assay; CBA: cell-based assay; SCLC: small cell lung cancer; FACS: fluorescence-activated cell-sorting assay; NMOSD: neuromyelitis optica spectrum disorder; CASPR2: contactin-associated protein-like 2; DPPX: dipeptidyl-peptidase-like protein; PERM: progressive encephalomyelitis with rigidity and myoclonus; GABAA: gamma-aminobutyric acid-A receptor; GABABR: gamma-aminobutyric acid-B receptor; LGI1: leucine-rich glioma inactivated protein 1;; mGluR: metabotropic glutamate receptor; MOG: myelin oligodendrocyte glycoprotein; NMDAR: N-methyl-d-aspartate receptor; PCA: Purkinje cell antibody; VGCC: P/Q type voltage-gated calcium channel; NHL: Non-Hodgkin lymphoma.
3.5. Antibody titers
Overall, the usefulness of antibody values (such as those derived from titration to endpoint) is uncertain in the diagnosis and management of autoimmune CNS disorders. Exceptions include the interpretation of GAD65 IgG positivity, where only serum levels above 20 nmol/L using RIPA and any positive CSF value are associated with neurologic manifestations [71]. Similarly, in contactin-associated protein like (CASPR)-2 IgG cell-based assay, titers of 1:100 have higher positive predictive values than values of 1:10. [72]. Other clinical laboratories have chosen different clinical cut-offs for CASPR2-IgG positivity [73]. In general, serial titer measurement in autoimmune CNS disorders is not useful, especially when the neural antibody is not considered pathogenic [59]. However, it has been shown that in NMDA-R encephalitis, elevated serum or CSF titers associate with poor outcome (modified Rankin Score [mRS] > 2) and higher likelihood of an underlying tumor. On serial measurements, an increase in CSF titers (but not serum) in NMDA-R encephalitis may confer a higher risk of disease relapse [63]. In many autoimmune neurologic disorders, patients may remain seropositive for many years after diagnosis, though in remission, without any prognostic relevance. However, in MOGAD, persistent seropositivity, and high MOG-IgG titers, appear to be associated with an increased likelihood of disease relapse when compared to transient seropositivity and therefore repeated testing can influence immunotherapeutic clinical decision making[48,74]. In clinical practice, low MOG-IgG titers detected by live cell fluorescence-activated cell sorting (FACS) assay have been reported to have a lower positive predictive value for MOGAD [75].
4. Diagnostic testing modalities
4.1. General principles
Tissue-based immunofluorescence assays (or peroxidase-baed immunohistochemistry) in combination with a confirmatory protein-specific assay is our recommended approach for lab-based testing of antibodies associated with CNS autoimmunity [58]. Tissue-based indirect immunofluorescence assay (TIFA) operates as an excellent screening tool for antibodies targeting both intracellular and extracellular antigens, but significant interpretative expertise is required [76] For confirmatory, protein-specific assays, the cellular location of the target protein is reflected in the choice of immunoassay. In general, antibodies which target intracellular antigens can be detected using western blots or line blots which use linear, denatured proteins. For antibodies which target extracellular, or cell-membrane proteins, assays which retain native protein structure such as cell-based assays are preferred [77]. For some neural antibodies (eg; antibodies targeting extracellular proteins), testing by one established modality (eg; CBA) may be sufficient, whereas it is not recommended to use commercial line blots in isolation due to the potential for false positive results [58,78]. Diagnostic assays for detecting individual analytes are considered below.
4.2. Tissue-based assays
Tissue-based immunofluorescence assays (TIFA) are performed using cryosections of adult murine tissue immobilized on glass slides. Sections of neural tissue (cerebral cortex, hippocampus cerebellum, midbrain) in addition to non-neural tissue (kidney, smooth muscle and gut) are fixed and permeabilized. The presence of non-neural tissue acts as an intra-assay control allowing the reader to discern whether antibody binding is neural-specific. However, in some instances non-neural tissue such as the glomerular podocytes may demonstrate characteristic staining patterns (eg; IgLON-5 or ANNA-3 IgG) which can aid in neural antibody identification. A blocking step is performed prevent non-specific binding. Patient serum or CSF specimens are incubated with murine tissue cryosections and patient antibody-binding to neural tissue is detected by probing with an anti-human secondary antibody conjugated with fluorescent dye (or an enzymatic conjugate such as horseradish peroxidase in tissue-based immunohistochemistry). Results are interpreted by visualization of the stained tissue section using an indirect fluorescence microscope. The tissue staining characteristics associated with specific neural antibodies can reliably detect classified neural antibodies when interpreted by an experienced reader [77]. Examples of common TIFA patterns for classified neural antibodies are shown in Fig. 1. Advantages of TIFA for the detection of neural antibodies include the ability to screen for a wide range of neural antibodies (both those targeting intracellular and extracellular antigens) within a single assay [76]. In contrast to protein-specific assays (where the result is a binary positive or negative for a single analyte) TIFA interpretation can detect rare antibodies or antibodies not requested by the ordering physician, and sometimes coexisting antibodies (e.g. ANNA-1 and amphiphysin-IgG in a patient with paraneoplastic encephalomyelitis). One study demonstrated that positive TIFA results changed management in 48 % of patients who had negative commercial cell-based assay and line-blot testing performed for a limited number of analytes. TIFA successfully detected rare, classified antibodies (such as KLHL-11, mGluR1) which were subsequently confirmed by protein-specific assays. Furthermore, TIFA can detect the presence of multiple antibodies in a single patient, such as the co-occurrence of NMDA-R antibody with GFAP IgG [21]. Due to its unbiased nature, TIFA can detect unclassified antibodies for which the antigenic target is unknown and no commercially available diagnostic test exists. Although such results are not formally reportable, in the authors’ experience the communication of this finding to the requesting physician can change patient management in the correct clinical context. Disadvantages of TIFA include the degree of reader experience required for proficient interpretation of a wide range of antibody staining patterns. Furthermore, the presence of additional antibodies (such as antinuclear antibody [ANA]), particularly in serum, may obscure the immunofluorescent staining pattern and impact visual interpretation [79].
Fig. 1.
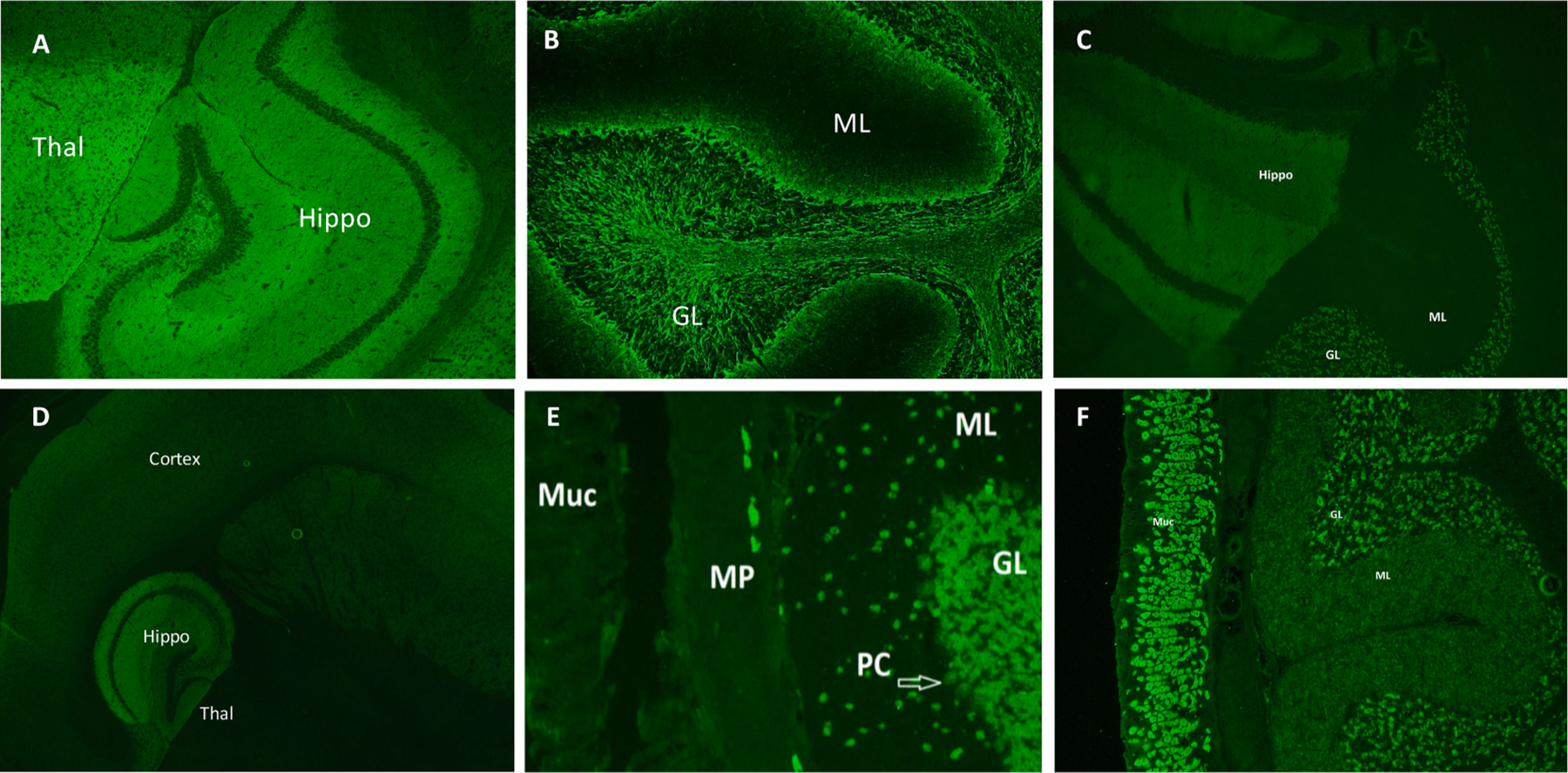
Tissue-based indirect immunofluorescence assay staining features of selected neural antibodies. Synaptic staining of the hippocampus and thalamus (image A, shown) alongside staining of the molecular layer of the cerebellum is characteristic of GABA-B receptor IgG. Neuronal intermediate filament (NIF) IgG demonstrates a classic ‘arborization’ pattern in the granular layer of the cerebellum with a dark molecular layer (image B, shown). NMDA-R IgG stains the hippocampus and the granular layer of the cerebellum (image C). A striking characteristic of AMPA-R IgG is the intense staining of the hippocampus compared to the adjacent thalamus (image D). ANNA-1 IgG demonstrates a nuclear staining pattern with staining of the nucleus and cytoplasm, with sparing of the nucleolus. The nerves of the myenteric plexus also stain which is its sole differing staining feature from ANNA-2 IgG (image E). GAD-65 IgG is visible on TIFA at high titers with punctate staining of the granular layer of the cerebellum (shown), with prominent globus pallida pars interna staining (not shown). It frequently co-exists with gastric parietal cell (GPC) IgG which stains the gastrointestinal mucosa (shown, image F) Thal: thalamus; Hippo: hippocampus; ML: molecular layer; GL: granular layer; Muc: mucosa; MP: myenteric plexus; PC: Purkinje cells.
4.3. Western blot
Antibodies that bind linear, denatured epitopes can be detected by protein-specific western blot. The substrate for western blot is typically denatured recombinant protein electrophoresed onto a polyacrylamide gel. The proteins are then transferred onto a nitrocellulose of polyvinylidene difluoride (PVDF) membrane, and following overnight blocking, probed with patient specimen. After washing away unbound antibody, secondary anti-human antibody conjugated with an enzyme (typically horseradish peroxidase) is added to detect for the presence of bound patient antibody. In the presence of the enzyme substrate, bound patient IgG results in a chemiluminescent color change which is detected either on X-ray film or digitally. Line blots are a related technique without electrophoretic step, more efficient and less labor intensive, which allow for multiple highly purified proteins to be coated on a single nitrocellulose strip [77]. However, line blots should not be used in isolation because the false positivity rate has been reported to be high in clinical practice, reflecting their suboptimal specificity [80]. Our preference (and that of others) is to use a commercially available line blot to confirm antibody positivity detected by TIFA [19]. Sensitivity and specificity of western blot or line blot diagnostic assays depends on the accurate characterization of the target antigen(s). For example, the pattern of TIFA staining for PCA-2 (Fig. 1) was first described in 2001 but the target antigen was not identified until 2017, which enabled the development of a confirmatory line blot assay [81]. Similarly, the commercial line blot for PCA-1 IgG detects binding to the cerebellar degeneration related protein 2 (CDR2) antigen. However, the use of another major antigen in PCA-1 autoimmunity, CDR2-Like, has demonstrated additional specificity for serum samples when tested alongside CDR2 [82].
4.4. Radioimmunoprecipitation assay
Radioimmunoprecipitation assays (RIPA) are a sensitive quantitative method for rapidly assessing for presence and positive values of antigen-specific antibodies. As such, they are particularly useful for detection of antibodies where quantitative nanomolar values are clinically informative, as in the case of GAD65 autoimmunity [71]. A radioactive ligand may be conjugated to recombinant human protein and incubated with patient specimen (eg; 125I-labelled recombinant GAD65 protein). Alternatively, a source of the antigen of interest (eg; porcine cerebral cortical membranes when testing for P/Q-type voltage gated calcium channel [P/Q-VGCC] IgG, or proteins solubilized from human limb muscle when testing for acetylcholine receptor [AChR] IgG) may be complexed with a radiolabeled toxin that binds specifically to the protein of interest (125I-labelled omega-conopeptide-MVIIC in the case of P/Q-VGCC; 125I-α-bungarotoxin in the case of AChR IgG) and then incubated with patient specimen. Goat anti-human IgG is then added to form an immune complex which is precipitated by centrifugation. Unbound radiolabeled protein remains in the supernatant and is discarded. The radioactivity of the precipitate is measured using a gamma counter. In negative patients, only background levels of radioactivity will be observed. When a sample is positive, the results can be extrapolated to nanomoles per liter by reference to a standard curve, and serial dilutions and can be performed in order to determine accurate titers [83]. RIPA has the advantage of detecting even low concentrations of antibody which is useful in the detection of GAD65 in the CSF, where very low concentrations (greater than 0.02 nmol/l) carry clinical significance. TIFA testing only demonstrates staining of GAD65 IgG at values which are significantly higher than the upper limit of normal and therefore does not represent a sensitive test for GAD65 IgG [84]. Disadvantages of RIPA include the use of radioactive reagents which have a short half-life and require technical expertise and safety training, and the potential for false positives owing to nonspecific IgG binding to proteins complexed with the antigen of interest [85–87].
4.5. ELISA
Enzyme-linked immunosorbent assay (ELISA) are commonly used in laboratory medicine generally and will not be described in detail herein. For autoimmune CNS disorders, ELISA is commonly used for detecting GAD65 antibody. The cut-off values may differ between laboratories depending on the kit used, but a positive value of > 10,000 international units(IU)/ml in serum and > 100 IU/ml in CSF have been proposed for GAD-65 IgG-associated neurological disorders [86]. Difficulties encountered with commercial ELISA kits include the potential for false positive results, particularly in the low positive range[77]. For antibodies which bind extracellular epitopes, such as AQP4 and MOG, ELISA is not our preferred diagnostic test. A comparison of ELISA and CBA for AQP4-IgG found a higher rate of false positivity in ELISA compared with CBA [88]. Similarly, ELISA is known to be inferior to CBA for detection of MOG-IgG [89].
4.6. Cell-based assay
Cell-based assays (CBAs) are now in common use. These are usually commercially available fixed CBAs which can be transported and stored easily. Eukaryotic cell lines (typically human-embryonic-kidney [HEK]-293 cells) are transfected with plasmids which encode for the protein of interest. Patient sample is incubated with cells (either live or fixed) which express the target protein and anti-human secondary antibody conjugated with a fluorophore is introduced. Transfection methods are either transient or stable with higher levels of the protein of interest expressed following transient transfections [77]. An additional well containing cells not expressing the protein of interest can also be used as a negative control. For neural antibodies which target extracellular antigens, CBA often provides higher sensitivity with retained specificity when compared to both TIFA and other protein-specific assays[90–93]. For some neural antibodies, live CBAs (where patient antibody is incubated with live cells at 4 degrees) appear to be more sensitive than fixed CBAs. However, live CBAs are more labor intensive, have a shorter time window for use, and are usually performed only in specialized research centers [90,94]. In a comparison of the detection methods for MOG IgG, live cell-based assays have been reported as offering increased positive-predictive value (PPV) when compared to fixed cell-based assay [89]. Live cell-based assays may also allow for insights into the pathophysiological function of antibodies which target cell-surface proteins, such as the observation that glycine receptor IgG from patient samples leads to heat-dependent receptor endocytosis [95]. This led to the development of a glycine receptor modulating antibody assay using live HEK-293 cells expressing the α1 subunit of the glycine receptor. Demonstration of glycine receptor internalization on live-cell based assay offers superior specificity for SPS than glycine receptor binding without receptor modulation [55].
4.7. Live-cell based flow cytometry
This technique has proven especially useful for the detection of aquaporin 4 and MOG IgG. HEK-293 cells are transfected with plasmid encoding both GFP and the protein-of-interest. A mixed population of cells (eg; AQP4, GFP-transfected and non-transfected) are harvested, trypsinized and brought into solution. Cells are incubated with patient specimen, and goat anti-human secondary antibody with a fluorescent conjugate (usually Alexafluor 647) is added. Cells are washed, fixed in 4 % PFA and analyzed by flow cytometry. Two populations are gated based upon GFP expression: positive (high AQP4 expression) and negative (low or no AQP4 expression). The median fluorescence intensity (MFI) of the bound anti-human secondary antibody for each cell population is resulted. MFI for the GFP-positive population indicates the relative abundance of patient IgG bound to AQP4 whereas the MFI for the GFP-negative population indicates non-specific IgG binding. The ratio of average MFI for each cell population (MFI GFP positive/MFI GFP negative) is calculated to determine the IgG binding index [96]. This method has proven to have a greater clinical sensitivity to other assays for AQP4 IgG, and also offers a specificity of 100 % [93].This technique has greater sensitivity when compared to fixed cell-based assay for the detection of MOG IgG, but has comparable sensitivity to live direct-visualization CBA [89].
4.8. Research-based novel antibody discovery tools
Novel molecular technologies such as Phage-display immunoprecipitation sequencing (PhIP-Seq) and protein microarrays have shown the potential to detect of novel antibodies in autoimmune CNS disorders [6,34,97] PhIP-Seq technology utilizes a phage-library which collectively express over 600,000 human peptides. Patient sample is incubated with the phage-library and bound-phage is immunoprecipitated using magnetic beads. DNA of the precipitated phage is sequenced to identify (in rank order) immunoreactive antigens. PhIP-Seq has been employed to identify KLHL-11 IgG (associated with paraneoplastic rhombencephalitis) and, more recently, Sloan-Kettering-Virus-Family-Transcriptional-Corepressor (SKOR)-2 autoimmunity (associated with a paraneoplastic encephalopathy) [34,97]. Protein microarrays have also demonstrated utility in identifying rare, classified antibodies which are typically not tested for outside of specialized centers, such as seizure related 6 homolog like 2 (SEZ6L2) and Neurexin3a [7]. Full-length GST-tagged recombinant human proteins are pre-printed in duplicate on nitrocellulose-coated glass slides. Patient sample is incubated with the slide, and anti-human secondary antibody is used to detect bound patient antibody, with the results expressed as a ranked list of gene names for proteins. Currently, both protein microarray and PhIP-Seq technology appear to perform better in detecting antibodies to linear epitopes of intracellular antigens and are less effective at identifying antibodies to cell-surface confirmational epitopes [6,7,34]. However, it is possible that future iterations of protein microarray or phage-display technology will overcome these limitations, enabling high-throughput screening patient samples for immunoreactivity against thousands of CNS auto-antigens in real time.
5. Utility of neural antibody testing
Neural antibodies for autoimmune CNS disorders communicate important clinical, prognostic and therapeutic information to the ordering physician. The detection of an antibody biomarker in an appropriate clinical context may provide additional diagnostic certainty. The detection of IgG biomarkers or positive TIFA assays informs management and may lead to changes in treatment [76].
5.1. Cancer screening
In cases where a high-risk neural antibody is detected, this may prompt a search for an underlying associated cancer (Tables 1 and 2). Given that the neurologic disorder typically precedes cancer diagnosis, the presence of a high-risk antibody justifies repeat surveillance for the emergence of an associated malignancy to facilitate early detection and treatment [98]. The most recent guidelines recommend screening for cancer every 4–6 month for 2 years in a patient with a high-risk neural antibody and a compatible clinical syndrome[58]. For example, a physician may screen a patient with rhombencephalitis and KLHL-11 IgG positivity every six months for two years, whereas in a patient with rhombencephalitis and MOG-IgG positivity (low risk) such surveillance would not be indicated [58]. Where a neural antibody is not detected (either due to lack of testing availability or in seronegative autoimmune encephalopathies), there is no possibility of oncologic risk stratification, further emphasizing the utility of neural antibody biomarkers in clinical practice.
5.2. Management of autoimmune CNS disorders
5.2.1. Treatment
The treatment of autoimmune CNS disorders centers on early initiation of immune therapy which is known to associate with better outcomes [14,99]. Therefore, it is not recommended to await antibody results before initiating immunotherapy in a patient with a suspected autoimmune CNS disorder. Prior to treatment initiation, baseline neurological assessments should be recorded in order to ascertain subsequent treatment response. Ideally serum and CSF samples for neural antibody testing should be obtained prior to treatment initiation as administration of intravenous immunoglobulin (IVIG) can cause both false positive and false negative results [100]. Similarly, B-cell depleting therapies (such as Rituximab) are known to affect neural antibody titers which may affect the sensitivity of diagnostic neural antibody assays [101,102].
First-line immunotherapy regimens typically consist of IV methylprednisolone 1 g and/or intravenous immunoglobulin (0.4 g/kg) once daily for 5 days. This may be followed by once-weekly doses for a period of 3 months or a slow steroid taper. Second-line therapies (eg; rituximab, cyclophosphamide) are often employed in instances of treatment resistance or may be used early in severe cases[99,103]. Plasmapheresis may be trialed as a first-line therapy or used as a second-line therapy in cases of treatment resistance. In relapsing disease, maintenance immunotherapy (eg; mycophenolate mofetil, azathioprine) may be warranted. There are no established guidelines for the duration of maintenance immunotherapy. For paraneoplastic disorders, oncologic management should occur in parallel to immunotherapy, and cancer treatment decisions guided by expert oncology input.
5.2.2. Prognosis
In general, patients who harbor antibodies against extracellular epitopes fare better than patients with antibodies targeting intracellular antigens where neurologic stabilization rather than reversal of symptoms is often the goal of care [2]. Neural antibodies may therefore guide physicians on the likelihood of neurological improvement following initiation of immunotherapy. Prognostication and treatment rationale may vary depending on the natural history of the underlying biomarker-defined disorder. For example, ataxia associated with PCA-1 IgG (antibody against intracellular target) is almost universally associated with poor neurologic outcome, whereas treatment responses can be robust in patients with ataxia associated with mGluR1 IgG (which targets an extracellular epitope) [26,28]. Similarly, in NMDA-R encephalitis, it is well established that recovery may take as long as 18 months, and that use of second-line agents is associated with better outcome in patients who do not respond to first-line immunotherapy [99].
5.2.3. Targeted therapies
Some neural antibodies exert a directly pathogenic effect on neurons (usually antibodies against extracellular antigens) and the pathomechanisms of these antibody-mediated disorders continue to be defined [13,104–106]. The most successful example of tailored treatment for a biomarker-defined autoimmune CNS disorder is the case of AQP4-IgG positive NMOSD. The observation that AQP4 antibodies mediate neuronal damage through complement activation led to the hypothesis that complement inhibition may represent an effective therapeutic strategy [104,107]. Eculizumab (a terminal C5a inhibitor) was shown in a recent randomized controlled trial to significantly reduce relapse risk compared to placebo in patients taking concomitant standard immune therapies [108]. Clinical trials are currently ongoing for other IgG biomarker-defined autoimmune CNS disorders, and in the future neural-antibody testing may inform the use of tailored disease-specific immune therapy regimens.
6. Conclusion
Neural antibody testing in the correct clinical context facilitates the diagnosis of IgG biomarker-defined autoimmune CNS disorders. Comprehensive antibody evaluations differ according to neurologic phenotype and the appropriate diagnostic assay is antibody dependent. For most analytes, we recommend initial screening by TIFA followed by confirmation with protein-specific assays. Exceptions exist such and some analytes (eg; AQP4 IgG) are not sensitively detected by TIFA. Use of a commercial line blots in isolation is not recommended due to the potential for false positive results. Neural antibody detection provides useful diagnostic and therapeutic associations to aid in the management of patients with autoimmune CNS disorders, including cancer risk-stratification and likelihood of immunotherapy response. In the future, improved understanding of pathomechanisms for pathogenic neural antibodies may inform the development of targeted therapies for autoimmune CNS disorders.
Footnotes
CRediT authorship contribution statement
Michael Gilligan: Writing – original draft, Writing – review & editing. Christopher McGuigan: Supervision, Writing – review & editing. Andrew McKeon: Conceptualization, Writing – review & editing, Supervision.
Declaration of competing interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: Dr Gilligan is funded by the Irish Clinical Academic Training (ICAT) Programme, supported by the Wellcome Trust and the Health Research Board (Grant Number 203930/B/16/Z), the Health Service Executive National Doctors Training and Planning and the Health and Social Care, Research and Development Division, Northern Ireland. Dr Gilligan has a patent pending for CAMKV-IgG. Dr. McGuigan reports no disclosures. Dr. McKeon is funded by grants from NIH (RO1NS126227, U01NS120901), and has consulted for Janssen and Roche Pharmaceuticals without personal compensation. Dr. McKeon has patents for Septin-5-IgG and Septin-7-IgG licensed to Euroimmun, a patent for GFAP-IgG issued, a patent for MAP1B-IgG with royalties paid to himself and licensed to Ravo Diagnostika, and patents for CAMKV-IgG, KLCHL11-IgG and PDE10A-IgG pending.
References
- [1].Lopez-Chiriboga AS, Clardy SL, Emerging subspecialties in neurology: autoimmune neurology, Neurology 89 (11) (2017) e129–e133. [DOI] [PubMed] [Google Scholar]
- [2].McKeon A, Pittock SJ, Paraneoplastic encephalomyelopathies: pathology and mechanisms, Acta Neuropathol. 122 (4) (2011) 381–400. [DOI] [PubMed] [Google Scholar]
- [3].Linnoila JJ, Binnicker MJ, Majed M, Klein CJ, McKeon A, CSF herpes virus and autoantibody profiles in the evaluation of encephalitis, Neurol Neuroimmunol Neuroinflamm. 3 (4) (2016) e245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Guedes BF, Ribeiro AF, Pinto LF, Vidal JE, de Oliveira FG, Sztajnbok J, et al. , Potential autoimmune encephalitis following yellow fever vaccination: a report of three cases, J. Neuroimmunol. 355 (2021) 577548. [DOI] [PubMed] [Google Scholar]
- [5].Dubey D, David WS, Reynolds KL, Chute DF, Clement NF, Cohen JV, et al. , Severe neurological toxicity of immune checkpoint inhibitors: growing Spectrum, Ann. Neurol. 87 (5) (2020) 659–669. [DOI] [PubMed] [Google Scholar]
- [6].Larman HB, Zhao Z, Laserson U, Li MZ, Ciccia A, Gakidis MA, et al. , Autoantigen discovery with a synthetic human peptidome, Nat. Biotechnol. 29 (6) (2011) 535–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].McKeon A, Lesnick C, Vorasoot N, Buckley MW, Dasari S, Flanagan EP, et al. , Utility of protein microarrays for detection of classified and novel antibodies in autoimmune neurologic disease, Neurol Neuroimmunol Neuroinflamm. 10 (5) (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dubey D, Pittock SJ, Kelly CR, McKeon A, Lopez-Chiriboga AS, Lennon VA, et al. , Autoimmune encephalitis epidemiology and a comparison to infectious encephalitis, Ann. Neurol. 83 (1) (2018) 166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Simard C, Vogrig A, Joubert B, Muniz-Castrillo S, Picard G, Rogemond V, et al. , Clinical spectrum and diagnostic pitfalls of neurologic syndromes with ri antibodies, Neurol Neuroimmunol. Neuroinflamm. 7 (3) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Bien CG, Limbic encephalitis, Handb. Clin. Neurol. 187 (2022) 467–487. [DOI] [PubMed] [Google Scholar]
- [11].Graus F, Titulaer MJ, Balu R, Benseler S, Bien CG, Cellucci T, et al. , A clinical approach to diagnosis of autoimmune encephalitis, Lancet Neurol. 15 (4) (2016) 391–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Graus F, Keime-Guibert F, Reñe R, Benyahia B, Ribalta T, Ascaso C, et al. , Anti-hu-associated paraneoplastic encephalomyelitis: analysis of 200 patients, Brain 124 (6) (2001) 1138–1148. [DOI] [PubMed] [Google Scholar]
- [13].Lancaster E, Lai M, Peng X, Hughes E, Constantinescu R, Raizer J, et al. , Antibodies to the GABA(B) receptor in limbic encephalitis with seizures: case series and characterisation of the antigen, Lancet Neurol. 9 (1) (2010) 67–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Irani SR, Stagg CJ, Schott JM, Rosenthal CR, Schneider SA, Pettingill P, et al. , Faciobrachial dystonic seizures: the influence of immunotherapy on seizure control and prevention of cognitive impairment in a broadening phenotype, Brain 136 (Pt 10) (2013) 3151–3162. [DOI] [PubMed] [Google Scholar]
- [15].Gadoth A, Pittock SJ, Dubey D, McKeon A, Britton JW, Schmeling JE, et al. , Expanded phenotypes and outcomes among 256 LGI1/CASPR2-IgG–positive patients, Ann. Neurol. 82 (1) (2017) 79–92. [DOI] [PubMed] [Google Scholar]
- [16].Quek AM, Britton JW, McKeon A, So E, Lennon VA, Shin C, et al. , Autoimmune epilepsy: clinical characteristics and response to immunotherapy, Arch. Neurol. 69 (5) (2012) 582–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Honorat JA, McKeon A, Autoimmune movement disorders: a clinical and laboratory approach, Curr. Neurol. Neurosci. Rep. 17 (1) (2017) 4. [DOI] [PubMed] [Google Scholar]
- [18].Flanagan EP, McKeon A, Lennon VA, Boeve BF, Trenerry MR, Tan KM, et al. , Autoimmune dementia: clinical course and predictors of immunotherapy response, Mayo Clin. Proc. 85 (10) (2010) 881–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Gilligan M, McGuigan C, McKeon A, Paraneoplastic neurologic disorders, Curr. Neurol. Neurosci. Rep. 23 (3) (2023) 67–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].O’Connor K, Waters P, Komorowski L, Zekeridou A, Guo CY, Mgbachi VC, et al. , GABA(A) receptor autoimmunity: a multicenter experience, Neurol. Neuroimmunol. Neuroinflamm. 6 (3) (2019) e552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Flanagan EP, Hinson SR, Lennon VA, Fang B, Aksamit AJ, Morris PP, et al. , Glial fibrillary acidic protein immunoglobulin G as biomarker of autoimmune astrocytopathy: analysis of 102 patients, Ann. Neurol. 81 (2) (2017) 298–309. [DOI] [PubMed] [Google Scholar]
- [22].Gravier-Dumonceau A, Ameli R, Rogemond V, Ruiz A, Joubert B, Muniz-Castrillo S, et al. , Glial fibrillary acidic protein autoimmunity: a french cohort study, Neurology 98 (6) (2022) e653–e668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Budhram A, Sharma M, Young GB, Seizures in anti-hu-associated extra-limbic encephalitis: characterization of a unique disease manifestation, Epilepsia 63 (12) (2022) e172–e177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Guasp M, Dalmau J, Encephalitis associated with antibodies against the NMDA receptor, Med. Clin. (Barc.) 151 (2) (2018) 71–79. [DOI] [PubMed] [Google Scholar]
- [25].Hadjivassiliou M, Graus F, Honnorat J, Jarius S, Titulaer M, Manto M, et al. , Diagnostic criteria for primary autoimmune cerebellar ataxia-guidelines from an international task force on immune-mediated cerebellar ataxias, Cerebellum 19 (4) (2020) 605–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Jones AL, Flanagan EP, Pittock SJ, Mandrekar JN, Eggers SD, Ahlskog JE, et al. , Responses to and outcomes of treatment of autoimmune cerebellar ataxia in adults, JAMA Neurol. 72 (11) (2015) 1304–1312. [DOI] [PubMed] [Google Scholar]
- [27].Honorat JA, Lopez-Chiriboga AS, Kryzer TJ, Fryer JP, Devine M, Flores A, et al. , Autoimmune septin-5 cerebellar ataxia, Neurol Neuroimmunol Neuroinflamm. 5 (5) (2018) e474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lopez-Chiriboga AS, Komorowski L, Kumpfel T, Probst C, Hinson SR, Pittock SJ, et al. , Metabotropic glutamate receptor type 1 autoimmunity: clinical features and treatment outcomes, Neurology 86 (11) (2016) 1009–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dalmau JGF, Autoimmune brainstem encephalitis, in: Dalmau JGF (Ed.), Autoimmune Encephalitis and Related Disorders of the Nervous System, Cambridge University Press, Cambridge, 2022, pp. 368–390. [Google Scholar]
- [30].Gruter T, Mollers FE, Tietz A, Dargvainiene J, Melzer N, Heidbreder A, et al. , Clinical, serological and genetic predictors of response to immunotherapy in anti-IgLON5 disease, Brain 146 (2) (2023) 600–611. [DOI] [PubMed] [Google Scholar]
- [31].Orozco E, Valencia-Sanchez C, Britton J, Dubey D, Flanagan EP, Lopez-Chiriboga AS, et al. , Autoimmune encephalitis criteria in clinical practice, Neurol Clin Pract. 13 (3) (2023) e200151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Odaka M, Yuki N, Yamada M, Koga M, Takemi T, Hirata K, et al. , Bickerstaff’s brainstem encephalitis: clinical features of 62 cases and a subgroup associated with guillain-Barre syndrome, Brain 126 (Pt 10) (2003) 2279–2290. [DOI] [PubMed] [Google Scholar]
- [33].Pittock SJ, Parisi JE, McKeon A, Roemer SF, Lucchinetti CF, Tan KM, et al. , Paraneoplastic jaw dystonia and laryngospasm with antineuronal nuclear autoantibody type 2 (anti-ri), Arch. Neurol. 67 (9) (2010) 1109–1115. [DOI] [PubMed] [Google Scholar]
- [34].Mandel-Brehm C, Dubey D, Kryzer TJ, O’Donovan BD, Tran B, Vazquez SE, et al. , Kelch-like protein 11 antibodies in seminoma-associated paraneoplastic encephalitis, N. Engl. J. Med. 381 (1) (2019) 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Blattner MS, Day GS, Sleep disturbances in patients with autoimmune encephalitis, Curr. Neurol. Neurosci. Rep. 20 (7) (2020) 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lana-Peixoto MA, Talim N, Neuromyelitis optica Spectrum disorder and anti-MOG syndromes, Biomedicines 7 (2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Tisavipat N, Chang BK, Ali F, Pittock SJ, Kammeyer R, Declusin A, et al. , Subacute horizontal diplopia, jaw dystonia, and laryngospasm, Neurol Neuroimmunol Neuroinflamm. 10 (4) (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Orozco E, Guo Y, Chen JJ, Dubey D, Howell B, Moutvic M, et al. , Clinical reasoning: a 43-year-old man with subacute onset of vision disturbances, jaw spasms, and balance and sleep difficulties, Neurology 99 (9) (2022) 387–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Flanagan EP, Autoimmune myelopathies, Handb. Clin. Neurol. 133 (2016) 327–351. [DOI] [PubMed] [Google Scholar]
- [40].Valencia-Sanchez C, Flanagan EP, Uncommon inflammatory/immune-related myelopathies, J. Neuroimmunol. 361 (2021) 577750. [DOI] [PubMed] [Google Scholar]
- [41].Banwell B, Bennett JL, Marignier R, Kim HJ, Brilot F, Flanagan EP, et al. , Diagnosis of myelin oligodendrocyte glycoprotein antibody-associated disease: international MOGAD panel proposed criteria, Lancet Neurol. 22 (3) (2023) 268–282. [DOI] [PubMed] [Google Scholar]
- [42].Ciron J, Cobo-Calvo A, Audoin B, Bourre B, Brassat D, Cohen M, et al. , Frequency and characteristics of short versus longitudinally extensive myelitis in adults with MOG antibodies: a retrospective multicentric study, Mult. Scler. 26 (8) (2020) 936–944. [DOI] [PubMed] [Google Scholar]
- [43].Flanagan EP, Weinshenker BG, Krecke KN, Lennon VA, Lucchinetti CF, McKeon A, et al. , Short myelitis lesions in aquaporin-4-IgG-positive neuromyelitis optica spectrum disorders, JAMA Neurol. 72 (1) (2015) 81–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Flanagan EP, McKeon A, Lennon VA, Kearns J, Weinshenker BG, Krecke KN, et al. , Paraneoplastic isolated myelopathy: clinical course and neuroimaging clues, Neurology 76 (24) (2011) 2089–2095. [DOI] [PubMed] [Google Scholar]
- [45].Dubey D, Pittock SJ, Krecke KN, Morris PP, Sechi E, Zalewski NL, et al. , Clinical, radiologic, and prognostic features of myelitis associated with myelin oligodendrocyte glycoprotein autoantibody, JAMA Neurol. 76 (3) (2019) 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Graus F, Delattre JY, Antoine JC, Dalmau J, Giometto B, Grisold W, et al. , Recommended diagnostic criteria for paraneoplastic neurological syndromes, J. Neurol. Neurosurg. Psychiatry 75 (8) (2004) 1135–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Cole J, Evans E, Mwangi M, Mar S, Acute disseminated encephalomyelitis in children: an updated review based on current diagnostic criteria, Pediatr. Neurol. 100 (2019) 26–34. [DOI] [PubMed] [Google Scholar]
- [48].Lopez-Chiriboga AS, Majed M, Fryer J, Dubey D, McKeon A, Flanagan EP, et al. , Association of MOG-IgG serostatus with relapse after acute disseminated encephalomyelitis and proposed diagnostic criteria for MOG-IgG-associated disorders, JAMA Neurol. 75 (11) (2018) 1355–1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Klaas JP, Ahlskog JE, Pittock SJ, Matsumoto JY, Aksamit AJ, Bartleson JD, et al. , Adult-onset opsoclonus-myoclonus syndrome, Arch. Neurol. 69 (12) (2012) 1598–1607. [DOI] [PubMed] [Google Scholar]
- [50].Antunes NL, Khakoo Y, Matthay KK, Seeger RC, Stram DO, Gerstner E, et al. , Antineuronal antibodies in patients with neuroblastoma and paraneoplastic opsoclonus-myoclonus, J. Pediatr. Hematol. Oncol. 22 (4) (2000) 315–320. [DOI] [PubMed] [Google Scholar]
- [51].Armangue T, Sabater L, Torres-Vega E, Martinez-Hernandez E, Arino H, Petit-Pedrol M, et al. , Clinical and immunological features of opsoclonus-myoclonus syndrome in the era of neuronal cell surface antibodies, JAMA Neurol. 73 (4) (2016) 417–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].McKeon A, Robinson MT, McEvoy KM, Matsumoto JY, Lennon VA, Ahlskog JE, et al. , Stiff-man syndrome and variants: clinical course, treatments, and outcomes, Arch. Neurol. 69 (2) (2012) 230–238. [DOI] [PubMed] [Google Scholar]
- [53].De Camilli P, Thomas A, Cofiell R, Folli F, Lichte B, Piccolo G, et al. , The synaptic vesicle-associated protein amphiphysin is the 128-kD autoantigen of stiff-man syndrome with breast cancer, J. Exp. Med. 178 (6) (1993) 2219–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Hutchinson M, Waters P, McHugh J, Gorman G, O’Riordan S, Connolly S, et al. , Progressive encephalomyelitis, rigidity, and myoclonus: a novel glycine receptor antibody, Neurology 71 (16) (2008) 1291–1292. [DOI] [PubMed] [Google Scholar]
- [55].Hinson SR, Lopez-Chiriboga AS, Bower JH, Matsumoto JY, Hassan A, Basal E, et al. , Glycine receptor modulating antibody predicting treatable stiff-person spectrum disorders, Neurol Neuroimmunol Neuroinflamm. 5 (2) (2018) e438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].McEvoy KM, Stiff-man syndrome, Mayo Clin. Proc. 66 (3) (1991) 300–304. [DOI] [PubMed] [Google Scholar]
- [57].Budhram A, Irani SR, Flanagan EP, Looking beyond syndrome-based criteria for autoimmune encephalitis-the need for complementary neural antibody-based diagnostic criteria, JAMA Neurol. (2023). [DOI] [PubMed] [Google Scholar]
- [58].Graus F, Vogrig A, Muniz-Castrillo S, Antoine JG, Desestret V, Dubey D, et al. , Updated diagnostic criteria for paraneoplastic neurologic syndromes, Neurol Neuroimmunol Neuroinflamm. 8 (4) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Budhram A, Dubey D, Sechi E, Flanagan EP, Yang L, Bhayana V, et al. , Neural antibody testing in patients with suspected autoimmune encephalitis, Clin. Chem. 66 (12) (2020) 1496–1509. [DOI] [PubMed] [Google Scholar]
- [60].Pittock SJ, Palace J, Paraneoplastic and idiopathic autoimmune neurologic disorders: approach to diagnosis and treatment, Handb. Clin. Neurol. 133 (2016) 165–183. [DOI] [PubMed] [Google Scholar]
- [61].Cellucci T, Van Mater H, Graus F, Muscal E, Gallentine W, Klein-Gitelman MS, et al. , Clinical approach to the diagnosis of autoimmune encephalitis in the pediatric patient, Neurol Neuroimmunol Neuroinflamm. 7 (2) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].McKeon A, Pittock SJ, Lennon VA, CSF complements serum for evaluating paraneoplastic antibodies and NMO-IgG, Neurology 76 (12) (2011) 1108–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Gresa-Arribas N, Titulaer MJ, Torrents A, Aguilar E, McCracken L, Leypoldt F, et al. , Antibody titres at diagnosis and during follow-up of anti-NMDA receptor encephalitis: a retrospective study, Lancet Neurol. 13 (2) (2014) 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Mackay G, Ahmad K, Stone J, Sudlow C, Summers D, Knight R, et al. , NMDA receptor autoantibodies in sporadic creutzfeldt-Jakob disease, J. Neurol. 259 (9) (2012) 1979–1981. [DOI] [PubMed] [Google Scholar]
- [65].Hammer C, Stepniak B, Schneider A, Papiol S, Tantra M, Begemann M, et al. , Neuropsychiatric disease relevance of circulating anti-NMDA receptor autoantibodies depends on blood-brain barrier integrity, Mol. Psychiatry 19 (10) (2014) 1143–1149. [DOI] [PubMed] [Google Scholar]
- [66].Flanagan E, Dubey D, Hinson S, Lennon V, Fang B, Aksamit A, et al. , Specificity of glial fibrillary acidic protein IgG autoantibody (GFAP-IgG) for autoimmune meningoencephalomyelitis diagnosis (S20.002), Neurology (16 Supplement) (2017) S20.002. [Google Scholar]
- [67].Majed M, Fryer JP, McKeon A, Lennon VA, Pittock SJ, Clinical utility of testing AQP4-IgG in CSF: guidance for physicians, Neurol Neuroimmunol Neuroinflamm. 3 (3) (2016) e231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Matsumoto Y, Kaneko K, Takahashi T, Takai Y, Namatame C, Kuroda H, et al. , Diagnostic implications of MOG-IgG detection in sera and cerebrospinal fluids, Brain 146 (9) (2023) 3938–3948. [DOI] [PubMed] [Google Scholar]
- [69].Carta S, Cobo Calvo A, Armangue T, Saiz A, Lechner C, Rostasy K, et al. , Significance of myelin oligodendrocyte glycoprotein antibodies in CSF: a retrospective multicenter study, Neurology 100 (11) (2023) e1095–e1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Muniz-Castrillo S, Haesebaert J, Thomas L, Vogrig A, Pinto AL, Picard G, et al. , Clinical and prognostic value of immunogenetic characteristics in anti-LGI1 encephalitis, Neurol Neuroimmunol Neuroinflamm. 8 (3) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].McKeon A, Tracy JA, GAD65 neurological autoimmunity, Muscle Nerve 56 (1) (2017) 15–27. [DOI] [PubMed] [Google Scholar]
- [72].Garrido Sanabria ER, Zahid A, Britton J, Kraus GJ, López-Chiriboga AS, Zekeridou A, et al. , CASPR2-IgG-associated autoimmune seizures, Epilepsia 63 (3) (2022) 709–722. [DOI] [PubMed] [Google Scholar]
- [73].Bien CG, Mirzadjanova Z, Baumgartner C, Onugoren MD, Grunwald T, Holtkamp M, et al. , Anti-contactin-associated protein-2 encephalitis: relevance of antibody titres, presentation and outcome, Eur. J. Neurol. 24 (1) (2017) 175–186. [DOI] [PubMed] [Google Scholar]
- [74].Hennes EM, Baumann M, Schanda K, Anlar B, Bajer-Kornek B, Blaschek A, et al. , Prognostic relevance of MOG antibodies in children with an acquired demyelinating syndrome, Neurology 89 (9) (2017) 900–908. [DOI] [PubMed] [Google Scholar]
- [75].Sechi E, Buciuc M, Pittock SJ, Chen JJ, Fryer JP, Jenkins SM, et al. , Positive predictive value of myelin oligodendrocyte glycoprotein autoantibody testing, JAMA Neurol. 78 (6) (2021) 741–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Gadoth A, Segal Y, Paran Y, Aizenstein O, Alcalay Y, The importance of tissue-based assay in the diagnosis of autoimmune encephalitis, J. Neurol. 269 (7) (2022) 3588–3596. [DOI] [PubMed] [Google Scholar]
- [77].Waters P, Pettingill P, Lang B, Detection methods for neural autoantibodies, Handb. Clin. Neurol. 133 (2016) 147–163. [DOI] [PubMed] [Google Scholar]
- [78].Ruiz-García R, Muñoz-Sánchez G, Naranjo L, Guasp M, Sabater L, Saiz A, et al. , Limitations of a commercial assay as diagnostic test of autoimmune encephalitis, Front. Immunol. 12 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Ricken G, Schwaiger C, De Simoni D, Pichler V, Lang J, Glatter S, et al. , Detection methods for autoantibodies in suspected autoimmune encephalitis, Front. Neurol. 9 (2018) 841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Budhram A, Yang L, Bhayana V, Mills JR, Dubey D, Clinical sensitivity, specificity, and predictive value of neural antibody testing for autoimmune encephalitis, J. Appl. Lab. Med. 7 (1) (2022) 350–356. [DOI] [PubMed] [Google Scholar]
- [81].Gadoth A, Kryzer TJ, Fryer J, McKeon A, Lennon VA, Pittock SJ, Microtubule-associated protein 1B: novel paraneoplastic biomarker, Ann. Neurol. 81 (2) (2017) 266–277. [DOI] [PubMed] [Google Scholar]
- [82].Vorasoot N, Scharf M, Miske R, Thakolwiboon S, Dubey D, Mills JR, et al. , CDR2 and CDR2L line blot performance in PCA-1/anti-yo paraneoplastic autoimmunity, Front. Immunol. 14 (2023) 1265797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Walikonis JE, Lennon VA, Radioimmunoassay for glutamic acid decarboxylase (GAD65) autoantibodies as a diagnostic aid for stiff-man syndrome and a correlate of susceptibility to type 1 diabetes mellitus, Mayo Clin. Proc. 73 (12) (1998) 1161–1166. [DOI] [PubMed] [Google Scholar]
- [84].Pittock SJ, Yoshikawa H, Ahlskog JE, Tisch SH, Benarroch EE, Kryzer TJ, et al. , Glutamic acid decarboxylase autoimmunity with brainstem, extrapyramidal, and spinal cord dysfunction, Mayo Clin. Proc. 81 (9) (2006) 1207–1214. [DOI] [PubMed] [Google Scholar]
- [85].Mirian A, Nicolle MW, Edmond P, Budhram A, Comparison of fixed cell-based assay to radioimmunoprecipitation assay for acetylcholine receptor antibody detection in myasthenia gravis, J. Neurol. Sci. 432 (2022) 120084. [DOI] [PubMed] [Google Scholar]
- [86].Budhram A, Freeman E, Bhayana V, Yang L, Positive predictive value of anti-GAD65 ELISA cut-offs for neurological autoimmunity, Can. J.. Neurol. Sci. 50 (5) (2023) 766–768. [DOI] [PubMed] [Google Scholar]
- [87].Zalewski NL, Lennon VA, Lachance DH, Klein CJ, Pittock SJ, McKeon A, P/Q- and N-type calcium-channel antibodies: oncological, neurological, and serological accompaniments, Muscle Nerve 54 (2) (2016) 220–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Pittock SJ, Lennon VA, Bakshi N, Shen L, McKeon A, Quach H, et al. , Seroprevalence of aquaporin-4-IgG in a northern California population representative cohort of multiple sclerosis, JAMA Neurol. 71 (11) (2014) 1433–1436. [DOI] [PubMed] [Google Scholar]
- [89].Waters PJ, Komorowski L, Woodhall M, Lederer S, Majed M, Fryer J, et al. , A multicenter comparison of MOG-IgG cell-based assays, Neurology 92 (11) (2019) e1250–e1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Thouin A, Gastaldi M, Woodhall M, Jacobson L, Vincent A, Comparison of N-methyl-d-aspartate receptor antibody assays using live or fixed substrates, J. Neurol. 268 (5) (2021) 1818–1826. [DOI] [PubMed] [Google Scholar]
- [91].Reindl M, Schanda K, Woodhall M, Tea F, Ramanathan S, Sagen J, et al. , International multicenter examination of MOG antibody assays, Neurol Neuroimmunol Neuroinflamm. 7 (2) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Waters P, Reindl M, Saiz A, Schanda K, Tuller F, Kral V, et al. , Multicentre comparison of a diagnostic assay: aquaporin-4 antibodies in neuromyelitis optica, J. Neurol. Neurosurg. Psychiatry 87 (9) (2016) 1005–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Fryer JP, Lennon VA, Pittock SJ, Jenkins SM, Fallier-Becker P, Clardy SL, et al. , AQP4 autoantibody assay performance in clinical laboratory service, Neurol Neuroimmunol Neuroinflamm. 1 (1) (2014) e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Gastaldi M, Scaranzin S, Jarius S, Wildeman B, Zardini E, Mallucci G, et al. , Cell-based assays for the detection of MOG antibodies: a comparative study, J. Neurol. 267 (12) (2020) 3555–3564. [DOI] [PubMed] [Google Scholar]
- [95].Carvajal-Gonzalez A, Leite MI, Waters P, Woodhall M, Coutinho E, Balint B, et al. , Glycine receptor antibodies in PERM and related syndromes: characteristics, clinical features and outcomes, Brain 137 (Pt 8) (2014) 2178–2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].McKeon A Autoimmunity: Diseases and Testing. In: Clift I, editor. Clinical Immunodiagnostics: Jones & Bartlett Learning; 2021. p. 295–332. [Google Scholar]
- [97].Rezk M, Pittock SJ, Kapadia RK, Knight AM, Guo Y, Gupta P, et al. , Identification of SKOR2 IgG as a novel biomarker of paraneoplastic neurologic syndrome, Front. Immunol. 14 (2023) 1243946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Titulaer MJ, Soffietti R, Dalmau J, Gilhus NE, Giometto B, Graus F, et al. , Screening for tumours in paraneoplastic syndromes: report of an EFNS task force, Eur. J. Neurol. 18 (1) (2011) 19–e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Titulaer MJ, McCracken L, Gabilondo I, Armangue T, Glaser C, Iizuka T, et al. , Treatment and prognostic factors for long-term outcome in patients with anti-NMDA receptor encephalitis: an observational cohort study, Lancet Neurol. 12 (2) (2013) 157–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Gruter T, Ott A, Meyer W, Jarius S, Kinner M, Motte J, et al. , Effects of IVIg treatment on autoantibody testing in neurological patients: marked reduction in sensitivity but reliable specificity, J. Neurol. 267 (3) (2020) 715–720. [DOI] [PubMed] [Google Scholar]
- [101].Hou Y, Zhang C, Yu X, Wang W, Zhang D, Bai Y, et al. , Effect of low-dose rituximab treatment on autoimmune nodopathy with anti-contactin 1 antibody, Front. Immunol. 13 (2022) 939062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Zhou Y, Yan C, Gu X, Zhou L, Lu J, Zhu W, et al. , Short-term effect of low-dose rituximab on myasthenia gravis with muscle-specific tyrosine kinase antibody, Muscle Nerve 63 (6) (2021) 824–830. [DOI] [PubMed] [Google Scholar]
- [103].Nosadini M, Eyre M, Molteni E, Thomas T, Irani SR, Dalmau J, et al. , Use and safety of immunotherapeutic management of N-methyl-d-aspartate receptor antibody encephalitis: a meta-analysis, JAMA Neurol. 78 (11) (2021) 1333–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Hinson SR, Romero MF, Popescu BF, Lucchinetti CF, Fryer JP, Wolburg H, et al. , Molecular outcomes of neuromyelitis optica (NMO)-IgG binding to aquaporin-4 in astrocytes, Proc. Natl. Acad. Sci. U S A. 109 (4) (2012) 1245–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Kornau HC, Kreye J, Stumpf A, Fukata Y, Parthier D, Sammons RP, et al. , Human cerebrospinal fluid monoclonal LGI1 autoantibodies increase neuronal excitability, Ann. Neurol. 87 (3) (2020) 405–418. [DOI] [PubMed] [Google Scholar]
- [106].Moscato EH, Peng X, Jain A, Parsons TD, Dalmau J, Balice-Gordon RJ, Acute mechanisms underlying antibody effects in anti-N-methyl-d-aspartate receptor encephalitis, Ann. Neurol. 76 (1) (2014) 108–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Saadoun S, Waters P, Bell BA, Vincent A, Verkman AS, Papadopoulos MC, Intra-cerebral injection of neuromyelitis optica immunoglobulin G and human complement produces neuromyelitis optica lesions in mice, Brain 133 (Pt 2) (2010) 349–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Pittock SJ, Berthele A, Fujihara K, Kim HJ, Levy M, Palace J, et al. , Eculizumab in Aquaporin-4-positive neuromyelitis optica Spectrum disorder, N. Engl. J. Med. 381 (7) (2019) 614–625. [DOI] [PubMed] [Google Scholar]