

Summary
Stereotactic radiosurgery (SRS) has been shown to be efficacious for the treatment of limited brain metastasis (BM); however, the effects of SRS on human brain metastases have yet to be studied. We performed genomic analysis on resected brain metastases from patients whose resected lesion was previously treated with SRS. Our analyses demonstrated for the first time that patients possess a distinct genomic signature based on type of treatment failure including local failure, leptomeningeal spread, and radio-necrosis. Examination of the center and peripheral edge of the tumors treated with SRS indicated differential DNA damage distribution and an enrichment for tumor suppressor mutations and DNA damage repair pathways along the peripheral edge. Furthermore, the two clinical modalities used to deliver SRS, LINAC and GK, demonstrated differential effects on the tumor landscape even between controlled primary sites. Our study provides, in human, biological evidence of differential effects of SRS across BM’s.
Subject areas: Genomics, Genomic analysis, Cancer systems biology, Cancer
Graphical abstract
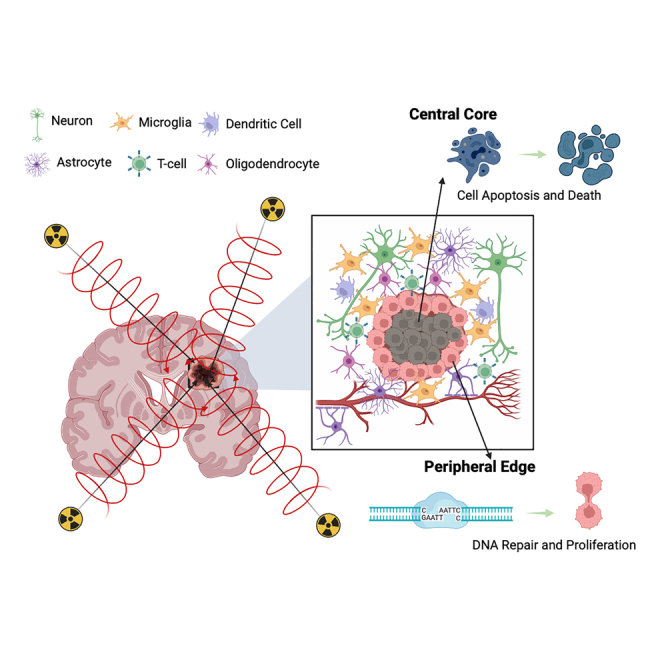
Highlights
-
•
Stereotactic Radiosurgery produces significant genomic damage in human tumors
-
•
DNA damage landscape varies between the peripheral edge and center core of tumors
-
•
Type of treatment failure corresponds to unique genomic signatures
-
•
LINAC and Gamma Knife treatment modalities enrich for different genomic signatures
Genomics; Genomic analysis; Cancer systems biology; Cancer
Introduction
Paradoxically, the most commonly diagnosed brain tumor does not originate from the brain itself but is a consequence of systemic cancers1,2 with roughly 200,000 brain metastases (BM) diagnosed annually.2 Stereotactic radiosurgery (SRS) has emerged as the leading treatment modality for limited BMs (1–4 brain lesions), due to its ability to target radiation dose directly around the tumor, allowing for both the sparing of normal tissue as well as the delivery of a more concentrated radiation dose.2,3,4,5
Currently, SRS is being used both as a front-line therapy for smaller lesions as well as adjuvant therapy following surgical resection for symptomatic BMs. SRS delivered to the resection cavity has been shown to significantly lower local recurrence.6,7 Preclinical studies have established that radiation therapy exerts its effects through the physical accumulation of DNA damage, specifically the introduction of both single and double-strand DNA breaks.8,9 Furthermore, the lethal effects of radiation can be amplified through the distribution of reactive oxygen species throughout the tumor through the tumor vasculature.10 There are two primary delivery modalities of SRS, Gamma-Knife (GK) and linear accelerator (LINAC), clinically they are considered comparable and are used equally for BM treatment,11,12 although some studies have hypothesized dosing differences between the two modalities.13
To date, research into SRS has been conducted exclusively in pre-clinical models and as such there are no direct studies analyzing the radiobiological effects of SRS in vivo within humans. To address the knowledge gap in the literature about the in situ effect of radiation in human brain, we analyzed our unique set of samples of BMs that were previously treated with SRS and then surgically resected. These samples included all major types of cancers that generally metastasize to the brain. The samples were treated with either GK or LINAC and within these samples tissue was obtained from the isodose center of the SRS field as well as along the peripheral edge of the SRS dosing field. This rare set of samples allows for the investigation of radiation’s effects directly on human BMs in the native central nervous system (CNS) microenvironment. In addition, it is possible to compare patients who received SRS through LINAC or GK and their underlying DNA damage profiles.
Results
Human sample characteristics
Between March 2018 and May 2022, we identified 34 patients who were initially treated with SRS for limited brain metastases and subsequently underwent surgical resection for at least one of the treated BMs. Tumor samples were flash frozen and saved as biopsies being taken from the center and peripheral edges of the tumors. To understand the effects of stereotactic radiation at the genomic level on tumors growing within the human CNS all samples were analyzed by whole exome sequencing (WES) and RNA sequencing (RNAseq)). Out of the 34 samples 50% were female. Age ranged from 36 to 85 years (median: 59), and 85% of patients had a KPS of 70 or higher. Out of 34, 20 patients had a previous diagnosis of cancer and 14 presented with precocious brain metastases (Table S1). Primary cancer type included lung, breast, melanoma, renal, GI, and endometrial (Figures 1A and 1B). SRS dosing was delivered either by LINAC or Gamma Knife (GK) as a single fraction. Of the patient samples analyzed for biological corelates, 3 out of 34 (8%) samples were from patients who developed local failure (LF), 3 out of 34 (8%) samples were from patients who developed radiation necrosis (RN) and 3 out of 34 (8%) samples were from patients with leptomeningeal failure (LMF). There was no overlap between patients with LF, LMF, or RN. All cases of RN were treated with LINAC and all cases of LMF were in patients with cerebellar metastases. Decisions on tumor resection were determined in consultation with the treating neurosurgeon and clinical decision-making was undertaken by the patient-specific treatment team. Patients were treated with SRS prior to surgical resection, a diagnosis of LM or RN was obtained post-surgical resection upon regular patient follow-up. Sufficient good-quality RNA and DNA were obtained from both peripheral and center-matched tumors in 13 patients, with the other 21 trial patients lacking at least one component of RNA or DNA from a central or peripheral sample. In total, all 34 patients had at least one component of usable sequencing data (Figure 1C), which was determined by extensive quality control metrics (Figures S1A–S1C). In addition, previously archived unirradiated resected lung cancer BMs were obtained separately and used as a control for comparison.
Figure 1.

Sample characteristics
(A) Characteristics of the 34 patients analyzed.
(B) Timeline of sample collection and processing.
(C) Breakdown of type of primary tumor location across all samples collected and analyzed.
Stereotactic radiosurgery incudes DNA damage resulting in genome wide transcriptomic changes in treated tumor tissue
The biological impact of radiation to halt the spread, or in some case eliminate, brain metastases has been well documented clinically,2,3,14 however, there is no available data analyzing radiation-induced damage at the genomic level within the human brain. Although the goal of radiation is to induce irreversible DNA damage in tumor cells resulting in tumor cell death, radiation can have other secondary effects on the tumor cells that can alter tumor cell biology in vivo.15,16,17,18,19,20,21 To accurately profile the DNA damage resulting from SRS to BMs, as well as understand the possible functional outcomes of this cellular damage, we compared WES and RNAseq data from the resected irradiated BMs (Figure 2A). Previously archived unirradiated resected lung cancer BMs were used as a control for comparison. WES quantification of DNA damage (SNP, DNP, TNP, insertion, deletions) demonstrated a sharp increase in overall DNA damage in the tumors that received SRS (Figure 2B) (non-radiated mean 841 marks vs. Radiated mean 1849 marks, p < 0.001), with the majority of damage coming from SNP’s within intronic regions (87.24%) (Figures S2A and S2B). High-impact variants annotated by SNPEFF22 demonstrated that between type-matched non-small cell lung cancer (NSCLC) tumors, high-impact deletions and insertions were more prevalent in radiated samples (Rad (12) vs. non-rad (5) mean high-impact variants detected; Deletion: 36 vs. 18 p < 0.01, Insertion: 17 vs. 9, p < 0.05) (Figure 2C). The total variant count per chromosome was visualized on a per sample basis indicating unique patterns of DNA damage were present among all samples (Figure 2D). Hierarchical clustering of high-impact mutations on a per sample basis also demonstrated heterogeneity between patient samples (Figure S2C). BCFtools23 vcfR,24 and plotVCF were used to create gene summary plots for total radiated and non-radiated samples, revealing the differential distribution of variants along chromosomes (Figure 2E). The C>T single base substitution (SBS), a hallmark of radiation damage,15,25 was the most prevalent SBS in our sample (Figure 2F), while BM’s arising from melanoma primary tumors contained the highest fraction of SBS marks per radiated tumor type analyzed, likely due to the lesions propensity for prior UV induced radiation damage20,26,27,28 (Figure 2G). Waterfall plots were created from gene ontology analysis of differentially expressed genes present in the radiosurgery cohort using ALLEZ29 (p < 0.05 significance level with previous term exclusion). Results indicated DNA damage repair as the top enriched term along with other cellular growth processes such as ATP-dependent activity, centriole formation, and regulation of DNA replication (Figures 2H and S2D). DeSeq230 was used for differential expression analysis between type-matched tumors receiving SRS or no SRS, 143 terms were enriched (α-threshold <0.01) (Figure 2I). Genes contributing to the global ALLEZ GSEA enrichment from terms double-strand break and DNA repair genes were then isolated to visualize expression changes confirming upregulation among the radiated tumors across both LINAC and GK delivery modality (Figures 2J and 2K). Finally, dimensionality reduction followed by UMAP based clustering was performed on samples revealing moderate clustering of samples from the same tumor (center vs. periphery) but not across patients (Figure S2E). Collectively, these data show that in humans SRS induces measurable and significant cellular damage across a range of metastatic tumors from differing primary locations. The functional consequences of the induction of cellular damage seems to be enrichment for cellular repair which is not surprising given cancer cells have proven adept at proliferating despite higher than baseline levels of cellular damage or mutations.19,31,32,33,34,35
Figure 2.

SRS incudes DNA damage resulting in genome wide transcriptomic changes in treated tumor tissue
(A) Schematic depicting analysis of samples with and without radiosurgery compared between common primary tumor locations (lung) or across primary tumor locations.
(B) Quantification of total damage marks and type of damage detected by variant calling between radiated and non-radiated samples.
(C) Comparison of high-impact deletions or insertions among radiated and non-radiated lung cancer samples.
(D) 3D plot of all samples organized by chromosome (x), number of variants detected (y), and patient (z).
(E) Chromosome visualization plot of mutated genes across non-radiated and radiated samples.
(F) Single base substitutions present in radiated samples.
(G) Single base substitutions present grouped by primary tumor location across radiated samples.
(H) ALLEZ GSEA waterfall plot with previous term exclusion applied to genes differentially expressed among tumors metastasized from lung.
(I) Heatmap visualization of differentially expressed genes among non-radiated samples (left/blue) and radiated samples from tumors metastasized from lung (right/brown).
(J) Heatmap visualizing the expression of genes associated with GO term “DNA Repair” among non-radiated and radiated tumors metastasized from lung.
(K) Heatmap visualizing the expression of genes associated with GO term “Double-Strand Break” among non-radiated and radiated tumors metastasized from lung. Error bars indicate SD. Comparison between samples done using students t-test (B), differential expression analysis was done within DESeq2 or ALLEZ using adjusted p value <0.05 (H, I, J, K). ∗∗∗∗p < 0.0001.
Peripheral edges of brain metastasis show signs of DNA damage repair and enrichment of cellular growth genes, likely contributing to treatment failure
Brain metastases originate from a malignancy growing within a different organ and therefore must embed and integrate into a foreign CNS microenvironment to grow.36,37,38 Because of the nature of this growth the center of the tumor, that is primarily made of metastatic cancer cells, has a different surrounding microenvironment compared to the periphery of the tumor where the metastatic cancer cells interact with the surrounding CNS microenvironment. Currently, there is no data in humans examining the comparative genomic effect of radiation between the center and periphery of a metastatic lesion that by nature has different microenvironmental interactions (Figure 3A). We examined tumor tissue resected from the center, and peripheral edges, of BM’s using both WES and RNA sequencing. Quantification of shared versus unique variants indicated most variants were common among the center and peripheral samples, however, unique variants still comprised roughly 20% of the variants detected (Figures 3B and 3C). Classification of variant type with SNPEFF and COSMIC39 indicated similar numbers of low, modifier, moderate, and high impacts across the center and peripheral samples while also demonstrating many variants detected (∼2/3) were not present within the existing COSMIC database (Figures S3A and S3B). Visualization of variants across chromosomal location between the the center and peripheral samples shows globally very similar overlap patters with different variant location visible on an individual chromosome level (bottom vs. top) (Figure 3D). Analysis of variants per gene across the samples demonstrated consistency within sample pairs but unique variation across primary tumor types (Figures 3E and S3C). DIFFUSE40 was utilized to perform isoform enrichment between the center and peripheral samples allowing for a more in-depth transcriptional picture of the underlying biology. Isoform enrichment revealed significant transcriptional variation can exist within the same tumor sample (Figures 3F and S3D) between center and periphery, something seen in high-grade glioma biology41,42,43,44 but not yet described in the BM population. Gene ontology enrichment terms included leukocyte activation, extracellular matrix, and angiogenesis demonstrating a microenvironmental interaction with the peripheral edge of the BM (Figure 3G). Furthermore, significant enrichment of DNA double-strand break processing was enriched among peripheral samples while apoptotic signaling was enriched among center samples (Figure 3H), possibly resulting from the differential effect of SRS on the biology of the cancer cell based on its interaction with the microenvironment. Peripheral samples also had higher numbers of mutations present with annotated tumor suppressor genes (Figure 3I), possibly increasing the proliferative capacity of the cells within this region, which might contribute to treatment failure. To examine interactions with surrounding brain and immune cells FARDEEP45 devolution analysis was conducted which demonstrated peripheral sample enrichment for excitatory neuronal cells (Figure 3J) and immune invasion dominated by the macrophage and plasma cell lineage (Figure 3K). IHC of central and peripheral samples corroborated our in silico finding demonstrating robust CD45+ immune cell invasion (Figure 3L) (Lung: center vs. peripheral CD45+ counts, n = min 100 counts, p < 0.05). These data demonstrate for the first time that radiation received by peripheral edges of brain metastasis results in distinct genomic and transcriptomic signatures. This differential impact of SRS in combination with the unique microenvironmental differences between the center and periphery of the tumor likely play some role in allowing for increased cellular recovery, upregulated DNA damage repair, as well as increased proliferation capacity. These biological phenomena together may lay the foundation for treatment failure.
Figure 3.

Peripheral edges of brain metastasis show signs of DNA damage repair and enrichment of cellular growth genes, likely contributing to treatment failure
(A) Schematic depicting the isolation of central and peripheral tumor samples as well as the components of the CNS microenvironment seen by the peripheral BM cells.
(B) Quantification of total somatic variants detected among central and peripheral biopsy locations.
(C) Quantification of shared or unique somatic variants detected among central and peripheral biopsy locations.
(D) Chromosome level visualization of variants from central or peripheral biopsy locations summed (top) or individualized (later in discussion).
(E) Heatmap visualization of variants per gene per sample among all central and peripheral biopsy locations across primary tumor location and SRS delivery modality.
(F) Heatmap of differential isoform enrichment analyses conducted by DIFFUSE across central and peripheral biopsy locations.
(G) ALLEZ GSEA waterfall plot with previous term exclusion conducted on differentially expressed isoforms between central and peripheral biopsy locations.
(H) GSEA of differentially expressed genes using ClusterProfiler between central and peripheral biopsy locations restricted to terms included in DNA damage or repair.
(I) Quantification of mutations on genes annotated to be tumor suppressors across central and peripheral biopsy locations.
(J) FARDEEP deconvolution analysis using brain cell type references conducted on central and peripheral biopsy locations.
(K) FARDEEP deconvolution analysis using immune cell type references conducted on central and peripheral biopsy locations.
(L) in vitro visualization of CD45+ cell invasion using IHC on central and peripheral tumor biopsy locations. Error bars indicate SD. Statistical comparisons between groups were conducted with ANOVA with Bonferroni correction (L). Differential expression analysis was done within DESeq2, ALLEZ, ClusterProfiler, and DIFFUSE using adjusted p value <0.05 (E, F, G, H, I). ∗p < 0.05.
Gamma Knife and Linear Accelerator treatment modalities induce differential genomic signatures and DNA damage across matched primary tumor types
Clinically, GK and LINAC are considered equal in terms of disease control efficacy. Both systems are routinely used interchangeably depending on the facilities present at the location of care.46 However, there are known technical differences between the two delivery types such as dose conformality, treatment times, fractionation, and single isocenter multitargeting.12,13 There is some dosing difference in the core vs. periphery of an SRS-treated lesion by GK compared to LINAC, where the dose delivery is more homogeneous.12,13,46 Since we discovered different biological processes in the core vs. periphery of the SRS-treated lesions, to delineate the role of differential radiation dosing in the core vs. periphery of the treated lesion we analyzed GK and LINAC treated primary lung lesions against one and other (Figure 4A). Using PlotVCF to merge all variants across chromosomes we observed a greater distribution of damage across peripheral lesions when treated with LINAC as opposed to GK while the centers of BM’s were similar (Figure 4B) Quantifying the total of high-impact variants across all of our samples showed no statistical difference between GK and LINAC (mean of variants called: GK: 707, LINAC: 776, p < 0.15), however, when restricted to only BM’s arising from lung primary tumors there was a statistically significant increase in variants in LINAC treated samples (mean of variants called: LINAC: 1142, GK: 387, p = 0.02) (Figure 4C). Differential expression analysis across all patients with GK and LINAC revealed 172 differentially expressed genes at p value <0.05 with FDR <0.10 (Figure 4D). Patients with GK were most enriched for NXPE4 and NOX1 (Log2FC: −26.82, −7.83 p < 0.0001) while patients with LINAC were most enriched for ACTN3, CT83, and MAGEA10 (Log2FC: 22.36, 22.36, 21.79 p < 0.0001). GSEA indicated significant enrichment for terms including lipid transport and localization among GK treated samples, while hormone-mediated signaling and androgen receptor signaling pathway terms were enriched in LINAC samples (Figure 4E). Driving these hormone-linked LINAC enrichments were several cancer testis antigen (CTA) genes such as TSPY3, PAGE5 and DAZ1 (p-adj <0.00001 all genes). The Human Protein Atlas database was utilized to obtain IHC sectioning of these genes demonstrating significant expression only within the male reproductive system in healthy individuals (Figure 4F). While their function is still not well understood CTA genes may be involved in tumor proliferation and immune response, with a number of antigens being trialed as vaccine candidates due to their limited expression in normal tissues outside of reproductive organs.47,48,49,50,51 This may explain the observed synergistic effect of radiation and immunotherapy seen in the clinical and pre-clinical setting.21,52
Figure 4.

Gamma Knife and LINAC treatment modalities induce differential genomic signatures and DNA damage across matched primary tumor types
(A) Schematic depicting comparison between GK and LINAC treated samples with representative images for dose contouring on individual patients.
(B) Chromosome visualization of mutations between GK and LINAC treated samples across central and peripheral biopsy locations.
(C) Quantification of total variants detected among peripheral samples between GK and LINAC SRS delivery.
(D) Volcano plot of DESeq2 determined differentially expressed genes between GK and LINAC treated samples.
(E) GSEA enrichment of DE genes when compared across GK and LINAC dosing modality.
(F) Visualization of enriched CTA genes using Human Protein Atlas data. Error bars indicate SD. Differential expression analysis was done within DESeq2 using adjusted p value <0.05 ∗p < 0.05.
Primary tumor location and types of treatment failure display distinct genomic profiles
As current research,53,54,55 as well as our data has demonstrated, metastases are not all created equal. To explore this question further we examined primary tumor location by visualizing the average frequency of high-impact deletions or insertions present across primary tumor types. Results demonstrated that lung BMs carried the most deletions while melanoma samples carried the most insertions (Figure 5A). Further underscoring primary tumor type differences breast primary BM’s and lung primary BM’s (the two most common sample types in our study) exhibited differential expression at the RNA level (1477 genes DE at α < 0.01) (Figure 5B). Clustering of all patient samples based on gene expression utilizing UMAP of defined principal components was also conducted which illustrated within patient similarity but between patient differences with even similar typed tumors failing to reliably cluster together (Figures S4A–S4C). Examination of genes that were frequently mutated as well as differentially expressed revealed a cohort of 38 genes (1% of our sample results) that fit both categories, interestingly one commonality CENPB, a protein that facilitates centromere formation, occurred in these results as well as was mutated in all our patient samples that experienced local treatment failure. To validate the in-silico analysis findings in vitro QPCR from either irritated (50gy) A549 primary lung cancer cells or isolated peripheral and central tumor samples was conducted on top hits). Enrichment for CENPB, PSG1, and POM12 (p < 0.0001, 0.5, 0.01) was confirmed in radiated A549 cells as well as the enrichment of POM121 and SPINK13 (p < 0.001, 0.001) within peripheral samples compared to central samples (Figures S4D–S4F). Mutated status of upregulated genes (obscurin, & usherin) was also visualized indicating enrichment among central samples (Figures S4G and S4H). Together these genes all belong to families highly enriched for cell division and replication further confirming that within human samples SRS delivers detectible genomic damage that disrupts cellular division and proliferation which may ultimately lead to treatment failure.
Figure 5.

Primary tumor location and types of treatment failure display distinct genomic profiles
(A) Visualization of insertion or deletion high-impact variants detected among all primary tumor locations.
(B) Heatmap displaying differentially expressed genes between BM” s of breast of lung primary tumor origin.
(C) Overlap between DE gene sets found in patients with LF/LMF/RN.
(D) GSEA enrichment conducted on DE genes in patients with LF.
(E) GSEA enrichment conducted on DE genes in patients with RN.
(F) GSEA enrichment conducted on DE genes in patients with LMF. Error bars indicate SD. Differential expression analysis and visualization was done within DESeq2 andClusterProfiler, and using adjusted p value <0.05.
SRS treatment failure can take the form of LF of the treated lesion, loco-regional LMF, and RN. Of all the samples analyzed there were 3 samples each from patients with LF, LMF and RN. All populations were compared using Deseq2 differential expression (DE) analysis and any DE genes which passed p < 0.05 and FDR<0.10 cutoffs were retained. 1677 genes were identified in LF, 500 genes were identified in RN, and 339 genes were identified in LMF (all α < 0.10). Between the 3 groups 7 genes were common (MAGEA10, KRT17, COL22A1, ACSM2A, TSPY3, BCAR4, and CT45A3) (Figure 5C). Over-representation analysis yielded no significant enrichments of this small gene set hinting that a pan gene signature between these conditions is unlikely, instead pointing to unique signatures for each molecular abnormality. To explore each condition individually GSEA was conducted on each of the DE gene sets across all 3 conditions and demonstrated unique signatures for each treatment complication (Figures 5D–5F). Patients with LF showed high enrichment for RNA biological processes as well as the suppression of synaptic signaling and formation (Figure 5D). Patients with RN enriched for metabolic pathways while showing the suppression of cell differentiation and development (Figure 5E). Finally, patients with LMF enriched for cell cycle processes, RNA splicing and phosphorylation, while suppressing autophagy and enzyme binding (Figure 5F). Collectively, these data indicate although some common pathways are overlapping between these conditions (cell cycle processing, ribosome activation, and biogenesis) most of the gene expression changes are unique likely necessitating targeted rather than broad therapeutic intervention.
Discussion
This genomic analysis of SRS-treated BM’s provides important insights into the effect of radiation on tumors within the human CNS. Although there is a large body of literature analyzing the genomic landscape of brain metastases, human genetic data from radiated tumors has yet to be examined or published. In this study, we provide the first in human dataset examining the genomic effects of SRS on BM. Consistent with the findings from prior studies using in vitro human cells,16,19,20,35 and mouse tumor models,18,56,57 we show that SRS induces significant genomic changes post-treatment in patients with BM. Our results clarify that these genetic changes, driven mainly by SNP’s, can be detected in both RNA and DNA as early as 1 and up to 4 days between dose and resection, although damage likely persists longer. Interestingly, patients who eventually had some form of treatment failure had a distinct genomic signature right after SRS treatment. Examination of our results in the context of other large genomic analyses of BMs58,59 that were not treated with radiotherapy identified both common and unique mutations (TP53 commonality, CENPB radiation specific) indicating that biologically SRS provides a unique evolutionary selection pressure, possibly being exploited through further mutation of tumor suppressor genes.
In our samples we also discovered similar upregulations in genes such as MYC and AKT that are reported in other studies examining BM’s before and after treatment.58 It is also noted in this study that there are not large genomic shifts in the BM’s before and after treatment which may explain our similar results of nominal genomic changes between the center and peripheral tumor locations.58 Additionally, because the majority of patients presenting with BM’s have undergone some type of prior systemic cancer therapy it can be difficult to classify mutations or genomic alterations specifically to a single therapy. As the field moves forward this will necessitate the examination of the tumors at all stages of therapy along with comparison between similar standard of care regiments across patients.
Our findings also highlight the heterogeneity between BM’s derived from primary tumors from different systemic locations. Although some systemic tumors such as lung or breast cancers are more likely to metastasize to the brain, we still found significant genomic differences between them in our dataset. This heterogeneity is likely underrepresented in our sample and will require larger studies in the future to fully tease out if shared genomic programs drive all BM’s or if specific primary tumors rely on specific growth signals to complete their metastasis to the brain. This will likely require multi-institution collaboration to gain large sample size, especially of metastases of more rare primary cancers (i.e., renal, GI).
Our results demonstrate for the first time in humans that clinically delivered SRS to both the center and periphery of BM’s have differential genetic effects. This differential effect of radiation-induced damage results in the upregulation of DNA damage repair pathways in the periphery of the lesion that may contribute to both the development of leptomeningeal failure from dissemination of viable tumor cells during surgery and local failure post SRS dosing and clinical resection. We believe it is likely that the complex CNS microenvironment which is in direct contact with the periphery of the BM lesions plays a large role in this unique genetic effect. In addition, we showed that radiation effects different cancer types differently and this will most likely drive the future combination of targeted therapies and treatment modalities for different cancer types. Our results provide important biological context currently lacking in the radiobiology field and highlight the need for designing more nuanced and targeted clinical trials considering the genomics of patient tumors.
One of the most thought-provoking findings of our study is the difference in genomic landscape across GK and LINAC, which are clinically considered interchangeable in terms of their ability to treat BM, across matched primary tumors. We found enrichment for CTA genes, that are becoming an increasing common target for immunotherapy, in lesions treated with LINAC but not lesions treated with GK. This data begs the question: could LINAC be a better radiotherapy delivery modality when used synergistically in a combination immunotherapy trial? Our data provides the biological foundation to ask these critical questions that can not only improve clinical trials for patients with BM but could also be extrapolated to patients with primary brain tumor.
Overall, our samples generate a unique human dataset of radiated BMs, which to date has not yet been collected or studied. Furthermore, the profiling of both DNA and RNA through advanced sequencing technology of SRS-treated human cancer cells in vivo is a novel contribution to the literature and provides insight into human brain radiobiology.
In summary, we conclude that the radiation effect is evident both at the DNA and RNA levels. The biological effect of radiation differs based on the location of the cancer cell and its proximity to the CNS microenvironment which may in turn drive treatment failure. Each type of treatment failure within our sample showed a distinct genomic signature. Further validation of these signatures in larger patient cohorts from the ongoing larger trials could allow for clinical stratification among patients and development of targeted combination therapeutics specifically for a predicted failure type. GK and LINAC SRS delivery modalities produce differential genomic signatures and LINAC generates CTAs which may provide better synergy with immunotherapy than GK. These results provide the support to ask novel clinical questions surrounding the application of SRS to BM’s as well as lay a foundation for more informed design of the next generation of clinical trials.
Limitations of the study
Despite its novelty, the study has limitations. Our overall sample size is small. The brain metastasis sample came from patients with several different types of primary cancers and therefore each cancer sub-type was not represented equally causing a bias toward lung cancer. Therefore, the generalizability of these findings for less common primary cancers needs to be further explored. The lack of germline genomic control samples, and samples from the primary cancer in our exome analysis also limits our ability to draw conclusions relating to direct clonal tracking and genomic evolution of the cancer.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CD45 | Abcam | Cat # 58-0459-42; RRID: AB_2747795 |
| Biological samples | ||
| Human Brain Tumor Samples | Indiana University Department of Neurological Surgery | Biobank |
| Deposited data | ||
| Raw and analyzed data WES GEO | This paper | GSE256463 |
| Raw and analyzed data RNA GEO | This paper | GSE259267 |
| Experimental models: Cell lines | ||
| A549 | University of Wisconsin Madison | ATCC: CCL-185 |
| Oligonucleotides | ||
| CENPB Primer F/R: AATGGCAGCAGAATCTCGTCGG CTTCACCTGTTGGACCACTCCT |
This Paper | Thermo Custom Oligos |
| PSG1 Primer F/R: GAGGAGTAACTGGACGTTTCACC TGGAGTCTCAGGGTCACAGGTT |
This Paper | Thermo Custom Oligos |
| POM121 Primer F/R: ATCACTGCCGAGGACCTAGACT GGTCTCAGTGACAGAGTTCGAG |
This Paper | Thermo Custom Oligos |
| Spink13 Primer F/R CGTGACTTCACTAGGTGGCCTA GAGGCACAAACAGGTGCTGTCA |
This Paper | Thermo Custom Oligos |
| Software and algorithms | ||
| Allez | Newton et al.29 | https://github.com/wiscstatmaN/Allez |
| GATK | Van Der Auwera60,61 | https://gatk.broadinstitute.org/hc/en-us |
| Deseq2 | Love, Huber, Anders30 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RSEM | Li and Dewey62 | https://github.com/deweylab/RSEM |
| ClusterProfiler | Wu et al.63 | https://guangchuangyu.github.io/software/clusterProfiler/ |
| Fardeep | Hao et al.45 | https://cran.r-project.org/web/packages/FARDEEP/index.html |
| plotVCF | No published paper | https://github.com/cccnrc/plot-VCF |
| Diffuse | Chen et al.40 | https://github.com/chenhcs/DIFFUSE |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mahua Dey (dey@neurosurgery.wisc.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
-
•
Whole exome and RNA sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed within the key resources table.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reporting in this paper is available from the lead contact upon request.
Experimental model and study participants
No experimental models or standardized patient datasets were used in this study. All human tumors were collected under the supervision of both the UW and IU IRB’s under approved biosafety protocols. All pertinent data on patients studied that is allowed to be provided is included within the supplemental material. Race and socioeconomic status combined with other data points are considered to be identifying information and cannot be released publicly due to HIPPA privacy concerns. Reasonable requests for inquiry regarding these data points will be honored by the lead contact.
Method details
Sample collection and isolation
During surgical resection of the SRS treated BM’s the center and peripheral portions of the lesion (as determined by the operating surgeon) were collected, removed from the OR, and then flash frozen using liquid nitrogen. Samples were then stored at −80 until isolation of DNA and RNA was able to be completed.
The AllPrep DNA and RNA isolation kit from Quiagen was utilized according to manufactures instructions to obtain viable DNA and RNA from patient samples. Samples were pre-minced with a tissue homogenizer and then run through a Quiagen shredder column to ensure homogenization before proceeding to AllPrep Once obtained, genomic material was QC’d and quantified using a thermo fisher Nanodrop system as well as separately QC’d by sequencing facility using Qubit and Bioanalyzer. Once DNA and RNA were obtained for patients the resulting material was stored at −80 until submission for sequencing. All patients (34) were able to provide some viable genetic material (peripheral/center DNA or RNA), however, due to the nature of the samples being dose with radiation and often necrotic isolation of high-quality RNA proved difficult for some samples, especially within the dosing center of the lesion. Control non-radiated BMs were separately obtained from the Northwestern University clinical tissue bank and were processed in the same manner as above samples.
Sample next generation sequencing
All sequencing for samples was performed by Novogene in accordance with company standards and protocols. Briefly, WES was performed using the Aligent Sure Select V6 kit and sequenced with 150bp paired end reads yielding a target of 12GB per sample run. RNA sequencing was performed using poly-A tail enrichment with NEBNext Ultra II RNA kit sequenced with 150bp paired end reads with a total target of 30M reads per sample (9GB data per sample). All sequencing was run at Novogene facilities on an Illumina NovaSeq 6000. At the completion of sequencing raw FASTQ files were delivered to us for downstream bioinformatic analysis. Upon receipt of FASTQ files MutliQC64 was utilized to ensure all files passed quality control metrics (detailed in Figure S1).
WES analysis
WES analysis for all samples was completed using the well-established GATK pipeline in order to entourage reproducibility of the project.60,61 Due to the nature of the clinical trial genomic control from whole blood was not able to be obtained from patients so a panel of normal variant calling approach was utilized. In this approach, documented well in the GATK pipeline, the panel of normal created by broad for hg38 (our reference genome) was utilized for calling of short variants (SNP and Indel) across both our non-radiated controls as well as our SRS treated samples. The rest of the GATK pipeline was followed as standard, with the output VCF files being annotated by SNPEFF.22 The COSMIC database was also used for functional classification of mutations across and between samples. Quantification of variants and eventual visualization was completed in R v 4.30 using GGPlot, PlotVCF, and pheatmap, hosted in Bioconductor. For visualizations in PlotVCF variants were combined into a single annotated file using BCF tools and then imaged separated by color indicating group or location. For comparisons between central and peripheral samples across GK and LINAC dosing modalities center to center or peripheral to peripheral comparisons were always made.
RNA-seq analysis
RNA sequencing analysis started with all FASTQ files passing QC via MultiQC (with adaptor trimming being completed when necessary) and began with alignment using STAR.65 After alignment normalization was completed with RSEM62 and the resulting normalized count matrix was supplied to DESeq2,30 and differential expression was analyzed according to recommended methods by package developer. ALLEZ29 and ClusterProfiler63 were used for GSEA analysis on differentially expressed genes within our samples and FARDEEP45 was employed for deconvolutional analyses. For isoform enrichment Diffuse40 was utilized after confirming convergence at the 0.001 level and ensuring model fit parameters using QQ and density plots. Isoform enrichment was considered significant at the α < 0.05 level.
QPCR
To validate in-silico differential expression hits we utilized QCPR to measure gene upregulation (relative to gapdh control) in both our biopsy samples as well as the lung cancer cell line A549. Samples were run on an Adaptive Biotechnologies QPCR machine utilizing SYBR green dye from Bio-Rad. Expression changes were calculated using delta-delta CT and visualized in Graphpad Prism. At least technical triplicates were run for each experiment.
Immunohistochemistry
Immunohistochemistry staining for CD45+ cells in our peripheral and central samples was completed by the TRIPP pathology lab at UW-Madison Carbone Cancer Center. CD45 antibody staining was validated on non-precious control samples and was then stained on cut sections from our patient biopsy samples. Quantification of CD45+ cell invasion was obtained using IGV as well as verified by two separate and experimentally blinded human counters. Prism was used for visualization of results.
Quantification and statistical analysis
Throughout the manuscript p < 0.05 was considered as statistically significant and is reported using ∗ within the figures. All statistical tests and their results are reported within the results secretion of the manuscript. For multiple comparisons adjusted p values are reported and utilized for the classification of statistical significance. Differential expression statistics were calculated in DeSeq2 and ALLEZ for their respective analyses and adjusted p values were used to correct for multiple comparisons. For the comparisons between radiated and non-radiated samples central biopsy location was used to represent radiated samples. Within ALLEZ previous term exclusion was utilized for GSEA comparisons and waterfall plot readouts. For comparison between two groups without multiple comparisons students T-test was utilized while comparison between 2 and 4 groups was handled with ANOVA using Bonferroni correction for multiple comparisons. All error bars within the manuscript indicate SD of the data within the bar. Cartoon illustrations were created using Biorender.
Acknowledgments
The authors thank the patients who participated in the trial and contributed invaluable samples for this research and whom all this work is dedicated.
Funding: This work was supported by the NIH K08NS092895 grant (MD), IU Value Research grant (MD). JMS is partly supported by NIH/NINDS T32 NS105602.
Author contributions
JMS/QW/ZN/GC/CM conducted bioinformatic analysis, LB/YZ conducted the clinical trial statistical analysis, JMS/LZ/XW/AP/LM/VK/NG conducted in vitro experiments and isolated/prepared samples for sequencing, AA/GP contributed to the article formulation and provided sequencing samples and cell lines, NA/JM/CK/ACG/TP/CL/JS/BL/KS/AK/MS/GW/MD provided clinical consultation and/or operated on patients on trial, JMS/MD/CK wrote the article, MD supervised the project and the clinical trial.
Declaration of interests
The authors declare no competing interests.
Published: March 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109601.
Supplemental information
References
- 1.Ostrom Q.T., Cioffi G., Gittleman H., Patil N., Waite K., Kruchko C., Barnholtz-Sloan J.S. CBTRUS Statistical Report: Primary Brain and Other Central Nervous System Tumors Diagnosed in the United States in 2012–2016. Neuro Oncol. 2019;21:v1–v100. doi: 10.1093/neuonc/noz150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Suh J.H., Kotecha R., Chao S.T., Ahluwalia M.S., Sahgal A., Chang E.L. Current approaches to the management of brain metastases. Nat. Rev. Clin. Oncol. 2020;17:279–299. doi: 10.1038/s41571-019-0320-3. [DOI] [PubMed] [Google Scholar]
- 3.Moravan M.J., Fecci P.E., Anders C.K., Clarke J.M., Salama A.K.S., Adamson J.D., Floyd S.R., Torok J.A., Salama J.K., Sampson J.H., et al. Current multidisciplinary management of brain metastases. Cancer. 2020;126:1390–1406. doi: 10.1002/cncr.32714. [DOI] [PubMed] [Google Scholar]
- 4.Brown P.D., Ballman K.V., Cerhan J.H., Anderson S.K., Carrero X.W., Whitton A.C., Greenspoon J., Parney I.F., Laack N.N.I., Ashman J.B., et al. Postoperative stereotactic radiosurgery compared with whole brain radiotherapy for resected metastatic brain disease (NCCTG N107C/CEC·3): a multicentre, randomised, controlled, phase 3 trial. Lancet Oncol. 2017;18:1049–1060. doi: 10.1016/s1470-2045(17)30441-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Soliman H., Das S., Larson D.A., Sahgal A. Stereotactic radiosurgery (SRS) in the modern management of patients with brain metastases. Oncotarget. 2016;7:12318–12330. doi: 10.18632/oncotarget.7131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mahajan A., Ahmed S., McAleer M.F., Weinberg J.S., Li J., Brown P., Settle S., Prabhu S.S., Lang F.F., Levine N., et al. Post-operative stereotactic radiosurgery versus observation for completely resected brain metastases: a single-centre, randomised, controlled, phase 3 trial. Lancet Oncol. 2017;18:1040–1048. doi: 10.1016/s1470-2045(17)30414-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bander E.D., Yuan M., Reiner A.S., Panageas K.S., Ballangrud Å.M., Brennan C.W., Beal K., Tabar V., Moss N.S. Durable 5-year local control for resected brain metastases with early adjuvant SRS: the effect of timing on intended-field control. Neurooncol. Pract. 2021;8:278–289. doi: 10.1093/nop/npab005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward J.F. DNA Damage Produced by Ionizing Radiation in Mammalian Cells: Identities, Mechanisms of Formation, and Reparability. Prog. Nucleic Acid Res. Mol. Biol. 1988;35:95–125. doi: 10.1016/s0079-6603(08)60611-x. [DOI] [PubMed] [Google Scholar]
- 9.Jonathan E.C., Bernhard E.J., McKenna W.G. How does radiation kill cells? Curr. Opin. Chem. Biol. 1999;3:77–83. doi: 10.1016/s1367-5931(99)80014-3. [DOI] [PubMed] [Google Scholar]
- 10.Ward J.F. The Complexity of DNA Damage: Relevance to Biological Consequences. Int. J. Radiat. Biol. 1994;66:427–432. doi: 10.1080/09553009414551401. [DOI] [PubMed] [Google Scholar]
- 11.Mantovani C., Gastino A., Cerrato M., Badellino S., Ricardi U., Levis M. Modern Radiation Therapy for the Management of Brain Metastases From Non-Small Cell Lung Cancer: Current Approaches and Future Directions. Front. Oncol. 2021;11 doi: 10.3389/fonc.2021.772789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tuleasca C., Negretti L., Faouzi M., Magaddino V., Gevaert T., von Elm E., Levivier M. Radiosurgery in the management of brain metastasis: a retrospective single-center study comparing Gamma Knife and LINAC treatment. J. Neurosurg. 2018;128:352–361. doi: 10.3171/2016.10.jns161480. [DOI] [PubMed] [Google Scholar]
- 13.Sebastian N.T., Glenn C., Hughes R., Raval R., Chu J., DiCostanzo D., Bell E.H., Grecula J., Arnett A., Gondal H., et al. Linear accelerator-based radiosurgery is associated with lower incidence of radionecrosis compared with gamma knife for treatment of multiple brain metastases. Radiother. Oncol. 2020;147:136–143. doi: 10.1016/j.radonc.2020.03.024. [DOI] [PubMed] [Google Scholar]
- 14.Loi M., Caini S., Scoccianti S., Bonomo P., De Vries K., Francolini G., Simontacchi G., Greto D., Desideri I., Meattini I., et al. Stereotactic reirradiation for local failure of brain metastases following previous radiosurgery: Systematic review and meta-analysis. Crit. Rev. Oncol. Hemat. 2020;153 doi: 10.1016/j.critrevonc.2020.103043. [DOI] [PubMed] [Google Scholar]
- 15.Grosovsky A.J., de Boer J.G., de Jong P.J., Drobetsky E.A., Glickman B.W. Base substitutions, frameshifts, and small deletions constitute ionizing radiation-induced point mutations in mammalian cells. Proc. Natl. Acad. Sci. USA. 1988;85:185–188. doi: 10.1073/pnas.85.1.185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Belyakov O.V., Mitchell S.A., Parikh D., Randers-Pehrson G., Marino S.A., Amundson S.A., Geard C.R., Brenner D.J. Biological effects in unirradiated human tissue induced by radiation damage up to 1 mm away. Proc. Natl. Acad. Sci. USA. 2005;102:14203–14208. doi: 10.1073/pnas.0505020102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Azzam E.I., de Toledo S.M., Little J.B. Direct evidence for the participation of gap junction-mediated intercellular communication in the transmission of damage signals from α-particle irradiated to nonirradiated cells. Proc. Natl. Acad. Sci. USA. 2001;98:473–478. doi: 10.1073/pnas.98.2.473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Castle K.D., Kirsch D.G. Establishing the Impact of Vascular Damage on Tumor Response to High-Dose Radiation Therapy. Cancer Res. 2019;79:5685–5692. doi: 10.1158/0008-5472.can-19-1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mirzayans R., Andrais B., Scott A., Wang Y.W., Murray D. Ionizing Radiation-Induced Responses in Human Cells with Differing TP53 Status. Int. J. Mol. Sci. 2013;14:22409–22435. doi: 10.3390/ijms141122409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rieger K.E., Chu G. Portrait of transcriptional responses to ultraviolet and ionizing radiation in human cells. Nucleic Acids Res. 2004;32:4786–4803. doi: 10.1093/nar/gkh783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Camphausen K., Moses M.A., Ménard C., Sproull M., Beecken W.-D., Folkman J., O’Reilly M.S. Radiation abscopal antitumor effect is mediated through p53. Cancer Res. 2003;63:1990–1993. [PubMed] [Google Scholar]
- 22.Cingolani P., Platts A., Wang L.L., Coon M., Nguyen T., Wang L., Land S.J., Lu X., Ruden D.M. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff. Fly. 2012;6:80–92. doi: 10.4161/fly.19695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H. A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics. 2011;27:2987–2993. doi: 10.1093/bioinformatics/btr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knaus B.J., Grünwald N.J. vcfr: a package to manipulate and visualize variant call format data in R. Mol. Ecol. Resour. 2017;17:44–53. doi: 10.1111/1755-0998.12549. [DOI] [PubMed] [Google Scholar]
- 25.Brandenburger A., Godson G.N., Radman M., Glickman B.W., van Sluis C.A., Doubleday O.P. Radiation-induced base substitution mutagenesis in single-stranded DNA phage M13. Nature. 1981;294:180–182. doi: 10.1038/294180a0. [DOI] [PubMed] [Google Scholar]
- 26.Mao P., Brown A.J., Esaki S., Lockwood S., Poon G.M.K., Smerdon M.J., Roberts S.A., Wyrick J.J. ETS transcription factors induce a unique UV damage signature that drives recurrent mutagenesis in melanoma. Nat. Commun. 2018;9:2626. doi: 10.1038/s41467-018-05064-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pfeifer G.P., Besaratinia A. UV wavelength-dependent DNA damage and human non-melanoma and melanoma skin cancer. Photochem. Photobiol. Sci. 2012;11:90–97. doi: 10.1039/c1pp05144j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Setlow R.B., Grist E., Thompson K., Woodhead A.D. Wavelengths effective in induction of malignant melanoma. Proc. Natl. Acad. Sci. USA. 1993;90:6666–6670. doi: 10.1073/pnas.90.14.6666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newton M.A., Quintana F.A., Den Boon J.A., Sengupta S., Ahlquist P. Random-set methods identify distinct aspects of the enrichment signal in gene-set analysis. Ann. Appl. Stat. 2007;1 doi: 10.1214/07-aoas104. [DOI] [Google Scholar]
- 30.Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Roos W.P., Kaina B. DNA damage-induced cell death: From specific DNA lesions to the DNA damage response and apoptosis. Cancer Lett. 2013;332:237–248. doi: 10.1016/j.canlet.2012.01.007. [DOI] [PubMed] [Google Scholar]
- 32.Su M., Wang H., Wang W., Wang Y., Ouyang L., Pan C., Xia L., Cao D., Liao Q. LncRNAs in DNA damage response and repair in cancer cells. Acta Biochim. Biophys. Sin. 2018;50:433–439. doi: 10.1093/abbs/gmy022. [DOI] [PubMed] [Google Scholar]
- 33.Hosoya N., Miyagawa K. Targeting DNA damage response in cancer therapy. Cancer Sci. 2014;105:370–388. doi: 10.1111/cas.12366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Singhapol C., Pal D., Czapiewski R., Porika M., Nelson G., Saretzki G.C. Mitochondrial Telomerase Protects Cancer Cells from Nuclear DNA Damage and Apoptosis. PLoS One. 2013;8 doi: 10.1371/journal.pone.0052989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mirzayans R., Andrais B., Scott A., Murray D. New Insights into p53 Signaling and Cancer Cell Response to DNA Damage: Implications for Cancer Therapy. J. Biomed. Biotechnol. 2012;2012 doi: 10.1155/2012/170325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang C., Yu D. Microenvironment Determinants of Brain Metastasis. Cell Biosci. 2011;1:8. doi: 10.1186/2045-3701-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schulz M., Salamero-Boix A., Niesel K., Alekseeva T., Sevenich L. Microenvironmental Regulation of Tumor Progression and Therapeutic Response in Brain Metastasis. Front. Immunol. 2019;10:1713. doi: 10.3389/fimmu.2019.01713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Berghoff A.S., Preusser M. The inflammatory microenvironment in brain metastases: potential treatment target? Chin. Clin. Oncol. 2015;4:21. doi: 10.3978/j.issn.2304-3865.2015.06.03. [DOI] [PubMed] [Google Scholar]
- 39.Tate J.G., Bamford S., Jubb H.C., Sondka Z., Beare D.M., Bindal N., Boutselakis H., Cole C.G., Creatore C., Dawson E., et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 2019;47:D941–D947. doi: 10.1093/nar/gky1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen H., Shaw D., Zeng J., Bu D., Jiang T. DIFFUSE: predicting isoform functions from sequences and expression profiles via deep learning. Bioinformatics. 2019;35:i284–i294. doi: 10.1093/bioinformatics/btz367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puchalski R.B., Shah N., Miller J., Dalley R., Nomura S.R., Yoon J.-G., Smith K.A., Lankerovich M., Bertagnolli D., Bickley K., et al. An anatomic transcriptional atlas of human glioblastoma. Science. 2018;360:660–663. doi: 10.1126/science.aaf2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Venteicher A.S., Tirosh I., Hebert C., Yizhak K., Neftel C., Filbin M.G., Hovestadt V., Escalante L.E., Shaw M.L., Rodman C., et al. Decoupling genetics, lineages, and microenvironment in IDH-mutant gliomas by single-cell RNA-seq. Science. 2017;355 doi: 10.1126/science.aai8478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L., et al. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang L., Jung J., Babikir H., Shamardani K., Jain S., Feng X., Gupta N., Rosi S., Chang S., Raleigh D., et al. A single-cell atlas of glioblastoma evolution under therapy reveals cell-intrinsic and cell-extrinsic therapeutic targets. Nat. Can. (Ott.) 2022;3:1534–1552. doi: 10.1038/s43018-022-00475-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hao Y., Yan M., Heath B.R., Lei Y.L., Xie Y. Fast and robust deconvolution of tumor infiltrating lymphocyte from expression profiles using least trimmed squares. PLoS Comput. Biol. 2019;15 doi: 10.1371/journal.pcbi.1006976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Park H.S., Wang E.H., Rutter C.E., Corso C.D., Chiang V.L., Yu J.B. Changing practice patterns of Gamma Knife versus linear accelerator-based stereotactic radiosurgery for brain metastases in the US. J. Neurosurg. 2016;124:1018–1024. doi: 10.3171/2015.4.jns1573. [DOI] [PubMed] [Google Scholar]
- 47.Scanlan M.J., Gure A.O., Jungbluth A.A., Old L.J., Chen Y.T. Cancer/testis antigens: an expanding family of targets for cancer immunotherapy. Immunol. Rev. 2002;188:22–32. doi: 10.1034/j.1600-065x.2002.18803.x. [DOI] [PubMed] [Google Scholar]
- 48.Caballero O.L., Chen Y.-T. 2011. Innate Immune Regulation and Cancer Immunotherapy; pp. 347–369. [DOI] [Google Scholar]
- 49.Djureinovic D., Hallström B.M., Horie M., Mattsson J.S.M., La Fleur L., Fagerberg L., Brunnström H., Lindskog C., Madjar K., Rahnenführer J., et al. Profiling cancer testis antigens in non–small-cell lung cancer. JCI Insight. 2016;1 doi: 10.1172/jci.insight.86837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fratta E., Coral S., Covre A., Parisi G., Colizzi F., Danielli R., Nicolay H.J.M., Sigalotti L., Maio M. The biology of cancer testis antigens: Putative function, regulation and therapeutic potential. Mol. Oncol. 2011;5:164–182. doi: 10.1016/j.molonc.2011.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li X.-F., Ren P., Shen W.-Z., Jin X., Zhang J. The expression, modulation and use of cancer-testis antigens as potential biomarkers for cancer immunotherapy. Am. J. Transl. Res. 2020;12:7002–7019. [PMC free article] [PubMed] [Google Scholar]
- 52.Reynders K., Illidge T., Siva S., Chang J.Y., De Ruysscher D. The abscopal effect of local radiotherapy: using immunotherapy to make a rare event clinically relevant. Cancer Treat Rev. 2015;41:503–510. doi: 10.1016/j.ctrv.2015.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Minn A.J., Kang Y., Serganova I., Gupta G.P., Giri D.D., Doubrovin M., Ponomarev V., Gerald W.L., Blasberg R., Massagué J. Distinct organ-specific metastatic potential of individual breast cancer cells and primary tumors. J. Clin. Invest. 2005;115:44–55. doi: 10.1172/jci22320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Morikawa K., Walker S.M., Nakajima M., Pathak S., Jessup J.M., Fidler I.J. Influence of organ environment on the growth, selection, and metastasis of human colon carcinoma cells in nude mice. Cancer Res. 1988;48:6863–6871. [PubMed] [Google Scholar]
- 55.Fischer G.M., Jalali A., Kircher D.A., Lee W.-C., McQuade J.L., Haydu L.E., Joon A.Y., Reuben A., de Macedo M.P., Carapeto F.C.L., et al. Molecular Profiling Reveals Unique Immune and Metabolic Features of Melanoma Brain Metastases. Cancer Discov. 2019;9:628–645. doi: 10.1158/2159-8290.cd-18-1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Rivina L., Davoren M.J., Schiestl R.H. Mouse models for radiation-induced cancers. Mutagenesis. 2016;31:491–509. doi: 10.1093/mutage/gew019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yang L., Yang J., Li G., Li Y., Wu R., Cheng J., Tang Y. Pathophysiological Responses in Rat and Mouse Models of Radiation-Induced Brain Injury. Mol. Neurobiol. 2017;54:1022–1032. doi: 10.1007/s12035-015-9628-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brastianos P.K., Carter S.L., Santagata S., Cahill D.P., Taylor-Weiner A., Jones R.T., Van Allen E.M., Lawrence M.S., Horowitz P.M., Cibulskis K., et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015;5:1164–1177. doi: 10.1158/2159-8290.cd-15-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shih D.J.H., Nayyar N., Bihun I., Dagogo-Jack I., Gill C.M., Aquilanti E., Bertalan M., Kaplan A., D’Andrea M.R., Chukwueke U., et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet. 2020;52:371–377. doi: 10.1038/s41588-020-0592-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Auwera G.A., Carneiro M.O., Hartl C., Poplin R., Del Angel G., Levy-Moonshine A., Jordan T., Shakir K., Roazen D., Thibault J., et al. From FastQ Data to High-Confidence Variant Calls: The Genome Analysis Toolkit Best Practices Pipeline. Curr Protoc Bioinform. 2013;43 doi: 10.1002/0471250953.bi1110s43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Auwera G.A., O’Connor B.D. 2020. Genomics in the Cloud: Using Docker, GATK, and WDL in Terra (O’Reilly Media) [Google Scholar]
- 62.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu T., Hu E., Xu S., Chen M., Guo P., Dai Z., Feng T., Zhou L., Tang W., Zhan L., et al. clusterProfiler 4.0: A universal enrichment tool for interpreting omics data. Innovation. 2021;2 doi: 10.1016/j.xinn.2021.100141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ewels P., Magnusson M., Lundin S., Käller M. MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics. 2016;32:3047–3048. doi: 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Dobin A., Davis C.A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T.R. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Whole exome and RNA sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed within the key resources table.
-
•
The paper does not report original code.
-
•
Any additional information required to reanalyze the data reporting in this paper is available from the lead contact upon request.