

Summary
Knowledge of cell signaling pathways that drive human neural crest differentiation into craniofacial chondrocytes is incomplete, yet essential for using stem cells to regenerate craniomaxillofacial structures. To accelerate translational progress, we developed a differentiation protocol that generated self-organizing craniofacial cartilage organoids from human embryonic stem cell-derived neural crest stem cells. Histological staining of cartilage organoids revealed tissue architecture and staining typical of elastic cartilage. Protein and post-translational modification (PTM) mass spectrometry and snRNA-seq data showed that chondrocyte organoids expressed robust levels of cartilage extracellular matrix (ECM) components: many collagens, aggrecan, perlecan, proteoglycans, and elastic fibers. We identified two populations of chondroprogenitor cells, mesenchyme cells and nascent chondrocytes, and the growth factors involved in paracrine signaling between them. We show that ECM components secreted by chondrocytes not only create a structurally resilient matrix that defines cartilage, but also play a pivotal autocrine cell signaling role in determining chondrocyte fate.
Subject area: Natural sciences, Biological sciences, Biochemistry, Cell biology, Stem cells research, Specialized functions of cells
Graphical abstract
Highlights
-
•
Craniofacial cartilage organoids were grown from human neural crest stem cells
-
•
These organoids exhibited elastic cartilage architecture and characteristic markers
-
•
Paracrine signaling drove chondrogenesis in mesenchyme cells and nascent chondrocytes
-
•
ECM components cemented chondrocyte cell fate through autocrine signaling
Natural sciences; Biological sciences; Biochemistry; Cell biology; Stem cells research; Specialized functions of cells
Introduction
Craniofacial reconstruction is necessary to treat several conditions, including craniofacial birth defects, damage caused by head and neck cancer treatment, and traumatic facial injuries. These cases not only affect function but may also cause or contribute to psychological and social difficulties. It is difficult to reconstruct natural features with plastic surgery techniques, however, and transplanted tissue is often rejected by the recipient without immunosuppressants. The use of stem cells to grow craniofacial cartilage would revolutionize the effective regeneration and repair of birth defects and injuries to the head and face. To harness this differentiation for clinical translation, we require an in-depth understanding of human-specific cell signaling mechanisms involved in the differentiation of this tissue.
Craniofacial cartilage is comprised of hyaline cartilage for the nose and developing bones of the head, elastic cartilage for the larynx, epiglottis, eustachian tube, external ear, and fibrocartilage for temporomandibular joints. Unlike articular cartilage, craniofacial cartilage originates during development from chondrocytes that are derived from the neural crest. Neural crest cells (NCCs) are a developmental cell lineage restricted to vertebrates and are sometimes considered the fourth germ layer. During neurulation, NCCs are specified at the neural plate border between the ectoderm and developing neuroepithelium. At the crest of the neural tube, NCCs undergo an epithelial-to-mesenchymal transition, delaminate, and migrate throughout the developing embryo.1 During this process, NCCs begin differentiating into a number of distinct cell types, including cells of the peripheral nervous system, melanocytes, smooth muscle, and craniofacial cartilage and bone. Depending on their positioning and migratory patterns along the rostrocaudal axis of the developing embryo, NCCs can be further categorized into four main populations: cranial neural crest cells (CNCCs), cardiac neural crest cells (CaNCCs), vagal neural crest cells (VNCCs), and trunk neural crest cells (TNCCs). CNCCs eventually differentiate into craniofacial cartilage and bone, as well as glia, Schwann cells, and cranial sensory glia.1 Therefore, craniofacial cartilage has a different developmental origin from the articular hyaline cartilage and fibrocartilage that compose the intervertebral disks and tendon insertions in the rest of the body. In body tissues, the skeleton is first made of cartilage by chondrocytes derived from the mesoderm, which ossifies into bone; and some cartilage remains in selective locations, for example at the interface of joints. Thus, to develop techniques to specifically generate craniofacial cartilage, different procedures from those used for other types of cartilage will be required.2,3,4 This motivates a greater understanding of the cellular processes that underlie craniofacial cartilage development.
In general, chondrogenesis is under the control of several major developmental signaling pathways including WNT, BMP/TGFβ, and FGF, among others.2,4,5,6 While insights can be gained from animal models about growth factor signaling pathways that drive craniofacial cartilage differentiation,7 details about these pathways in humans are not well defined, which motivates this study. To model human craniofacial cartilage development, we generated craniofacial cartilage organoids from human embryonic stem cells (hESCs) and induced pluripotent stem cells (iPSCs) via a neural crest cell intermediate. Our protocol for neural crest differentiation integrated and streamlined several existing protocols from the recent literature.8,9,10,11 Craniofacial cartilage organoids were generated from neural crest stem cells (NCSCs) that were self-organizing, did not require a scaffold or embedding in extracellular matrices, and were amenable to handling and histological techniques. We focused on the specific cell types and cell signaling pathways that regulate human craniofacial cartilage formation by comprehensively interrogating gene expression patterns in differentiating cells using single nuclei RNA sequencing (snRNA-seq). We also employed mass spectrometry to analyze total protein expression and clustering patterns in protein post-translational modifications (PTMs) to outline cell signaling pathways in organoids.12,13 These data provide a resource for further studies of human craniofacial development with an eye toward the improvement of stem cell models for craniofacial regeneration and repair.14,15
Results
Neural crest stem cells differentiated into cartilage organoids
To differentiate craniofacial cartilage, we first directed the differentiation of human embryonic stem cells (hESCs) into a neural crest stem cell (NCSC) intermediate (Figure 1A). We integrated several previously published protocols with the goal of shortening this stage of differentiation and generating the NCSC intermediate as quickly as possible while optimizing the yield. To obtain neural crest cells, it was first necessary to induce neuroectoderm formation using the protocol previously published by Kreitzer et al., which utilizes dual SMAD inhibition via the small molecules LDN193189 and SB431542, inhibiting BMP and TGF-β signaling, respectively.8 However, we fit the timeline of this protocol to the research by Mica et al., which showed that early removal of TGF-β inhibitors, followed by the prolonged addition of the small molecule WNT signaling activator CHIR99021 (GSK3β inhibitor), increased anterior NCSC yield.9,11 Finally, we added Shh, FGF8, ascorbic acid, and BDNF to the last stage of NCSC induction, following the neural crest specification protocol outlined by Zeltner et al., to further increase the number of NCSCs over a short time period.10 After neural crest specification (Figure 1A), following the re-plating stages of the Zeltner et al. protocol, we then sorted NCSCs via magnetic-activated cell sorting based on low-affinity nerve growth factor receptor (p75/NGFR) expression and re-plated cells as a monolayer on poly-L-ornithine, laminin, and fibronectin. Over the course of NCSC differentiation, hESCs acquired NCSC morphology and migrated outwards from hESC colonies (Figure 1B, Day 1–9).
Figure 1.

Neural crest differentiation protocol and cartilage organoids
(A) Neural crest stem cell (NCSC) differentiation protocol. hESCs were differentiated using the stepwise replacement of knockout serum replacement (KSR) media with N2 for 5 days, followed by the addition of NCSC specification media containing BDNF, FGF8, SHH, and ROCKi. Small molecule inhibitors were added to guide differentiation (see STAR Methods). NCSCs and organoid-producing cells were maintained in N2 media containing FGF2 and EGF.
(B) Representative light microscopy images of cell morphology throughout differentiation and after prolonged culture. Day = number of days after the original differentiation protocol. Organoids continued to spontaneously form in cell culture dishes containing differentiated NCSCs. Three images at day 68 show highly plastic morphology of cells as they migrate individually and begin to assemble into an organoid.
(C) Histology and stain of human ear tissue (top) and representative organoid (bottom). H&E, hematoxylin and eosin. Saffranin O and Toluidine Blue stain cartilage ECM components red-orange and purple, respectively; Weigert’s resorcin-fuchsin stains elastic fibers violet. (N = 5). Scale bar = 50 μm.
When left undisturbed for approximately 52 h, confluent NCSCs migrated together and self-organized into roughly spherical growths on the surface of dishes (Figure 1B). Once formed, these organoids detached from the dish surface and floated in the cell culture media. Over time, cells remaining on the surface of the dish exhibited the morphology of migrating chondrocyte precursors (Figure 1B, Day 68). Floating organoids were easily harvested with a spatula, and remaining cells on the dish surface continued to produce new organoids. Cell confluence typically remained around 50% at this stage, with localized dense patches of cells that begin to migrate into new growths. The largest harvested organoid was one centimeter in diameter (Figure 1B, Day 162), and it was possible to maintain organoid-producing culture dishes for up to one year. Organoids had physical characteristics of cartilage, appearing opaque and glassy, robust to handling, and resisting deformation in response to applied pressure.
Histological analysis using hematoxylin/eosin (H & E) staining of organoid paraffin sections revealed typical morphology of cartilaginous tissues – small cell nuclei in the center of lacunae embedded in a glassy gray matrix (Figure 1C). Saffranin O and Toluidine Blue revealed basophilic staining likely due to the presence of negatively charged aggrecan and other proteoglycans (Figure 1C). The tissue morphology and Weigert’s resorcin-fuchsin staining for elastic fibers suggest that elastic cartilage similar to human ear tissue is forming in these organoids (Figure 1C).
Organoids were positive for collagens and other extracellular matrix proteins
To explore the developmental pathways that lead to human craniofacial cartilage, we profiled gene expression at the single-cell level during the formation of neural crest-derived cartilage organoids. We employed single nuclei RNA sequencing (snRNA-seq) because of the difficulty of extracting intact cells from the cartilage organoid matrix.16 NCSCs were differentiated and sorted as described above, and a representative sample of all cells was taken using a cell scraper. After merging and pruning data (see STAR Methods and Figure S1) from cells harvested directly the day after sorting (NCSCs) and a sample of adherent cells and organoids harvested at day 87, six clusters representing different populations of cells were obtained (Figures 2A and 2B; Table S1). NCSCs harvested one day after sorting formed a distinct large cluster in both t-SNE and UMAP projections (Figures 2A, 2B, S1A, and S1B). One cluster expressed high amounts of collagens, proteoglycans, and markers for the synthesis and modification of elastic fibers17(Figures 2C–2E, S2, and S3), so we provisionally labeled them chondrocytes. Because the cells in other clusters have not been previously described, automated annotation was unsuccessful; we therefore investigated the cell clusters manually (see STAR Methods). Based on marker and pathway analysis (see later in discussion), these other populations of cells resembled migratory neural crest (mesenchyme), immature neurons (neuroblasts), and a small population of melanocytes (Figure 2A).
Figure 2.

snRNA-seq data from cells in differentiating cultures and craniofacial cartilage organoids
(A and B) UMAP projections of snRNA-seq data from day 1 (NCSC) and day 87 (after differentiation) cultures, merged, after a cluster of artifactual objects was removed (see Figure S1).
(C–E) In A different clusters are identified by cell type. In B the cells from day 1 and day 87 are identified. Several features confirmed that the green cluster in A contained chondrocytes that expressed the main components of elastic cartilage: collagen genes (C), proteoglycan genes (D), and genes reads involved in elastic fiber metabolism (E), all expressed as percentile per cell.
To further investigate the composition of organoids, we performed tandem mass tag mass spectrometry (TMT-MS) analysis comparing the parent stem cell line (WA09), neural stem cells (NGFR-negative population after magnetic sorting, NSCs), the NGFR-positive NCSCs, and cartilage organoids. TMT-MS analysis identified 11,064 proteins, 10,729 of which were detected in at least three samples, which provided an in-depth view of protein expression in NCSCs and craniofacial cartilage organoids. hESCs (WA09) expressed the highest amounts of the pluripotency markers OCT4 (POU5F1 gene) and SOX2 (Figure 3A). NCSCs had higher amounts of the neural crest marker NGFR, which was used to sort this cell population. Craniofacial cartilage organoid samples retained SOX9, which is involved in originating neural crest and chondrogenesis,18,19 and had low expression of OCT4 and SOX2 (Figure 3A).
Figure 3.

Markers in cells in differentiating cultures and craniofacial cartilage organoids
(A and B) Heatmaps show the expression of stem cell markers (A) and cartilage markers (B) in hESCs (WA09), neural stem cells (NSCs), neural crest stem cells (NCSCs), and organoids (ORGs). Median and maximum values were calculated from the row Z score values, with increased expression represented in yellow and decreased expression in blue (inset in A; black represents missing values). In B, data were ordered by hierarchical clustering (dendrogram).
(C) Immunoflourescence of markers (labeled) in representative organoids (red or green according to the secondary antibody used) counterstained with DAPI (blue). Scale bar = 50 μm.
Twenty-five markers commonly associated with chondrocytes were elevated in cartilage organoids compared to NCSCs in our proteomic data (Figure 3B; p = 0.026, Fisher’s chi-square method; p = 0.0078, Fisher’s truncated method). Furthermore, 131 markers including a variety of generic collagens and ECM components were even more significantly elevated in organoids (p = 3.202 × 10–11, Fisher’s truncated method; Figure S4A). Notably, collagen X (COL10A1), a marker for chondrocyte hypertrophy,20 was detected only in one experiment by mass spectrometry and in lower amounts in organoids than in WA09 stem cells and NCSCs. COL10A1 also had low expression in snRNA-seq data and was not enriched in chondrocytes (Figure 4C). Accordingly, we saw no evidence of bone formation (hypertrophy) in organoids.
Figure 4.

BioPlanet pathways enriched in craniofacial cartilage organoids
(A) Pathway crosstalk network (PCN). BioPlanet pathway-pathway relationships were determined from protein and PTM data, and by Jaccard similarity, which means they have genes in common (purple, orange, and green edges, respectively). Pathway enrichment was determined from t-SNE clusters of protein and PTM data in organoids and NCSCs (see STAR Methods). The network shown is the difference between organoid- and NCSC-enriched pathways, in other words pathways that were enriched in organoids and not in NCSCs. These pathways intersected with those identified by pooled p values from: proteomic data in Figure S1A (nodes circled in pink); snRNA-seq data comparing chondrocytes to mesenchyme cells (nodes circled in blue); and snRNA-seq data comparing chondrocytes to NCSCs (yellow nodes).
(B) Pathways identified by EnrichR in Chondrocytes snRNA-seq cell cluster. Only the BioPlanet pathways with an adjusted p value smaller than 0.05 are shown. The pathways overlapping with the pathways identified in the proteomics analysis (A and B) are highlighted in yellow. Some pathway names are abbreviated for legibility.
(C and D) Volcano plots show the RNA expression of genes in the chondrocyte cluster in pathways Collagen biosynthesis and modifying enzymes (C) and TGFβ regulation of extracellular matrix (D). Fold change (log2FC) is expression in day 87 chondrocytes vs. day 1 NCSCs.
There was variability in protein expression as determined by mass specrometry, however, likely due to the extent of differentiation and variation in different populations of cells that make up organoids. For example, two of four organoid samples highly expressed cartilage markers COL2A1 and Aggrecan (ACAN), and only one had elevated levels of SOX5, SOX6, and SOX9 (Figures 3B and S4A). Similarly, matrilins (MATN genes), Hyaluronan And Proteoglycan Link proteins (HAPLN genes), Elastin Microfibril Interfacer proteins (EMILIN genes), which are involved in cell-matrix attachments and hyaluronan and proteoglycan binding,21,22 were high in some organoids but exhibited variable expression (Figures 3B and S4A). High but variable expression was also observed for perlecan (HSPG2) and many other collagen family members, some of which are involved in pericellular matrix induction (Figure S4A).
Immunofluorescence identified distinct patterns of protein expression in individual organoids (Figure 3C). Transcription factors SOX2, SOX9, and neural cell adhesion molecule (NCAM, a marker for immature neurons and early neural crest lineage), were expressed in a few isolated cells dispersed throughout organoids (Figure 3C, top row). In mature organoids, COL2A1 staining was bright and diffuse throughout the central portion, and COL1A1 staining was predominantly on the outer edge (Figure 3C, middle). In immature organoids, COL1A1-positive regions surrounded cells with lower amounts of COL2A1 (Figure 3C, middle right). COMP and MATN1 staining was observed throughout organoids, while doublecortin (DCX), a migratory neural crest and chondrocyte cell marker23,24,25 showed more robust expression in the outer layer of organoids (Figure 3C, bottom). These data indicate that organoids were composed of more than one cell type, as was seen in differentiating cultures (Figure 2A).
Pathways involved in differentiation
We used several different approaches to reveal cell signaling and metabolic pathways that were activated during NCSC differentiation into chondrocytes. Enrichr (45) identified the following pathways from the 106 genes most significantly up-regulated in organoids compared to NCSCs from which they were derived (Figures S4B and S4C): TGF-β regulation of extracellular matrix (BioPlanet), extracellular matrix organization (Reactome), ECM-receptor interaction (KEGG), and inhibition of matrix metalloproteinases (BioCarta). Comparing total protein expression, 11 of 1657 curated pathways from NCATS BioPlanet26 were significantly changed in organoids vs. NCSCs (Fishers truncated p ≤ 0.05 using pooled p values for gene sets from median expression; Figure S5A). The top five pathways that were enriched in organoids over NCSCs were: Inhibition of matrix metalloproteinases; Glycosphingolipid biosynthesis: ganglio series; Chondroitin sulfate biosynthesis; Dermatan sulfate biosynthesis; and Matrix metalloproteinases. Other enriched pathways identified from these markers in chondrocytes reflected changes in the metabolism of carbohydrates and lipids, and membrane traffic (Figure S5).
Since post-translational modifications (PTMs) reflect the activation of cell signaling pathways more directly than mRNA and protein expression,12 we also profiled protein phosphorylation, acetylation, and methylation in cartilage organoids and NCSCs. We analyzed the data using network models of data structure,13 which allow the interrogation of protein and multi-PTM proteomics data with three levels of granularity at the pathway, protein-protein interaction, and PTM levels. The highest-level network, called the Pathway Crosstalk Network (PCN),13 connects biological pathways from BioPlanet based on the extent to which each pathway’s proteins (and their PTMs) co-cluster using t-SNE. To focus on pathways that are active in NCSCs and differentiated organoids, we constructed versions of the PCN based on clusters that were enriched for proteins or PTMs whose expression/abundance was significantly different in these samples relative to SH-SY5Y neuroblastoma cells.13 Figure 4A shows the subnetwork from the organoid PCN that was different from the NCSC PCN. Several pathways related to the construction of ECM components (collagens, chondroitin sulfate, heparin sulfate, and glycosaminoglycans) were linked by PTM and protein cluster evidence (purple and orange edges, respectively, Figure 4A). Many of these pathways have genes in common, as indicated by Jaccard similarity (green edges, Figure 4A). Most of these pathways identified by the PCN analysis were also identified by comparing snRNA-seq data from chondrocytes vs. NCSCs (using pooled p values and EnrichR; Figures 4A and 4B, highlighted in yellow). These pathway-pathway interactions provide an overview of how cell signaling mechanisms drive specific transcriptional differentiation programs that activate many pathways to build cartilage ECM.
The mesenchyme cluster identified by snRNA-seq expressed some collagens, but marker and Enricher pathway analysis suggests that this cell population is a previously uncharacterized cell type at an intermediate state of differentiation (Figures S2 and S5C). Using snRNA-seq data to identify pathways enriched in chondrocytes relative to mesenchyme cells, 8 pathways were identified (Figure 4A, nodes circled in blue). Because the mesenchyme cell population expressed some genes in common with both chondrocytes and neuroblasts, we hypothesized that these cells may represent progenitors to both of these populations. To test this, we examined markers from characteristic pathways of chondrocyte and neuroblast cell clusters in snRNA-seq data. The chondrocyte cell cluster had many genes whose expression was significantly up-regulated in pathways such as collagen synthesis and modifying enzymes; TGF-β regulation of extracellular matrix; and extracellular matrix organization, (Figures 4B–4D and S6A). These pathways had a subset of genes up-regulated in mesenchyme cells (Figure S6B) but very few in neuroblasts (Figure S6C). In contrast, pathways neural system and neurotransmitter release cycle, shared markers between neuroblast and mesenchyme cells but few with chondrocytes (Figures S6B and S6C). These data suggest that mesenchyme cells represent a distinct cell type not differentiated into chondrocytes or neuroblasts.
Ligand and receptor pairs that the direct differentiation of craniofacial cartilage organoids
We next examined protein-protein interactions among the pathways identified above with an eye toward the question, what are the key cell signaling mechanisms that play a role in driving human NCSC differentiation into chondrocytes? Receptor and ligand pairs that are known to be involved in neural crest formation or cartilage differentiation were present in the proteomic dataset using the R package connectome27 (Figure 5). Notably, ligands enriched in organoids compared to hESCs and NCSCs included tenascin C (TNC); midkine (MDK); pleiotrophin (PTN); transforming growth factor β3 (TGFB3); bone morphogenic proteins (BMPs) 5 and 7; WNT ligands 3, 4, 5A, 5B, 7A and 11; fibroblast growth factors 1 and 2 (FGF1, FGF2); platelet-derived growth factors C and D (PDGFC, PDGFD); delta-like non-canonical notch ligand 1 (DLK1); and VGF (Figures 5A and 5D). Receptors corresponding to these ligands were also expressed, including ALK, NOTCH1 and 3, PTPRS, PTPRZ1, BMPR2; LRP1; FZD1, 2, and 7; TGFBR3; and PDGFRB (Figures 5C and 5D). With the exception of FGF2, ligand-receptor pairs enriched in organoid samples were different from the set of ligands added to NCSC cultures, suggesting that the organoids may have established autocrine and/or paracrine signaling patterns during differentiation.
Figure 5.

Ligands and receptor protein expression in NCSCs and organoids
(A–C) Heatmaps show the expression of growth factor ligands (A), ECM ligands (B), and receptors (C) in stem cells, NCSs, NCSCs and craniofacial cartilage organoids from mass spectrometry data. Data are expressed as row z-scores (see legend in C); black represents missing data.
(D and E) Selected non-ECM (D) and ECM (E) ligand-receptor pairs derived from protein data. Node size and color is proportional to the maximum row Z score (inset). Edges are from connectome.27 Node border and shape and edge colors are defined in Figure S7C.
ECM components are also ligands for cell signaling receptors (Figures 5B and 5E).28,29,30,31,32 Those enriched in organoids included fibrillin 1 (FBN1); fibronectin 1 (FN1); fibulins (FBLN1, EFEMP2); tissue inhibitor of metalloproteinases 1 (TIMP1); proteoglycans perlecan, versican (HSPG2, VCAN); laminins (LAMA4, LAMC1, LAMB1); thrombospondins (THBS1, THBS2); and several collagens (Figures 5B and 5E). Receptors for these proteins were also expressed (Figures 5C and 5E), including integrins (ITGAV, ITGB5, ITGA11, ITGB1, ITGA8); tetraspanins (CD81, CD63); and discoidin domain receptor tyrosine kinase 1 and 2 (DDR1, DDR2).
From snRNA-seq data, similar ligand-receptor pairs were identified (Figure 6A). Several collagen transcripts were abundant; these collagens interact with integrins, and with DDR2, which was up-regulated in chondrocytes (Figures 6A and 6C).28,29,30,31,32 Also specifically up-regulated in chondrocytes were PDGFD and its receptors PDGFRA, PDGFRB; DLK1, which interacts with NOTCH3; and VCAN and DCN, which interact with EGFR (Figures 6A and 6C). In contrast, the main ligand-receptor pair up-regulated in mesenchyme cells was PTN-PTPRZ1; the collagen receptor, DDR1 was also highly expressed in this population (Figure 6B).
Figure 6.

Receptor-ligand expression in cell populations
(A) Ligand-receptor network derived from snRNA-seq data and connectome.27 Ligands are on the outer circle; receptors in the inner circle. Node size and color are proportional to mRNA expression; edge width is proportional to the weight of evidence.
(B and C) Violin plots of selections from this network enriched in mesenchyme cells (B) and chondrocytes (C) show expression levels.
To understand mechanisms of crosstalk between cell signaling pathways, we interrogated a protein-protein interaction (PPI) network that was filtered to retain only interactions between proteins that show correlated abundance in our proteomic data, which we call a cluster-filtered network (CFN).12,13 We focused on pathways such as Wnt signaling pathway and neural crest differentiation because they are likely upstream of differentiation (Figure 4A). Figure S7A shows the complex CFN interactions between all proteins in these pathways. From these, we selected ligand-receptor pairs identified from proteomic and snRNA-seq data in neural-crest-derived chondrocytes, and their nearest neighbors (Figure S8A). Impressively, crosstalk between these pathways involves ECM components and their receptors (e.g., collagens, integrins, matrilins, and fibronectin), which appear to be downstream effectors of WNTs, NOTCH1, PTPRZ1, and receptor tyrosine kinases (ALK, PDGFRA, ROR2, and several EPH receptors). Many of these proteins are up-regulated in craniofacial cartilage organoids (large yellow nodes, Figure S8A).
We also examined connections between pathways TGF-β regulation of extracellular matrix and Glycosaminoglycan metabolism because they are likely central to cell fate specification due to autocrine signaling, as mentioned above, and cartilage biophysical and biochemical properties (Figures 4A and S7B).33,34,35 “Mutual friends,” defined as proteins that directly connect to at least one member of each pathway in the CFN (Figure S8B), were examined. Consistent with our analysis above, these mutual friends include many collagens and other ECM components, along with receptors and several transcription factors (Figure S8B).
These data suggest the hypothesis that, in addition to being components of the structurally resilient matrix that defines cartilage, ECM components play a profound autocrine signaling role in chondrocyte differentiation though a feedforward mechanism mediated by receptors that respond to collagens, proteoglycans, and other ECM components (Figures 5E and 6A).
Cell types involved in differentiation
Based on the data above, we hypothesized that more than one cell type plays a role in craniofacial chondrocyte differentiation. To test this hypothesis, we analyzed differentiating cells at an earlier stage of differentiation 28 days after NCSC sorting. At this stage, cells are beginning to migrate together but have not yet formed organoids or discernible cartilage tissue. After removing artifactual data as described above, snRNA-seq data from day 28 cells sorted into five cell types (Figure 7A). Nascent chondrocytes were clearly distinguished by the expression of collagens and other ECM components similar to chondrocytes at day 87 (Figure S9A). Analysis of cell cycle gene expression revealed that a subset of nascent chondrocytes and mesenchyme cells expressed genes indicating they were in G2 and S phase (Figure S9B). These actively proliferating cells were labeled Dividing Chondrocytes and Dividing Mesenchyme (Figures 7A and 7B).
Figure 7.

Clusters of cells identified by snRNA-seq data 28 days after NCSC sorting
(A) UMAP projections of snRNASeq data from differentiating cultures day 28 after NCSC sorting. Mesencyme, Chondrocyte, and Neuroblast cell clusters were subdivided by cell cycle analysis and by Seurat based on marker expression (B). A fraction of chondrocytes (Dividing Chondrocytes, dark green) and a population mesenchyme cells (Dividing Mesenchyme, brown) share cell cycle markers of proliferating cells (B; see Figure S9).
(C and D) Paracrine (C, graphed as in Figure 6A except with ligands arranged vertically and receptors arranged horizontially) and autocrine (D) receptor-ligand interactions from day 28 cells (identified by connectome). Mesenchyme and Dividing Mesenchyme cells had 11-12 autocrine signaling interactions almost identical to each other, and secrete EGF, PTN, PDGFC, WNT3, and WNT7B. Day 28 Chondrocytes (including Dividing Chondrocytes) had 47 autocrine interactions and secrete ECM components that include 10 different collagen proteins, three laminins, FN1, FBN1, VCAN, DCN, and ligands TNC, TGFB3, WNT5A, PDGFD, and INHBA. Mesenchyme cells had 27 paracrine interactions with the entire chondrocyte population (including Dividing Chondrocytes). N = 3.
We hypothesized that mesenchyme cells that do not express cartilage ECM components may influence chondrocyte differentiation by paracrine signaling. To test this, we analyzed ligand-receptor expression grouped by sources and targets using connectome27 (Figure 7C). Dividing chondrocytes and dividing mesenchyme cells had the most paracrine interactions (46) among all cell types, which include ligands and receptors from each cell type (Figure 7C). Dividing chondrocytes at day 28 also had the most autocrine interactions among all cell types (96 edges compared to 43 in the remaining chondrocytes, and 4–12 for other cell populations; Figure 7D). Many of these autocrine interactions were also noted in older organoids (Figures 5D, 5E, and 6A).
The large number of paracrine signaling interactions suggests a functional relationship between nascent chondrocytes and mesenchyme cells, such that both cell types could be considered chondroprogenitors. We asked if these two cell populations were physically close to one another in organoids by examining markers specific to each population at different stages of differentiation (Figure 8A). Immunostaining of PTPRZ1, a mesenchyme cell marker (Figure 6B), was observed in isolated cells at 28 days post sorting, with more prominent expression in areas where cells had migrated together to form clumps. In mature differentiated organoids, PTPRZ1 was mainly found at the outermost surface, with a few isolated PTPRZ1-positive cells in the interior (Figure 8A, top right). DDR1, another marker for mesenchyme cells (Figure 6B), showed a similar pattern of expression (Figure 8, DDR1).
Figure 8.

Spatial distribution of mesenchyme cells and chondroprogenitors in organoids
(A) UMAP projections of snRNA-seq data and immunofluorescence of markers for mesenchyme cells (PTPRZ1, DDR1, TNC - day 87) and chondroprogenitors (TNC - day 28, PDGFRA, DCN) at different stages of differentiation: day 28 cells and organoids, and organoids at immature (day 87), and mature stages, where the morphology is that of elastic cartilage as in Figure 1C.
(B) Expression of COL1A1 and collagen receptor DDR2 at day 28, day 87 (UMAP projections, left) and mature organoids (immunofluorescence, right). Scale bar = 50 μM.
(C) Comparison of expression of mesenchyme marker PTPRZ1, and chondrocyte markers COL1A1 and DDR2 in individual cells at day 28 and 87. Blended colors (right) indicate co-expression in individual cells as yellow points.
(D) Day 87 clusters and cell cycle assignments for individual cells. Cells and organoids at day 28 and 87 were differentiated from hESCs; mature organoids were differentiated from iPSCs (H20961, 120 days). N ≧ 2 for all IF results. iPS cells produced craniofacial cartilage in two experiments each from two different iPS lines.
Nascent chondrocytes at day 28 expressed tenascin C (TNC) as shown by snRNA-seq; by immunofluoresce expression was seen at lower levels in isolated cells as well as in clumps (Figure 8A, TNC). At later times, however, TNC was more highly expressed in mesenchyme cells, and in mature organoids TNC expression was exclusively restricted to the outer layer (Figure 8A, TNC). In contrast, chondrocyte markers PDGFRA and DCN (Figure 6C) showed consistent expression in early chondrocytes, in border regions, and throughout mature organoids (Figure 8A; PDGFRA, DCN).
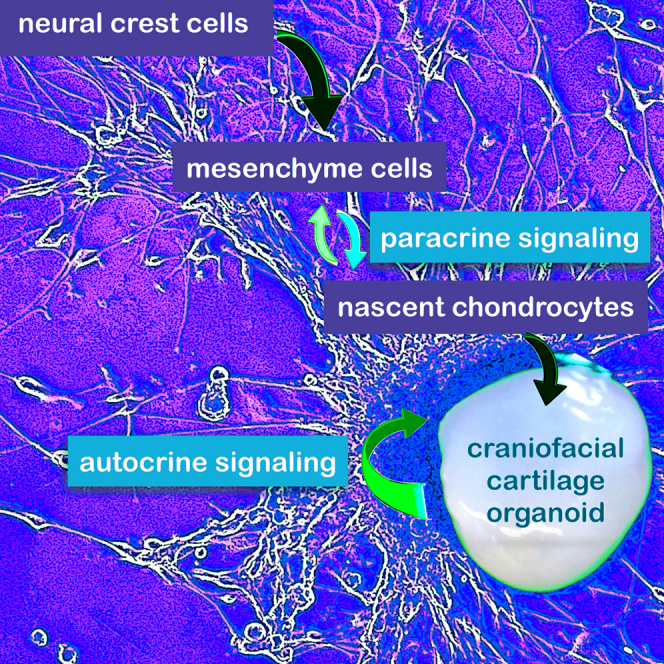
These data indicate that proliferating mesenchyme cells and nascent chondrocytes have functional paracrine interactions at the border regions of mature organoids. To ask whether a few cells could express high levels of both mesenchyme and chondrocyte markers simultaneously, we examined co-expression in individual cells comparing the mesenchyme marker, PTPRZ1 with the chondrocyte markers, COL1A1 and DDR2, which, such as PTPRZ1, are expressed in border regions of mature organoids (Figures 3C and 8). Fewer than five cells were observed to express high levels of both PTPRZ1 and chondrocyte markers (Figure 8C, yellow points). That very few cells concomitantly express both mesenchyme and chondrocyte markers indicates that these cell types largely retain their distinct identities. Gene expression patterns in UMAP embeddings at day 28 (Figures 7A and 7B) and day 87 (Figures 8C and 8D) suggest the possibility that dividing mesenchyme cells may differentiate into nascent chondrocytes (Figure S9C). In any case, our data indicate that not only do mesenchyme and chondrocyte cell populations signal to each other in a paracrine fashion in early differentiating cultures, but both cell types maintain their identities and functional interactions at the outer surface of growing organoids to generate craniofacial cartilage within (Figure 9).36,37,38,39,40
Figure 9.

Model of craniofacial chondrocyte differentiation from stem cells
hESCs and iPSCs were differentiated into NCSCs by the manipulation of several signaling mechanisms and magnetic bead sorting (Figure 1A). Based on the data in this article, we hypothesize that there are two intermediate cell populations that generate cartilage organoids, represented in the figure by nascent chondrocytes and mesenchyme cells, both of which act as chondroprogenitors. Horizontal arrows indicate differentiation steps; vertical arrows indicate paracrine cell signaling (Figure 7C); curved arrows represent autocrine signaling: secretion of ECM components (collagens, proteoglycans) that interact with integrins, DDRs and other signaling receptors sets up a feedforward mechanism (Figures 5E, 6A, and 7D). Organoids recapitulate human elastic craniofacial cartilage formation in vivo, with an outer layer of proliferating chondroprogenitors surrounding cartilage tissue (Figure 8). (Images are from Biorender and EasyNoteCards).
Discussion
Here we have described the differentiation of craniofacial cartilage organoids from hESCs and iPSCs via an NCSC intermediate. We first differentiated the neural crest lineage from specified neuroectoderm, merging several previously published protocols (Figure 1A).8,9,10,11,41,42 We undertook a thorough analysis of gene expression and proteomics in craniofacial chondrocyte organoids with an eye toward identifying cell signaling pathways that drive differentiation. Two points stand out from this work. First, by characterizing craniofacial chondrocyte differentiation from human NCSCs, we discerned paracrine interactions between mesenchyme cells and nascent chondrocytes at early stages of differentiation and in border regions of organoids; both of these cell types cooperate to act as chondroprogenitors. Second, ECM components not only define the physical characteristics of cartilage but also act as feedforward signals that play a profound autocrine signaling role in specifying and cementing cartilage cell fate. The emerging picture is that growth factor ligand-receptor pathways initiate chondrogenesis, and ECM components are induced to further drive it forward (Figure 9).
Cell populations that generate cartilage
There is great interest in regenerating cartilage throughout the body to repair birth defects and injuries.2,4,43 Our study adds to the characterization of pathways that lead to craniofacial cartilage, which is distinct from skeletal cartilage at the tips of bones.2,4,44 The presence of chondrocyte progenitor cells (chondroprogenitors) has been reported on the surface of articular cartilage45 and nasal septum.46 Current approaches for facial reconstruction usually involve autologous chondrocyte implantation (ACI) to the damaged area, which involves harvesting patient cartilage from an uninjured location in the face, typically from nasal cartilage.47 The regeneration of ear cartilage tissue is much more successful if perichondrium is employed, due to the presence of chondroprogenitor cells.36,39,48 Rhinoplasty grafts with perichondria components also look promising from animal studies, but evidence is lacking for humans.48 Nasal chondrocytes are neural crest-lineage chondrocytes that retain some proliferative potential and have the ability to repair articular cartilage with even greater chondrogenic capacity than mesenchyme-derived chondrocytes.44,49,50,51 For example, isolated human nasal septal chondrocytes have been successfully grafted to mice, expanded, applied to synthetic polymers to facilitate injection, and then used for reconstruction.52 Auricular perichondrial chondroprogenitor cells have been cultured in various ways, including by differential adhesion to fibronectin or cell migration, and successfully transplanted to produce elastic cartilage.39,53,54 NCSCs have been identified within isolated dental pulp stem cell (DPSC) populations,55 which have been successfully used to regenerate mandibular bone.15,56,57 Isolated DPSCs were limited to bone re-growth, but clonal DPSCs could differentiate into other neural crest-derived cell lineages.55 Together, these studies highlight the overlapping applications of different approaches to cartilage regeneration and reconstruction.
How different neural crest lineages colonize and differentiate within facial structures, and how this is affected by different signaling pathways is an open area of research. Shen et al.,58 recently reported the induction of cartilage from an NCSC intermediate that also had a propensity to differentiate into chondrocytes, and different types of chondrocytes were identified. These cells became hypertrophic, however, as indicated by collagen X and MMP-13 expression.58 Kaucka et al.59 tracked different neural crest lineages throughout mouse cranial development and found that ectomesenchymal neural crest cells could contribute equally to osteogenic, chondrogenic, odontogenic, and adipogenic lineages within a clonal niche in the developing facial structure. Our neural crest cells produced expected lineages (e.g., neuroblasts, melanocytes; Figure 2), but our cartilage organoids were composed of elastic cartilage that did not differentiate into bone. Determining the differentiation potential of neural crest-derived chondrocytes, and other cells within organoids, will be critical for experiments exploring organoid growth for transplantations and cartilage repair.
Neural crest-derived cartilage may have good regenerative capacity due to the ability to produce mesenchymal cells like those seen in this work. It will be interesting to compare our data to intermediate cell populations obtained using other methods of chondrogenesis that go through mesoderm.15,60,61,62,63 Comparison of our snRNA-seq data to that from Wu et al.,63 shows many common ECM components (as well as TWIST1, PRRX1, and DDR2) in chondrocytes, and another cell population that has markers in common with our mesenchyme cells (e.g., PTPRZ1, DDR1). It is possible to generate cartilage from mesenchymal-lineage cells from patient-derived adult bone marrow stem cells and adipose-derived stem cells, which can then be differentiated into articular cartilage via the application of specific growth factors.15,64 Cells called mesenchymal stem cells, or mesenchymal stromal cells (MSCs), have been previously characterized, but it is controversial whether or not they are multipotent stem cells.54,65,66 MSCs were found to induce chondrocytes and stimulate their chondrogenic capacity, and preserving the pericellular matrix enhanced this interaction with MSCs.67,68 Whether MSCs can themselves differentiate into chondrocytes, or stimulate chondrocyte proliferation through paracrine signaling (or both), remains to be determined.68 Embryoid body (EB) formation from hESCs, pellet cell culture, and micromass culture have been employed to derive MSCs, although, because the terminal differentiation stage of mesenchymal tissue is bone, these cell lineages often suffer from hypertrophy and osteogenic differentiation.69,70,71,72,73,74 In fact, transplanted cartilage derived from a mesenchymal origin can express heterogeneous markers for cartilaginous, fibrous, and hypertrophic tissues simultaneously.75,76 Notably, cartilage produced using embryoid body or mesoderm formation also has the potential to retain stem-like characteristics in the differentiated tissue, which could contribute to tumor formation.60,77,78,79 The characterization of these processes will aid approaches such as the transplantation of MSCs, isolation and expansion of chondrocytes, or recruitment from subchondral bone (microfracturing) as a means to regenerate damaged joint and intervertebral cartilage.64,76,80 It should be noted that cartilage designed for joint repair has a different function from craniofacial cartilage because it has to withstand applied stress,76,81,82 and microfracturing techniques are not applicable for craniofacial cartilage that has no adjacent bone.
ECM-receptor interactions
A distinctive feature of human ear elastic cartilage is the organization in two distinct regions, perichondrium and chondrium.36 This spatial dichotomy is associated with different ECM components. The perichondrium ECM is rich in collagen 1 proteins, while the chondrium ECM is composed of, among other things, collagen 2 proteins and elastic fibers.39 We indeed observed such dichotomy in our cartilage organoids using immunostaining (Figures 3C and 8A). A study of microtia using scRNA-seq revealed several populations of cells in auricular cartilage, including two cell types that were called chondral and stromal stem/progenitor stem cells,83 the latter of which expressed COL1A1 on edge regions of human auricular cartilage tissue, similar to our organoids. Another study comparing nonsyndromic microtia to normal total RNA and protein found 17 genes downregulated in microtia enriched in the ECM−receptor interaction, focal adhesion, cholesterol metabolism, and peroxisome proliferator-activated receptors (PPAR) signaling pathways.84 This is consistent with our data and other proteomic analyses, emphasizing the role of the ECM and integrin-mediated chondrocyte–matrix interactions in chondrocyte development.85,86
We noted robust expression of a large number of ECM ligand-receptor pairs in our data (Figures 5 and 6). Receptors that bind to ECM components, including integrins, syndecan, and DDRs, play a role in cell signaling, migration, and attachment to the matrix.87 The many collagens expressed in craniofacial cartilage organoids compel the consideration of collagens as both ligands and structural components.32 The activation of DDR2, a receptor tyrosine kinase that binds collagens to initiate signaling, which is a transcriptional target of Twist1 in the cranial neural crest,88,89 leads to more ECM deposition and is required for craniofacial development in mice.90 DDR2 is known to interact directly with COL1A1 and COL3A1,27,91 but may bind other collagens in the fibril-forming group, given similarities among these collagen types.92,93 Collagens also activate integrins (Figures 5E and 6A).31,32 Collagens and other ECM components also bind other ligands such as the TGFβ family, BMPs, PDGFs, Wnts and FGFs,87,94,95 which contributes to specifying cartilage tissue as part of the feedforward mechanism that combines signaling by growth factors and ECM components (Figure 8).
Growth factors and receptors
Concomitant expression of growth factors and their receptors suggesting that autocrine and/or paracrine loops form to drive chondrocyte differentiation (Figures 5, 6, and 9). Both pleiotrophin (PTN) and midkine (MDK) activate signaling pathways that are well known to be involved in chondrogenesis during mouse development and bone repair.96,97,98,99,100 PTN and MDK attach to extracellular heparan sulfate proteoglycans and cell surface syndecans and activate signaling proteins ALK and PTPRZ1.101 Signaling pathways initiated by fibroblast growth factor (FGF) family members have also been shown to be important for both bone and cartilage formation in mice. Specifically, FGF8 has been shown to promote chondrogenesis over osteogenesis, both in the mouse skull and during the thickening of the palate,102 and FGF18 has been known to induce cell proliferation and extracellular matrix synthesis in porcine and human cartilage.103 The neuroendocrine specific peptide VGF is induced by the receptor tyrosine kinase BDNF (NTRK2), both of which were present in our dataset.104
Transforming growth factor β (TGFβ) family members have also been implicated in chondrogenesis, although the impact of different ligands to the formation of cartilage seems to be variable.105,106 All three isoforms (TGF-β1,2, and 3) are represented in MSC populations and TGFβ-I and TGFβ-III have been used to increase chondrogenesis during the in vitro differentiation of MSCs.107 In particular, TGF-β1 increases collagen production in embryonic stem cells, while TGF-β2 and -β3 increase glycosaminoglycan production in human bone marrow mesenchymal stem cells.106,108 Pre-seeding of TGFβ ligands on scaffolding materials is being investigated as a method to increase the mechanical strength of transplanted cartilage tissues.109,110
In mice, bone morphogenetic protein 2 (BMP2) and BMP4 are expressed in the cranial neural crest-derived mesenchyme during E10.5–13.5 and signaling via the BMPRIa receptor is essential for proper palate, tooth, and temporomandibular joint formation.111,112,113,114,115,116 Accordingly, mice lacking functional BMP receptors in the neural crest lineage exhibit hypoplastic mandibles, cleft palates, and die shortly after birth.117,118 BMP7 has been shown to be able to induce extracellular matrix synthesis, prevent chondrocyte hypertrophy and maintain chrondrogenic potential.103 BMP signaling crosstalk with the p53 apoptotic pathway controls nasal cartilage formation and fusion of the nasal septum in mice.119 There is less evidence in the literature for BMP5 involvement during these processes, especially in human models, and we were surprised to see it more highly represented in our dataset compared to the canonical cartilage growth factors BMP2 and BMP4. However, in rats, BMP5 expression has been shown to increase during tibia growth plate development, and inhibition of BMP5 signaling significantly decreased extracellular matrix production in primary rat chondrocytes.120 Kingsley et al. found that mice harboring mutations and deletions in the short ear gene locus also exhibited deletions in the BMP5 coding region, which were a likely cause for the skeletal defects seen in short ear mice.121,122 Guenther et al. then found that specific BMP5 enhancer regions controlled cartilage and bone growth zones induced by BMP5 expression, which included cartilage deposits in the mouse nasal cavity.123 In zebrafish, BMP5 is required for neural crest progenitor survival, and a BMP5 morpholino significantly reduced Alcian blue cartilage staining, though this could be due to the loss of progenitor NC populations and not specifically NC derived chondrocytes.124 To our knowledge, a thorough analysis of the contribution of BMP5 signaling to human craniofacial development has not been done, although insights from animal models, and our present study, suggest that BMP5 plays a role in both neural crest differentiation and in elastic cartilage formation. Gremlin proteins (GREM1, GREM2) are BMP antagonists; GREM1 suppresses cartilage hypertrophy,125,126,127 and is expressed in craniofacial cartilage organoids (Figure 5A). It may be that there are more intermediate states of differentiation to discover in which there is sequential expression of BMPs and Gremlins.
Limitations of this study
These data make a substantial contribution toward understanding the complex mechanisms of craniofacial cartilage differentiation in humans.
The discussion above highlights the emerging challenge of defining different populations of differentiating chondrogenic cells by gene expression, and giving them appropriate names. Proliferating cells that express chondrocyte markers are one class of chondroprogenitors. Though the less differentiated cells that we call mesenchyme lack ECM component expression, their paracrine interactions with chondrocytes, and colocalization on the surface of organoids, indicate that they play a key role in chondrogenesis, and therefore deserve to be grouped with nascent chondrocytes in the class of cells named chondroprogenitors. Our data is consistent with recent work showing that the cell cycle plays a role in early cranial neural crest differentiation.128 The gene expression patterns in dividing mesenchyme and dividing chondrocytes (Figures 7 and 8) suggest the interesting possibility that dividing mesenchyme cells can differentiate into chondrocytes (Figure S9C), but our data do not rigorously establish this.129,130 Proteomics does not distinguish different cell populations within organoids; snRNA-seq does not determine the spatial distribution of different cell types within organoids; and immunofluorescence microscopy does not elucidate the entire proteome at a single-cell level. Nevertheless, our work and that of others noted above strongly suggests that building cartilage is a cooperative endeavor between two or more cell types that communicate in a paracrine fashion. It will be important to further define in future studies intermediate populations of cells that emerge at earlier stages of differentiation, and cell-cell interactions within the perichondrium at later stages of tissue formation. If stem cell-generated cartilage is going to be used clinically, then it will be necessary to learn how to specifically generate specific subtypes of cartilage (e.g., elastic cartilage for ears and hyaline cartilage for the nose) and define conditions that initiate or prevent hypertrophy.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Sox 2 (D9B8N) Rabbit mAb | Cell Signaling Technology | Cat# 23064, RRID: AB_2714146 |
| Oct – 4A (C52G3) Rabbit mAb | Cell Signaling Technology | Cat# 2890, RRID: AB_2167725 |
| NCAM1 (CD56) (123C3) Mouse mAb | Cell Signaling Technology | Cat# 3576, RRID: AB_2149540 |
| Doublecortin Antibody Rabbit polyclonal antibody | Cell Signaling Technology | Cat# 4604, RRID: AB_561007 |
| Anti-Collagen Type II Antibody, clone 6B3 | Millipore Sigma | Cat# MAB8880, RRID: AB_2260779 |
| COL1A1 (E6A8E) Rabbit mAb | Cell Signaling Technology | Cat# 39952, RRID: AB_2895124 |
| Recombinant Anti-Elastin antibody (EPR20603) | Abcam | Cat# ab213720 |
| COMP Polyclonal Antibody | ThermoFisher | Cat# PA5-95547, RRID: AB_2807349 |
| Human Matrilin-1 Antibody (Mouse) | R&D Systems | Cat# MAB7878; RRID: AB_3096006 |
| PTPRZ1 (E7C8Z) Rabbit mAb | Cell Signaling Technology | Cat# 15641; RRID: AB_3096003 |
| DDR1 (D1G6) XP Rabbit mAb | Cell Signaling Technology | Cat# 5583, RRID: AB_10694842 |
| Anti-DDR2 antibody (3B11E4) | Abcam | Cat# ab63337, RRID: AB_955827 |
| Sox9 (D8G8H) Rabbit mAB | Cell Signaling Technology | Cat# 82630, RRID: AB_2665492 |
| PDGF Receptor alpha (D1E1E XP Rabbit mAb | Cell Signaling Technology | Cat# 3174, RRID: AB_2162345 |
| Tenascin C (E5J3B) Rabbit mAb | Cell Signaling Technology | Cat# 33352; RRID: AB_3096005 |
| Anti-Decorin antibody (Rabbit) | Abcam | Cat#175404, RRID: Ab_2890261 |
| Prolong Gold Antifade Reagent with DAPI | Cell Signaling Technology | Cat# 8961 |
| Anti-mouse IgG (H + L), F(ab’)2 Fragment (Alexa Fluor 594 Conjugate) raised in goat | Cell Signaling Technology | Cat# 8890, RRID: AB_2714182 |
| Anti-Rabbit IgG (H + L), F(ab’)2 Fragment (Alexa Fluor 488 Conjugate) raised in goat | Cell Signaling Technology | Cat# 4412, RRID: AB_1904025 |
| PE anti-human CD271 (NGFR) | Biolegend | Cat# 345105, RRID: AB_2282827 |
| Chemicals, peptides, and recombinant proteins | ||
| Chroman - 1 | MedChem Express | Cat# HY-15392, CAS: 1273579-40-0 |
| trans-ISRIB | Tocris | Cat# 5284, CAS: 1597403-47-8 |
| Emricasan (IDN-6556) | Selleckchem | Cat# S7775, CAS: 254750-02-2 |
| Polyamine Supplement (1000X) | Millipore Sigma | P8483 |
| Antioxidant Supplement (1000X) | Millipore Sigma | A1345 |
| CHIR99021 | Millipore Sigma | SML1046, CAS: 252917-06-9 |
| SB 431542 hydrate | Millipore Sigma | S4317, CAS: 301836-41-9 |
| SB 431542 | Tocris | Cat# 1614, CAS: 301836-41-9 |
| LDN193189 | VWR | Cat# 1995-5, 25, CAS: 1062368-24-4 |
| Poly – L -ornithine hydrobromide | Millipore Sigma | P3655, CAS: 27378-49-0 |
| Laminin Mouse Protein, Natural | ThermoFisher | Cat# 23017015, |
| Fibronectin, Human | Corning | Cat# 356008 |
| Matrigel Growth Factor Reduced (GFR) Basement Membrane Matrix, LDEV-free | Corning | Cat# 354230 |
| L- Glutamine | ThermoFisher | Cat# 25030081 |
| MEM Non-Essential Amino Acids Solution | ThermoFisher | Cat# 11140050 |
| Β – Mercaptoethanol | ThermoFisher | Cat# 21985023 |
| Insulin from bovine pancreas | Millipore Sigma | I6634, CAS: 11070-73-8 |
| apo – Transferrin Human | Millipore Sigma | T1147, CAS: 11096-37-0 |
| Sodium selenite | Millipore Sigma | S9133, CAS: 10102-18-8 |
| Putrescine dihydrochloride | Millipore Sigma | P5780, CAS: 333-93-7 |
| Progesterone | Millipore Sigma | P6149, CAS: 57-83-0 |
| (+) – Sodium L-ascorbate | Millipore Sigma | A4034, CAS: 134-03-2 |
| Human Brain-Derived Neurotrophic Factor (BDNF) Recombinant Protein | Cell Signaling Technology | Cat# 3897, UniProt ID: #P23560 |
| Recombinant Human/Murine FGF-8b | Peprotech | Cat# 100-25, UniProt ID: P55075 |
| Recombinant Murine Sonic Hedgehog (Shh) | Peprotech | Cat# 315-22, UniProt ID: Q62226 |
| Y- 27632 (Dihydrochloride) | Stemcell Technologies | Cat# 72304, CAS: 129830-38-2 |
| Recombinant Human FGF-basic | Peprotech | Cat# 100-18b, UniProt ID: P09038 |
| Human Epidermal Growth Factor (hEGF) | Cell Signaling Technology | Cat# 8916, UniProt ID: P01133 |
| Deposited data | ||
| snRNA-seq | Mendeley Data | https://data.mendeley.com/datasets/49gxpjrcn9/1 |
| Proteomic data | PRIDE | http://www.ebi.ac.uk/pride/archive/projects/PXD050668 |
| Networks (CFN and CFN plus CCCN) | NDEx |
https://www.ndexbio.org/viewer/networks/78c8f03d-d592-11ee-8a40-005056ae23aa https://www.ndexbio.org/viewer/networks/fa2c707c-d653-11ee-8a40-005056ae23aa |
| Experimental models: Cell lines | ||
| WA09/H9 Human | WiCell | RRID: CVCL_9773 |
| H20961 iPS Cells | Weizmann Institute of Science | RRID: CVCL_HA53 |
| Software and algorithms | ||
| Seurat | CRAN | RRID: SCR_007322 |
| Connectome | GitHub | https://github.com/msraredon/Connectome |
| RCy3 | Bioconductor | RRID: SCR_006442 |
| enrichR | CRAN | RRID: SCR_001575 |
| Other | ||
| Robosep Buffer 2 | Stemcell Technologies | Cat# 20164 |
| EasySep Magnet | Stemcell Technologies | Cat# 18000 |
| EasySep Release Human PSC-derived Neural Crest Cell Positive Selection Kit | Stemcell Technologies | Cat# 100-0047 |
| Anti-FITC Microbeads | Miltenyi Biotec | Cat# 130-048-܁701 |
| MACS Multistand | Miltenyi Biotec | Cat# 130-042-303 |
| MS Columns | Miltenyi Biotec | Cat# 130-042-201 |
| DMEM F-12 Media | ThermoFisher | Cat# 12500-096 |
| mTeSR1 Media | StemcellTechnologies | Cat# 85850 |
| mTeSR1 Plus Media | StemcellTechnologies | Cat# 100-0483 |
| ReLeSR | StemcellTechnologies | Cat# 100-0276 |
| Knockout DMEM | ThermoFisher | Cat# 10829018 |
| Knockout Serum Replacement | ThermoFisher | Cat# 108280 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Mark Grimes, (mark.grimes@mso.umt.edu).
Materials availability
Commercial sources for reagents and cell lines are indicated in STAR Methods above, except H20961 cells, which were provided by Dr. David Gokhman.
Data and code availability
-
•
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner131 repository (https://www.ebi.ac.uk/pride/ ) with the dataset identifier PXD050668.
-
•
The CFN (https://www.ndexbio.org/viewer/networks/78c8f03d-d592-11ee-8a40-005056ae23aa) and CCCN (https://www.ndexbio.org/viewer/networks/fa2c707c-d653-11ee-8a40-005056ae23aa) networks from total protein and PTM proteomic data have been deposited in the NDEx Network Data Exchange.132
-
•
snRNA-seq data has been deposited with Mendeley Data (https://data.mendeley.com/datasets/49gxpjrcn9/1).
Experimental model and study participant details
Cell culture
WA09 hESCs and AICS-5 iPSCs were purchased from the WiCell Institute and H20961 iPCs were provided by our collaborators at The Weizmann Institute of Science. Both the cell lines were expanded and maintained in mTeSR™1 and mTeSR™ Plus (Stem Cell Technologies), passaged with Versene, and cultured on plates coated with growth factor reduced Matrigel (Corning). Small molecule cocktail (CEPT) (Chen et al., 2021) consisting of 50 nm Chroman-1 (Medchem Express), 0.7 μM trans-ISRIB (Tocris), 5 μM Emricasan (Selleckchem), Polyamine Supplement (Sigma) diluted 1:1000 and Antioxidant Supplement (Sigma) diluted 1:1000 was added during passaging and sorting to promote cell viability. All cell lines were grown in a humidified incubator at 37°C, 5% CO2.
Media for NCSC differentiation and maintenance was prepared in the following way:
KSR Medium (1L): 820 mL Knockout DMEM, 150 mL Knockout Serum Replacement medium, 10 mL L-glutamine (200 mM), 1 mL β-mercaptoethanol.
N2 Base Medium (1L): DMEM f-12 medium, 25 mg/L Insulin, 10 μM apo-Transferrin, 30 nM Sodium Selenite, 100 μM Putrescine, 20 nM Progesterone
NCSC Specification medium (1L): Base N2 medium (prepared as above), 200 μM Sodium L-ascorbate, 20 ng/mL BDNF, 100 ng/mL FGF-8b, 20 ng/mL Shh, 10 μM Y-27632
NCSC Maintenance Medium (1L): Base N2 medium (prepared as above), 10 ng/mL FBF basic, 20 ng/mL EGF.
Neural crest differentiation
Neural Crest Stem Cells (NCSCs) and Neural Stem Cells (NSCs) were derived by integrating the approaches of several previously published protocols.8,9,10,11,41,42,133 Fluorescence-activated cell sorting (FACS) of NCSC cultures provided a more uniform NCSC population, but contributed to high amounts of cell death and severely decreased the NCSC yield. Therefore, we fit the NCSC differentiation to a shorter timeline, and purified NCSCs via magnetic-activated cell sorting (MACS) to increase cell survival. WA09 hESCs were passaged in Matrigel coated dishes to 25–40% confluency prior to differentiation. Daily changes with different media compositions and the addition of growth factors and small molecule inhibitors were used to direct differentiation. Day 1: media was changed from mTeSR1 to KSR containing 0.1 μM LDN193189 and 10 μM SB431542. Day 2: 75% KSR, 25% N2 containing 10 μM SB431542 and 3 μM CHIR99021. Day 3: 50% KSR, 50% N2 containing 3 μM CHIR99021. Day 4: 25% KSR, 75% N2 containing 3 μM CHIR99021. Day 5: 100% N2 containing 3 μM CHIR99021. Day 6–9: media was changed daily with NC Specification media containing 10 μM ROCK inhibitor Y-27632. On day 10 after differentiation, NCSCs were sorted via magnetic cell sorting. p75+ NCSCs were positively selected following the MACS MS protocol (Miltenyi Biotech) or Stemcell Technolgies’ EasySep Release Human PSC-derived Neural Crest Cell Positive Selection Kit. p75-flow-through was collected as “NSCs”, although this population represents a multitude of cell types. NCSCs were plated and maintained by passaging on Poly-L-Ornithine/Laminin/Fibronectin coated dishes in NCSC Maintenance media (N2 media containing FGF2 and EGF; alternatively for Org 3: FGF2, EGF, PTN [R&D, 500 pg/mL], MDK [Preprotech, 100 ng/mL], TGFB2 [Cell Signaling Technology, 10 ng/mL] and BMP5 [Abcam, 10 ng/mL]). NSCs were plated and maintained by passaging on Matrigel coated dishes in NSC media. Overly confluent NCSC cultures spontaneously formed and grew free-floating cartilage organoids when fed regularly with NCSC maintenance media. Organoids were harvested with a sterile spatula for cryosectioning.
Method details
Tissue organoid processing and sectioning
Cartilage organoids were originally fixed in fresh 4% PFA in 1X PBS, rotated at 4°C overnight. Organoids were dehydrated in the following stepwise sucrose washes at 4°C: 5% sucrose - 2 h, 10% sucrose - 2 h, 30% sucrose - overnight, 1:1 ratio of 30% sucrose:OCT - 2 h, 100% OCT - 2 h. Organoids were embedded in OCT and snap frozen in a dry ice and ethanol bath. Samples were stored at −80°C before sectioning. Cryosectioning was performed using a Leica CM1950 cryostat. 16 μm thick slices were collected at −25°C on Superfrost plus slides (Fisherbrand). Slices were air dried before storage at −80°C.
Alternatively, cartilage organoids were fixed in 10% Neutral Buffered Saline, rotated overnight at room temperature. Organoids were dehydrated in the following stepwise manner using automated Leica ASP300: Alcoholic formalin for 15 min, 95% ethanol for 10 min, 2 changes of absolute ehtanol for 20 min, absolute ethanol for 10 min, 2 changes of xylene for 15 min each, Paraffin Wax 1 for 10 min at 62°C, Wax 2 for 20 min at 62°C and Wax 3 for 20 min at 62°C. The organoids were then embedded in paraffin using AutoSette Cassettes and embedding mold. 5 μm sections were collected on Superfrost plus slides (Fisherbrand) using Leica RM 2235 microtome.
Immunofluorescence
Before staining, frozen slides were dried at room temperature overnight. Slices were rehydrated at room temperature in 1X TBS for 20 min, and permeabilized for 45 min (Permeabilization solution: 0.1% saponin, 1% BSA, 2% Goat serum in 1X TBS). Primary antibodies were diluted in Permeabilization solution, added to samples, and incubated overnight at 4°C. Slides were washed with Perm. four times. Secondary antibody diluted in Perm. was added to samples and incubated for 1 h at room temperature. Sections were mounted with SlowFade Gold antifade reagent with DAPI (Invitrogen) and imaged on an Olympus FV1000 confocal microscope and alternatively on Zeiss Laser-Scanning Microscope 880.
Paraffin sections were rehydrated using 2 washes of xylene and single wash of 100%, 70% and 50% ethanol each. The staining protocol was then carried out similar to cryosections with an additional step of antigen retrieval using citrate buffer. For immunofluorescence of cells at day28, p75+ NCSCs were grown on coverslips coated with poly-L-ornithine, fibronectin, and laminin in 6-well plates. Methanol was used to fix the cells at day 28 and immunofluorescence was carried out using the above mentioned protocol.
Simple stains
Safranin O staining: Frozen slides were rehydrated in distilled water, stained with Weigert’s iron hematoxylin for 10 min, washed in running tap water for 10 min, stained with 0.05% fast green for 5 min, rinsed with 1% acetic acid for 10 s, stained in 0.1% safranin for 5 minutes, dehydrated in 95% ethanol for 2 minutes (2X), 100% ethanol for 2 minutes (2X), and finally xylene for 2 minutes (2X).
Toluidine Blue staining: Frozen slides were rehydrated in distilled water, stained in toluidine blue solution (3.5 mM in 1% NaCl, 7.7% ethanol, pH 2.38), washed 3X with distilled water, dehydrated: 10 dips in 95% ethanol, 10 dips in 100% ethanol, 10 dips in 100% ethanol, cleared in xylene 2 × 3 minutes each, and mounted with mounting media and coverslip media.
Hematoxylin and Eosin: H&E stained slides of organoid cryosections were prepared using standard conditions at The Department of Pathology, Community Medical Center, 2827 Fort Missoula Road, Missoula, Montana 59804.
Weigert’s Resorcin-Fuchsin staining for elastic fibers: Sections were rehydrated using 2 washes of xylene and single wash of 100%, 70% and 50% ethanol to distilled water. The sections were stained in Weigert’s hematoxylin working solution (50% Weigert’s hematoxylin A, 50% Weigert’s hematoxylin B) for 10 min, stained with Resorcin-Fuchsin solution () (slides periodically checked under microscope ever 30 min) for 120 min, rinsed in 95% ethanol, counter stained with Van Gieson’s stain () for 60s, dehydrated using 3 changes of absolute ethanol and 2 changes of xylene, mounted using Permount and imaged using Nikon TE 300 inverted microscope.
Mass spectrometry sample preparation
TMT data were obtained in collaboration with Bluefin Biomedicine (Beverly, MA) comparing H9 human embryonic stem cells (WA09); neural stem cells (NSCs); neural crest stem cells (NCSCs); and neuroblastoma cell lines (SH-SY5Y, SMS-KCN) to chondrocyte organoids.
Sample collection
Cells were washed and harvested in either phosphate-buffered saline or versene. Organoids were harvested with a sterile spatula. Cell pellets/organoids were stored at −80°C. Proteins were extracted in two separate groups of samples.
Lysis and protein extraction for group 1
Samples from the first batch were lysed in a 10:1 (v/w) volume of lysis buffer [5% SDS, 100 mM NaCl, 20 mM Hepes (pH 8.5), 5 mM dithiothreitol, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, and leupeptin (1 mg/mL)], and proteins were reduced at 60°C for 30 min. Proteins were then alkylated by the addition of 10 mM iodoacetamide (Sigma-Aldrich) for 45 minutes at room temperature in the dark, and methanol/chloroform precipitation was performed. Protein pellets were resuspended in urea lysis buffer (8 M urea, 20 mM Hepes (pH 8.5), 1 mM sodium orthovanadate, 2.5 mM sodium pyrophosphate, and 1 mM β-glycerolphosphate) and sonicated.
Lysis and protein extraction for group 2
Samples from a second batch were resuspended and probe tip sonicated in 8M urea with 200 mM HEPES pH 8.5 with 1× Protease and Phosphatase inhibitors (CST #5872). Samples were then reduced for 50 minutes at 58°C with 5 mM DTT. Samples were alkylated in the dark for 40 minutes at room temperature with 10 mM iodoacetamide. Alkylation was quenched with 5 mM DTT.
Protein digestion
Samples were diluted to 2M urea with 20 mM HEPEs pH 8.5 with 1 mM CaCl2, and digested overnight at 37°C with LysC (Wako-Chem). Samples were then diluted to 1M urea with 20 mM HEPEs pH 8.5 with 1 mM CaCl2 and digested for 6 hours with Trypsin (Pierce). Samples were acidified with trifluoroacetic acid (TFA) de-salted with a Waters SepPak C18 column. Peptide concentrations were estimated with a microBCA kit from Pierce (#23235).
Total protein analysis sample preparation
For total protein analysis, peptides were TMT-labeled in 200 mM HEPES pH 8.5 with 30% acetonitrile for 1 hour, combined, dried in a speedvac, and desalted with a SepPak C18 cartridge. 100 μg of combined peptide material was resuspended in buffer A (10 mM NH4HCO2, pH10, 5% ACN), and fractionated on a Zorbax Extended C18 column (2.1 × 150 mm, 3.5 μm, no. 763750-902, Agilent) using a gradient of 10–40% bRP buffer B (10 mM NH4HCO2, pH10, 90% ACN). 96 fractions were collected and concatenated into 24 fractions. Fractions were dried, de-salted with stop and go extraction tips (STAGE-tips) and dried for LC-MS analysis.
Enrichment of post-translationally modified peptides
To enrich for post-translationally modified peptides, 1 mg of peptides from each sample were TMT labeled as described above, combined, dried, and SepPak purified. The TMT labeled material was subjected to sequential immunoaffinity purifications using PTM-scan kits from CST, including anti phospho-tyrosine (#8803), AKT substrate (#5561, #5563), ATM/ATR (#12267), acetyl-lysine (#13416), pan-methyl lysine (#14809), and mono-methyl arginine (#12235). AKT and ATM/ATR substrate specific pulldowns were not performed on group 1. Immunoaffinity purified fractions were STAGE-tip purified prior to LC-MS analysis. The flowthrough from immunoaffinity purifications was desalted on a SepPak, and remaining phosphopeptides were purified using High-SelectTM Fe-NTA Phosphopeptide Enrichment Kits (Thermo Fisher Scientific, San Jose, CA). The resulting phosphopeptide elution from IMAC enrichment was fractionated as described above, using a gradient from 5 to 40% Buffer B. 96 fractions were collected and concatenated into 24 fractions. Fractions were dried, de-salted with STAGE-tips, and dried for LC-MS analysis.
LC-MS analysis of total protein
Samples were analyzed on an Orbitrap Fusion Lumos mass spectrometer (Thermo Fisher Scientific, San Jose, CA) coupled with a Proxeon EASY-nLC 1200 liquid chromatography (LC) pump (Thermo Fisher Scientific, San Jose, CA). Peptides were separated on a 100 μm inner diameter microcapillary column packed with ∼40 cm of Accucore150 resin (2.6 μm, 150 Å, ThermoFisher Scientific, San Jose, CA). For each analysis, we loaded approximately 1 μg onto the column. Peptides were separated using either a 2.5 h gradient of 6–30% acetonitrile in 0.125% formic acid with a flow rate of 550 nL/min. Each analysis used an SPS-MS3-based TMT method,134,135 which has been shown to reduce ion interference compared to MS2 quantification.136 The scan sequence began with an MS1 spectrum (Orbitrap analysis, resolution 120,000; 350–1400 m/z, automatic gain control (AGC) target 4.0 × 105, maximum injection time 50 ms). Precursors for MS2/MS3 analysis were selected using a Top10 method. MS2 analysis consisted of collision-induced dissociation (quadrupole ion trap; AGC 2.0 × 104; normalized collision energy (NCE) 35; maximum injection time 120 ms). Following acquisition of each MS2 spectrum, we collected an MS3 spectrum a method in which multiple MS2 fragment ions are captured in the MS3 precursor population using isolation waveforms with multiple frequency notches.135 MS3 precursors were fragmented by HCD and analyzed using the Orbitrap (NCE 65, AGC 3.5 × 105, maximum injection time 150 ms, isolation window 1.2 Th, resolution was 50,000 at 200 Th).
LC-MS analysis of PTM enriched fractions
For analysis of PTM enriched samples, the same MS and HPLC instruments were used as described above. For each analysis approximately 500 ng of enriched peptides were loaded on the column and run over a 120-min gradient of 2–32% acetonitrile in 0.125% formic acid with a flow rate of 400 nL/min. MS1 spectra were collected in the Orbitrap at a resolution of 60,000 with a scan range of 300–1500 m/z using a AGC target of 4.0e5 with a maximum inject time of 25 ms. Peptides for MS2 analysis were isolated using the quadrupole with an isolation window of 0.5 m/z. MS2 spectra were generated using Higher-energy collision dissociation (HCD) with a collision energy of 40%. Fragments were collected in the Orbitrap at a resolution of 50,000 with a first mass of 110 m/z, an AGC target of 5.0e4 and a maximum injection time of 200 ms.
Total protein data processing and analysis
Mass Spectra were processed using a Comet-base software pipeline.137,138 Resulting data were searched with a fully tryptic database containing human Swissprot consensus entries plus isoforms allowing for a static modification of lysine and N-termini with TMT (229.1629 Da) and carbamidomethylation (57.0215 Da) of cysteine, along with variable oxidation (15.9949 Da) of methionine. Searches were performed using a 20 ppm precursor ion tolerance, the product ion tolerance was set to 1.0 Th. Peptide-spectrum matches (PSMs) were adjusted to a 2% false discovery rate (FDR) using previously described linear discriminant analysis.139,140 Filtered PSMs were collapsed to a final protein-level FDR of <2%. Protein assembly was guided by principles of parsimony to produce the smallest set of proteins necessary to account for all observed peptides. MS3 spectra with TMT reporter ion summed signal-to-noise ratios less than 100 were excluded from quantitation.141
PTM MS data analysis
PTM data was analyzed as described above, with a fragment ion tolerance of 0.02 Th instead. In the appropriate fractions, variable modifications were considered for phosphorylation on serine, threonine, and tyrosine (79.9663 Da), acetylation on lysine (42.0106), mono-methylation of lysine (14.0157), and mono-methylation of arginine (14.0157). PTM sites with A-score values >13 were considered as localized for downstream analysis.137,142
Single nuclei RNA sequencing (snRNA-seq)
Preliminary experiments differentiating NCSCs in multi-well plates showed inconsistent cartilage formation in individual wells, presumably due to the stochastic feedforward autocrine/paracrine loops that tipped the balance of differentiation differently in different wells. Therefore, we designed the snRNA-seq experiment to ensure that the early time points contained cells that we could confirm made cartilage at later stages. NCSCs were differentiated and sorted and plated in 10 cm dishes as described above. A representative sample of all cells was taken a cell scraper across the diameter of the dish at different times. Cells harvested directly the day after sorting represent NCSCs. Adherent cells and self-organized 3D organoids were harvested at day 87. The cells were then pelleted and frozen at −80°C before being shipped to our collaborators for single nuclei RNA sequencing and proteomics analysis.
Nuclei extraction
The nuclei isolation protocol was adapted from Slyper, M. et al. 2020.143 Samples were placed in a 6 well flat bottom plate with 1 mL of TST buffer on ice. If the sample was a dense tissue, then it was chopped with Noyes Spring Scissors (FST cat# 15514-12) for 5 min then gently pipetted up and down for an additional 5 min. If the sample predominantly consisted of single cells, the sample was pipetted up and down gently for 10 min.
Isolated nuclei were filtered using a 30 μm MACS SmartStrainer (Miltenyi Biotec cat# 130-110-915). 1 mL of TST was added to the filter to recover remaining nuclei. 3 mL of 1X ST was added to the filtered nuclei and spun down in a 15 mL tube at 4°C for 5 min at 500g. The supernatant was discarded, and the pellet was resuspended with 200 μL to 500 μL of 1X ST based on the size of the pellet then filtered with the 30 μm MACS SmartStrainer again. Isolated filtered nuclei were counted before loading them on the 10X Genomics controller.
snRNA data generation and processing
Extracted nuclei were loaded onto a 10X Genomics chip, targeting ∼5,000 nuclei per sample, and snRNA-seq libraries were prepared following the v3.1 Chemistry Dual Index with Feature Barcoding technology for Cell Surface Protein protocol, Rev C. Pooled libraries were sequenced using an Illumina NextSeq 2000, 100-cycle P3 Kit. Fastq files were generated using the NextSeq 2000’s on-board Dragen software. Fastq files were then analyzed using cellranger-7.1.0.144
snRNA-seq analysis methods
Analysis of snRNA-seq data was done in an R using Seurat v4.3.145 The sequencing data from day 1 and day 87 were merged into a Seurat object and processed via Seurat clustering stages (https://satijalab.org/seurat/articles/pbmc3k_tutorial.html). When analyzing the basic characteristics of the six clusters, including genes counts, reads counts, nuclear marker MALAT-1 and ribosomal protein percentile expressions per cell, one cluster from day 87 cells exhibited abnormalities (labeled Artifact, Figure S1). All cells from this cluster showed a low number of genes detected, RNA reads, a high level of ribosomal proteins, and a very low MALAT-1 expression, which is unexpected for nuclei. We therefore removed the cells from this abnormal cluster for subsequent analysis, re-ran the clustering algorithms, and obtained five clusters (Figures 2A and 2B).
Characterization of cell types associated with the clusters - Marker analysis
The identity of the largest cluster as Neural Crest Stem cells (NCSCs) was straightforward since it was composed of nuclei from NCSCs one day after sorting (Table S1). The other four clusters were formed by day 87 cells and we identified them principally by analyzing their specific markers using the Seurat FindAllMarkers function and the Wilcoxon Rank-Sum test. The cluster labeled Chondrocytes contained cells with several very distinctive pattern of gene expression. Six of the top 10 markers were collagen genes (Figure S2). A wider analysis with more markers (using violin plots) showed that cluster Chondrocytes had the highest level of expression for collagens, proteoglycans, and genes involved in the metabolism of elastin-based fibers17 (Figures 2C–2E) and many proteins that we detected in cartilage organoids using proteomics.
Cluster Melanocytes was composed of only 31 cells. Nevertheless, the cell type of cluster Melanocytes was well-defined because among the top 10 markers were many markers defining mature melanocytes (DCT, PMEL, TYR, TYRP1, TRPM1). The presence of 31 melanocytes wasn’t a surprise, since it’s one of the cell types that NCSCs may differentiate into.146
Cluster Neuroblasts was called as such because of the presence, among the top 10 markers, of genes involved in axon and synapse formation like DCC, LINGO2, SYN2, a neuronal transcription factor (MYT1L), and several members of the neurexin family (CNTNAP5, NRXN1, and NRXN3). Cluster Mesenchyme was named as such because it had markers of NCSCs, chondrocytes, and neuroblasts, and appeared to be at an intermediate state of differentiation.
Characterization of BioPlanet pathways associated with the chondrocytes cluster markers
We used the R package enrichR to find the BioPlanet pathways linked to the Chondrocytes cluster. Since we are interested in the pathways changes between the NCSCs (cluster NCSCs, day 1) and the Chondrocytes cluster composed of day 87 cells (Figure 2B), we used the Seurat function FindMarkers to find the markers specific to the day 87 Chondrocytes cluster versus the day 1 NCSCs cluster. We then ran the enrichR function with the "BioPlanet_2019" database on the top 45 markers from the Chondrocytes versus NCSCs FindMarkers list. We chose 45 as the numbers of markers since it was the mean of the length of BioPlanet pathway gene lists. There were a total of 35 BioPlanet pathways enrichR-identified with an adjusted p value smaller than 0.05 (Figure S4C). A very large majority of these pathways were linked to the extracellular matrix or a component of it (collagen, glycosaminoglycan, chondroitin/dermatan/heparan sulfates). Finally, we looked at the intersection of these 35 BioPlanet pathways associated with the Chondrocytes cluster from the single nuclei RNA sequencing analysis with the BioPlanet pathways identified during the proteomics/PTM analysis (see below). Nine BioPlanet pathways overlapped between the two different analysis methods (yellow bars in Figure S4C), which underscored the complementary nature of the techniques presented in this work.
Analysis of proteomic data
We used several approaches to analyze mass spectrometry data. TMT analysis identified 30,802 PTMs including tyrosine, serine, and threonine phosphorylation, lysine acetylation, and lysine and arginine methylation. TMT data is reliably expressed as ratios within a single experimental run. To rank expression levels, we calculated the row Z score for each protein in each TMT run, then determined the median expression for all experiments in NCSCs (N = 2) and organoids (N = 4). We calculated from the raw data the ratio (fold change) of protein signals in organoids vs. NCSCs, and p values using the Welch two-sample t-test. To combine p values for groups of proteins (e.g., BioPlanet pathway genes or chondrocyte markers) we employed Fisher’s sum of logs (chi-square) method,147 and the truncated version of Fisher’s method148,149 (Table S2).
Clustering of PTMs and construction of CCCN and CFN
To compare different experiments for clustering, a common denominator was chosen to be neuroblastoma SH-SY5Y cells, which had the best correlations between different experiments. Ratios were limited to +/−100 to avoid statistical bias and transformed into log2 values as described previously.12 Using log2-transformed data for clustering produced the best results for t-SNE clustering, and TMT ratio data contains extreme values, which may skew correlation analyses, and ratios beyond 100-fold are not functionally different.12 Construction of the CCCN and CFN were done as described.12,150,151 Briefly, t-SNE embeddings were created using Rtsne, the Barnes-Hut implementation of t-Distributed Stochastic Neighbor Embedding. Embeddings were created from Euclidean distance; Spearman dissimilarity defined as 1 - abs(cor); and hybrid Spearman-Euclidean dissimilarity (SED) defined as the mean of normalized Euclidean and Spearman dissimilarity as 1 - cor. Clusters were identified from embeddings using the minimum spanning tree, single linkage method from three three-dimensional t-SNE embeddings. Each cluster represents the intersection of all three t-SNE embeddings. An adjacency matrix was constructed by pairing co-clustered modifications to each other. To construct co-cluster correlation networks (CCCNs), the adjacency matrix was used to filter Spearman correlations. Based on this filtering, co-clustered Spearman correlations were used as weight for CCCN edges. Additionally, negative correlations (<-0.5) between different PTM types on the same protein were added as CCCN edges to examine reciprocally antagonistic relationships when graphing PTM CCCN edges. In the graph based on the PTM data, the expression ratio assigned to each, each gene node represents the sum of all PTM ratios for that gene. Experiments where total proteins were identified were treated separately from those where PTMs were identified, yielding separate CCCNs and CFNs (available on NDeX,132 see key resources table).
The resulting protein CCCN was used to create a cluster-filtered network (CFN) of known PPI interactions that were filtered by excluding all interactions save those from proteins with co-clustered modifications. To construct the CFN from protein data, PPI edges were filtered by protein co-clustering. The PPI dataset was composed of curated physical interactions with a focus on direct interactions as described12 from STRING,91 GeneMANIA,152,153 BioPlex,154,155 Pathway Commons,156 and the kinase-substrate data from PhosphositePlus.157 Networks were graphed in Cytoscape using RCy3.158,159
Pathway Crosstalk Network (PCN) construction
The PCN was constructed using R as described.13 BioPlanet pathways were downloaded from the NCATS website (https://tripod.nih.gov/bioplanet/) as gene sets and converted into a list of pathway names whose elements are the genes in each pathway. In the PCN, each BioPlanet pathway was represented as a node. Pathways were joined by edges weighted according to the protein or PTM Cluster Weight (pathway interaction based on co-clustering of proteins or PTMs), or Jaccard Similarity (pathway interaction based on shared protein members), which are described in detail below. When only pathway interactions with non-zero PTM Cluster Weight are included, the PCN contained 1623/1657 BioPlanet pathways and 764,470 edges out of 1,316,253 possible edges (density = 0.5807926). The version of the PCN that only includes interactions with non-zero PTM Cluster Weight and zero Jaccard Similarity contained 1621 pathways, but only 562,735 edges (density = 0.4285839). PCN graphs and sub-networks of the PCN that were selected by thresholding the various edge weights were graphed in Cytoscape using RCy3.158,159
Cluster enrichment of pathways
CCCN clusters of proteins and PTMs that were significantly enriched in organoids or NCSCs were identified as described.13 Briefly, proteins (or PTMs) whose median expression/abundance values were changed at least 2.25-fold relative to neuroblastoma SH-SY5Y cells were identified and mapped to their respective CCCN clusters. Clusters that were statistically enriched for changed proteins/PTMs were then identified using a one-sided Fisher Test13. For the protein CCCN, 109 enriched clusters were identified using organoid data and 63 enriched clusters were identified using NCSC data, including 37 clusters enriched using both organoid and NCSC data. For the PTM CCCN, 202 enriched clusters were identified using organoid data and 104 enriched clusters were identified using NCSC data, including 39 clusters enriched using both organoid and NCSC data. To build the organoid- and NCSC-specific PCNs, PCNs were constructed as described above using the subsets of clusters from the full CCCN that were enriched in organoids or NCSCs, respectively.
Acknowledgments
M.G. was supported by NIH R15DE028434, the NIH LINCS program U54 RFA-HG-14-001, and University of Montana Center for Translational Medicine Pilot Grants, and generous donations from Craig Wilkinson. L.F. was supported by Centers of Biomedical Research Excellence (COBRE) NIH NCRR Grant P20GM103546. We thank David Gokhman for iPS cells, and Karen Ross and Katherine Stayner for comments on the article.
Author contributions
LF, NA - cell culture, organoid characterization, IF, stains. BP - histological analysis. JML, MG - proteomic and snRNA-seq data analysis. LF, MG, JML, NA, - writing, experimental design. MA, TL - proteomics and snRNA-seq.
Declaration of interests
Tyler Levy, Anthony Possemato, and Majd Ariss are employees of Cell Signaling Technology, Danvers, MA 01923, USA, and listed as such on the title page.
Published: March 28, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109585.
Supplemental information
References
- 1.Bronner M.E., LeDouarin N.M. Development and evolution of the neural crest: An overview. Dev. Biol. 2012;366:2–9. doi: 10.1016/j.ydbio.2011.12.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Green J.D., Tollemar V., Dougherty M., Yan Z., Yin L., Ye J., Collier Z., Mohammed M.K., Haydon R.C., Luu H.H., et al. Multifaceted signaling regulators of chondrogenesis: Implications in cartilage regeneration and tissue engineering. Genes Dis. 2015;2:307–327. doi: 10.1016/j.gendis.2015.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chen S., Fu P., Wu H., Pei M. Meniscus, articular cartilage and nucleus pulposus: a comparative review of cartilage-like tissues in anatomy, development and function. Cell Tissue Res. 2017;370:53–70. doi: 10.1007/s00441-017-2613-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Humphreys P.A., Mancini F.E., Ferreira M.J.S., Woods S., Ogene L., Kimber S.J. Developmental principles informing human pluripotent stem cell differentiation to cartilage and bone. Semin. Cell Dev. Biol. 2022;127:17–36. doi: 10.1016/j.semcdb.2021.11.024. [DOI] [PubMed] [Google Scholar]
- 5.Mishina Y., Snider T.N. Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell Res. 2014;325:138–147. doi: 10.1016/J.YEXCR.2014.01.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhong L., Huang X., Karperien M., Post J.N. The Regulatory Role of Signaling Crosstalk in Hypertrophy of MSCs and Human Articular Chondrocytes. Int. J. Mol. Sci. 2015;16:19225–19247. doi: 10.3390/ijms160819225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Van Otterloo E., Williams T., Artinger K.B. The old and new face of craniofacial research: How animal models inform human craniofacial genetic and clinical data. Dev. Biol. 2016;415:171–187. doi: 10.1016/j.ydbio.2016.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kreitzer F.R., Salomonis N., Sheehan A., Huang M., Park J.S., Spindler M.J., Lizarraga P., Weiss W.A., So P.-L., Conklin B.R. A robust method to derive functional neural crest cells from human pluripotent stem cells. Am. J. Stem Cells. 2013;2:119–131. [PMC free article] [PubMed] [Google Scholar]
- 9.Mica Y., Lee G., Chambers S.M., Tomishima M.J., Studer L. Modeling neural crest induction, melanocyte specification, and disease-related pigmentation defects in hESCs and patient-specific iPSCs. Cell Rep. 2013;3:1140–1152. doi: 10.1016/j.celrep.2013.03.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeltner N., Lafaille F.G., Fattahi F., Studer L. Feeder-free derivation of neural crest progenitor cells from human pluripotent stem cells. J. Vis. Exp. 2014:51609. doi: 10.3791/51609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chambers S.M., Mica Y., Lee G., Studer L., Tomishima M.J. Dual-SMAD Inhibition/WNT Activation-Based Methods to Induce Neural Crest and Derivatives from Human Pluripotent Stem Cells. Methods Mol. Biol. 2015;1307:329–343. doi: 10.1007/7651. [DOI] [PubMed] [Google Scholar]
- 12.Grimes M., Hall B., Foltz L., Levy T., Rikova K., Gaiser J., Cook W., Smirnova E., Wheeler T., Clark N.R., et al. Integration of protein phosphorylation, acetylation, and methylation data sets to outline lung cancer signaling networks. Sci. Signal. 2018;11 doi: 10.1126/scisignal.aaq1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ross K.E., Zhang G., Akcora C., Lin Y., Fang B., Koomen J., Haura E.B., Grimes M. Network models of protein phosphorylation, acetylation, and ubiquitination connect metabolic and cell signaling pathways in lung cancer. PLoS Comput. Biol. 2023;19 doi: 10.1371/journal.pcbi.1010690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Drubin D.G., Hyman A.A. Stem cells: the new “model organism”. Mol. Biol. Cell. 2017;28:1409–1411. doi: 10.1091/mbc.E17-03-0183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Oliver J.D., Madhoun W., Graham E.M., Hendrycks R., Renouard M., Hu M.S. Stem Cells Regenerating the Craniofacial Skeleton: Current State-Of-The-Art and Future Directions. J. Clin. Med. 2020;9 doi: 10.3390/jcm9103307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu H., Kirita Y., Donnelly E.L., Humphreys B.D. Advantages of Single-Nucleus over Single-Cell RNA Sequencing of Adult Kidney: Rare Cell Types and Novel Cell States Revealed in Fibrosis. J. Am. Soc. Nephrol. 2019;30:23–32. doi: 10.1681/ASN.2018090912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yanagisawa H., Davis E.C. Unraveling the mechanism of elastic fiber assembly: The roles of short fibulins. Int. J. Biochem. Cell Biol. 2010;42:1084–1093. doi: 10.1016/j.biocel.2010.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simões-Costa M., Bronner M.E. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lefebvre V., Dvir-Ginzberg M. SOX9 and the many facets of its regulation in the chondrocyte lineage. Connect. Tissue Res. 2017;58:2–14. doi: 10.1080/03008207.2016.1183667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Miao Z., Lu Z., Wu H., Liu H., Li M., Lei D., Zheng L., Zhao J. Collagen, agarose, alginate, and Matrigel hydrogels as cell substrates for culture of chondrocytes in vitro: A comparative study. J. Cell. Biochem. 2018;119:7924–7933. doi: 10.1002/jcb.26411. [DOI] [PubMed] [Google Scholar]
- 21.Spicer A.P., Joo A., Bowling R.A. A Hyaluronan Binding Link Protein Gene Family Whose Members Are Physically Linked Adjacent to Chrondroitin Sulfate Proteoglycan Core Protein Genes. J. Biol. Chem. 2003;278:21083–21091. doi: 10.1074/jbc.M213100200. [DOI] [PubMed] [Google Scholar]
- 22.Chen S., Birk D.E. The regulatory roles of small leucine-rich proteoglycans in extracellular matrix assembly. FEBS J. 2013;280:2120–2137. doi: 10.1111/febs.12136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang Y., Ryan J.A., Di Cesare P.E., Liu J., Walsh C.A., You Z. Doublecortin is expressed in articular chondrocytes. Biochem. Biophys. Res. Commun. 2007;363:694–700. doi: 10.1016/j.bbrc.2007.09.030. [DOI] [PubMed] [Google Scholar]
- 24.Ge D., Zhang Q.S., Zabaleta J., Zhang Q., Liu S., Reiser B., Bunnell B.A., Braun S.E., O'Brien M.J., Savoie F.H., You Z. Doublecortin may play a role in defining chondrocyte phenotype. Int. J. Mol. Sci. 2014;15:6941–6960. doi: 10.3390/ijms15046941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vermillion K.L., Lidberg K.A., Gammill L.S. Expression of actin-binding proteins and requirement for actin depolymerizing factor in chick neural crest cells. Dev. Dyn. 2014;243:730–738. doi: 10.1002/dvdy.24105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang R., Grishagin I., Wang Y., Zhao T., Greene J., Obenauer J.C., Ngan D., Nguyen D.-T., Guha R., Jadhav A., et al. The NCATS BioPlanet – An Integrated Platform for Exploring the Universe of Cellular Signaling Pathways for Toxicology, Systems Biology, and Chemical Genomics. Front. Pharmacol. 2019;10:445. doi: 10.3389/fphar.2019.00445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Raredon M.S.B., Yang J., Garritano J., Wang M., Kushnir D., Schupp J.C., Adams T.S., Greaney A.M., Leiby K.L., Kaminski N., et al. Computation and visualization of cell-cell signaling topologies in single-cell systems data using Connectome. Sci. Rep. 2022;12:4187. doi: 10.1038/s41598-022-07959-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hynes R.O. The extracellular matrix: not just pretty fibrils. Science. 2009;326:1216–1219. doi: 10.1126/science.1176009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hynes N.E., Ingham P.W., Lim W.A., Marshall C.J., Massagué J., Pawson T. Signalling change: signal transduction through the decades. Nat. Rev. Mol. Cell Biol. 2013;14:393–398. doi: 10.1038/nrm3581. [DOI] [PubMed] [Google Scholar]
- 30.Hynes R.O. Stretching the boundaries of extracellular matrix research. Nat. Rev. Mol. Cell Biol. 2014;15:761–763. doi: 10.1038/nrm3908. [DOI] [PubMed] [Google Scholar]
- 31.Zeltz C., Gullberg D. The integrin-collagen connection - a glue for tissue repair? J. Cell Sci. 2016;129:1284. doi: 10.1242/jcs.188672. [DOI] [PubMed] [Google Scholar]
- 32.Elango J., Hou C., Bao B., Wang S., Maté Sánchez de Val J.E., Wenhui W. The Molecular Interaction of Collagen with Cell Receptors for Biological Function. Polymers. 2022;14 doi: 10.3390/polym14050876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Horiguchi M., Ota M., Rifkin D.B. Matrix control of transforming growth factor-beta function. J. Biochem. 2012;152:321–329. doi: 10.1093/jb/mvs089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Muncie J.M., Weaver V.M. The Physical and Biochemical Properties of the Extracellular Matrix Regulate Cell Fate. Curr. Top. Dev. Biol. 2018;130:1–37. doi: 10.1016/bs.ctdb.2018.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu M., Ling Z., Ren X. Extracellular matrix dynamics: tracking in biological systems and their implications. J. Biol. Eng. 2022;16:13. doi: 10.1186/s13036-022-00292-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kobayashi S., Takebe T., Inui M., Iwai S., Kan H., Zheng Y.W., Maegawa J., Taniguchi H. Reconstruction of human elastic cartilage by a CD44+ CD90+ stem cell in the ear perichondrium. Proc. Natl. Acad. Sci. USA. 2011;108:14479–14484. doi: 10.1073/pnas.1109767108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Somoza R.A., Welter J.F., Correa D., Caplan A.I. Chondrogenic differentiation of mesenchymal stem cells: challenges and unfulfilled expectations. Tissue Eng. Part B Rev. 2014;20:596–608. doi: 10.1089/ten.TEB.2013.0771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Li J., Dong S. The Signaling Pathways Involved in Chondrocyte Differentiation and Hypertrophic Differentiation. Stem Cells Int. 2016;2016 doi: 10.1155/2016/2470351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Oba T., Okamoto S., Ueno Y., Matsuo M., Tadokoro T., Kobayashi S., Yasumura K., Kagimoto S., Inaba Y., Taniguchi H. In vitro elastic cartilage reconstruction using human auricular perichondrial chondroprogenitor cell-derived micro 3D spheroids. J. Tissue Eng. 2022;13 doi: 10.1177/20417314221143484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schwarzl T., Keogh A., Shaw G., Krstic A., Clayton E., Higgins D.G., Kolch W., Barry F. Transcriptional profiling of early differentiation of primary human mesenchymal stem cells into chondrocytes. Sci. Data. 2023;10:758. doi: 10.1038/s41597-023-02686-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lee G., Chambers S.M., Tomishima M.J., Studer L. Derivation of neural crest cells from human pluripotent stem cells. Nat. Protoc. 2010;5:688–701. doi: 10.1038/nprot.2010.35. [DOI] [PubMed] [Google Scholar]
- 42.Leung A.W., Murdoch B., Salem A.F., Prasad M.S., Gomez G.A., García-Castro M.I. WNT/β-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development. 2016;143:398–410. doi: 10.1242/dev.130849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vinod E., Kachroo U., Rebekah G., Thomas S., Ramasamy B. In vitro chondrogenic differentiation of human articular cartilage derived chondroprogenitors using pulsed electromagnetic field. J. Clin. Orthop. Trauma. 2021;14:22–28. doi: 10.1016/j.jcot.2020.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Li T., Chen S., Pei M. Contribution of neural crest-derived stem cells and nasal chondrocytes to articular cartilage regeneration. Cell. Mol. Life Sci. 2020;77:4847–4859. doi: 10.1007/s00018-020-03567-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dowthwaite G.P., Bishop J.C., Redman S.N., Khan I.M., Rooney P., Evans D.J.R., Haughton L., Bayram Z., Boyer S., Thomson B., et al. The surface of articular cartilage contains a progenitor cell population. J. Cell Sci. 2004;117:889–897. doi: 10.1242/jcs.00912. [DOI] [PubMed] [Google Scholar]
- 46.Shafiee A., Kabiri M., Ahmadbeigi N., Yazdani S.O., Mojtahed M., Amanpour S., Soleimani M. Nasal septum-derived multipotent progenitors: a potent source for stem cell-based regenerative medicine. Stem Cells Dev. 2011;20:2077–2091. doi: 10.1089/scd.2010.0420. [DOI] [PubMed] [Google Scholar]
- 47.Watson D., Reuther M.S. Tissue-engineered cartilage for facial plastic surgery. Curr. Opin. Otolaryngol. Head Neck Surg. 2014;22:300–306. doi: 10.1097/MOO.0000000000000068. [DOI] [PubMed] [Google Scholar]
- 48.Akkina S.R., Most S.P. The effect of perichondrium on cartilage graft properties. Curr. Opin. Otolaryngol. Head Neck Surg. 2022;30:215–218. doi: 10.1097/MOO.0000000000000812. [DOI] [PubMed] [Google Scholar]
- 49.Candrian C., Vonwil D., Barbero A., Bonacina E., Miot S., Farhadi J., Wirz D., Dickinson S., Hollander A., Jakob M., et al. Engineered cartilage generated by nasal chondrocytes is responsive to physical forces resembling joint loading. Arthritis Rheum. 2008;58:197–208. doi: 10.1002/art.23155. [DOI] [PubMed] [Google Scholar]
- 50.Mumme M., Barbero A., Miot S., Wixmerten A., Feliciano S., Wolf F., Asnaghi A.M., Baumhoer D., Bieri O., Kretzschmar M., et al. Nasal chondrocyte-based engineered autologous cartilage tissue for repair of articular cartilage defects: an observational first-in-human trial. Lancet. 2016;388:1985–1994. doi: 10.1016/S0140-6736(16)31658-0. [DOI] [PubMed] [Google Scholar]
- 51.Pelttari K., Mumme M., Barbero A., Martin I. Nasal chondrocytes as a neural crest-derived cell source for regenerative medicine. Curr. Opin. Biotechnol. 2017;47:1–6. doi: 10.1016/j.copbio.2017.05.007. [DOI] [PubMed] [Google Scholar]
- 52.Dobratz E.J., Kim S.W., Voglewede A., Park S.S. Injectable Cartilage Using Alginate and Human Chondrocytes. Arch. Facial Plast. Surg. 2009;11:40–47. doi: 10.1001/archfacial.2008.509. [DOI] [PubMed] [Google Scholar]
- 53.Gvaramia D., Kern J., Jakob Y., Zenobi-Wong M., Rotter N. Regenerative Potential of Perichondrium: A Tissue Engineering Perspective. Tissue Eng. Part B Rev. 2022;28:531–541. doi: 10.1089/ten.TEB.2021.0054. [DOI] [PubMed] [Google Scholar]
- 54.Vinod E., Padmaja K., Ramasamy B., Sathishkumar S. Systematic review of articular cartilage derived chondroprogenitors for cartilage repair in animal models. J. Orthop. 2023;35:43–53. doi: 10.1016/j.jor.2022.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Janebodin K., Horst O.V., Ieronimakis N., Balasundaram G., Reesukumal K., Pratumvinit B., Reyes M. Isolation and Characterization of Neural Crest-Derived Stem Cells from Dental Pulp of Neonatal Mice. PLoS One. 2011;6:27526. doi: 10.1371/journal.pone.0027526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gronthos S., Mankani M., Brahim J., Robey P.G., Shi S. Postnatal human dental pulp stem cells (DPSCs) in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 2000;97:13625–13630. doi: 10.1073/pnas.240309797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.d'Aquino R., Graziano A., Sampaolesi M., Laino G., Pirozzi G., De Rosa A., Papaccio G. Human postnatal dental pulp cells co-differentiate into osteoblasts and endotheliocytes: A pivotal synergy leading to adult bone tissue formation. Cell Death Differ. 2007;14:1162–1171. doi: 10.1038/sj.cdd.4402121. [DOI] [PubMed] [Google Scholar]
- 58.Shen P., Chen L., Zhang D., Xia S., Lv Z., Zou D., Zhang Z., Yang C., Li W. Rapid induction and long-term self-renewal of neural crest-derived ectodermal chondrogenic cells from hPSCs. NPJ Regen. Med. 2022;7:69. doi: 10.1038/s41536-022-00265-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kaucka M., Ivashkin E., Gyllborg D., Zikmund T., Tesarova M., Kaiser J., Xie M., Petersen J., Pachnis V., Nicolis S.K., et al. Analysis of neural crest-derived clones reveals novel aspects of facial development. Sci. Adv. 2016;2:e1600060. doi: 10.1126/sciadv.1600060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Oldershaw R.A., Baxter M.A., Lowe E.T., Bates N., Grady L.M., Soncin F., Brison D.R., Hardingham T.E., Kimber S.J. Directed differentiation of human embryonic stem cells toward chondrocytes. Nat. Biotechnol. 2010;28:1187–1194. doi: 10.1038/nbt.1683. [DOI] [PubMed] [Google Scholar]
- 61.Umeda K., Oda H., Yan Q., Matthias N., Zhao J., Davis B.R., Nakayama N. Long-term expandable SOX9+ chondrogenic ectomesenchymal cells from human pluripotent stem cells. Stem Cell Rep. 2015;4:712–726. doi: 10.1016/j.stemcr.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nakayama N., Pothiawala A., Lee J.Y., Matthias N., Umeda K., Ang B.K., Huard J., Huang Y., Sun D. Human pluripotent stem cell-derived chondroprogenitors for cartilage tissue engineering. Cell. Mol. Life Sci. 2020;77:2543–2563. doi: 10.1007/s00018-019-03445-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wu C.L., Dicks A., Steward N., Tang R., Katz D.B., Choi Y.R., Guilak F. Single cell transcriptomic analysis of human pluripotent stem cell chondrogenesis. Nat. Commun. 2021;12:362. doi: 10.1038/s41467-020-20598-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burnsed O.A., Schwartz Z., Marchand K.O., Hyzy S.L., Olivares-Navarrete R., Boyan B.D. Hydrogels derived from cartilage matrices promote induction of human mesenchymal stem cell chondrogenic differentiation. Acta Biomater. 2016;43:139–149. doi: 10.1016/j.actbio.2016.07.034. [DOI] [PubMed] [Google Scholar]
- 65.Caplan A.I. Mesenchymal Stem Cells: Time to Change the Name! Stem Cells Transl. Med. 2017;6:1445–1451. doi: 10.1002/sctm.17-0051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.de Windt T.S., Vonk L.A., Saris D.B.F. Response to: Mesenchymal Stem Cells: Time to Change the Name. Stem Cells Transl. Med. 2017;6:1747–1748. doi: 10.1002/sctm.17-0120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bekkers J.E.J., Tsuchida A.I., van Rijen M.H.P., Vonk L.A., Dhert W.J.A., Creemers L.B., Saris D.B.F. Single-stage cell-based cartilage regeneration using a combination of chondrons and mesenchymal stromal cells: comparison with microfracture. Am. J. Sports Med. 2013;41:2158–2166. doi: 10.1177/0363546513494181. [DOI] [PubMed] [Google Scholar]
- 68.de Windt T.S., Hendriks J.A.A., Zhao X., Vonk L.A., Creemers L.B., Dhert W.J.A., Randolph M.A., Saris D.B.F. Concise review: unraveling stem cell cocultures in regenerative medicine: which cell interactions steer cartilage regeneration and how? Stem Cells Transl. Med. 2014;3:723–733. doi: 10.5966/sctm.2013-0207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pelttari K., Winter A., Steck E., Goetzke K., Hennig T., Ochs B.G., Aigner T., Richter W. Premature induction of hypertrophy during in vitro chondrogenesis of human mesenchymal stem cells correlates with calcification and vascular invasion after ectopic transplantation in SCID mice. Arthritis Rheum. 2006;54:3254–3266. doi: 10.1002/art.22136. [DOI] [PubMed] [Google Scholar]
- 70.Chen S., Fu P., Cong R., Wu H., Pei M. Strategies to minimize hypertrophy in cartilage engineering and regeneration. Genes Dis. 2015;2:76–95. doi: 10.1016/J.GENDIS.2014.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dexheimer V., Gabler J., Bomans K., Sims T., Omlor G., Richter W. Differential expression of TGF-β superfamily members and role of Smad1/5/9-signalling in chondral versus endochondral chondrocyte differentiation. Sci. Rep. 2016;6:1–14. doi: 10.1038/srep36655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Suchorska W.M., Augustyniak E., Richter M., Trzeciak T. Comparison of Four Protocols to Generate Chondrocyte-Like Cells from Human Induced Pluripotent Stem Cells (hiPSCs) Stem Cell Rev. Rep. 2017;13:299–308. doi: 10.1007/s12015-016-9708-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Suchorska W.M., Augustyniak E., Richter M., Łukjanow M., Filas V., Kaczmarczyk J., Trzeciak T. Modified methods for efficiently differentiating human embryonic stem cells into chondrocyte-like cells. Postepy Hig. Med. Dosw. 2017;71:500–509. doi: 10.5604/01.3001.0010.3831. [DOI] [PubMed] [Google Scholar]
- 74.Van de Walle A., Faissal W., Wilhelm C., Luciani N. Role of growth factors and oxygen to limit hypertrophy and impact of high magnetic nanoparticles dose during stem cell chondrogenesis. Comput. Struct. Biotechnol. J. 2018;16:532–542. doi: 10.1016/j.csbj.2018.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Steck E., Fischer J., Lorenz H., Gotterbarm T., Jung M., Richter W. Mesenchymal Stem Cell Differentiation in an Experimental Cartilage Defect: Restriction of Hypertrophy to Bone-Close Neocartilage. Stem Cells Dev. 2009;18:969–978. doi: 10.1089/scd.2008.0213. [DOI] [PubMed] [Google Scholar]
- 76.Huey D.J., Hu J.C., Athanasiou K.A. Unlike bone, cartilage regeneration remains elusive. Science. 2012;338:917–921. doi: 10.1126/science.1222454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Maguire L.H., Thomas A.R., Goldstein A.M. Tumors of the neural crest: Common themes in development and cancer. Dev. Dyn. 2015;244:311–322. doi: 10.1002/dvdy.24226. [DOI] [PubMed] [Google Scholar]
- 78.Fu Q., Su D., Wang K., Zhao Y. Tumorigenesis of nuclear transfer-derived embryonic stem cells is reduced through differentiation and enrichment following transplantation in the infarcted rat heart. Mol. Med. Rep. 2016;13:4659–4665. doi: 10.3892/mmr.2016.5092. [DOI] [PubMed] [Google Scholar]
- 79.Soto J., Ding X., Wang A., Li S. Neural crest-like stem cells for tissue regeneration. Stem Cells Transl. Med. 2021;10:681–693. doi: 10.1002/sctm.20-0361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Guo T., Noshin M., Baker H.B., Taskoy E., Meredith S.J., Tang Q., Ringel J.P., Lerman M.J., Chen Y., Packer J.D., Fisher J.P. 3D printed biofunctionalized scaffolds for microfracture repair of cartilage defects. Biomaterials. 2018;185:219–231. doi: 10.1016/j.biomaterials.2018.09.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Panadero J.A., Lanceros-Mendez S., Ribelles J.L.G. Differentiation of mesenchymal stem cells for cartilage tissue engineering: Individual and synergetic effects of three-dimensional environment and mechanical loading. Acta Biomater. 2016;33:1–12. doi: 10.1016/J.ACTBIO.2016.01.037. [DOI] [PubMed] [Google Scholar]
- 82.Choi J.R., Yong K.W., Choi J.Y. Effects of mechanical loading on human mesenchymal stem cells for cartilage tissue engineering. J. Cell. Physiol. 2018;233:1913–1928. doi: 10.1002/jcp.26018. [DOI] [PubMed] [Google Scholar]
- 83.Ma J., Zhang Y., Yan Z., Wu P., Li C., Yang R., Lu X., Chen X., He A., Fu Y., et al. Single-cell transcriptomics reveals pathogenic dysregulation of previously unrecognised chondral stem/progenitor cells in children with microtia. Clin. Transl. Med. 2022;12 doi: 10.1002/ctm2.702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Chen X., Xu Y., Li C., Lu X., Fu Y., Huang Q., Ma D., Ma J., Zhang T. Key Genes Identified in Nonsyndromic Microtia by the Analysis of Transcriptomics and Proteomics. ACS Omega. 2022;7:16917–16927. doi: 10.1021/acsomega.1c07059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dong W., Jiang H., He L., Pan B., Lin L., Song Y., Yang Q. Protein profile of ear auricle cartilage and the important role of ITGB1/PTK2 in microtia. Int. J. Pediatr. Otorhinolaryngol. 2020;137 doi: 10.1016/j.ijporl.2020.110235. [DOI] [PubMed] [Google Scholar]
- 86.Dong W., Jiang H., He L., Pan B., Yang Q. Changes in the Transcriptome and Proteome of Cartilage in Microtia. J. Craniofac. Surg. 2021;32:2301–2304. doi: 10.1097/SCS.0000000000007660. [DOI] [PubMed] [Google Scholar]
- 87.Arseni L., Lombardi A., Orioli D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018;19 doi: 10.3390/ijms19051407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vogel W., Gish G.D., Alves F., Pawson T. The Discoidin Domain Receptor Tyrosine Kinases Are Activated by Collagen. Mol. Cell. 1997;1:13–23. doi: 10.1016/S1097-2765(00)80003-9. [DOI] [PubMed] [Google Scholar]
- 89.Bildsoe H., Fan X., Wilkie E.E., Ashoti A., Jones V.J., Power M., Qin J., Wang J., Tam P.P.L., Loebel D.A.F. Transcriptional targets of TWIST1 in the cranial mesoderm regulate cell-matrix interactions and mesenchyme maintenance. Dev. Biol. 2016;418:189–203. doi: 10.1016/J.YDBIO.2016.08.016. [DOI] [PubMed] [Google Scholar]
- 90.Mohamed F.F., Ge C., Hallett S.A., Bancroft A.C., Cowling R.T., Ono N., Binrayes A.A., Greenberg B., Levi B., Kaartinen V.M., Franceschi R.T. Control of craniofacial development by the collagen receptor, discoidin domain receptor 2. Elife. 2023;12 doi: 10.7554/eLife.77257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P., et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45:D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Vuorio E., de Crombrugghe B. The family of collagen genes. Annu. Rev. Biochem. 1990;59:837–872. doi: 10.1146/annurev.bi.59.070190.004201. [DOI] [PubMed] [Google Scholar]
- 93.Nassa M., Anand P., Jain A., Chhabra A., Jaiswal A., Malhotra U., Rani V. Analysis of human collagen sequences. Bioinformation. 2012;8:26–33. doi: 10.6026/97320630008026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Luo Y., Sinkeviciute D., He Y., Karsdal M., Henrotin Y., Mobasheri A., Önnerfjord P., Bay-Jensen A. MINI-REVIEW The minor collagens in articular cartilage. Protein Cell. 2017;8:560–572. doi: 10.1007/s13238-017-0377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Somasundaram R., Schuppan D. Type I, II, III, IV, V, and VI collagens serve as extracellular ligands for the isoforms of platelet-derived growth factor (AA, BB, and AB) J. Biol. Chem. 1996;271:26884–26891. doi: 10.1074/jbc.271.43.26884. [DOI] [PubMed] [Google Scholar]
- 96.Dreyfus J., Brunet-de Carvalho N., Duprez D., Raulais D., Vigny M. HB-GAM/pleiotrophin but not RIHB/midkine enhances chondrogenesis in micromass culture. Exp. Cell Res. 1998;241:171–180. doi: 10.1006/excr.1998.4040. [DOI] [PubMed] [Google Scholar]
- 97.Ohta S., Muramatsu H., Senda T., Zou K., Iwata H., Muramatsu T. Midkine Is Expressed During Repair of Bone Fracture and Promotes Chondrogenesis. J. Bone Miner. Res. 1999;14:1132–1144. doi: 10.1359/jbmr.1999.14.7.1132. [DOI] [PubMed] [Google Scholar]
- 98.Zhang Z.H., Li H.X., Qi Y.P., Du L.J., Zhu S.Y., Wu M.Y., Lu H.L., Yu Y., Han W. Recombinant human midkine stimulates proliferation of articular chondrocytes. Cell Prolif. 2010;43:184–194. doi: 10.1111/j.1365-2184.2010.00668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Bouderlique T., Henault E., Lebouvier A., Frescaline G., Bierling P., Rouard H., Courty J., Albanese P., Chevallier N. Pleiotrophin commits human bone marrow mesenchymal stromal cells towards hypertrophy during chondrogenesis. PLoS One. 2014;9 doi: 10.1371/journal.pone.0088287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Haffner-Luntzer M., Heilmann A., Rapp A.E., Beie S., Schinke T., Amling M., Ignatius A., Liedert A. Midkine-Deficiency Delays Chondrogenesis during the Early Phase of Fracture Healing in Mice. PLoS One. 2014;9:e116282. doi: 10.1371/journal.pone.0116282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Pufe T., Groth G., Goldring M.B., Tillmann B., Mentlein R. Effects of pleiotrophin, a heparin-binding growth factor, on human primary and immortalized chondrocytes. Osteoarthritis Cartilage. 2007;15:155–162. doi: 10.1016/j.joca.2006.07.005. [DOI] [PubMed] [Google Scholar]
- 102.Xu J., Huang Z., Wang W., Tan X., Li H., Zhang Y., Tian W., Hu T., Chen Y.P. FGF8 Signaling Alters the Osteogenic Cell Fate in the Hard Palate. J. Dent. Res. 2018;97:589–596. doi: 10.1177/0022034517750141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mariani E., Pulsatelli L., Facchini A. Signaling pathways in cartilage repair. Int. J. Mol. Sci. 2014;15:8667–8698. doi: 10.3390/ijms15058667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Bozdagi O., Rich E., Tronel S., Sadahiro M., Patterson K., Shapiro M.L., Alberini C.M., Huntley G.W., Salton S.R.J. The Neurotrophin-Inducible Gene Vgf Regulates Hippocampal Function and Behavior through a Brain-Derived Neurotrophic Factor-Dependent Mechanism. J. Neurosci. 2008;28:9857–9869. doi: 10.1523/JNEUROSCI.3145-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Chimal-Monroy J., Díaz de León L. Differential effects of transforming growth factors β-1, β-2, β-3 and β-5 on chondrogenesis in mouse limb bud mesenchymal cells. Int. J. Dev. Biol. 1997;41:91–102. [PubMed] [Google Scholar]
- 106.Yang Z., Sui L., Toh W.S., Lee E.H., Cao T. Stage-Dependent Effect of TGF-β1 on Chondrogenic Differentiation of Human Embryonic Stem Cells. Stem Cells Dev. 2009;18:929–940. doi: 10.1089/scd.2008.0219. [DOI] [PubMed] [Google Scholar]
- 107.Wang W., Rigueur D., Lyons K.M. TGF beta signaling in cartilage development and maintenance. Birth Defects Res. C Embryo Today. 2015;102:37–51. doi: 10.1002/bdrc.21058.TGF. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Barry F., Boynton R.E., Liu B., Murphy J.M. Chondrogenic Differentiation of Mesenchymal Stem Cells from Bone Marrow: Differentiation-Dependent Gene Expression of Matrix Components. Exp. Cell Res. 2001;268:189–200. doi: 10.1006/excr.2001.5278. [DOI] [PubMed] [Google Scholar]
- 109.Deng Y., Sun A.X., Overholt K.J., Yu G.Z., Fritch M.R., Alexander P.G., Shen H., Tuan R.S., Lin H. Enhancing chondrogenesis and mechanical strength retention in physiologically relevant hydrogels with incorporation of hyaluronic acid and direct loading of TGF-β. Acta Biomater. 2019;83:167–176. doi: 10.1016/j.actbio.2018.11.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Pfeifer C.G., Karl A., Kerschbaum M., Berner A., Lang S., Schupfner R., Koch M., Angele P., Nerlich M., Mueller M.B. TGF- β Signalling is Suppressed under Pro-Hypertrophic Conditions in MSC Chondrogenesis Due to TGF- β Receptor Downregulation. Int. J. Stem Cells. 2019;12:139–150. doi: 10.15283/ijsc18088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Bennett J.H., Hunt P., Thorogood P. Bone morphogenetic protein-2 and -4 expression during murine orofacial development. Arch. Oral Biol. 1995;40:847–854. doi: 10.1016/0003-9969(95)00047-S. [DOI] [PubMed] [Google Scholar]
- 112.Liu W., Sun X., Braut A., Mishina Y., Behringer R.R., Mina M., Martin J.F. Distinct functions for Bmp signaling in lip and palate fusion in mice. Development. 2005;132:1453–1461. doi: 10.1242/dev.01676. [DOI] [PubMed] [Google Scholar]
- 113.Li Z., Chen Y.-G. Functions of BMP signaling in embryonic stem cell fate determination. Exp. Cell Res. 2013;319:113–119. doi: 10.1016/j.yexcr.2012.09.016. [DOI] [PubMed] [Google Scholar]
- 114.Gu S., Wu W., Liu C., Yang L., Sun C., Ye W., Li X., Chen J., Long F., Chen Y. BMPRIA Mediated Signaling Is Essential for Temporomandibular Joint Development in Mice. PLoS One. 2014;9:e101000. doi: 10.1371/journal.pone.0101000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Graf D., Malik Z., Hayano S., Mishina Y. Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev. 2016;27:129–139. doi: 10.1016/j.cytogfr.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mimura S., Suga M., Okada K., Kinehara M., Nikawa H., Furue M.K. Bone morphogenetic protein 4 promotes craniofacial neural crest induction from human pluripotent stem cells. Int. J. Dev. Biol. 2016;60:21–28. doi: 10.1387/ijdb.160040mk. [DOI] [PubMed] [Google Scholar]
- 117.Dudas M., Sridurongrit S., Nagy A., Okazaki K., Kaartinen V. Craniofacial defects in mice lacking BMP type I receptor Alk2 in neural crest cells. Mech. Dev. 2004;121:173–182. doi: 10.1016/J.MOD.2003.12.003. [DOI] [PubMed] [Google Scholar]
- 118.Komatsu Y., Fukuda T., Scott G., Kamiya N., Yamamura K.-I., Mishina Y. Enhanced BMP signaling through a type I BMP receptor ALK2 shows ectopic cartilage formation in mouse craniofacial portion. Dev. Biol. 2006;295:399. doi: 10.1016/j.ydbio.2006.04.223. [DOI] [Google Scholar]
- 119.Hayano S., Komatsu Y., Pan H., Mishina Y. Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development. 2015;142:1357–1367. doi: 10.1242/DEV.118802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Mailhot G., Yang M., Mason-Savas A., Mackay C.A., Leav I., Odgren P.R. BMP-5 expression increases during chondrocyte differentiation in vivo and in vitro and promotes proliferation and cartilage matrix synthesis in primary chondrocyte cultures. J. Cell. Physiol. 2008;214:56–64. doi: 10.1002/jcp.21164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kingsley D.M., Bland A.E., Grubber J.M., Marker P.C., Russell L.B., Copeland N.G., Jenkins N.A., Jenkins N.A. The Mouse short ear Skeletal Morphogenesis Locus Is Associated with Defects in a Bone Morphogenetic Member of the TGFP Superfamily. Cell. 1992;71:399–410. doi: 10.1016/0092-8674(92)90510-J. [DOI] [PubMed] [Google Scholar]
- 122.Nie X., Luukko K., Kettunen P. BMP signalling in craniofacial development. Int. J. Dev. Biol. 2006;50:511–521. doi: 10.1387/ijdb.052101xn. [DOI] [PubMed] [Google Scholar]
- 123.Guenther C., Pantalena-Filho L., Kingsley D.M. Shaping Skeletal Growth by Modular Regulatory Elements in the Bmp5 Gene. PLoS Genet. 2008;4 doi: 10.1371/journal.pgen.1000308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shih H.-Y., Hsu S.-Y., Ouyang P., Lin S.-J., Chou T.-Y., Chiang M.-C., Cheng Y.-C. Bmp5 Regulates Neural Crest Cell Survival and Proliferation via Two Different Signaling Pathways. Stem Cell. 2017;35:1003–1014. doi: 10.1002/stem.2533. [DOI] [PubMed] [Google Scholar]
- 125.Merino R., Rodriguez-Leon J., Macias D., Gañan Y., Economides A.N., Hurle J.M. The BMP antagonist Gremlin regulates outgrowth, chondrogenesis and programmed cell death in the developing limb. Development. 1999;126:5515–5522. doi: 10.1242/dev.126.23.5515. [DOI] [PubMed] [Google Scholar]
- 126.Yeung C.Y.C., Gossan N., Lu Y., Hughes A., Hensman J.J., Bayer M.L., Kjær M., Kadler K.E., Meng Q.J. Gremlin-2 is a BMP antagonist that is regulated by the circadian clock. Sci. Rep. 2014;4:5183. doi: 10.1038/srep05183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Díaz-Payno P.J., Browe D.C., Freeman F.E., Nulty J., Burdis R., Kelly D.J. Gremlin-1 Suppresses Hypertrophy of Engineered Cartilage In Vitro but Not Bone Formation In Vivo. Tissue Eng. Part A. 2022;28:724–736. doi: 10.1089/ten.TEA.2021.0176. [DOI] [PubMed] [Google Scholar]
- 128.Zhao R., Moore E.L., Gogol M.M., Uhruh J.R., Yu Z., Scott A., Wang Y., Rajendran N.K., Trainor P.A. Identification and characterization of intermediate states in mammalian neural crest cell epithelial to mesenchymal transition and delamination. Elife. 2024;13:RP92844. doi: 10.7554/eLife.92844.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kalhor R., Kalhor K., Mejia L., Leeper K., Graveline A., Mali P., Church G.M. Developmental barcoding of whole mouse via homing CRISPR. Science. 2018;361 doi: 10.1126/science.aat9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Leeper K., Kalhor K., Vernet A., Graveline A., Church G.M., Mali P., Kalhor R. Lineage barcoding in mice with homing CRISPR. Nat. Protoc. 2021;16:2088–2108. doi: 10.1038/s41596-020-00485-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Perez-Riverol Y., Bai J., Bandla C., García-Seisdedos D., Hewapathirana S., Kamatchinathan S., Kundu D.J., Prakash A., Frericks-Zipper A., Eisenacher M., et al. The PRIDE database resources in 2022: a hub for mass spectrometry-based proteomics evidences. Nucleic Acids Res. 2022;50:D543–D552. doi: 10.1093/nar/gkab1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Pratt D., Chen J., Welker D., Rivas R., Pillich R., Rynkov V., Ono K., Miello C., Hicks L., Szalma S., et al. NDEx, the Network Data Exchange. Cell Syst. 2015;1:302–305. doi: 10.1016/j.cels.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Leung A.W., Murdoch B., Salem A.F., Prasad M.S., Gomez G.A., García-Castro M.I. WNT/-catenin signaling mediates human neural crest induction via a pre-neural border intermediate. Development. 2016;143:398–410. doi: 10.1242/dev.130849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ting L., Rad R., Gygi S.P., Haas W. MS3 eliminates ratio distortion in isobaric multiplexed quantitative proteomics. Nat. Methods. 2011;8:937–940. doi: 10.1038/nmeth.1714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.McAlister G.C., Nusinow D.P., Jedrychowski M.P., Wühr M., Huttlin E.L., Erickson B.K., Rad R., Haas W., Gygi S.P. MultiNotch MS3 enables accurate, sensitive, and multiplexed detection of differential expression across cancer cell line proteomes. Anal. Chem. 2014;86:7150–7158. doi: 10.1021/ac502040v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Paulo J.A., O'Connell J.D., Gygi S.P. A Triple Knockout (TKO) Proteomics Standard for Diagnosing Ion Interference in Isobaric Labeling Experiments. J. Am. Soc. Mass Spectrom. 2016;27:1620–1625. doi: 10.1016/j.physbeh.2017.03.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Huttlin E.L., Jedrychowski M.P., Elias J.E., Goswami T., Rad R., Beausoleil S.A., Villén J., Haas W., Sowa M.E., Gygi S.P. A tissue-specific atlas of mouse protein phosphorylation and expression. Cell. 2010;143:1174–1189. doi: 10.1016/j.cell.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Eng J.K., Jahan T.A., Hoopmann M.R. Comet: an open-source MS/MS sequence database search tool. Proteomics. 2013;13:22–24. doi: 10.1002/pmic.201200439. [DOI] [PubMed] [Google Scholar]
- 139.Elias J.E., Gygi S.P. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat. Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 140.Elias J.E., Gygi S.P. Target-decoy search strategy for mass spectrometry-based proteomics. Methods Mol. Biol. 2010;604:55–71. doi: 10.1007/978-1-60761-444-9_5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.McAlister G.C., Huttlin E.L., Haas W., Ting L., Jedrychowski M.P., Rogers J.C., Kuhn K., Pike I., Grothe R.A., Blethrow J.D., Gygi S.P. Increasing the multiplexing capacity of TMT using reporter ion isotopologues with isobaric masses. Anal. Chem. 2012;84:7469–7478. doi: 10.1021/ac301572t.Increasing. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Beausoleil S.A., Villén J., Gerber S.A., Rush J., Gygi S.P. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat. Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 143.Slyper M., Porter C.B.M., Ashenberg O., Waldman J., Drokhlyansky E., Wakiro I., Smillie C., Smith-Rosario G., Wu J., Dionne D., et al. A single-cell and single-nucleus RNA-Seq toolbox for fresh and frozen human tumors. Nat. Med. 2020;26:792–802. doi: 10.1038/s41591-020-0844-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Zheng G.X.Y., Terry J.M., Belgrader P., Ryvkin P., Bent Z.W., Wilson R., Ziraldo S.B., Wheeler T.D., McDermott G.P., Zhu J., et al. Massively parallel digital transcriptional profiling of single cells. Nat. Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Hao Y., Hao S., Andersen-Nissen E., Mauck W.M., 3rd, Zheng S., Butler A., Lee M.J., Wilk A.J., Darby C., Zager M., et al. Integrated analysis of multimodal single-cell data. Cell. 2021;184:3573–3587.e29. doi: 10.1016/j.cell.2021.04.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Simões-Costa M., Bronner M.E. Establishing neural crest identity: a gene regulatory recipe. Development. 2015;142:242–257. doi: 10.1242/dev.105445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Fisher R.A. 14th Edition. Oliver and Boyd; 1970. Statistical Methods for Research Workers. [Google Scholar]
- 148.Dudbridge F., Koeleman B.P.C. Rank truncated product of P-values, with application to genomewide association scans. Genet. Epidemiol. 2003;25:360–366. doi: 10.1002/gepi.10264. [DOI] [PubMed] [Google Scholar]
- 149.Zaykin D.V., Zhivotovsky L.A., Czika W., Shao S., Wolfinger R.D. Combining p-values in large-scale genomics experiments. Pharm. Stat. 2007;6:217–226. doi: 10.1002/pst.304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Grimes M.L., Lee W.J., van der Maaten L., Shannon P. Wrangling phosphoproteomic data to elucidate cancer signaling pathways. PLoS One. 2013;8 doi: 10.1371/journal.pone.0052884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Palacios-Moreno J., Foltz L., Guo A., Stokes M.P., Kuehn E.D., George L., Comb M., Grimes M.L. Neuroblastoma tyrosine kinase signaling networks involve FYN and LYN in endosomes and lipid rafts. PLoS Comput. Biol. 2015;11 doi: 10.1371/journal.pcbi.1004130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Montojo J., Zuberi K., Rodriguez H., Kazi F., Wright G., Donaldson S.L., Morris Q., Bader G.D. GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics. 2010;26:2927–2928. doi: 10.1093/bioinformatics/btq562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Mostafavi S., Morris Q. Combining many interaction networks to predict gene function and analyze gene lists. Proteomics. 2012;12:1687–1696. doi: 10.1002/pmic.201100607. [DOI] [PubMed] [Google Scholar]
- 154.Huttlin E.L., Ting L., Bruckner R.J., Gebreab F., Gygi M.P., Szpyt J., Tam S., Zarraga G., Colby G., Baltier K., et al. The BioPlex Network: A Systematic Exploration of the Human Interactome. Cell. 2015;162:425–440. doi: 10.1016/j.cell.2015.06.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Huttlin E.L., Bruckner R.J., Navarrete-Perea J., Cannon J.R., Baltier K., Gebreab F., Gygi M.P., Thornock A., Zarraga G., Tam S., et al. Dual proteome-scale networks reveal cell-specific remodeling of the human interactome. Cell. 2021;184:3022–3040.e28. doi: 10.1016/j.cell.2021.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Cerami E.G., Gross B.E., Demir E., Rodchenkov I., Babur O., Anwar N., Schultz N., Bader G.D., Sander C. Pathway Commons, a web resource for biological pathway data. Nucleic Acids Res. 2011;39:D685–D690. doi: 10.1093/nar/gkq1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Hornbeck P.V., Kornhauser J.M., Tkachev S., Zhang B., Skrzypek E., Murray B., Latham V., Sullivan M. PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res. 2012;40:D261–D270. doi: 10.1093/nar/gkr1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Kohl M., Wiese S., Warscheid B. Cytoscape: software for visualization and analysis of biological networks. Methods Mol. Biol. 2011;696:291–303. doi: 10.1007/978-1-60761-987-1_18. [DOI] [PubMed] [Google Scholar]
- 159.Gustavsen J.A., Pai S., Isserlin R., Demchak B., Pico A.R. RCy3: Network biology using Cytoscape from within R. F1000Res. 2019;8:1774. doi: 10.12688/f1000research.20887.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the PRIDE partner131 repository (https://www.ebi.ac.uk/pride/ ) with the dataset identifier PXD050668.
-
•
The CFN (https://www.ndexbio.org/viewer/networks/78c8f03d-d592-11ee-8a40-005056ae23aa) and CCCN (https://www.ndexbio.org/viewer/networks/fa2c707c-d653-11ee-8a40-005056ae23aa) networks from total protein and PTM proteomic data have been deposited in the NDEx Network Data Exchange.132
-
•
snRNA-seq data has been deposited with Mendeley Data (https://data.mendeley.com/datasets/49gxpjrcn9/1).