

Abstract
Depression is a mood disorder characterized by abnormal thoughts. The pathophysiology of depression is related to the deficiency of serotonin (5HT), which is derived from tryptophan (Trp). Mitochondrial dysfunction, oxidative stress, and neuroinflammation are involved in the pathogenesis of depression. Notably, the renin–angiotensin system (RAS) is involved in the pathogenesis of depression, and different findings revealed that angiotensin‐converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) may be effective in depression. However, the underlying mechanism for the role of dysregulated brain RAS‐induced depression remains speculative. Therefore, this review aimed to revise the conceivable role of ACEIs and ARBs and how these agents ameliorate the pathophysiology of depression. Dysregulation of brain RAS triggers the development and progression of depression through the reduction of brain 5HT and expression of brain‐derived neurotrophic factor (BDNF) and the induction of mitochondrial dysfunction, oxidative stress, and neuroinflammation. Therefore, inhibition of central classical RAS by ARBS and ACEIs and activation of non‐classical RAS prevent the development of depression by regulating 5HT, BDNF, mitochondrial dysfunction, oxidative stress, and neuroinflammation.
Keywords: angiotensin, angiotensin receptor blockers, angiotensin‐converting enzyme inhibitors, depression, renin–angiotensin system
Brain renin‐angiotensin system (RAS).
1. INTRODUCTION
Depression is a mental state of low mood and aversion to activity characterized by abnormal mood and thoughts. 1 Depression is associated with low extraversion, and people who have high levels of neuroticism are more likely to experience depressive symptoms and to receive a diagnosis of a depressive disorder. 1 Depression affects 3.5% of the general population. 2 Depression involves a triad of symptoms, including depressed mood, fatigue, and anhedonia, as well as other symptoms such as sleep disorders and autonomic dysfunction‐mediated gastrointestinal disturbances. 3 Depression is more common in women than men and can happen at any age, leading to disruption of life. If untreated, it can be fatal. 4 , 5 In relation to the etiology, depression is classified as endogenous depression due to genetic factors or reactive depression due to external stimuli. 6 The risk factors for development are multifactorial, including stressful life events, 7 borderline personality disorders, 8 chronic use of sedatives and hypnotics, and adverse effects from long‐term use of β‐blockers, antipsychotic drugs, isotretinoin, and antimigraine medications. 9 , 10 Moreover, psychiatric disorders such as bipolar disorders, major depressive disorder (MDD) episodes, and seasonally affected disorders increase the risk of the development of depression. 11 Additionally non‐psychiatric disorders, including nutritional deficiencies, 12 infectious diseases, 13 Addison disease, 14 hypothyroidism, 15 Cushing disease, 16 hyperparathyroidism, 17 Parkinson's disease (PD) 18 and multiple sclerosis, 19 trigger the incidence of depression.
There is evidence for a link between inflammation and depression. Inflammatory processes can be triggered by negative cognitions or their consequences, such as stress, violence, or deprivation. Thus, negative cognitions can cause inflammation that can, in turn, lead to depression. In addition, there is increasing evidence that inflammation can cause depression because of the increase of cytokines, setting the brain into a “sickness mode”. 13 Classical symptoms of being physically sick, such as lethargy, show a large overlap in behaviors that characterize depression. Levels of cytokines tend to increase sharply during the depressive episodes of people with bipolar disorder and drop off during remission. Inflammations that lead to serious depression could be caused by common infections caused by viruses, bacteria, or even parasites. 13
Depressive mood might be a symptom of other mood disorders, including dysthymia and MDD. 1 , 2 The depression term was derived from the Latin word deprimere, which means to press down. This term was used previously by English authors Richard Baker and Samuel Johnson in 1665 and 1753, respectively. 4 Changes in brain neurotransmitter theory were postulated in the 1950s when anti‐tuberculosis medications isoniazid and reserpine were used, causing depressive symptoms. 5 In the late 1960s and early 1970s, depression was categorized as a unipolar and bipolar mood disorders. Unipolar and bipolar mood disorders were primarily described by German psychiatrist Karl Kleist. 3
The pathophysiology of depression is related to the deficiency of serotonin (5HT), which is derived from tryptophan (Trp). 5HT is released into the synaptic cleft to act on the post‐synaptic 5HT and on the presynaptic 5HT1A, which acts as an autoreceptor. 20 Consequently, the increase of 5HT1A autoreceptors and the reduction of 5HT1A heteroreceptors are intricate in the pathogenesis of depression by reducing glutamate. 20 , 21 This effect induces upregulation of N‐methyl‐d‐aspartate (NMDA) receptors and upregulation of amino‐methyl propionic acid (AMPA) receptors, leading to the reduction of brain‐derived neurotrophic factor (BDNF) and inhibition of neuronal plasticity 20 , 21 (Figure 1).
FIGURE 1.
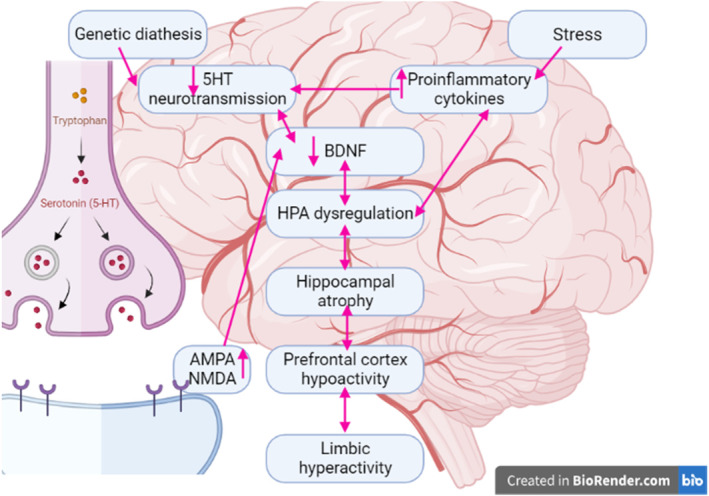
Pathophysiology of depression.
Alongside the well‐known deficiency in 5HT neurotransmission as a pathophysiological correlate of depression, different evidence points to a pivotal role of increased glutamate receptor activation as well. 22 An immune activation, including increased production of pro‐inflammatory cytokines such as interleukin‐2 (IL‐2), interferon‐gamma (INF‐Y), and tumor necrosis factor‐alpha (TNF‐α), activate the tryptophan and 5HT‐degrading enzyme indoleamine 2,3‐dioxygenase (IDO). 23 Depressive states during inflammatory somatic disorders are also associated with increased pro‐inflammatory cytokines and increased consumption of tryptophan via activation of IDO. 22 , 23 Enhanced consumption of 5HT and its precursor tryptophan through IDO activation could well explain the reduced availability of 5HT neurotransmission in depression. An increased activation of IDO and its subsequent enzyme kynurenine monooxygenase by pro‐inflammatory cytokines, moreover, leads to an enhanced production of quinolinic acid, a strong agonist of the NMDA. In inflammatory states, IDO is mainly activated in microglial cells, which preferentially metabolize tryptophan to the NMDA receptor agonist quinolinic acid, whereas astrocytes counteract this metabolism due to the lack of an enzyme for this metabolism, which has been observed to be reduced in depression. 22 , 23 Therefore, the type 1/type 2 immune response imbalance, associated with an astrocyte/microglia imbalance, leads to 5HT deficiency and glutamatergic overproduction. 24 Astrocytes are further strongly involved in the reuptake and metabolic conversion of glutamate. The reduced number of astrocytes could contribute to both a diminished counterregulation of IDO activity in microglia and an altered glutamatergic neurotransmission. 24 Further search for antidepressant agents should take into account anti‐inflammatory drugs; for example, cyclooxygenase‐2 (COX2) inhibitors might exert antidepressant effects by acting on 5HT deficiency, glutamatergic hyperfunction, and the antagonizing neurotoxic effects of quinolinic acid. 25
Despite these findings, the pathophysiology of depression is still elusive, and existing treatments that mainly focus on monoamine alterations are only partially effective. In addition, the effectiveness of antidepressant agents is only 30%. 26 Therefore, focusing on mitochondrial dysfunction, oxidative stress, and neuroinflammation could be a novel avenue in the management of depression. These cellular mechanisms are mainly regulated by the renin–angiotensin system (RAS). 27 Notably, RAS is involved in the pathogenesis of depression, 26 and different studies revealed that angiotensin‐converting enzyme inhibitors (ACEIs) and angiotensin receptor blockers (ARBs) may be effective in depression. 28 Thus, this review aimed to revise the possible role of ACEIs and ARBs and how these agents ameliorate the pathophysiology of depression.
2. A BRIEF OVERVIEW OF RAS
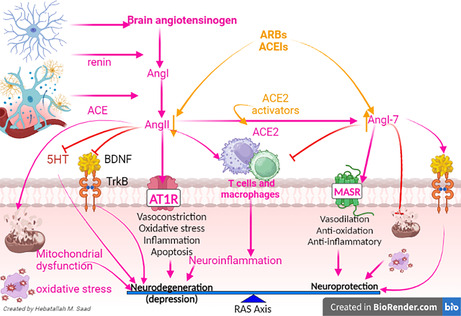
RAS was primarily regarded as a humoral system involved in regulating water and sodium homeostasis and controlling blood pressure. 29 Of note, RAS is phylogenetically one of the important hormone systems intricate in human evolution. 29 RAS consists of various peptides and enzymes. Angiotensinogen from the liver is regarded as the main precursor for the formation of angiotensin I (AngI) by the effect of renin, which is released from the kidney. 30 AngI is converted by the effect of ACE to AngII, which acts on AngII receptors AT1 and AT2. AT1 induces vasoconstriction and pro‐inflammatory effects, though AT2 mediates the effect of vasodilation and anti‐inflammatory effects of AngII. Besides, ACE2 promotes the conversion of AngI to Ang1‐9 and AngII to Ang1‐7, which, via activation of the MAS receptor, induces an anti‐inflammatory effect. 31
Furthermore, in addition to its systemic effects, RAS can produce local and paracrine effects. 32 Both systemic and local RAS act mutually in different tissues. 32 Importantly, the upregulation of AngII and activation of AT1 trigger the development of oxidative stress and inflammation by activating NADPH oxidase and inducing mitochondrial dysfunction. 33 Notably, NADPH oxidase is upregulated during the aging process and aging‐related disorders such as diabetes, hypertension, and atherosclerosis. However, stimulation of AT2 by AngII leads to an opposing effect characterized by inhibition of inflammation and oxidative stress mediated by AT1. 34 AT2 inhibits the release of pro‐inflammatory cytokines, chemokines, and expression of adhesion molecules. 34 Therefore, RAS has a dual inflammatory effect via the AngII/AT1 axis and an anti‐inflammatory effect via the AngII/AT2 axis.
3. BRAIN RAS
The potential role of RAS on the brain was attributed to its local effect in the area engaged with the regulation of water/sodium homeostasis and blood pressure that lacks the blood–brain barrier (BBB). 35 Later on, all components of RAS were recognized in various brain regions, and the AngII level was higher in the brain than in the peripheral circulation. 36 Interestingly, angiotensinogen, pro‐renin, and renin cannot cross the BBB. 37 However, pro‐renin has been detected in the brain, but its origin has not been identified. 38 Neuronal pro‐renin, via activation of the pro‐renin receptor, promotes cleavage of angiotensinogen and activation of RAS. 38 Surprisingly, neuronal pro‐renin/renin may be involved in many neurological functions not related to RAS, like neurogenic hypertension. 38 , 39
Besides, brain angiotensinogen is not correlated with the circulating one. 40 It has been shown that 90% of brain angiotensinogen is mainly produced by astrocytes and little from neurons and glial cells. 41 In addition, components of RAS as well as pro‐renin are also expressed in the basal ganglion, mainly in the nigrostriatal pathway. 42 AT1, AT2, and pro‐renin receptors are highly expressed in glial cells, suggesting the role of RAS in inflammatory and oxidative stress in the brain. 43 In particular, RAS components are expressed in intracellular parts of the neurons. 32 Of note, brain AngI is an inactive peptide and is found at a low level. 44 However, AngII is highly abundant in the brain and intricately involved in different physiological and neuropathological alterations. 45 AngII is further converted to AngIII by the aminopeptidase enzyme; AngIII activates AT1 and AT2 receptors as well as non‐Ang receptors. 46 AngIII is further converted to AngIV by the aminopeptidase enzyme AngIV, which acts on the AT4 receptor and has a neuroprotective effect. 47
Furthermore, ACE, which is the primary enzyme in RAS, is expressed in the astrocytes, choroid plexus, and cerebral vascular endothelium. 48 Brain ACE is mainly present in regions lacking BBB, though it is also expressed in the neurons and glial cells. 48 However, ACE2 is chiefly expressed in brain regions intricately involved in the regulation of blood pressure. 49 ACE2 promotes the expression of neuroprotective Ang1‐7 and Ang1‐9, particularly the ACE2/Ang1‐7/MASR axis, which has anti‐inflammatory and antioxidant effects. 50 In the brain, the AT2 receptor is less expressed than the AT1 receptor and restricted to some brain regions. 51 However, binding of AngII to the AT1 receptor induces oxidative stress and inflammation by activating mitogen‐activated protein kinase (MAPK) and the JNK signaling pathway. 52 These changes may lead to neurotoxicity by activating NMDA receptors and inducing BBB impairment. 52 In addition, the AngII/AT1 receptor complex is translocated into the nucleus, triggering the activation of RAS components. 53 Specifically, mitochondrial AT1 via activation of NADPH oxidase and the respiratory chain promotes the generation of reactive oxygen species (ROS) and the development of oxidative stress. 54 NADPH oxidase is highly expressed in glial cells and neurons. 55 However mitochondrial AT2 produces the opposite effect, as the AngII/AT2 receptor complex enhances the expression of nuclear sirtuin 1 (SIRT1), which has a neuroprotective effect. 56 , 57
It has been shown that over‐activation of brain RAS is associated with cognitive impairment and the development of neurodegenerative diseases. 58 ACE over‐activity in the dopaminergic neurons of the substantia nigra increases vulnerability to damage to these neurons and the development of PD. 59 , 60 Therefore, ACEIs and ARBs are effective against motor dysfunction and cognitive impairment in PD. 56 , 61 Similarly, ACEIs and ARBs are effective against Alzheimer's disease (AD) neuropathology by inhibiting inflammation and oxidative stress‐induced neuronal injury. 62 , 63 Moreover, over‐activity of RAS is correlated with disease severity in different neurodegenerative disorders, including dementia, multiple sclerosis, amyotrophic lateral sclerosis, Huntington's disease, stroke, and traumatic brain injury. 64 , 65
Furthermore, dysregulation of brain RAS is also implicated in different mental and psychiatric disorders, such as bipolar disorder. 66 Bipolar disorder, previously known as manic depression, is a mental disorder characterized by periods of depression and periods of abnormally elevated mood that each last from days to weeks. It has been observed that brain RAS activity is augmented and correlated with the severity of bipolar disorder. 66 Recent studies have linked RAS not only with neuro‐immunological processes but also with psychiatric conditions like mood and anxiety disorders. Anxiety disorders represent a heterogeneous group of illnesses that are characterized by excessive fear and anxiety, hypervigilance, and related behavioral disturbances. Anxiety is one of the most common comorbid disorders with MDD. A large psychiatric cohort study has reported that depression preceded anxiety in 18% of such comorbid cases, while in 57% of the cases, anxiety preceded depression. 67 It has been shown that anxiety associated with depression often leads to reduced responses and decreased compliance with depression pharmacotherapy. 68 Of note, increased Ang II level is linked with depression, anxiety, hyperactivity of the HPA axis, and stress. 69 Moreover, hyperactivation of AT1 receptors is associated with promoting anxiety‐like behaviors in the brain. Deletion of AT1 receptors from the paraventricular nucleus (PVN) reduced anxiety‐like behaviors in mice. 70
Clinical studies show associations between RAS and mood disorders. Animal studies on mood disorder models, either depression or mania, were focused on the reversal of behavioral and/or cognitive symptoms through the inhibition of RAS components like the ACE, AT1, or Mas receptors. 71 ACE polymorphisms are linked to mood disorders and suicidal behavior. Hypertension was associated with neurocognitive deficits in mood disorders, which were reversed with RAS inhibition. Low levels of RAS components and mood symptoms improved with ACE inhibitors or AT1 blockers were also observed in mood disorders. 71 Epidemiological, genetic, and biochemical findings hypothesize the possible relationship between RAS and suicide, as the RAS is involved in the neurobiology of suicide, 72 although the exact mechanisms underlying this involvement are still unidentified. On the other hand, some epidemiological studies have raised alarms about conceivable associations between the use of medications targeting the RAS and an increased risk of suicide. In particular, the association between the use of ARBs and suicide has been discussed not only in the specialized literature but also in the mass media due to the potential implications and the widespread use of these medications in the treatment of hypertension and other cardiovascular conditions. 73 , 74 Moreover, the central RAS affects inflammation, glutamate, dopamine, gamma‐aminobutyric acid (GABA), and peroxisome proliferator‐activated receptor (PPAR)‐γ, all of which are associated with schizophrenia etiology. In addition, it has been demonstrated the therapeutic potential of RAS modulators, especially ARBs, as adjunctive therapy to the currently available antipsychotic medications for schizophrenia treatment. 75 With a greater understanding of how RAS inhibition directly modulates neurotransmitter balance in the brain, it is possible that compounds with RAS‐inhibiting properties could be used to optimize physiological levels of glutamate, dopamine, and GABA and the balance among the three neurotransmitters, analogously to how antipsychotic medications mediate the dopaminergic pathways. It can be hoped that a novel approach based on this concept, such as adjunctive telmisartan therapy, may offer practical interventional strategies to address currently unmet therapeutic needs in patients with schizophrenia, especially those with treatment‐resistant schizophrenia. A case–control study that involved 25 patients with schizophrenia and 20 healthy controls revealed that plasma levels of ACE were reduced in schizophrenic patients compared to healthy controls; no significant differences were found regarding ACE2, Ang‐1–7, and Ang II levels. There were no associations between the measured molecules and clinical parameters, 76 suggesting that the RAS is involved in the pathophysiology of schizophrenia. These findings indicated that brain RAS is implicated in the pathogenesis of mental and psychiatric disorders.
Collectively, brain RAS has two arms: the harmful one is mediated by the AngII/AT1 receptor, and the protective arm is mediated by the AngII/AT2 receptor and the ACE2/Ang1‐7/MASR axis (Figure 2).
FIGURE 2.
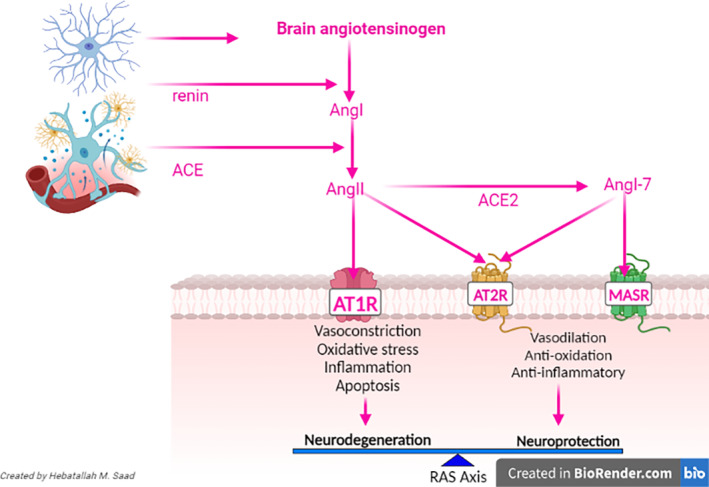
Brian renin–angiotensin system.
4. HYPERTENSION AND DEPRESSION
It has been shown that depression increases the risk of hypertension, and the high prevalence of depression among hypertensive patients suggests a potential association between them. 77 One possible link between depression and hypertension is sympathetic over‐activity. 77 Besides, the use of antidepressant agents may interfere with blood pressure control by inducing circadian alteration in the blood pressure. 78 A cross‐sectional, population‐based study that involved 5000 subjects with hypertension showed that subjects with unaware and uncontrolled hypertension were inversely correlated with depression risk, though controlled hypertension was positive for depression risk. 78 This finding suggests that depression risk in hypertensive patients is related to hypertensive treatments, which may affect mood and cognitive functions. Conversely, low adherence to antihypertensive drugs and uncontrolled hypertension increase the risk of depression. 78 However, a large population‐based study revealed that 16.7% of hypertensive patients have 12‐month depressive and anxiety disorders. After adjusting for confounding factors, there was no association between hypertension and 12‐month depressive disorders. 79 These observations proposed a controversial verdict regarding the association between depression and hypertension. A systematic review and meta‐analysis included 41 clinical studies observed that 26.8% of hypertensive patients have depression. 80 This association seems to be similar with other chronic diseases, as depression risk is 20.3% in chronic kidney disease and 19.3% in patients with heart failure. 81 , 82 Furthermore, depressive symptoms are common in hypertensive patients, even without comorbidities. 83 Remarkably, depression augments the risk of the development of hypertension. 84 A meta‐analysis illustrated that depression is regarded as an independent risk factor for the development and progression of hypertension. 84 Depression increases hypertension risk by inducing dysregulation of Ca2+/cylic adenosine monophosphate (cAMP) signaling, which is intricate in the pathogenesis of hypertension. Furthermore, oxidative stress, inflammation, mitochondrial dysfunction, and endoplasmic reticulum stress, which are common in both depression and hypertension, could be the possible links between them. 85 , 86 Therefore, early management of depression and hypertension prevents the development and progression of a pathogenic link between them. One of the most commonly used antihypertensive agents are ARBs and ACEIs, which modulate peripheral and central RAS. Thus, brain RAS is implicated in the pathogenesis of depression, and modulation of this system may ameliorate the development and progression of depression.
5. BRIAN RAS AND DEPRESSION
Neuroimaging has been a powerful tool to map actual changes in the brain structure of depressed patients that might be directly related to their symptoms of depression. 87 Some imaging studies of brain structure have shown smaller hippocampal volume, with the chronicity of depression correlating to a reduction in hippocampal volume. Though the meaning of these findings is unclear, other studies have shown increased amygdala volume. Studies have found reductions in the volume of the frontal cortex, with some studies showing specific reductions in sub‐regions of the frontal cortex, including the orbitofrontal cortex. 87 Findings of an increase in white matter lesions in elderly patients with depression have been replicated and correlated with late‐onset depression, as well as impairments in social and cognitive function. These findings point to alterations in a circuit of brain regions hypothesized to include the frontal cortex, hippocampus, amygdala, striatum, and thalamus, which underlie symptoms of depression. 87 , 88 These structural changes in depression affect connections between certain brain regions, or nuclei.
It has been shown that brain RAS affects these brain regions and nuclei differentially. 89 A smaller hippocampus is associated with gene variants of the RAS. Genetic variants at three AGTR1 SNPs (rs2638363, rs1492103, and rs2675511) were independently associated with accelerated hippocampal volume loss over the 4‐year follow‐up period in the right but not left hemisphere. 89 Intriguingly, these AGTR1 risk alleles also predicted worse episodic memory performance but were not related to other cognitive measures. Risk genetic variants of the RAS may accelerate memory decline in older adults, an effect that may be conferred by accelerated hippocampal volume loss. 89 Molecules involved in this system may hold promise as early therapeutic targets for late‐life neuropsychiatric disorders. In addition, brain RAS regulates the functional and structural integrity of the amygdala. 90
Furthermore, brain RAS integrates the connections among different brain nuclei and regions. AT1 receptors are primarily distributed in areas that control stress responses, such as the median eminence in the hypothalamic PVN, anterior pituitary gland, amygdala, septal nuclei, and hippocampus. 91 Stress results in the formation of Ang II in the thalamus and other parts of the brain that contribute to catecholamine release. 91 As AT1 and AT2 receptors are found in the HPA axis, HPA plays an important role in stress and stress‐related behavior. 92 Activation of corticotropin‐releasing hormone (CRH) gene expression was found via AT1 receptor activation in immobilization‐induced stress. Treatment with ARB (telmisartan, candesartan, and valsartan) or ACE inhibitors reduces CRH‐induced adrenocorticotropic hormone (ACTH) and corticosterone release in spontaneously hypertensive rats. They also decreased pituitary sensitivity to CRH and reduced hypothalamic CRH expression, which ultimately led to a reduction in stress. 92
Taken together, brain RAS regulates the functional activity and structural integrity of different brain regions involved in depression neuropathology. Different preclinical and clinical studies have highlighted the possible role of brain RAS and RAS modulators in the pathogenesis and clinical manifestations of depression.
5.1. Clinical findings
Different clinical studies indicated that over‐activity of brain RAS is linked with the development of mood and depressive symptoms. 93 , 94 The possible role of the RAS in hypertension and emotional disorders is well proven. Evidence points to the relationship between exaggerated RAS activity and depression and anxiety, partly through the induction of neuroinflammation, stress, and oxidative stress. 94 Thus, blocking the RAS affords a theoretical basis for future treatment of anxiety and depression. 94 Hertzman et al. 93 revealed that ACEI lisinopril improves depressive symptoms in patients with MDD. Therefore, targeting brain RAS could reduce neuronal inflammation and oxidative stress. 94 A case–control study that involved 972 diabetic patients with newly diagnosed depression and 972 diabetic patients without depression showed that the use of ACEIs were more effective than other antihypertensive drugs in the reduction of depressive symptoms. 95 In addition, the use of calcium channel blockers and beta blockers in hypertensive patients increased the risk for the development of depression. 95 Analysis of hypertensive patients from the hospital database with follow‐up for 5 years showed those on ACEIs or ARBs had a low risk for mood disorders. 96 It has been shown that ACEIs such as enalopril and captopril reduce the negative emotional effects of hypertension. 97 In particular, different studies highlighted that captopril has an antidepressant effect mainly in hypertensive patients. 98 A previous clinical trial reported that ACEI captopril reduced depressive symptoms in hypertensive patients as compared to other hypertensive agents. 99 Likewise, ACEI enalopril is more effective than beta blockers in ameliorating stressful events in hypertensive patients. 100 Of interest, ACEI candesartan improves depressive symptoms in diabetic patients by regulating the HPA axis. 101 A population‐based study revealed that hypertensive patients on ACEIs or ARBs showed less depressive symptoms compared to other antihypertensive agents. 102 A meta‐analysis and systematic review confirmed that ACEIs and ARBs are more effective than other antihypertensive agents in reducing depressive and anxiety disorders in hypertensive patients. 103 It has been shown that ACEIs and ARBs reduce the use of antidepressant drugs in hypertensive patients with depression. 104 These clinical findings illustrated that ACEIs and ARBs are effective to reduce depressive symptoms in hypertensive patients with depression, though the underlying mechanism of their antidepressant effects was suggested to be mediated by anti‐inflammatory and antidepressant effects.
However, despite these findings, some studies proposed a link between prolonged use of ARBs and an increasing risk of suicide; nevertheless, the underlying mechanism was not fully elucidated. 72 , 73 Thus, preferential use of ACEIs instead of ARBs should be considered, especially in patients with mental disorders. 105 Prolonged use of ARBs triggers a compensatory increase of brain AngII, leading to alteration of neurotransmitters and suicidal thought; nonetheless, this effect was not seen in ACEIs, which reduce AngII. 106 Supporting this notion, ACE gene polymorphism, which increases AngII, is associated with suicide. 106 In addition, higher AngII triggers the release of substance P, which affects the hypothalamic–pituitary–adrenal (HPA) axis, leading to stress and depressive symptoms. 107 In addition, increasing circulating and central AngII in response to ARBs through activation of the AT1 receptor promotes inflammatory changes that induce mood and depressive disorders. 108 These considerations suggest that ACEIs are safer than ARBs in hypertensive patients with a risk history of depression and other mood disorders.
It has been shown that RAS plays a critical role in the pathogenesis of depression. 26 Furthermore, polymorphism and levels of ACE are increased in the Iranian population and increase the risk of depression. 109 A case–control study that involved 191 patients with depression and 104 healthy controls showed that plasma ACE activity was higher in patients with depression as compared to the controls. 109 Different studies highlighted that ACE polymorphisms affect antidepressant response, cognitive function, and suicidal behavior. 110 , 111 In relation to aldosterone, which is activated by AngII, it has been shown that patients with primary hyperaldosteronism experience depressive symptoms. 112 As well, the high salivary concentration of aldosterone is correlated with depression severity in patients with MDD in a sex‐dependent manner. 113 A cohort study on 60 patients with MDD (23 men and 37 women) showed that the salivary concentration of aldosterone was higher in women as compared to men. 113 However, MDD patients with suicidal behavior have low plasma concentrations of aldosterone as compared with suicidal behavior without MDD. 114 Besides, the aldosterone receptor antagonist spironolactone provokes depressive symptoms and reduces the efficacy of antidepressant drugs. 115 These findings indicated a potential controversy regarding the role of aldosterone in depression neuropathology. In addition, ACEIs seem to be more appropriate for the alleviation of depressive symptoms in patients with depression.
5.2. Preclinical findings
Various preclinical studies have highlighted that RAS is augmented in depression. The AngII level and its expression are more intricate in the pathogenesis of depression. Targeting RAS either by gene deletion or by pharmacological inhibition leads to an antidepressant effect. 116 , 117 Deletion of the Ang gene reduces depressive‐like behavior in mice, 116 and the use of ACEI captopril attenuates depression in rats. 117 Many preclinical findings proposed that ARBs lead to antidepressant effects. 118 , 119 The AT1 receptor blocker irbesartan has an antidepressant effect in a depression mouse model. 118 Similarly, the AT1 receptor blocker telmisartan reduces depression in diabetic rats. Besides, ACEIs such as candesartan produce an antidepressant effect in animal model study. 120 The underlying mechanisms for the antidepressant effects of ARBs and ACEIs might be related to the upregulation of ACE2/Ang1‐7/MASR. Different experimental studies have observed that overexpression of ACE2 and Ang1‐7 in transgenic rats promotes antidepressant and antianxiety effects that are reversed by using MASR antagonists. 121 , 122 Supporting this notion, central administration of Ang1‐7 attenuates oxidative stress in the amygdala, and the use of MASR antagonists reverses the antidepressant effect of ACEI captopril in hypertensive rats. 123 , 124
The neuroprotective role of AngII/AT2 and AngII/Ang1‐7/MASR is related to their anti‐inflammatory and vasodilatory effects by inducing the release of nitric oxide (NO). 125 The expression of AngII/AT2 and AngII/Ang1‐7/MASR in the brain is linked to the expression of AngII/AT1 and ACE. 126 ACE2 reduces ACE function by reducing AngII levels; therefore, ACE2 has a dual effect, inhibiting AngII/AT1 and activating AngII/Ang1‐7/MASR in the brain. 127 It has been shown that administration of ACE2 activator diminazen aceturate improves depressive and anxiety symptoms in mice by regulating HPA. 121 MASR is involved in the regulation of emotional disorders, and the use of MASR antagonist A799 increases anxiety behavior in mice, 124 and MASR‐deficient mice experience anxiety and depressive behaviors. 128 Furthermore, the central effects of RAS inhibitors are also mediated through anti‐inflammatory and antioxidant effects. For example, in comparison with the antidepressant fluoxetine, the AT1 receptor blocker irbesartan reduces inflammatory and oxidative stress biomarkers as well as increases the 5HT level in chronic stress‐induced depression in mice. 117 Likewise, ACEIs such as captopril, telmisartan, and candesartan attenuate central inflammation by inhibiting microglia, as evident by reducing inflammatory biomarkers such as tumor necrosis factor‐alpha (TNF‐α). 129 , 130 However, chronic captopril in mice provokes depressive‐like behavior by inhibiting regulatory T cells and induction of AngII‐mediated oxidative stress. 131
Collectively, these verdicts indicated that depression neuropathology is linked with RAS over‐activity, and selective inhibition of AngII/AT1 or activation of AngII/AT2 and AngII/Ang1‐7/MASR could be a therapeutic way for attenuating depression.
6. MECHANISTIC ROLE OF BRAIN RAS IN DEPRESSION
6.1. RAS and 5HT
According to the monoamine theory, 5HT is the main neurotransmitter that is reduced in depression. 132 The 5HT theory was based on the depressogenic activity of reserpine, which depletes 5HT and other monoamines in the presynaptic terminals, and the effects of antidepressants, which increase 5HT in the synaptic cleft. 133 A case–control study showed that subjects with suicidal attempts have low serum 5HT levels. Treatment with selective serotonin reuptake inhibitors (SSRIs) improves depressive symptoms but not serum 5HT levels. 134 A systematic review and meta‐analysis illustrated that brain 5HT is not associated with depression, and long‐term therapy with antidepressants reduced rather than increased 5HT. 135 Even so, 5HT is still implicated in the pathogenesis of depression.
It has been revealed that AngII can inhibit the synthesis and release of 5HT in the hippocampus. 136 In addition, AngII increases the turnover and metabolism of 5HT, as evidenced by an increase in its metabolite 5HIAA. 136 ARBs have antidepressant effects through modulation of 5HT metabolism. 136 Remarkably, AngII at a higher concentration promotes 5HT biosynthesis, though at a lower concentration, AngII inhibits 5HT biosynthesis. 137 However, AngII is negatively associated with 5HT biosynthesis. Back to this concept, many experimental studies confirmed that ARBs and ACEIs increase 5HT in the hippocampus and prefrontal cortex. 138 , 139 , 140 Furthermore, the neuroprotective Ang1‐7, which antagonizes AngII, improves brain 5HT and depressive symptoms in transgenic rats with low angiotensinogen levels. 141 Klempin et al. 142 showed that depletion of brain ACE2 reduces brain 5HT, leading to significant impairment of neurogenic response in mice. Of note, intestinal ACE2 enhances the absorption of tryptophan, which is a precursor of 5HT. 143 ARBs and ACEIs, via activation of the ACE2/Ang1‐7/MASR axis, improve neurogenesis and 5HT biosynthesis. 143 Therefore, dysregulated brain RAS is linked with the development and progression of depression by inhibiting 5HT. Thus, inhibition of the classical RAS pathway by ARBs and ACEIs mitigates brain 5HT biosynthesis and reduces depressive symptoms. As well, ACE2 activators or enhancement of non‐classical RAS pathways by ARBs and ACEIs also improve brain 5HT synthesis and release.
6.2. RAS and BDNF
BDNF is a member of the neurotrophins protein family that is concerned with neuronal injury resistance. 144 BDNF acts mainly on tyrosine kinase receptor B (TrkB) and, to a lesser extent, on the p75NT receptor (p75NTR). 144 BDNF is released from peripheral tissues and the CNS, mostly the hypothalamus, hippocampus, and limbic system. 145 It has been reported that the deregulation of BDNF is linked to the pathophysiology of depression and other mood disorders. 36 BDNF/TrkB signaling is essential for hippocampal long‐term potentiation (LTP) via activation of extracellular signal‐regulated protein kinase (ERK) and mitogen‐activated protein kinase (MAPK). 146 For example, increasing the activity of HPA and reducing estradiol cause dysfunction of the BDNF signaling pathway, which leads to depression. 147
Different preclinical studies established that BDNF expression in the hippocampus and cerebral cortex was reduced in animal model studies. 148 Decrease of BDNF and other neurotrophic factors in the prefrontal cortex and hippocampus and stimulation of neurotrophic factors, which is mediated by antidepressant agents, can attenuate neuronal atrophy. 148 Likewise, chronic administration of the antidepressant duloxetine in rats for 21 days improves BDNF levels. 149 In addition, acute stress episodes in rats treated with antidepressants activate increased BDNF levels. 149 Interestingly, reduction of BDNF expression in specific forebrain regions provokes the development of depression, and upregulation of BDNF expression in these areas is mediated by the action of antidepressant agents. 150 In addition, antidepressant effects mediated by BDNF are differential according to specific brain areas as well as in healthy and depressed animals. 45 Deletion of the BDNF gene in forebrain regions leads to hyperactivity in male mice, but depressive‐like behavior in female mice, 150 suggesting a sex‐dependent effect of BDNF.
Moreover, numerous clinical studies have exposed that BDNF levels are condensed in patients with depression. 151 , 152 , 153 , 154 A postmortem brain analysis of 21 MDD patients and 21 healthy controls showed that BDNF expression in the amygdala was abridged in MDD patients compared to healthy controls. 154 BDNF gene expression and BDNF/TrkB signaling in the anterior cingulate cortex are found in the brains of 51 postmortem patients with MDD compared to 102 healthy controls. 154 Additionally, low BDNF expression is correlated with depression severity. 155 A meta‐analysis that included 11 studies showed that BDNF serum level was reduced in patients with depression, and prolonged use of antidepressant drugs was linked with an increase in BDNF serum level. 155 A recent study observed that training exercise improves depressive symptoms in patients with depression by increasing circulating BDNF serum levels. 156 However, pro‐BDNF, which acts on p75NTR, is augmented in depression. 157 A case–control study on 42 patients with depression and 40 healthy controls revealed that pro‐BDNF and BDNF serum levels were augmented and decreased correspondingly in patients with depression 157 signifying an interruption in the conversion of pro‐BDNF to BDNF. These findings indicated that the BDNF/TrkB axis is extremely downregulated in patients with depression.
Importantly, RAS regulates the expression of the BDNF/TrkB axis in different ways. 158 For example, exaggerated RAS in experimental diabetic rats induces downregulation of the BDNF/TrkB axis, leading to depression. Inhibition of overactive RAS by AT1 receptor antagonist losartan restores BDNF/TrkB activity in astrocytes. 158 Furthermore, it has been observed that AngII reduced BDNF expression by inducing the expression of toll‐like receptor 4 (TLR4) and pro‐inflammatory NF‐κB. 159 Administration of a small non‐antihypertensive dose of AT1 receptor blocker candesartan (0.1 mg/kg) in mice for 15 days inhibits LPS‐induced memory impairment by increasing expression of the BDNF/TrkB axis, which inhibits TLR4 and NF‐κB. 159 Likewise, administration of the AT1 receptor antagonist valsartan promotes neurogenesis via a BDNF‐dependent pathway in mice. 118 Remarkably, valsartan triggers antidepressant effects in mice in a dose‐dependent manner. 118 It has been documented that inhibition of the central ACE and AT1 receptors by captopril and valsartan, respectively, improved cognitive function in rats with experimental AD by increasing BDNF expression. 160 As well, numerous experimental studies showed that activation of the Ang1‐7/MASR axis promotes expression of the BDNF/TrkB axis, thereby reducing cognitive dysfunction and memory impairment. 161 , 162 Notoriously, AngIV has a neuroprotective effect against memory impairment in diabetic rats by increasing the expression of neuronal BDNF. 163 Peripheral administration of AngIV reduces spatial memory impairments by inhibiting hippocampal oxidative stress and activating hippocampal BDNF. 163
These observations propose that dysregulation of central RAS may increase depression neuropathology by reducing the expression and functional activity of the BDNF/TrkB axis. Targeting of central RAS through inhibition of the AngII/AT1 pathway by ACEIs and ARBs or by activating AngII/AT2 and AngII/Ang1‐7/MASR by AT2 and MASR activators may attenuate depression by increasing BDNF (Figure 3).
FIGURE 3.
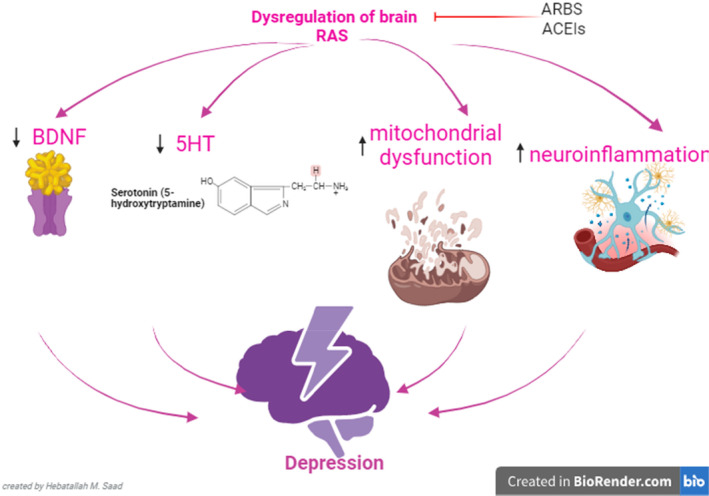
Role of serotonin (5HT) in depression. Dysregulation of brain renin–angiotensin system (RAS) triggers the development and progression of depression through the reduction of brain 5HT and expression of BDNF and the induction of mitochondrial dysfunction, oxidative stress, and neuroinflammation. Therefore, inhibition of central classical RAS by angiotensin‐ACEIs and ARBs and activation of non‐classical RAS prevent the development of depression by regulating 5HT, BDNF, mitochondrial dysfunction, oxidative stress, and neuroinflammation.
6.3. RAS, mitochondrial dysfunction and oxidative stress
It has been shown that ROS are produced constantly by all body tissues that are removed by endogenous antioxidant capacity. 164 When there is an imbalance between ROS generation and antioxidant capacity, oxidative stress is developed. 165 The mitochondria are the major site for the generation of ROS, which affect mitochondrial DNA, leading to more ROS generation. 166 Notably, the brain is extremely vulnerable to the effects of oxidative stress due to advanced metabolic activity, the generation of ROS, and its lower antioxidant capacity. 167 Chronic stressful condition in depression encourages oxidative stress and mitochondrial dysfunction. 168 Depression‐induced oxidative stress is involved in the development of structural and functional changes, including the reduction of the size of the hippocampus and frontal cortex and the dysfunction of synaptic plasticity. 168 Functional brain changes in depression are suggested to develop according to the oxidative stress hypothesis of depressive disorders. 168 , 169 Oxidative stress in mood disorders, including depression, may extend to the periphery, causing distant organ damage. A nested case–control study observed that peripheral biomarkers of oxidative stress were higher in patients with mood disorders compared to controls. 167 Pro‐oxidants and oxidants are advanced in depression, causing an alteration in the permeability of the BBB and the development of systemic oxidative complications. 170 Treatment with antioxidant and antidepressant agents overturned depressive symptoms in experimental diabetes. 171 , 172 Higher brain oxidative stress depletes 5HT, which has antioxidant effects, leading to the progression of depression. 173 , 174 Of interest, antidepressant drugs like fluoxetine and mirtazapine, which inhibit the reuptake of 5HT reduce the severity of oxidative stress. 175 , 176 Thus, depletion of 5HT exacerbates depressive symptoms through the induction of oxidative stress. 176
On the other hand, over‐activation of RAS is associated with the development and progression of mitochondrial dysfunction and oxidative stress. 177 Dysregulation of RAS and associated mitochondrial dysfunction and oxidative stress might be a possible link between obesity and insulin resistance. 177 Unbalanced and activated AngII can cause direct mitochondrial dysfunction and oxidative stress in the skeletal muscles of experimental mice. 178 It has been reported that AngII‐induced vascular injury is mediated through the induction of mitochondrial dysfunction and oxidative stress. 179 Similarly, exaggerated AngII and linked mitochondrial dysfunction and oxidative stress are associated with cognitive impairment and the development of neurodegenerative diseases. 180 However, Ang1‐7 reverses AngII‐induced cerebral endothelial dysfunction by inhibiting NADPH oxidase and the generation of ROS. 181 Therefore, inhibition of AngII/AT1 and activation of AngII/AT2 and Ang1‐7/MASR axes may reduce brain mitochondrial dysfunction and oxidative stress. Ketan et al. 182 proposed that ARBs attenuate brain mitochondrial dysfunction and oxidative stress. A preclinical study conducted by Gupta et al. 183 demonstrated that ARB azilsartan attenuates experimental stroke in rats by inhibiting mitochondrial dysfunction, oxidative stress, and associated neuroinflammation. Notoriously, AngII inhibits transcription of the ACE2 gene, leading to a lowering of ACE2 expression. Therefore, ACEIs and ARBs increase the expression of ACE2 and promote the Ang1‐7/MASR axis, which has a neuroprotective effect. 184 , 185
These findings revealed that dysregulated AngII, linked mitochondrial dysfunction, and oxidative stress may be involved in the pathogenesis of depression. Inhibition of brain RAS by ACEIs and ARBs not only mitigates AngII‐induced neuronal mitochondrial dysfunction and oxidative stress through suppression of AngII/AT1 but also increases the ACE2/Ang1‐7/MASR axis.
6.4. RAS and neuroinflammation
Neuroinflammation is an immune response of the CNS to exogenous infectious agents or endogenous neurological disorders, as in neurodegenerative diseases. 186 Microglia and astrocytes are involved in the development of neuroinflammation; nonetheless, peripheral immune cells that traverse injured BBB can involve the development of neuroinflammation in chronic inflammatory disorders. 165 Neuroinflammation in the acute phase is defended to eradicate the underlying cause, though chronic neuroinflammation may induce neuronal injury, synaptic dysfunction, and exacerbation of brain neuropathology. It has been shown that neuroinflammation affects synaptic plasticity, inhibits hippocampal neurogenesis, and dysregulates HPA. 187 As well, chronic stress triggers microglial activation and the release of pro‐inflammatory cytokines, which inhibit neurogenesis and induce neurodegeneration. 187 Of note, depression is associated with low‐grade inflammatory reactions due to the activation of microglia and the release of pro‐inflammatory cytokines. 188 Besides, exaggerated inflammatory reactions are linked with the progression of depression. 189 A higher level of CRP is regarded as a potential biomarker of depression severity. 190 A clinical study observed that inflammatory biomarkers were increased in both plasma and CSF in patients with depression. 191 Therefore, inhibitors of pro‐inflammatory cytokines such as etanercept have antidepressant effects. 192 Over‐activity of brain RAS is associated with the development and progression of neuroinflammation. 193 The classical RAS pathway (AngII/AT1) activates T cells and macrophages, leading to the release of pro‐inflammatory cytokines and the development of neuroinflammation. However, the non‐classical RAS pathway (ACE2/Ang1‐7/MASR axis) counteracts the activating effect of AngII/AT1 on the immune cells and the progression of neuroinflammation. 193 Impairment of BBB facilitates the entry of peripheral AngII into the CNS that normally does not pass. 193 Central AngII triggers the activation of brain NADPH oxidase, leading to the generation of ROS and the development of oxidative stress, which promotes the progression of neuroinflammation. 194 Targeting neuroinflammation by inhibiting RAS is an essential step, mainly in patients with resistance depression. 195 It has been reported that ACEIs and ARBs can attenuate neuroinflammation by blocking the expression of pro‐inflammatory cytokines. 196 An experiment showed that ARB losartan attenuates LPS‐induced neuroinflammation in mice through suppression of the expression of pro‐inflammatory cytokines. 196 Likewise, ACEI candesartan tempers LPS‐induced neuroinflammation by inhibiting microglial activation and expression of pro‐inflammatory cytokines and regulating neuronal insulin signaling in rats. 197 Interestingly, candesartan at a low dose increases the expression of the anti‐inflammatory cytokines IL‐10 and AT2. 197 Therefore, ARBS and ACEIs seem to have neuroprotective and antidepressant effects by attenuating neuroinflammation. Supporting this idea, experimental studies revealed that ARBS and ACEIs have antidepressant effects. 117 , 198 Losartan and irbesartan inhibit depressive‐like behavior in mice. 117 , 198 In addition, losartan and telmisartan attenuate diabetes‐induced depression in experimental animals. 119 , 158 Furthermore, ACEI captopril mitigates depressive disorders in experimental rats by inhibiting hypothalamic microglia. 199
These observations indicated that inhibition of the classical RAS pathway can reduce depression through attenuation of neuroinflammation.
7. DEPRESSION THERAPEUTIC MODALITIES AND BRAIN RAS
Antidepressant drugs are one of the most commonly used in the management of depression. 200 SSRIs are frequently used in the management of depression compared to tricyclic antidepressants (TCAs) due to their low incidence of adverse effects. 200 , 201 Previous preclinical studies indicated that TCAs had the ability to reduce the function activity of brain AngII. 202 , 203 Genetic polymorphisms in ACE affect the response to antidepressant agents. 202 A clinical trial on patients with depression showed that the G8790A genetic variant of ACE2 is linked with a better response to the efficacy of SSRIs. 204 It has been shown that the action of AngII is reduced by antidepressants, and this signifies the complex interplay of different mechanisms involved in response to therapy. Furthermore, TCA imipramine can attenuate AngII‐induced depressive‐like behaviors by inhibiting hippocampal microglial activation and HPA axis hyperactivation in mice. 205 A cohort study on 200 newly diagnosed depressed patients treated with fluoxetine or sertraline for 12 weeks showed that genetic variants of RAS may influence or be an indicator for a better response to sertraline but not fluoxetine, 206 suggesting a drug‐specific effect on brain RAS. These findings indicated that antidepressant agents may induce antidepressant effects through modulation of brain RAS.
In addition, psychotherapy is also involved in the management of depression. Patients in regional, rural, and remote communities experience perennial difficulties accessing mental health treatments in a timely manner, which contributes to inequitable outcomes. 207 However, the effect of psychotherapy on brain RAS is still not identified. In the treatment of depression, when pharmacotherapy, psychotherapy, and the oldest brain stimulation techniques are deadlocked, the emergence of new therapies is a necessary development. The field of neuromodulation is very broad and controversial for treatment‐resistant depression. Neuromodulatory techniques including magnetic seizure therapy; focal electrically administered seizure therapy, transcranial pulsed electromagnetic fields, transcranial direct current stimulation, epidural cortical stimulation, trigeminal nerve stimulation, transcutaneous vagus nerve stimulation, transcranial focused ultrasound, near‐infrared transcranial radiation, and closed‐loop stimulation could be effective in the management of depression. 208 , 209 Notably, transcranial direct current stimulation increased parasympathetic activity and decreased sympathetic activity by modulating brain RAS, suggesting the importance of this neuromodulatory technique in the management of stress‐related disorders and depression. 210 The underlying mechanisms of transcranial direct current stimulation in depression are related to the regulation of resting membrane potential, spontaneous neuronal firing rates, synaptic strength, cerebral blood flow, and neuronal metabolism. 210 However, the effects of neuromodulatory techniques on brain RAS are not fully elucidated.
Taken together, dysregulation of brain RAS triggers the development and progression of depression through the reduction of brain 5HT and expression of BDNF and the induction of mitochondrial dysfunction, oxidative stress, and neuroinflammation. Therefore, inhibition of central classical RAS by ARBS and ACEIs and activation of non‐classical RAS prevent the development of depression by regulating 5HT, BDNF, mitochondrial dysfunction, oxidative stress, and neuroinflammation (Figure 3).
8. CONCLUSIONS
Depression is a neuropsychiatric disorder characterized by abnormal thoughts and suicidal attempts. The pathophysiology of depression is related to the deficiency of 5HT, which is derived from Trp. Mitochondrial dysfunction, oxidative stress, and neuroinflammation are intricate in the pathogenesis of depression. Particularly, RAS is involved in the pathogenesis of depression, and all components of RAS have been recognized in various brain regions, and the AngII level was higher in the brain than in the peripheral circulation. Brain RAS has two arms: the harmful one is mediated by the AngII/AT1 receptor, and the protective arm is mediated by the AngII/AT2 receptor and the ACE2/Ang1‐7/MASR axis. Depression increases the risk of hypertension and increases the prevalence of depression among hypertensive patients, suggesting a potential association between them. Therefore, early management of depression and hypertension prevents the development and progression of a pathogenic link between them. One of the most commonly used antihypertensive agents are ARBs and ACEIs, which modulate peripheral and central RAS. Different findings revealed that ACEIs and ARBs may be effective in treating depression. Therefore, brain RAS is implicated in the pathogenesis of depression, and modulation of this system may ameliorate the development and progression of depression. Depression neuropathology is linked with RAS over‐activity, and selective inhibition of AngII/AT1 or activation of AngII/AT2 and AngII/Ang1‐7/MASR could be a therapeutic way of attenuating depression. Overall, dysregulation of brain RAS triggers the development and progression of depression through reduction of brain 5HT and expression of BDNF and induction of mitochondrial dysfunction, oxidative stress, and neuroinflammation. Therefore, inhibition of central classical RAS by ARBS and ACEIs and activation of non‐classical RAS prevent the development of depression by regulating 5HT, BDNF, mitochondrial dysfunction, oxidative stress, and neuroinflammation. This review suggests preclinical and clinical studies in this regard.
AUTHOR CONTRIBUTIONS
Naif H. Ali, Hayder M. Al‐Kuraishy, Ali I. Al‐Gareeb, and Ali K. Albuhadily conceptualized the manuscript, wrote, edited, and reviewed the main text, and approved the final edition of the manuscript. Rabab S. Hamad, Athanasios Alexiou, Marios Papadakis, Hebatallah M. Saad, and Gaber El‐Saber Batiha prepared the figures, wrote, corrected, amended, and approved the final edition of the manuscript.
FUNDING INFORMATION
Open Access funding enabled and organized by Projekt DEAL. This work was supported by the University of Witten‐Herdecke Germany.
CONFLICT OF INTEREST STATEMENT
The authors declare no conflict of interest.
ACKNOWLEDGMENTS
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia [Grant No. GRANT4,718]. Open Access funding enabled and organized by Projekt DEAL.
Ali NH, Al‐Kuraishy HM, Al‐Gareeb AI, et al. Role of brain renin–angiotensin system in depression: A new perspective. CNS Neurosci Ther. 2024;30:e14525. doi: 10.1111/cns.14525
The first two authors contributed equally to this work.
Contributor Information
Naif H. Ali, Email: dr.naif1989@gmail.com
Marios Papadakis, Email: marios_papadakis@yahoo.gr, Email: drmariospapadakis@gmail.com.
Hebatallah M. Saad, Email: heba.magdy@mau.edu.eg.
Gaber El‐Saber Batiha, Email: gaberbatiha@gmail.com.
DATA AVAILABILITY STATEMENT
Data sharing are not applicable to this article as no data sets were generated or analyzed during the current study.
REFERENCES
- 1. Gold SM, Köhler‐Forsberg O, Moss‐Morris R, et al. Comorbid depression in medical diseases. Nat Rev Dis Primers. 2020;6(1):69. [DOI] [PubMed] [Google Scholar]
- 2. Kocalevent R‐D, Hinz A, Brähler E. Standardization of the depression screener Patient Health Questionnaire (PHQ‐9) in the general population. Gen Hosp Psychiatry. 2013;35(5):551‐555. [DOI] [PubMed] [Google Scholar]
- 3. Heitmann H, Andlauer TF, Korn T, et al. Fatigue, depression, and pain in multiple sclerosis: how neuroinflammation translates into dysfunctional reward processing and anhedonic symptoms. Mult Scler J. 2022;28(7):1020‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Logan RW, McClung CA. Rhythms of life: circadian disruption and brain disorders across the lifespan. Nat Rev Neurosci. 2019;20(1):49‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Yoldemir T, Yassa M, Atasayan K. Comparison of depression between primary and secondary infertile couples. Gynecol Endocrinol. 2020;36(12):1131‐1135. [DOI] [PubMed] [Google Scholar]
- 6. Yang X‐J, Zhao B‐C, Li J, et al. Serum NLRP3 inflammasome and BDNF: potential biomarkers differentiating reactive and endogenous depression. Front Psych. 2022;13:814828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rippin I, Eldar‐Finkelman H. Mechanisms and therapeutic implications of GSK‐3 in treating neurodegeneration. Cell. 2021;10(2):262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Pan H‐Y, Valapala M. Regulation of autophagy by the glycogen synthase kinase‐3 (GSK‐3) signaling pathway. Int J Mol Sci. 2022;23(3):1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Manduca JD, Thériault R‐K, Perreault ML. Glycogen synthase kinase‐3: the missing link to aberrant circuit function in disorders of cognitive dysfunction? Pharmacol Res. 2020;157:104819. [DOI] [PubMed] [Google Scholar]
- 10. Kaidanovich‐Beilin O, Woodgett JR. GSK‐3: functional insights from cell biology and animal models. Front Mol Neurosci. 2011;4:40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jianing L, Shiqun S, Jia L, et al. NCAPG, mediated by miR‐378a‐3p, regulates cell proliferation, cell cycle progression, and apoptosis of oral squamous cell carcinoma through the GSK‐3β/β‐catenin signaling. Neoplasma. 2021;68(6):1201‐1211. [DOI] [PubMed] [Google Scholar]
- 12. Noori T, Dehpour AR, Sureda A, et al. The role of glycogen synthase kinase 3 beta in multiple sclerosis. Biomed Pharmacother. 2020;132:110874. [DOI] [PubMed] [Google Scholar]
- 13. Lee YH, Choi H‐J, Kim JY, et al. Ginsenoside Rg4 enhances the inductive effects of human dermal papilla spheres on hair growth via the AKT/GSK‐3β/β‐catenin signaling pathway. J Microbiol Biotechnol. 2021;31(7):933‐941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Feng J, Xie L, Yu X, et al. Perilipin 5 ameliorates high‐glucose‐induced podocyte injury via Akt/GSK‐3β/Nrf2‐mediated suppression of apoptosis, oxidative stress, and inflammation. Biochem Biophys Res Commun. 2021;544:22‐30. [DOI] [PubMed] [Google Scholar]
- 15. Oh DH, Park YC, Kim SH. Increased glycogen synthase kinase‐3β mRNA level in the hippocampus of patients with major depression: a study using the Stanley neuropathology consortium integrative database. Psychiatry Investig. 2010;7(3):202‐207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Shapira M, Licht A, Milman A, Pick CG, Shohami E, Eldar‐Finkelman H. Role of glycogen synthase kinase‐3β in early depressive behavior induced by mild traumatic brain injury. Mol Cell Neurosci. 2007;34(4):571‐577. [DOI] [PubMed] [Google Scholar]
- 17. Silva R, Mesquita A, Bessa J, et al. Lithium blocks stress‐induced changes in depressive‐like behavior and hippocampal cell fate: the role of glycogen‐synthase‐kinase‐3β. Neuroscience. 2008;152(3):656‐669. [DOI] [PubMed] [Google Scholar]
- 18. Karege F, Perroud N, Burkhardt S, et al. Alteration in kinase activity but not in protein levels of protein kinase B and glycogen synthase kinase‐3β in ventral prefrontal cortex of depressed suicide victims. Biol Psychiatry. 2007;61(2):240‐245. [DOI] [PubMed] [Google Scholar]
- 19. Tsai S, Liou Y, Hong C, Yu YW, Chen T. Glycogen synthase kinase‐3β gene is associated with antidepressant treatment response in Chinese major depressive disorder. Pharmacogenomics J. 2008;8(6):384‐390. [DOI] [PubMed] [Google Scholar]
- 20. Spellman T, Liston C. Toward circuit mechanisms of pathophysiology in depression. Am J Psychiatry. 2020;177(5):381‐390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Duman RS. Pathophysiology of depression and innovative treatments: remodeling glutamatergic synaptic connections. Dialogues Clin Neurosci. 2022:16(1):11‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dogra S, Conn PJ. Targeting metabotropic glutamate receptors for the treatment of depression and other stress‐related disorders. Neuropharmacology. 2021;196:108687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Savonije K, Meek A, Weaver DF. Indoleamine 2, 3‐dioxygenase as a therapeutic target for Alzheimer's disease and geriatric depression. Brain Sci. 2023;13(6):852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Yuan B, Li W, Liu H, et al. Correlation between immune response and self‐reported depression during convalescence from COVID‐19. Brain Behav Immun. 2020;88:39‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. He Y, Han Y, Liao X, Zou M, Wang Y. Biology of cyclooxygenase‐2: an application in depression therapeutics. Front Psych. 2022;13:1037588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vian J, Pereira C, Chavarria V, et al. The renin–angiotensin system: a possible new target for depression. BMC Med. 2017;15:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hinarejos I, Machuca C, Sancho P, Espinós C. Mitochondrial dysfunction, oxidative stress and neuroinflammation in neurodegeneration with brain iron accumulation (NBIA). Antioxidants. 2020;9(10):1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. van Sloten TT, Souverein PC, Stehouwer CD, Driessen JH. Angiotensin‐converting enzyme inhibitors and angiotensin receptor blockers and risk of depression among older people with hypertension. J Psychopharmacol. 2022;36(5):594‐603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Paz Ocaranza M, Riquelme JA, García L, et al. Counter‐regulatory renin–angiotensin system in cardiovascular disease. Nat Rev Cardiol. 2020;17(2):116‐129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Bhandari S, Mehta S, Khwaja A, et al. Renin–angiotensin system inhibition in advanced chronic kidney disease. N Engl J Med. 2022;387(22):2021‐2032. [DOI] [PubMed] [Google Scholar]
- 31. Verdecchia P, Cavallini C, Spanevello A, Angeli F. The pivotal link between ACE2 deficiency and SARS‐CoV‐2 infection. Eur J Intern Med. 2020;76:14‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Labandeira‐Garcia JL, Valenzuela R, Costa‐Besada MA, Villar‐Cheda B, Rodriguez‐Perez AI. The intracellular renin‐angiotensin system: friend or foe. Some light from the dopaminergic neurons. Prog Neurobiol. 2021;199:101919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chang CC, Cheng HC, Chou WC, et al. Sesamin suppresses angiotensin‐II‐enhanced oxidative stress and hypertrophic markers in H9c2 cells. Environ Toxicol. 2023;38:2165‐2172. [DOI] [PubMed] [Google Scholar]
- 34. Lugnier C, Al‐Kuraishy HM, Rousseau E. PDE4 inhibition as a therapeutic strategy for improvement of pulmonary dysfunctions in Covid‐19 and cigarette smoking. Biochem Pharmacol. 2021;185:114431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Xue B, Zhang Y, Johnson AK. Interactions of the brain renin‐angiotensin‐system (RAS) and inflammation in the sensitization of hypertension. Front Neurosci. 2020;14:650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Abiodun OA, Ola MS. Role of brain renin angiotensin system in neurodegeneration: an update. Saudi J Biol Sci. 2020;27(3):905‐912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Haron S, Kilmister EJ, Davis PF, et al. The renin‐angiotensin system in central nervous system tumors and degenerative diseases. Front Biosci. 2021;26(9):628‐642. [DOI] [PubMed] [Google Scholar]
- 38. Mohsin M, Souza LA, Aliabadi S, et al. Increased (pro) renin receptor expression in the hypertensive human brain. Front Physiol. 2020;11:606811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Worker CJ, Li W, Feng C‐y, et al. The neuronal (pro) renin receptor and astrocyte inflammation in the central regulation of blood pressure and blood glucose in mice fed a high‐fat diet. Am J Physiol‐Endocrinol Metabol. 2020;318(5):E765‐E778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Ren L, Lu X, Danser AJ. Revisiting the brain renin‐angiotensin system—focus on novel therapies. Curr Hypertens Rep. 2019;21:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Li S, Zhou C, Zhu Y, et al. Ferrostatin‐1 alleviates angiotensin II (ang II)‐induced inflammation and ferroptosis in astrocytes. Int Immunopharmacol. 2021;90:107179. [DOI] [PubMed] [Google Scholar]
- 42. Hoffmann N, Peters J. Functions of the (pro) renin receptor (Atp6ap2) at molecular and system levels: pathological implications in hypertension, renal and brain development, inflammation, and fibrosis. Pharmacol Res. 2021;173:105922. [DOI] [PubMed] [Google Scholar]
- 43. Cosarderelioglu C, Nidadavolu LS, George CJ, et al. Brain renin–angiotensin system at the intersect of physical and cognitive frailty. Front Neurosci. 2020;14:586314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen H, Ren M, Li H, et al. Neuroprotection of benzoinum in cerebral ischemia model rats via the ACE‐AngI‐VEGF pathway. Life Sci. 2020;260:118418. [DOI] [PubMed] [Google Scholar]
- 45. Rodrigues AF, Todiras M, Qadri F, Campagnole‐Santos MJ, Alenina N, Bader M. Increased angiotensin II formation in the brain modulates cardiovascular homeostasis and erythropoiesis. Clin Sci. 2021;135(11):1353‐1367. [DOI] [PubMed] [Google Scholar]
- 46. Krause LMH, Kemp BA, Tan ASJ, et al. Renal functional effects of the highly selective AT2R agonist, β‐Pro7 ang III, in normotensive rats. Clin Sci. 2020;134(7):871‐884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ramirez‐Exposito M, Carrera‐Gonzalez M, Martinez‐Martos J. Sex differences exist in brain renin‐angiotensin system‐regulating aminopeptidase activities in transplacental ethyl‐nitrosourea‐induced gliomas. Brain Res Bull. 2021;168:1‐7. [DOI] [PubMed] [Google Scholar]
- 48. Marquié M, Valero S, Castilla‐Marti M, et al. Association between retinal thickness and β‐amyloid brain accumulation in individuals with subjective cognitive decline: Fundació ACE healthy brain initiative. Alzheimers Res Ther. 2020;12:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Cui H, Su S, Cao Y, Ma C, Qiu W. The altered anatomical distribution of ACE2 in the brain with Alzheimer's disease pathology. Front Cell Develop Biol. 2021;9:684874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Barzegar M, Stokes KY, Chernyshev O, Kelley RE, Alexander JS. The role of the ACE2/MasR axis in ischemic stroke: new insights for therapy. Biomedicine. 2021;9(11):1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rodriguez‐Perez AI, Garrido‐Gil P, Pedrosa MA, et al. Angiotensin type 2 receptors: role in aging and neuroinflammation in the substantia nigra. Brain Behav Immun. 2020;87:256‐271. [DOI] [PubMed] [Google Scholar]
- 52. Cui C, Xu P, Li G, et al. Vitamin D receptor activation regulates microglia polarization and oxidative stress in spontaneously hypertensive rats and angiotensin II‐exposed microglial cells: role of renin‐angiotensin system. Redox Biol. 2019;26:101295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Dang R, Yang M, Cui C, et al. Activation of angiotensin‐converting enzyme 2/angiotensin (1–7)/mas receptor axis triggers autophagy and suppresses microglia proinflammatory polarization via forkhead box class O1 signaling. Aging Cell. 2021;20(10):e13480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Koju N, Taleb A, Zhou J, et al. Pharmacological strategies to lower crosstalk between nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and mitochondria. Biomed Pharmacother. 2019;111:1478‐1498. [DOI] [PubMed] [Google Scholar]
- 55. Gage MC, Thippeswamy T. Inhibitors of Src family kinases, inducible nitric oxide synthase, and NADPH oxidase as potential CNS drug targets for neurological diseases. CNS Drugs. 2021;35:1‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Diaz‐Ruiz C, Villar‐Cheda B, Dominguez‐Meijide A, Garrido‐Gil P, Guerra MJ, Labandeira‐Garcia JL. Aging‐related overactivity of the angiotensin/AT1 axis decreases sirtuin 3 levels in the substantia nigra, which induces vulnerability to oxidative stress and neurodegeneration. J Gerontol. 2020;75(3):416‐424. [DOI] [PubMed] [Google Scholar]
- 57. Batiha GE‐S, Al‐Kuraishy HM, Al‐Gareeb AI, Elekhnawy E. SIRT1 pathway in Parkinson's disease: a faraway snapshot but so close. Inflammopharmacology. 2022;31(1):37‐56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Wu H, Sun Q, Yuan S, et al. AT1 receptors: their actions from hypertension to cognitive impairment. Cardiovasc Toxicol. 2022;22(4):311‐325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Pedrosa MA, Labandeira CM, Valenzuela R, et al. AT1 receptor autoantibodies mediate effects of metabolic syndrome on dopaminergic vulnerability. Brain Behav Immun. 2023;108:255‐268. [DOI] [PubMed] [Google Scholar]
- 60. Al‐Kuraishy HM, Al‐Gareeb AI, Elewa YHA, et al. Parkinson's disease risk and hyperhomocysteinemia: the possible link. Cell Mol Neurobiol. 2023;43(6):2743‐2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yang J, Gao Y, Duan Q, et al. Renin‐angiotensin system blockers affect cognitive decline in Parkinson's disease: the PPMI dataset. Parkinsonism Relat Disord. 2022;105:90‐95. [DOI] [PubMed] [Google Scholar]
- 62. Lee HW, Kim S, Jo Y, Kim Y, Ye BS, Yu YM. Neuroprotective effect of angiotensin II receptor blockers on the risk of incident Alzheimer's disease: a nationwide population‐based cohort study. Front Aging Neurosci. 2023;15:1137197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Al‐kuraishy HM, Al‐Gareeb AI, Saad HM, Batiha GE‐S. Benzodiazepines in Alzheimer's disease: beneficial or detrimental effects. Inflammopharmacology. 2022;31:221‐230. doi: 10.1007/s10787-022-01099-4 [DOI] [PubMed] [Google Scholar]
- 64. Bild W, Vasincu A, Rusu R‐N, et al. Impact of the renin‐angiotensin system on the pathogeny and pharmacotherapeutics of neurodegenerative diseases. Biomolecules. 2022;12(10):1429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Szczepanska‐Sadowska E, Wsol A, Cudnoch‐Jedrzejewska A, Czarzasta K, Żera T. Multiple aspects of inappropriate action of renin–angiotensin, vasopressin, and oxytocin systems in neuropsychiatric and neurodegenerative diseases. J Clin Med. 2022;11(4):908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Barbosa IG, Ferreira GC, Andrade Júnior DF, et al. The renin angiotensin system and bipolar disorder: a systematic review. Protein Pept Lett. 2020;27(6):520‐528. [DOI] [PubMed] [Google Scholar]
- 67. Lamers F, van Oppen P, Comijs HC, et al. Comorbidity patterns of anxiety and depressive disorders in a large cohort study: The Netherlands study of depression and anxiety (NESDA). J Clin Psychiatry. 2011;72(3):341‐348. [DOI] [PubMed] [Google Scholar]
- 68. Babaev O, Piletti Chatain C, Krueger‐Burg D. Inhibition in the amygdala anxiety circuitry. Exp Mol Med. 2018;50(4):1‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Tashev R, Ivanova M. Involvement of hippocampal angiotensin 1 receptors in anxiety‐like behaviour of olfactory bulbectomized rats. Pharmacol Rep. 2018;70:847‐852. [DOI] [PubMed] [Google Scholar]
- 70. Wang L, Hiller H, Smith JA, de Kloet AD, Krause EG. Angiotensin type 1a receptors in the paraventricular nucleus of the hypothalamus control cardiovascular reactivity and anxiety‐like behavior in male mice. Physiol Genomics. 2016;48(9):667‐676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mohite S, Sanches M, Teixeira AL. Exploring the evidence implicating the renin‐angiotensin system (RAS) in the physiopathology of mood disorders. Protein Pept Lett. 2020;27(6):449‐455. [DOI] [PubMed] [Google Scholar]
- 72. Sanches M, Teixeira AL. The renin‐angiotensin system, mood, and suicide: are there associations? World J Psychiatry. 2021;11(9):581‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Mamdani M, Gomes T, Greaves S, et al. Association between angiotensin‐converting enzyme inhibitors, angiotensin receptor blockers, and suicide. JAMA Netw Open. 2019;2(10):e1913304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Katz IR. Concerns raised by a study of suicide as an adverse drug effect—replicating findings from real‐world data. JAMA Netw Open. 2019;2(10):e1913284. [DOI] [PubMed] [Google Scholar]
- 75. Oh SJ, Fan X. The possible role of the angiotensin system in the pathophysiology of schizophrenia: implications for pharmacotherapy. CNS Drugs. 2019;33:539‐547. [DOI] [PubMed] [Google Scholar]
- 76. Mohite S, de Campos‐Carli SM, Rocha NP, et al. Lower circulating levels of angiotensin‐converting enzyme (ACE) in patients with schizophrenia. Schizophr Res. 2018;202:50‐54. [DOI] [PubMed] [Google Scholar]
- 77. Scalco AZ, Scalco MZ, Azul JBS, Neto FL. Hypertension and depression. Clinics. 2005;60(3):241‐250. [DOI] [PubMed] [Google Scholar]
- 78. Michal M, Wiltink J, Lackner K, et al. Association of hypertension with depression in the community: results from the Gutenberg Health Study. J Hypertens. 2013;31(5):893‐899. [DOI] [PubMed] [Google Scholar]
- 79. Grimsrud A, Stein DJ, Seedat S, Williams D, Myer L. The association between hypertension and depression and anxiety disorders: results from a nationally‐representative sample of South African adults. PloS One. 2009;4(5):e5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li Z, Li Y, Chen L, Chen P, Hu Y. Prevalence of depression in patients with hypertension: a systematic review and meta‐analysis. Medicine. 2015;94(31):e2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Palmer S, Vecchio M, Craig JC, et al. Prevalence of depression in chronic kidney disease: systematic review and meta‐analysis of observational studies. Kidney Int. 2013;84(1):179‐191. [DOI] [PubMed] [Google Scholar]
- 82. Rutledge T, Reis VA, Linke SE, Greenberg BH, Mills PJ. Depression in heart failure: a meta‐analytic review of prevalence, intervention effects, and associations with clinical outcomes. J Am Coll Cardiol. 2006;48(8):1527‐1537. [DOI] [PubMed] [Google Scholar]
- 83. Rantanen AT, Korkeila JJA, Löyttyniemi ES, Saxén UKM, Korhonen PE. Awareness of hypertension and depressive symptoms: a cross‐sectional study in a primary care population. Scand J Prim Health Care. 2018;36(3):323‐328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Meng L, Chen D, Yang Y, Zheng Y, Hui R. Depression increases the risk of hypertension incidence: a meta‐analysis of prospective cohort studies. J Hypertens. 2012;30(5):842‐851. [DOI] [PubMed] [Google Scholar]
- 85. Zhou Y, Murugan DD, Khan H, Huang Y, Cheang WS. Roles and therapeutic implications of endoplasmic reticulum stress and oxidative stress in cardiovascular diseases. Antioxidants. 2021;10(8):1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Czarny P, Wigner P, Galecki P, Sliwinski T. The interplay between inflammation, oxidative stress, DNA damage, DNA repair and mitochondrial dysfunction in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2018;80:309‐321. [DOI] [PubMed] [Google Scholar]
- 87. Bremner JD. Structural changes in the brain in depression and relationship to symptom recurrence. CNS Spectr. 2002;7(2):129‐139. [DOI] [PubMed] [Google Scholar]
- 88. Drevets WC, Price JL, Furey ML. Brain structural and functional abnormalities in mood disorders: implications for neurocircuitry models of depression. Brain Struct Function. 2008;213:93‐118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Zannas AS, McQuoid DR, Payne ME, et al. Association of gene variants of the renin‐angiotensin system with accelerated hippocampal volume loss and cognitive decline in old age. Am J Psychiatry. 2014;171(11):1214‐1221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Xu T, Zhou X, Jiao G, et al. Modulation of the renin‐angiotensin system inhibits memory advantage for negative emotional material via decreasing hippocampus activation and its coupling with the amygdala. bioRxiv 2021.
- 91. Urmila A, Rashmi P, Nilam G, Subhash B. Recent advances in the endogenous brain renin‐angiotensin system and drugs acting on it. J Renin Angiotensin Aldosterone Syst. 2021;2021:1‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Pushpa V, Shetty P, Suresha R, Jayanthi M, Ashwini V, Vaibhavi P. Evaluation and comparison of anticonvulsant activity of telmisartan and olmesartan in experimentally induced animal models of epilepsy. J Clin Diagn Res. 2014;8(10):HC08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Hertzman M, Adler LW, Arling B, Kern M. Lisinopril may augment antidepressant response. J Clin Psychopharmacol. 2005;25(6):618‐620. [DOI] [PubMed] [Google Scholar]
- 94. Gong S, Deng F. Renin‐angiotensin system: the underlying mechanisms and promising therapeutical target for depression and anxiety. Front Immunol. 2023;13:1053136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Rathmann W, Haastert B, Roseman JM, Giani G. Cardiovascular drug prescriptions and risk of depression in diabetic patients. J Clin Epidemiol. 1999;52(11):1103‐1109. [DOI] [PubMed] [Google Scholar]
- 96. Boal AH, Smith DJ, McCallum L, et al. Monotherapy with major antihypertensive drug classes and risk of hospital admissions for mood disorders. Hypertension. 2016;68(5):1132‐1138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Karwowska‐Polecka W, Halicka D, Jakubów P, Braszko JJ. The effect of enalapril and captopril on emotional processes in hypertensive patients. Psychiatr Pol. 2002;36(4):591‐601. [PubMed] [Google Scholar]
- 98. Gard PR. The role of angiotensin II in cognition and behaviour. Eur J Pharmacol. 2002;438(1–2):1‐14. [DOI] [PubMed] [Google Scholar]
- 99. Hill J, Bulpitt C, Fletcher A. Angiotensin converting enzyme inhibitors and quality of life: the European trial. J Hypertens Suppl. 1985;3(2):S91‐S94. [PubMed] [Google Scholar]
- 100. Edmonds D, Vetter H, Vetter W. Angiotensin converting enzyme inhibitors in the clinic: quality of life. J Hypertens. 1987;5:S31‐S36. [PubMed] [Google Scholar]
- 101. Pavlatou MG, Mastorakos G, Lekakis I, et al. Chronic administration of an angiotensin II receptor antagonist resets the hypothalamic–pituitary–adrenal (HPA) axis and improves the affect of patients with diabetes mellitus type 2: preliminary results. Stress. 2008;11(1):62‐72. [DOI] [PubMed] [Google Scholar]
- 102. Kessing LV, Rytgaard HC, Gerds TA, Berk M, Ekstrøm C, Andersen P. New drug candidates for depression–a nationwide population‐based study. Acta Psychiatr Scand. 2019;139(1):68‐77. [DOI] [PubMed] [Google Scholar]
- 103. Brownstein DJ, Salagre E, Köhler C, et al. Blockade of the angiotensin system improves mental health domain of quality of life: a meta‐analysis of randomized clinical trials. Aust N Z J Psychiatry. 2018;52(1):24‐38. [DOI] [PubMed] [Google Scholar]
- 104. Nasr SJ, Crayton JW, Agarwal B, Wendt B, Kora R. Lower frequency of antidepressant use in patients on renin‐angiotensin‐aldosterone system modifying medications. Cell Mol Neurobiol. 2011;31:615‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Habl G, Zink M, Petroianu G, et al. Increased D‐amino acid oxidase expression in the bilateral hippocampal CA4 of schizophrenic patients: a post‐mortem study. J Neural Transm. 2009;116:1657‐1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Baghai T, Binder E, Schule C, et al. Polymorphisms in the angiotensin‐converting enzyme gene are associated with unipolar depression, ACE activity and hypercortisolism. Mol Psychiatry. 2006;11(11):1003‐1015. [DOI] [PubMed] [Google Scholar]
- 107. Wolf G, Wenzel U, Burns KD, Harris RC, Stahl RA, Thaiss F. Angiotensin II activates nuclear transcription factor‐κB through AT1 and AT2 receptors. Kidney Int. 2002;61(6):1986‐1995. [DOI] [PubMed] [Google Scholar]
- 108. Miklowitz DJ, Portnoff LC, Armstrong CC, et al. Inflammatory cytokines and nuclear factor‐kappa B activation in adolescents with bipolar and major depressive disorders. Psychiatry Res. 2016;241:315‐322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Firouzabadi N, Shafiei M, Bahramali E, Ebrahimi SA, Bakhshandeh H, Tajik N. Association of angiotensin‐converting enzyme (ACE) gene polymorphism with elevated serum ACE activity and major depression in an Iranian population. Psychiatry Res. 2012;200(2–3):336‐342. [DOI] [PubMed] [Google Scholar]
- 110. Zill P, Baghai TC, Schüle C, et al. DNA methylation analysis of the angiotensin converting enzyme (ACE) gene in major depression. PloS One. 2012;7(7):e40479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Sparks DL, Hunsaker JC III, Amouyel P, et al. Angiotensin I‐converting enzyme I/D polymorphism and suicidal behaviors. Am J Med Genet B Neuropsychiatr Genet. 2009;150(2):290‐294. [DOI] [PubMed] [Google Scholar]
- 112. Künzel H. Psychopathological symptoms in patients with primary hyperaldosteronism–possible pathways. Horm Metab Res. 2012;44(3):202‐207. [DOI] [PubMed] [Google Scholar]
- 113. Segeda V, Izakova L, Hlavacova N, Bednarova A, Jezova D. Aldosterone concentrations in saliva reflect the duration and severity of depressive episode in a sex dependent manner. J Psychiatr Res. 2017;91:164‐168. [DOI] [PubMed] [Google Scholar]
- 114. Murck H, Schüssler P, Steiger A. Renin‐angiotensin‐aldosterone system: the forgotten stress hormone system: relationship to depression and sleep. Pharmacopsychiatry. 2012;45(3):83‐95. [DOI] [PubMed] [Google Scholar]
- 115. Moreno‐Santos B, Marchi‐Coelho C, Costa‐Ferreira W, Crestani CC. Angiotensinergic receptors in the medial amygdaloid nucleus differently modulate behavioral responses in the elevated plus‐maze and forced swimming test in rats. Behav Brain Res. 2021;397:112947. [DOI] [PubMed] [Google Scholar]
- 116. Zhang S, He L. Captopril reverses chronic unpredictable mild stress‐induced depression‐like behavior in rats via bradykinin‐B2r signaling pathway. Trop J Pharmac Res. 2022;21(10):2131‐2137. [Google Scholar]
- 117. Ayyub M, Najmi A, Akhtar M. Protective effect of irbesartan an angiotensin (AT1) receptor antagonist in unpredictable chronic mild stress induced depression in mice. Drug Res. 2017;67(1):59‐64. [DOI] [PubMed] [Google Scholar]
- 118. Ping G, Qian W, Song G, Zhaochun S. Valsartan reverses depressive/anxiety‐like behavior and induces hippocampal neurogenesis and expression of BDNF protein in unpredictable chronic mild stress mice. Pharmacol Biochem Behav. 2014;124:5‐12. [DOI] [PubMed] [Google Scholar]
- 119. Aswar U, Chepurwar S, Shintre S, Aswar M. Telmisartan attenuates diabetes induced depression in rats. Pharmacol Rep. 2017;69(2):358‐364. [DOI] [PubMed] [Google Scholar]
- 120. Luo C, Fan H, Li S, Zou Y. Therapeutic of candesartan and music therapy in diabetic retinopathy with depression in rats. Evid Based Complement Alternat Med. 2021;2021:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Wang L, De Kloet AD, Pati D, et al. Increasing brain angiotensin converting enzyme 2 activity decreases anxiety‐like behavior in male mice by activating central mas receptors. Neuropharmacology. 2016;105:114‐123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Kangussu LM, Almeida‐Santos AF, Moreira FA, et al. Reduced anxiety‐like behavior in transgenic rats with chronically overproduction of angiotensin‐(1–7): role of the mas receptor. Behav Brain Res. 2017;331:193‐198. [DOI] [PubMed] [Google Scholar]
- 123. Bild W, Ciobica A. Angiotensin‐(1–7) central administration induces anxiolytic‐like effects in elevated plus maze and decreased oxidative stress in the amygdala. J Affect Disord. 2013;145(2):165‐171. [DOI] [PubMed] [Google Scholar]
- 124. Almeida‐Santos AF, Kangussu LM, Moreira FA, Santos RA, Aguiar DC, Campagnole‐Santos MJ. Anxiolytic‐and antidepressant‐like effects of angiotensin‐(1–7) in hypertensive transgenic (mRen2) 27 rats. Clin Sci. 2016;130(14):1247‐1255. [DOI] [PubMed] [Google Scholar]
- 125. Gironacci MM, Vicario A, Cerezo G, Silva MG. The depressor axis of the renin–angiotensin system and brain disorders: a translational approach. Clin Sci. 2018;132(10):1021‐1038. [DOI] [PubMed] [Google Scholar]
- 126. Fraga‐Silva RA, Pinheiro SVB, Gonçalves ACC, Alenina N, Bader M, Souza Santos RA. The antithrombotic effect of angiotensin‐(1‐7) involves mas‐mediated NO release from platelets. Mol Med. 2008;14:28‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Sriramula S, Cardinale JP, Lazartigues E, Francis J. ACE2 overexpression in the paraventricular nucleus attenuates angiotensin II‐induced hypertension. Cardiovasc Res. 2011;92(3):401‐408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Walther T, Balschun D, Jr‐P V, et al. Sustained long term potentiation and anxiety in mice lacking themas protooncogene. J Biol Chem. 1998;273(19):11867‐11873. [DOI] [PubMed] [Google Scholar]
- 129. Benicky J, Sánchez‐Lemus E, Honda M, et al. Angiotensin II AT1 receptor blockade ameliorates brain inflammation. Neuropsychopharmacology. 2011;36(4):857‐870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Torika N, Asraf K, Danon A, Apte RN, Fleisher‐Berkovich S. Telmisartan modulates glial activation: in vitro and in vivo studies. PloS One. 2016;11(5):e0155823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Park H‐S, Han A, Yeo H‐L, et al. Chronic high dose of captopril induces depressive‐like behaviors in mice: possible mechanism of regulatory T cell in depression. Oncotarget. 2017;8(42):72528‐72543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Liu Y, Zhao J, Guo W. Emotional roles of mono‐aminergic neurotransmitters in major depressive disorder and anxiety disorders. Front Psychol. 2018;9:2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Cowen PJ, Browning M. What has serotonin to do with depression? World Psychiatry. 2015;14(2):158‐160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Saldanha D, Kumar N, Ryali V, Srivastava K, Pawar A. Serum serotonin abnormality in depression. Med J Armed Forces India. 2009;65(2):108‐112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Moncrieff J, Cooper RE, Stockmann T, Amendola S, Hengartner MP, Horowitz MA. The serotonin theory of depression: a systematic umbrella review of the evidence. Mol Psychiatry. 2022;1‐14:3243‐3256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Bali A, Jaggi AS. Angiotensin as stress mediator: role of its receptor and interrelationships among other stress mediators and receptors. Pharmacol Res. 2013;76:49‐57. [DOI] [PubMed] [Google Scholar]
- 137. Tanaka J, Kariya K, Nomura M. Angiotensin II reduces serotonin release in the rat subfornical organ area. Peptides. 2003;24(6):881‐887. [DOI] [PubMed] [Google Scholar]
- 138. Jenkins TA. Effect of angiotensin‐related antihypertensives on brain neurotransmitter levels in rats. Neurosci Lett. 2008;444(2):186‐189. [DOI] [PubMed] [Google Scholar]
- 139. Nocito C, Lubinsky C, Hand M, et al. Centrally acting angiotensin‐converting enzyme inhibitor suppresses type I interferon responses and decreases inflammation in the periphery and the CNS in lupus‐prone mice. Front Immunol. 2020;11:573677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Tanaka J, Miyakubo H, Kawakami A, Hayashi Y, Nomura M. Involvement of NMDA receptor mechanisms in the modulation of serotonin release in the lateral parabrachial nucleus in the rat. Brain Res Bull. 2006;71(1–3):311‐315. [DOI] [PubMed] [Google Scholar]
- 141. Kangussu LM, Almeida‐Santos AF, Bader M, et al. Angiotensin‐(1‐7) attenuates the anxiety and depression‐like behaviors in transgenic rats with low brain angiotensinogen. Behav Brain Res. 2013;257:25‐30. [DOI] [PubMed] [Google Scholar]
- 142. Klempin F, Mosienko V, Matthes S, et al. Depletion of angiotensin‐converting enzyme 2 reduces brain serotonin and impairs the running‐induced neurogenic response. Cell Mol Life Sci. 2018;75:3625‐3634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Singer D, Camargo SM, Ramadan T, et al. Defective intestinal amino acid absorption in Ace2 null mice. Am J Physiol Gastrointest Liver Phys Ther. 2012;303(6):G686‐G695. [DOI] [PubMed] [Google Scholar]
- 144. Wang CS, Kavalali ET, Monteggia LM. BDNF signaling in context: from synaptic regulation to psychiatric disorders. Cell. 2022;185:62‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Notaras M, van den Buuse M. Neurobiology of BDNF in fear memory, sensitivity to stress, and stress‐related disorders. Mol Psychiatry. 2020;25(10):2251‐2274. [DOI] [PubMed] [Google Scholar]
- 146. Panja D, Kenney JW, D'Andrea L, et al. Two‐stage translational control of dentate gyrus LTP consolidation is mediated by sustained BDNF‐TrkB signaling to MNK. Cell Rep. 2014;9(4):1430‐1445. [DOI] [PubMed] [Google Scholar]
- 147. Numakawa T, Richards M, Nakajima S, et al. The role of brain‐derived neurotrophic factor in comorbid depression: possible linkage with steroid hormones, cytokines, and nutrition. Front Psych. 2014;5:136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Duman RS, Monteggia LM. A neurotrophic model for stress‐related mood disorders. Biol Psychiatry. 2006;59(12):1116‐1127. [DOI] [PubMed] [Google Scholar]
- 149. Molteni R, Calabrese F, Cattaneo A, et al. Acute stress responsiveness of the neurotrophin BDNF in the rat hippocampus is modulated by chronic treatment with the antidepressant duloxetine. Neuropsychopharmacology. 2009;34(6):1523‐1532. [DOI] [PubMed] [Google Scholar]
- 150. Monteggia LM, Luikart B, Barrot M, et al. Brain‐derived neurotrophic factor conditional knockouts show gender differences in depression‐related behaviors. Biol Psychiatry. 2007;61(2):187‐197. [DOI] [PubMed] [Google Scholar]
- 151. Dwivedi Y, Rizavi HS, Pandey GN. Antidepressants reverse corticosterone‐mediated decrease in brain‐derived neurotrophic factor expression: differential regulation of specific exons by antidepressants and corticosterone. Neuroscience. 2006;139(3):1017‐1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Guilloux J‐P, Douillard‐Guilloux G, Kota R, et al. Molecular evidence for BDNF‐and GABA‐related dysfunctions in the amygdala of female subjects with major depression. Mol Psychiatry. 2012;17(11):1130‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Thompson DR. Motivational interviewing improves patients' mood and reduces mortality 12 months poststroke. Evid Based Nurs. 2012;15(2):35. [DOI] [PubMed] [Google Scholar]
- 154. Tripp A, Oh H, Guilloux J‐P, Martinowich K, Lewis DA, Sibille E. Brain‐derived neurotrophic factor signaling and subgenual anterior cingulate cortex dysfunction in major depressive disorder. Am J Psychiatry. 2012;169(11):1194‐1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Sen S, Duman R, Sanacora G. Serum brain‐derived neurotrophic factor, depression, and antidepressant medications: meta‐analyses and implications. Biol Psychiatry. 2008;64(6):527‐532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Murawska‐Ciałowicz E, Wiatr M, Ciałowicz M, et al. BDNF impact on biological markers of depression—role of physical exercise and training. Int J Environ Res Public Health. 2021;18(14):7553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Gelle T, Samey RA, Plansont B, et al. BDNF and pro‐BDNF in serum and exosomes in major depression: evolution after antidepressant treatment. Prog Neuropsychopharmacol Biol Psychiatry. 2021;109:110229. [DOI] [PubMed] [Google Scholar]
- 158. Lenart L, Balogh DB, Lenart N, et al. Novel therapeutic potential of angiotensin receptor 1 blockade in a rat model of diabetes‐associated depression parallels altered BDNF signalling. Diabetologia. 2019;62:1501‐1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Goel R, Bhat SA, Hanif K, Nath C, Shukla R. Angiotensin II receptor blockers attenuate lipopolysaccharide‐induced memory impairment by modulation of NF‐κB‐mediated BDNF/CREB expression and apoptosis in spontaneously hypertensive rats. Mol Neurobiol. 2018;55:1725‐1739. [DOI] [PubMed] [Google Scholar]
- 160. Arjmand Abbassi Y, Eidi A. Protective effects of captopril and valsartan on memory function and gene expression of brain‐derived neurotrophic factor (BDNF) in experimental model of Alzheimer's disease in rats. J Adv Med Biomed Res. 2015;23(100):22‐35. [Google Scholar]
- 161. Kamel AS, Abdelkader NF, Abd El‐Rahman SS, Emara M, Zaki HF, Khattab MM. Stimulation of ACE2/ANG (1–7)/mas axis by diminazene ameliorates Alzheimer's disease in the D‐galactose‐ovariectomized rat model: role of PI3K/Akt pathway. Mol Neurobiol. 2018;55:8188‐8202. [DOI] [PubMed] [Google Scholar]
- 162. Wang X‐L, Iwanami J, Min L‐J, et al. Deficiency of angiotensin‐converting enzyme 2 causes deterioration of cognitive function. NPJ Aging Mechan Dis. 2016;2(1):1‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163. Kilic A, Ustunova S, Elibol B, Bulut H, Meral I, Sahin G. Angiotensin IV improves spatial memory in streptozotocin‐induced diabetic rats by reducing oxidative stress and altering BDNF levels. Acta Neurobiol Exp (Wars). 2021;81(2):161‐170. [PubMed] [Google Scholar]
- 164. Al‐Kuraishy HM, Al‐Gareeb AI, Al‐Niemi MS, et al. The prospective effect of allopurinol on the oxidative stress index and endothelial dysfunction in Covid‐19. Inflammation. 2022;1‐17:1651‐1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Alorabi M, Cavalu S, Al‐Kuraishy HM, et al. Pentoxifylline and berberine mitigate diclofenac‐induced acute nephrotoxicity in male rats via modulation of inflammation and oxidative stress. Biomed Pharmacother. 2022;152:113225. [DOI] [PubMed] [Google Scholar]
- 166. Abdul‐Hadi MH, Naji MT, Shams HA, et al. Oxidative stress injury and glucolipotoxicity in type 2 diabetes mellitus: the potential role of metformin and sitagliptin. Biomed Biotechnol Res J. 2020;4(2):166‐172. [Google Scholar]
- 167. Tobe EH. Mitochondrial dysfunction, oxidative stress, and major depressive disorder. Neuropsychiatr Dis Treat. 2013;9:567‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Bhatt S, Nagappa AN, Patil CR. Role of oxidative stress in depression. Drug Discov Today. 2020;25(7):1270‐1276. [DOI] [PubMed] [Google Scholar]
- 169. Salim S. Oxidative stress and psychological disorders. Curr Neuropharmacol. 2014;12(2):140‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Bailey DM, Brugniaux JV, Filipponi T, et al. Exaggerated systemic oxidative‐inflammatory‐nitrosative stress in chronic mountain sickness is associated with cognitive decline and depression. J Physiol. 2019;597(2):611‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Ceretta LB, Reus GZ, Stringari RB, et al. Imipramine treatment reverses depressive‐like behavior in alloxan‐diabetic rats. Diabetes Metab Res Rev. 2012;28(2):139‐144. [DOI] [PubMed] [Google Scholar]
- 172. Reus GZ, Dos Santos MAB, Abelaira HM, et al. Antioxidant treatment ameliorates experimental diabetes‐induced depressive‐like behaviour and reduces oxidative stress in brain and pancreas. Diabetes Metab Res Rev. 2016;32(3):278‐288. [DOI] [PubMed] [Google Scholar]
- 173. Muñoz‐Castañeda JR, Montilla P, Padillo FJ, et al. Role of serotonin in cerebral oxidative stress in rats. Acta Neurobiol Exp (Wars). 2006;66(1):1‐6. [DOI] [PubMed] [Google Scholar]
- 174. Correia AS, Cardoso A, Vale N. Oxidative stress in depression: the link with the stress response, neuroinflammation, serotonin, neurogenesis and synaptic plasticity. Antioxidants. 2023;12(2):470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Ribaudo G, Bortoli M, Witt CE, et al. ROS‐scavenging selenofluoxetine derivatives inhibit in vivo serotonin reuptake. ACS Omega. 2022;7(10):8314‐8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Correia AS, Cardoso A, Vale N. Significant differences in the reversal of cellular stress induced by hydrogen peroxide and corticosterone by the application of mirtazapine or L‐tryptophan. Int J Translat Med. 2022;2(3):482‐505. [Google Scholar]
- 177. Ramalingam L, Menikdiwela K, LeMieux M, et al. The renin angiotensin system, oxidative stress and mitochondrial function in obesity and insulin resistance. Biochim Biophys Acta Mol Basis Dis. 2017;1863(5):1106‐1114. [DOI] [PubMed] [Google Scholar]
- 178. Kadoguchi T, Kinugawa S, Takada S, et al. Angiotensin II can directly induce mitochondrial dysfunction, decrease oxidative fibre number and induce atrophy in mouse hindlimb skeletal muscle. Exp Physiol. 2015;100(3):312‐322. [DOI] [PubMed] [Google Scholar]
- 179. van Thiel BS, van der Pluijm I, te Riet L, Essers J, Danser AJ. The renin–angiotensin system and its involvement in vascular disease. Eur J Pharmacol. 2015;763:3‐14. [DOI] [PubMed] [Google Scholar]
- 180. Sunanda T, Ray B, Mahalakshmi AM, et al. Mitochondria‐endoplasmic reticulum crosstalk in Parkinson's disease: the role of brain renin angiotensin system components. Biomolecules. 2021;11(11):1669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Xiao X, Zhang C, Ma X, et al. Angiotensin‐(1–7) counteracts angiotensin II‐induced dysfunction in cerebral endothelial cells via modulating Nox2/ROS and PI3K/NO pathways. Exp Cell Res. 2015;336(1):58‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182. Ketan Prusty S, Kumar Sahu P, Bhusan SB. Angiotensin mediated oxidative stress and neuroprotective potential of antioxidants and AT1 receptor blockers. Mini Rev Med Chem. 2017;17(6):518‐528. [DOI] [PubMed] [Google Scholar]
- 183. Gupta V, Dhull DK, Joshi J, Kaur S, Kumar A. Neuroprotective potential of azilsartan against cerebral ischemic injury: possible involvement of mitochondrial mechanisms. Neurochem Int. 2020;132:104604. [DOI] [PubMed] [Google Scholar]
- 184. Ferrario CM, Ahmad S, Groban L. Mechanisms by which angiotensin‐receptor blockers increase ACE2 levels. Nat Rev Cardiol. 2020;17(6):378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185. Rabie MA, Abd El Fattah MA, Nassar NN, El‐Abhar HS, Abdallah DM. Angiotensin 1‐7 ameliorates 6‐hydroxydopamine lesions in hemiparkinsonian rats through activation of MAS receptor/PI3K/Akt/BDNF pathway and inhibition of angiotensin II type‐1 receptor/NF‐κB axis. Biochem Pharmacol. 2018;151:126‐134. [DOI] [PubMed] [Google Scholar]
- 186. Batiha GE‐S, Al‐kuraishy HM, Al‐Gareeb AI, et al. Targeting of neuroinflammation by glibenclamide in Covid‐19: old weapon from arsenal. Inflammopharmacology. 2022;31:1‐7. doi: 10.1007/s10787-022-01087-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. Troubat R, Barone P, Leman S, et al. Neuroinflammation and depression: a review. Eur J Neurosci. 2021;53(1):151‐171. [DOI] [PubMed] [Google Scholar]
- 188. Leonard B, Maes M. Mechanistic explanations how cell‐mediated immune activation, inflammation and oxidative and nitrosative stress pathways and their sequels and concomitants play a role in the pathophysiology of unipolar depression. Neurosci Biobehav Rev. 2012;36(2):764‐785. [DOI] [PubMed] [Google Scholar]
- 189. Porter GA, O'Connor JC. Brain‐derived neurotrophic factor and inflammation in depression: pathogenic partners in crime? World J Psychiatry. 2022;12(1):77‐97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Wium‐Andersen MK, Ørsted DD, Nordestgaard BG. Elevated C‐reactive protein, depression, somatic diseases, and all‐cause mortality: a mendelian randomization study. Biol Psychiatry. 2014;76(3):249‐257. [DOI] [PubMed] [Google Scholar]
- 191. Miller AH, Maletic V, Raison CL. Inflammation and its discontents: the role of cytokines in the pathophysiology of major depression. Biol Psychiatry. 2009;65(9):732‐741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192. Kappelmann N, Lewis G, Dantzer R, Jones P, Khandaker G. Antidepressant activity of anti‐cytokine treatment: a systematic review and meta‐analysis of clinical trials of chronic inflammatory conditions. Mol Psychiatry. 2018;23(2):335‐343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193. Hammer A, Stegbauer J, Linker RA. Macrophages in neuroinflammation: role of the renin‐angiotensin‐system. Pflügers Archiv‐Eur J Physiol. 2017;469(3–4):431‐444. [DOI] [PubMed] [Google Scholar]
- 194. Tran S, Kuruppu S, Rajapakse NW. Chronic renin‐angiotensin system activation induced neuroinflammation: common mechanisms underlying hypertension and dementia? J Alzheimers Dis. 2022;85(3):943‐955. [DOI] [PubMed] [Google Scholar]
- 195. Furtado M, Katzman MA. Examining the role of neuroinflammation in major depression. Psychiatry Res. 2015;229(1–2):27‐36. [DOI] [PubMed] [Google Scholar]
- 196. Salmani H, Hosseini M, Baghcheghi Y, Moradi‐Marjaneh R, Mokhtari‐Zaer A. Losartan modulates brain inflammation and improves mood disorders and memory impairment induced by innate immune activation: the role of PPAR‐γ activation. Cytokine. 2020;125:154860. [DOI] [PubMed] [Google Scholar]
- 197. Bhat SA, Goel R, Shukla R, Hanif K. Angiotensin receptor blockade modulates NFκB and STAT3 signaling and inhibits glial activation and neuroinflammation better than angiotensin‐converting enzyme inhibition. Mol Neurobiol. 2016;53:6950‐6967. [DOI] [PubMed] [Google Scholar]
- 198. Singh B, Mourya A, Sah SP, Kumar A. Protective effect of losartan and ramipril against stress induced insulin resistance and related complications: anti‐inflammatory mechanisms. Eur J Pharmacol. 2017;801:54‐61. [DOI] [PubMed] [Google Scholar]
- 199. Xue B, Yu Y, Wei S‐G, et al. Stress‐induced sensitization of angiotensin II hypertension is reversed by blockade of angiotensin‐converting enzyme or tumor necrosis factor‐α. Am J Hypertens. 2019;32(9):909‐917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200. Guerrera CS, Furneri G, Grasso M, et al. Antidepressant drugs and physical activity: a possible synergism in the treatment of major depression? Front Psychol. 2020;11:857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201. Alruwaili NS, Al‐Kuraishy HM, Al‐Gareeb AI, et al. Antidepressants and type 2 diabetes: highways to knowns and unknowns. Diabetol Metab Syndr. 2023;15(1):1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202. Bondy B, Baghai TC, Zill P, et al. Genetic variants in the angiotensin I‐converting‐enzyme (ACE) and angiotensin II receptor (AT1) gene and clinical outcome in depression. Prog Neuropsychopharmacol Biol Psychiatry. 2005;29(6):1094‐1099. [DOI] [PubMed] [Google Scholar]
- 203. Gard PR, Mandy A, Whiting JM, Nickels DP, Meakin AJ. Reduction of responses to angiotensin II by antidepressant drugs. Eur J Pharmacol. 1994;264(3):295‐300. [DOI] [PubMed] [Google Scholar]
- 204. Firouzabadi N, Farshadfar P, Haghnegahdar M, Alavi‐Shoushtari A, Ghanbarinejad V. Impact of ACE2 genetic variant on antidepressant efficacy of SSRIs. Acta Neuropsychiatrica. 2022;34(1):30‐36. [DOI] [PubMed] [Google Scholar]
- 205. Park H‐S, You M‐J, Yang B, et al. Chronically infused angiotensin II induces depressive‐like behavior via microglia activation. Sci Rep. 2020;10(1):22082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206. Bahramali E, Firouzabadi N, Yavarian I, et al. Influence of ACE gene on differential response to sertraline versus fluoxetine in patients with major depression: a randomized controlled trial. Eur J Clin Pharmacol. 2016;72:1059‐1064. [DOI] [PubMed] [Google Scholar]
- 207. Weightman M. Digital psychotherapy as an effective and timely treatment option for depression and anxiety disorders: implications for rural and remote practice. J Int Med Res. 2020;48(6):300060520928686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208. Vlaicu A, Bustuchina VM. New neuromodulation techniques for treatment resistant depression. Int J Psychiatry Clin Pract. 2020;24(2):106‐115. [DOI] [PubMed] [Google Scholar]
- 209. van Rooij SJ, Riva‐Posse P, McDonald WM. The efficacy and safety of neuromodulation treatments in late‐life depression. Curr Treat Options Psychiatry. 2020;7:337‐348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210. Gonçalves EM. Stress prevention by modulation of autonomic nervous system (heart rate variability): a preliminary study using transcranial direct current stimulation. Open Journal of Psychiatry. 2012. doi: 10.4236/ojpsych.2012.22016 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing are not applicable to this article as no data sets were generated or analyzed during the current study.