

Abstract
Proteolysis targeting chimera (PROTAC) is a state-of-the-art technology for ablating undruggable targets. A PROTAC degrader achieves targeted protein degradation (TPD) through the simultaneous binding of a protein of interest (POI) and an E3 ligase to form a ternary complex. Nanofibril-based PROTAC strategy to form a polynary (E3)m:PROTAC:(POI)n complex has not been reported in the TPD field. A recent innovation shows that a POI ligand and E3 ligase ligand don’t have to be within a fused degrader molecule. Instead, they can be recruited to cellular proximity by a self-assembly-driving peptide and click chemistry. The resulting nanofibril can recruit multiple POI and E3 ligase molecules to form a polynary complex as a degradation center. The Nano-PROTAC provides a novel approach for TPD in cancer therapy.
Keywords: PROTAC, targeted protein degradation, cancer, nanofibril, self-assembly
Graphical Abstract
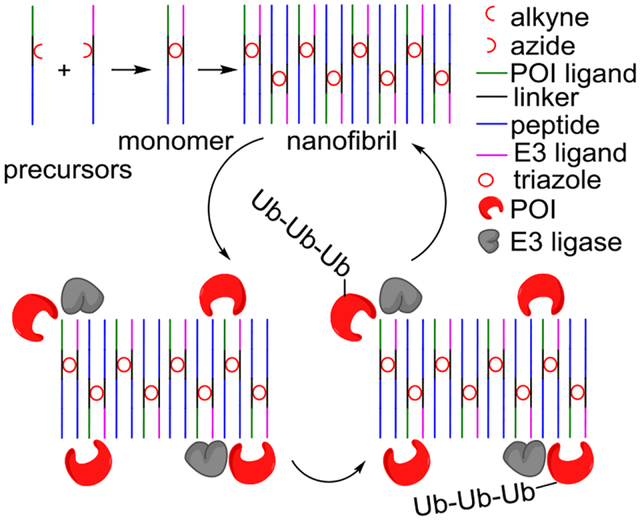
A new PROTAC (proteolysis targeting chimera) method reported by Zhang and co-workers uses self-assembling peptides as carriers of POI (protein of interest) and E3 ligase ligands. Cellular nanofibrils built upon the peptide assembly can serve as degradation centers (Nano-PROTACs) through the formation of polynary POI:Nano-PROTAC:E3 complexes. This strategy thus has the potential to overcome the hook effect of traditional PROTACs.
Proteolysis targeting chimera (PROTAC) and molecular glue-induced targeted protein degradation (TPD) has emerged as an appealing approach to chemically knockdown (degrade) a protein of interest (POI) through ubiquitin-proteosome system (UPS)[1]. Besides, alternative approaches including autophagy-targeting chimera (AUTAC), autophagosome-tethering compound (ATTEC), lysosome-targeting chimera (LYTAC), antibody-based PROTAC (AbTAC), AUTOphagy-TArgeting Chimera (AUTOTAC) using non-UPS systems (lysosome)[1–2] are emerging and have attracted many attentions from both academia and industry.
The development of these lysosome-focused TPD approaches is still in the infancy stage; it is not clear whether targeting lysosome, an important intracellular organelle, would cause damage to the body as a whole[2]. In contrast, two rationally designed PROTACs (ARV-110 and ARV-471) representing the most advanced clinical status of existing protein degraders have been in phase II clinical trials since 2020 providing the first proof-of-concept for the entire TPD field[1]. Encouraged by this advancement, the TPD field has witnessed rapid development in both protein-degradation technologies and heterobifunctional degraders in the past few years. Nevertheless, the PROTAC strategy still faces many layers of challenges including improving the drug properties of PROTAC molecules, reducing PROTAC toxicity, and expanding the repertoire of E3 ubiquitin ligases[2].
An essential step in the degradation efficiency of a PROTAC molecule is the formation of a cooperative ternary POI:PROTAC:E3 complex for UPS-mediated protein degradation.[3] Thus, the appropriate concentration window of PROTAC to form a ternary complex is a key factor in determining the success of a degrader.
There are often cases where high concentrations of PROTACs result in unproductive binary complexes POI:PROTAC or PROTAC:E3 instead of the desired ternary POI:PROTAC:E3 complex. This widely observed phenomenon has been termed the “hook” effect as an intrinsic property of any PROTACs[4]. The hook effect is associated with an inevitable entropic penalty from the amalgamation process at high concentrations. Possible ways to counter the hook effect are increasing the protein-protein interaction[5] or cooperativity of the ternary complex[6].
Extended from previous technology on in vivo peptide self-assembly[7], Zhang et al have reported a novel PROTAC strategy (Figure 1) that utilizes bifunctional nanofibril to achieve multivalent binding with POI and E3 ligase, resulting in improved cooperativity of polynary complex to counteract the hook effect[8]. A key innovation is to conjugate ligands of POI and E3 ligase with self-assembling GNNQQNY peptide (P), part of the prion-like domain of yeast Sup35 protein[9]. Through self-assembly, GNNQQNY peptides carrying ligands of both POI (PPOI) and E3 ligase (Pligase) enable the recruitment of POI and E3 ligase. However, PPOI-Pligase monomer only presents POI and E3 ligands in fixed angles and positions which may not efficiently induce a ubiquitination-degradation cascade. The authors thus have introduced the alkyne, azide, and GHK-CuII to the peptide for in-cell induction of conjugated PPOI-Pligase monomer via glutathione (GSH) triggered click chemistry[10]. It is a smart design strategy to take advantage of the higher GSH level in cancer than normal cells to achieve cell-type specificity. Covalent conjugation of PPOI and Pligase via click chemistry greatly accelerated their self-assembly behaviors to yield PPOI-Pligase monomer which further forms nanofibril assembly. Such fibrils equipped with both POI and E3 ligase ligands formed the chemical basis of the Nano-PROTAC. While it is possible to change ligands for broader applications, several questions arise: can the self-assembling peptide be changed into other peptides?[11] If so, will these precursors still work the same as current Nano-PROTACs? These questions warrant further exploration in subsequent studies.
Figure 1.
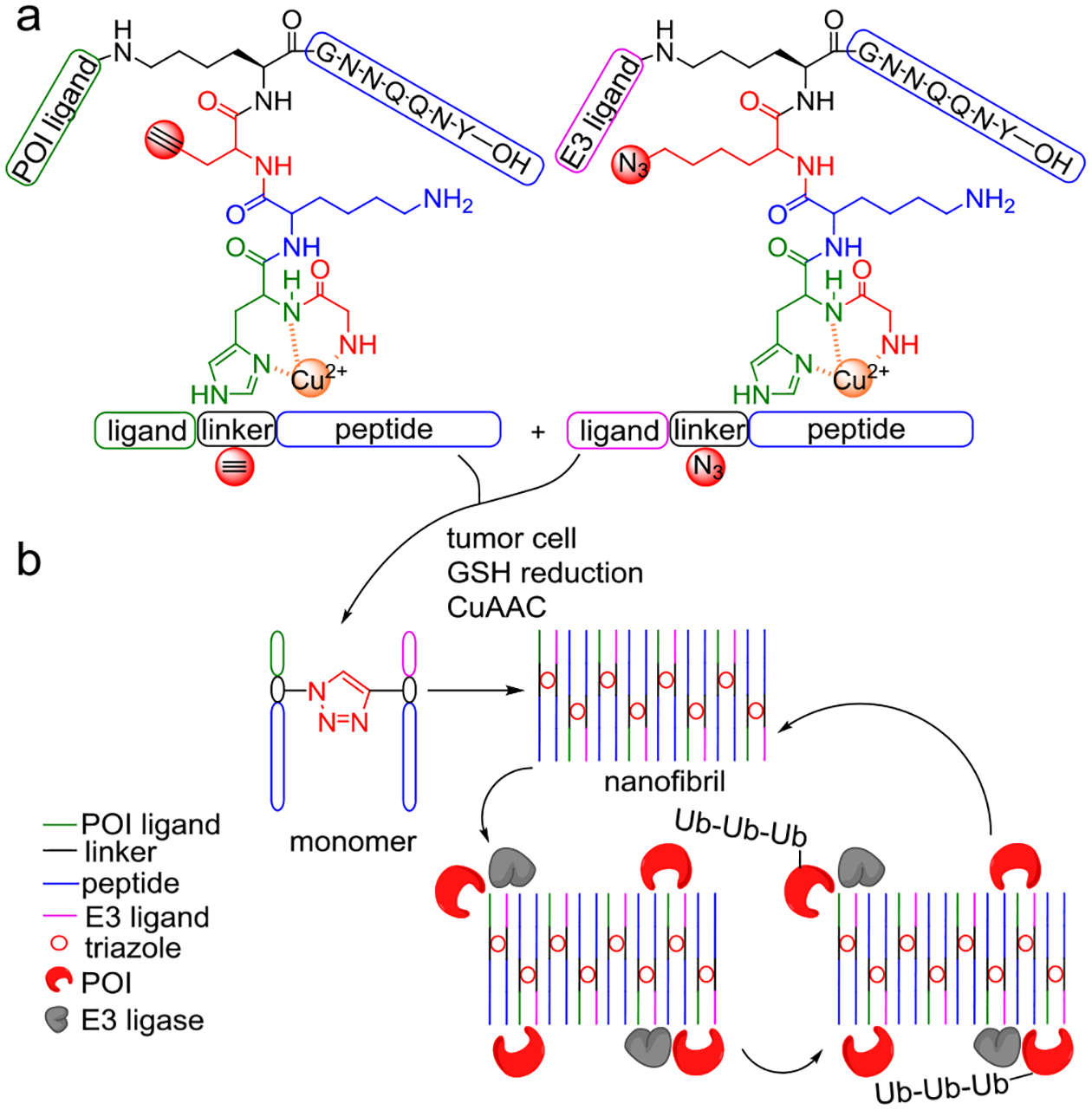
Chemical structures and mechanism of action of the new Nano-PROTACs. (a) Precursors of a Nano-PROTAC consist of three moieties: a ligand (POI or E3 ligase), a linker, and a self-assembling peptide. The linker is additionally functionalized with a shared GHK (Gly-His-Lys) coordinated copper and alkyne (or azide). (b) Mechanism of targeted protein degradation (TPD) achieved by Nano-PROTACs. High levels of glutathione (GSH) in tumor reduces Cu2+ into Cu+ which activates in-cell click chemistry between two precursors. While peptide precursors self-assemble through H-bonds into monomers, the resulting triazole accelerates the formation of monomers and bifunctional nanofibrils. Peptide fibrils induce cellular proximity of E3 ligase and POI for degradation through polynary complexes, thus have great potential to overcome the inherent hook effect during the drug development of existing PROTACs.
After Nano-PROTAC characterization, proof-of-concept data showed that Nano-PROTAC can form cooperative polynary complexes with desired POI and E3 ligase with a calculated ratio of POI:E3 = 2:1. In comparison with unassembled PPOI and Pligase, the binding of POI and ligase to Nano-PROTAC showed higher affinity when measured by microscale thermophoresis (MST). The ability of Nano-PROTAC to recruit POI and E3 ligase in vitro and in-cell provided the functional basis of the Nano-PROTAC. It is encouraging to see that this approach induced the in vitro degradation efficiency of two POIs in several cancer cells. Importantly, the Nano-PROTAC assembly showed great promise in its anti-hook effect with a wide concentration range and long-lasting protein degradation efficiency. The self-assembled Nano-PROTAC can efficiently form polynary complexes with multiple supramolecular interactions at high concentrations which directly contribute to anti-hook effect. Such property makes Nano-PROTAC distinct from recent nanoscale PROTAC advancements including split-and-mix PROTAC (SM-PROTAC)[12], Gold nanocluster PROTAC (GNC-PROTAC)[13], carbon-dot (CD)-based PROTAC (CDTAC)[14] and DNA framework-based PROTAC (DbTAC)[15].
The Nano-PROTACs were evaluated in vivo for their tumor-specific localization and retention using animal models. The injected Nano-PROTAC was able to quickly accumulate at tumor sites within 30 min and retain their localization for up to 72 h, such tumor enrichment and retention was time- and dose-dependent. Interestingly, nanofibrils were observed in bundles inside tumors, confirming the nanofibril formation in vivo, being consistent with in situ observations. Efficacy evaluation was carried out against two POIs in mouse xenograft models. Tumor growth was suppressed by Nano-PROTAC treatment in a dose-dependent manner. POI degradation was confirmed by measuring the POI level in tumor tissues. More importantly, no obvious toxicity was observed for Nano-PROTAC treatments when monitoring the body weights of animals. Histological and biochemical data on major tissues, organs, and blood, further confirmed the safety and biocompatibility of applying Nano-PROTAC to anti-cancer treatments.
This work has demonstrated an unprecedented PROTAC strategy to achieve oncoprotein degradation. GSH-dependent click chemistry accelerated the formation of Nano-PROTAC fibrils while achieving tumor specificity due to the presence of high GSH levels. Ligands of POI and E3 ligase on Nano-PROTAC enable the recruitment of POIs and E3 ligases to form polynary complexes with great potential to overcome the hook effect of traditional PROTACs. Nano-PROTAC demonstrated promising anti-cancer and anti-tumor efficacies, represents a significant progression in the TPD field, and is an emerging therapeutic modality in cancer therapy.
Acknowledgements
ZL thanks a WUSM BMB Seed Grant (PJ000027587). BAG thanks partial support from NIH NS111997 and NSF CHE 2127882 grants. DL thanks the support of Mays Cancer Center Early Career Pilot Award from CCSG (P30 CA054174).
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- [1].Bekes M, Langley DR, Crews CM, Nat Rev Drug Discov 2022, 21, 181–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Zhao L, Zhao J, Zhong K, Tong A, Jia D, Signal Transduct. Target Ther 2022, 7, 113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Gadd MS, Testa A, Lucas X, Chan KH, Chen W, Lamont DJ, Zengerle M, Ciulli A, Nat Chem Biol 2017, 13, 514–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Pettersson M, Crews CM, Drug Discov. Today Technol 2019, 31, 15–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Moreau K, Coen M, Zhang AX, Pachl F, Castaldi MP, Dahl G, Boyd H, Scott C, Newham P, Br. J. Pharmacol 2020, 177, 1709–1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Roy RD, Rosenmund C, Stefan MI, BMC Syst. Biol 2017, 11, 74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Zhang D, Qi GB, Zhao YX, Qiao SL, Yang C, Wang H, Adv. Mater 2015, 27, 6125–6130. [DOI] [PubMed] [Google Scholar]
- [8].Zhang NY, Hou DY, Hu XJ, Liang JX, Wang MD, Song ZZ, Yi L, Wang ZJ, An HW, Xu W, Wang H, Angew. Chem. Int. Ed 2023, 62, e202308049. [DOI] [PubMed] [Google Scholar]
- [9].Haratake M, Takiguchi T, Masuda N, Yoshida S, Fuchigami T, Nakayama M, Colloids Surf B Biointerfaces 2017, 149, 72–79. [DOI] [PubMed] [Google Scholar]
- [10].Ufnalska I, Drew SC, Zhukov I, Szutkowski K, Wawrzyniak UE, Wroblewski W, Fraczyk T, Bal W, Inorg. Chem 2021, 60, 18048–18057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Kim BJ, Yang D, Xu B, Trends Chem 2020, 2, 71–83. [Google Scholar]
- [12].Yang F, Luo Q, Wang Y, Liang H, Wang Y, Hou Z, Wan C, Wang Y, Liu Z, Ye Y, Zhu L, Wu J, Yin F, Li Z, J. Am. Chem. Soc 2023, 145, 7879–7887. [DOI] [PubMed] [Google Scholar]
- [13].Wang Z, Tan M, Su W, Huang W, Zhang J, Jia F, Cao G, Liu X, Song H, Ran H, J. Med. Chem 2023, 66, 6263–6273. [DOI] [PubMed] [Google Scholar]
- [14].Su W, Tan M, Wang Z, Zhang J, Huang W, Song H, Wang X, Ran H, Gao Y, Nie G, Angew. Chem. Int. Ed 2023, 62, e202218128. [DOI] [PubMed] [Google Scholar]
- [15].Zhou L, Yu B, Gao M, Chen R, Li Z, Gu Y, Bian J, Ma Y, Nat. Comm 2023, 14, 4510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.