

Abstract
The development of viral diversity during the course of human immunodeficiency virus type 1 (HIV-1) infection may significantly influence viral pathogenesis. The paradigm for HIV-1 evolution is based primarily on studies of male cohorts in which individuals were presumably infected with a single virus variant of subtype B HIV-1. In this study, we evaluated virus evolution based on sequence information of the V1, V2, and V3 portions of HIV-1 clade A envelope genes obtained from peripheral blood and cervical secretions of three women with genetically heterogeneous viral populations near seroconversion. At the first sample following seroconversion, the number of nonsynonymous substitutions per potential nonsynonymous site (dn) significantly exceeded substitutions at potential synonymous sites (ds) in plasma viral sequences from all individuals. Generally, values of dn remained higher than values of ds as sequences from blood or mucosa evolved. Mutations affected each of the three variable regions of the envelope gene differently; insertions and deletions dominated changes in V1, substitutions involving charged amino acids occurred in V2, and sequential replacement of amino acids over time at a small subset of positions distinguished V3. The relationship among envelope nucleotide sequences obtained from peripheral blood mononuclear cells, plasma, and cervical secretions was evaluated for each individual by both phylogenetic and phenetic analyses. In all subjects, sequences from within each tissue compartment were more closely related to each other than to sequences from other tissues (phylogenetic tissue compartmentalization). At time points after seroconversion in two individuals, there was also greater genetic identity among sequences from the same tissue compartment than among sequences from different tissue compartments (phenetic tissue compartmentalization). Over time, temporal phylogenetic and phenetic structure was detectable in mucosal and plasma viral samples from all three women, suggesting a continual process of migration of one or a few infected cells into each compartment followed by localized expansion and evolution of that population.
Genetic variation in the genome of human immunodeficiency virus type 1 (HIV-1) is a hallmark of viral infection. Diversity in the viral genome is due in part to the high error rate and lack of proofreading activity of the retroviral reverse transcriptase (13, 14, 27) and is influenced by the cellular (11, 21, 26) and immunological (1, 2, 7, 32, 43) environment of each host. In cohorts that have been most intensively studied, viral population complexity is generally low near the time of infection and increases over time (3, 19, 33, 60). The prognostic value of viral diversity at any time point in infection remains to be determined. Some theoretical models predicted that disease would ensue if a threshold of viral diversity was exceeded (47, 48). Clinical data, however, suggest that the development of viral diversity may be correlated with a prolonged asymptomatic phase (18, 37, 42, 71).
Previous studies of viral transmission and subsequent viral evolution have focused primarily on male cohorts infected with clade B HIV-1. The virus population at transmission or seroconversion in these individuals was homogeneous (44, 70, 74, 75). The earliest evidence of viral diversity in individuals infected with a single virus species was 5 to 12 months postseroconversion (18, 19, 71). In contrast, in cohorts of women from East Africa infected with HIV-1 subtypes A, C, and D, the virus population detectable at seroconversion was heterogeneous (28, 57). There have been no studies of HIV-1 evolution in women who harbor a diverse virus population at the time their HIV-1-positive status was determined.
The mutational history of an evolving viral genome is a reflection of two processes: genetic drift and selection for viral variants that are more fit for a given host environment. Studies of viral evolution frequently involve portions of the envelope gene, which encodes the envelope glycoprotein of HIV-1, gp120, because it contains determinants of cell tropism (9, 10, 16, 23, 25, 29, 49, 62) and is a target of the host immune system (2, 31, 32, 36, 77). In the absence of positive selection, substitutions that are silent (synonymous mutations) will proportionally exceed substitutions that lead to amino acid changes (nonsynonymous mutations), since most structural changes in a protein are deleterious. Indeed, in some individuals infected with a homogeneous virus population, substitutions that characterize viral sequences obtained near seroconversion are predominantly silent (36, 53). On the other hand, a high incidence of nonsynonymous site changes in sequences of the envelope gene would imply that forces which favor diversity in this gene are operative. At various times following infection, dn (the number of nonsynonymous substitutions per potential nonsynonymous site) does exceed ds (the number of synonymous substitutions per potential synonymous site) in the envelope gene in some individuals initially infected with a homogeneous virus population (5, 37, 71). Because viral populations in cohorts of African women were heterogeneous at seroconversion, viral evolution may proceed differently than that reported in longitudinal studies of individuals who harbor a single genotype at seroconversion.
Longitudinal studies on viral genetic diversity generally examine viral sequences obtained from cells or plasma of peripheral blood. However, variants that differ from those found in peripheral blood have been found in other tissues (4, 12, 21, 30, 58), including genital secretions (17, 50, 57, 76), at different times during infection. Tissue variants distinct from those in peripheral blood may arise because of selective migration of a subset of infected cells to a tissue or variants may arise independently in response to unique selection pressures that each tissue site may provide. There is evidence to support independent evolution of lung-specific HIV-1 variants in some symptomatic individuals (26). It will be important to determine if viral populations in different sites, such as genital mucosa, evolve in concert with, or independently of, variants in peripheral blood in order to design and evaluate effective antiretroviral therapies.
In this study, we analyzed the evolutionary relationships of envelope sequences in peripheral blood mononuclear cells (PBMCs), plasma, and cervical secretions during the first 1.5 to 2.5 years following seroconversion in three women infected with clade A HIV-1 (56). Two of these women (Q23 and Q47) had heterogeneous virus populations in cervical secretions and PBMCs at seroconversion, and the third (Q45) had viral heterogeneity in PBMCs but not in cervical samples (57). We applied phylogenetic and phenetic methods to gain insight into the dynamic events that characterized the period of asymptomatic infection in these individuals. Our results demonstrate that substitutions resulting in amino acid changes are dominant at seroconversion in these individuals and that, at any point in time, different tissues of women infected with clade A HIV-1 may harbor phylogenetically and phenetically distinct viral variants.
MATERIALS AND METHODS
Cohort information and samples.
The cohort for this study and the methods of sample collection have been previously described (57). Briefly, women were participants in an HIV-1 seroincidence study in Mombasa, Kenya (41). Blood samples and cervical swab samples were collected approximately 1 week following seroconversion and at 3- to 6-month intervals thereafter. All times in this report are referenced to the last negative serology (post-negative serology [PNS] time point) for each subject. None of the women exhibited clinical signs referable to their HIV-1 infection, and none received antiretroviral treatment during this study. Viral load in plasma samples was determined by a competitive PCR method as previously described (35, 45) and is shown in Table 1.
TABLE 1.
Summary of sample collection dates, plasma viral burden, and number of clones sequenced from each tissue for three women infected with clade A HIV-1
| Source of tissue | Date (mo/day/yr) | Particles/ ml | No. of clones from:
|
Total | ||
|---|---|---|---|---|---|---|
| Cervical swabs | PBMCs | Plasma viral RNA | ||||
| Q23 | 7/22/93 | 5,410,000 | 9 (5a) | 10 (7) | 12 (8) | |
| 11/09/93 | 574,320 | 0 | 12 (6) | 0 | ||
| 12/20/93 | 2,183,640 | 0 | 8 (6) | 11 (6) | ||
| 6/13/94 | 1,021,320 | 6 (4) | 12 (8) | 12 (5) | ||
| 11/25/94 | 1,774,920 | 9 (4) | 11 (5) | 12 (2) | ||
| 2/22/95 | 739,920 | 6 (4) | 9 (5) | 7 (4) | ||
| 6/06/95 | NDb | 0 | 11 (5) | 0 | ||
| 9/08/95 | ND | 8 (5) | 11 (5) | 9 (4) | ||
| Total | 38 | 84 | 63 | 185 | ||
| Q47 | 2/01/94 | 77,550 | 8 (6) | 12 (3) | 7 (4) | |
| 7/19/94 | 30,250 | 0 | 11 (5) | 0 | ||
| 11/08/94 | 126,280 | 10 (2) | 10 (5) | 4 (3) | ||
| 7/19/95 | ND | 5 (2) | 10 (6) | 5 (3) | ||
| Total | 23 | 43 | 16 | 82 | ||
| Q45 | 9/30/93 | 18,120 | 5 (3) | 3 (3) | 8 (3) | |
| 1/06/94 | 9,984 | 0 | 4 (3) | 10 (4) | ||
| 4/27/94 | ND | 8 (3) | 0 | 0 | ||
| 8/03/94 | 12,000 | 0 | 13 (6) | 9 (5) | ||
| 10/26/94 | 3,708 | 3 (1) | 9 (3) | 10 (3) | ||
| 2/01/95 | 1,702 | 0 | 6 (4) | 0 | ||
| Total | 16 | 35 | 37 | 88 | ||
Number of PCRs from which the clones were derived.
ND, not done.
Envelope gene cloning.
A 1.2-kb fragment that encompassed the majority of the HIV-1 surface glycoprotein-coding region was amplified by nested PCR from PBMCs and cervical secretions as previously described (57). Plasma viral RNA was isolated (6), and cDNA was prepared by using primer env 12 (CCTGGTGGGTGCTACTCCTA). Plasma viral cDNA was amplified under the same conditions as used for proviral DNA (57). For all samples, multiple PCRs were performed with serial dilutions of template. In most cases, envelope gene fragments could not be amplified from cervical swab samples if lysates were diluted. Fragments from positive PCRs were cloned into either M13r19 or pCRII (Invitrogen Corporation, San Diego, Calif.), and the V1, V2, and V3 domains were sequenced by the Sanger dideoxy-chain termination method (61).
Sequence alignment.
The nucleotide regions defining the V1, V2, and V3 loops of sequences from PBMC, plasma, and cervical swab samples from an individual were aligned to each other manually. Regions of ambiguous alignment were removed. If identical sequences were obtained from a single PCR, only one representative sequence was included in the final data set to avoid bias introduced from resampling the same template. The difference between values obtained for distance analysis using a representative data set with all sequences from Q23 versus the same data set with redundant sequences removed was, in general, less than 0.1%, with a maximum difference of 0.3% observed for one data point. The number of clones analyzed in the final data set for each individual and the number of PCRs from which they were derived are shown in Table 1.
Nucleotide distance analysis.
Pairwise nucleotide distances were calculated by the Kimura two-parameter model in MEGA (34), using the pairwise-deletion option. For each sample, ds or dn was computed in MEGA (34) with the Jukes-Cantor correction as described elsewhere (46).
Phylogenetic tree construction.
For each individual, phylogenetic trees were constructed by the neighbor-joining method using a Kimura two-parameter distance matrix and a transition-to-transversion ratio of 2. Analyses were performed with DNADIST and NEIGHBOR, part of the PHYLIP suite of programs (22). Bootstrap values were determined from 1,000 bootstrap resamplings of the original data by using SEQBOOT (22) and jumbled sequence addition order during tree building.
Phylogenetic compartmental structure analysis.
Phylogenetic evaluation of sequence data is used to determine ancestral relationships among sequences. We used a cladistic method (64, 65) to determine if HIV envelope gene sequences from within any one compartment (i.e., PBMCs, plasma, or cervical secretions) shared more common ancestry than to sequences derived from different compartments. The null hypothesis, in this case, would accept that assemblages of sequences were due to chance events and not to tissue compartmentalization of variants. A phylogenetic tree was constructed for sequences from the entire data set of each individual, and sequences from all three compartments were evaluated at each time point. A new, three-state character was created such that sequences were assigned the same character state based on compartment (e.g., PBMC = 1, plasma RNA = 2, cervical secretion = 3). The number of changes or evolutionary steps required to fit this character to the phylogenetic tree was computed by using MacClade 3.06 (40). If all sequences in a compartment from a time point were monophyletic, the maximum number of character state changes would equal the number of compartments (three, in this case) minus 1 (because there would be one source compartment to colonize the others). If, however, sequences from one compartment were more closely related to sequences from other compartments, the number of changes would increase. The number of changes represents a most parsimonious estimate of the minimum number of putative ancestral viruses (free or cell associated) that migrated from one compartment to another sufficient to explain the pattern of relationships on the phylogenetic tree.
Slatkin and Maddison (64, 65) did not address in depth the issue of phylogenetic uncertainty and how this translates into uncertainty in the estimation of s (the minimum number of migration events). Depending on the quality of the data, the relationships implied by the phylogenetic tree are subject to greater or lesser support. To incorporate this uncertainty into our estimate of s, we calculated the mean and variance of s (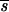 and ss2, respectively) for each of 100 bootstrap trees which were inferred from the nucleotide sequence data set of each individual. To test whether s was greater than expected from a random sample of sequences from a population with no compartmental structure, 100 randomly branching trees with the same number of sequences were generated by using the random joining/splitting option in MacClade 3.06 (40) to obtain
and ss2, respectively) for each of 100 bootstrap trees which were inferred from the nucleotide sequence data set of each individual. To test whether s was greater than expected from a random sample of sequences from a population with no compartmental structure, 100 randomly branching trees with the same number of sequences were generated by using the random joining/splitting option in MacClade 3.06 (40) to obtain 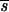 rand and srand2, the mean and variance of s, from the profile of random trees. Thus, we obtained a null distribution of the statistic, s, against which observed values could be compared. Results are shown as the ratio of the average length of the bootstrap trees to the average length of the randomly branching trees for each time point. The standard deviation of the ratio was calculated by standard methods (59).
rand and srand2, the mean and variance of s, from the profile of random trees. Thus, we obtained a null distribution of the statistic, s, against which observed values could be compared. Results are shown as the ratio of the average length of the bootstrap trees to the average length of the randomly branching trees for each time point. The standard deviation of the ratio was calculated by standard methods (59).
Phylogenetic temporal structure analysis.
Temporal relationships among viral sequences from each tissue compartment were evaluated in a manner similar to that described above. In this case, the new character contained character states that represented time points from which the sequences were obtained. Since it is not possible for sequences from a later time point to give rise to sequences obtained earlier, directionality was imposed on the character by constructing a step matrix in MacClade 3.06 (40). The step matrix weighted a particular character change to disallow sequences obtained later from giving rise to sequences obtained earlier in time. The step matrix was also ordered such that a change from time point 1 to time point 2 required one step, a change from time point 1 to time point 3 required two steps, and so on. Bootstrap and random trees were generated as described above and scored for the time state character for each tissue compartment individually up to and including the time point under consideration.
Phenetic compartmental and temporal analysis.
Whereas molecular phylogenetic studies determine ancestral relationships among sequences, phenetic analyses determine the degree of genetic similarity among sequences. To determine if sequences from any compartment (or time point) shared more genetic identity with each other than with sequences from other compartments (or time points), we used Mantel’s test (66, 67), a generalized regression permutation procedure which compares two distance matrices. One distance matrix consists of pairwise Kimura two-parameter distances of sequences from all compartments obtained at a given time point. The second matrix, Mc, is an idealized matrix of the same dimensions such that
Mc(i,j) = {0 if sequence i is from the same compartment as sequence j 1 otherwise
The test statistic is the square of the Pearson correlation coefficient, r2, computed over all pairs of elements, excluding the diagonal, of both matrices. If sequences from each compartment are more similar to other sequences in that compartment than to sequences from different compartments, then r2 will be high. The null distribution was constructed by permuting the rows and columns of the idealized matrix 1,000 times and counting the number of times the value of r2 is exceeded. The hypothesis that there is compartmental phenetic structure is rejected if more than 5% of the permutations exceed r2.
The same procedure was used to test for temporal phenetic structure in each compartment; however, the idealized matrix for temporal structure, Mt, was modified to reflect the ordinal nature of sampling time as follows:
Mt(i,j) = {0 if sequence i is from the same time point as sequence j|k −m| if sequence i is from time point m and sequence j is from time point k
Nucleotide sequence accession numbers.
Sequences have been deposited in GenBank with accession no. AF047979 to AF048685.
RESULTS
Nucleotide substitutions.
The relative magnitude of ds and dn can elucidate the selection pressures that operate on a viral population. A population is defined as being under positive selection if the selection coefficient is greater than 0, which occurs if dn is greater than ds. We determined average dn and ds for sequences obtained from tissue samples at every time point for each subject (Fig. 1). In plasma RNA sequences obtained at seroconversion from all individuals, dn was significantly greater than ds. Similarly, dn was also statistically greater than ds in sequences obtained from PBMC samples from Q23 and Q45 and from cervical swab samples from Q23 at seroconversion. At individual time points after seroconversion, however, dn was significantly greater than ds only for Q45 plasma viral sequences.
FIG. 1.

Average intra-time point ds and dn for sequences from Q23, Q47, and Q45. At each time point, ds and dn were determined for sequences of the V1, V2, and V3 portions of the HIV-1 envelope gene that were obtained from cervical (Cx), plasma RNA (Rn), and PBMC (Pb) samples from each subject. Significant differences between dn and ds are indicated by ∗ (P < 0.05) and ∗∗ (P < 0.01).
To determine if selection was acting on viral populations as they evolved from those detected at seroconversion, we estimated dn and ds for sequences obtained from samples at each time point compared with seroconversion sequences. Throughout the time period examined, dn exceeded ds, on average, for all tissue variants from all individuals (Fig. 2) except for sequences from Q45 cervical swab samples. Accumulation of nonsynonymous and synonymous substitutions, however, occurred at the same rate in Q23 plasma sequences (0.42 and 0.46% per year, respectively). There were insufficient data points for Q47 and Q45 samples to accurately estimate rates. Thus, substitutions involving nonsynonymous sites were dominant during the first two years of viral evolution in all subjects.
FIG. 2.

Average inter-time point frequencies of ds and dn for sequences from Q23, Q47, and Q45. Values of dn and ds were determined by pairwise comparison between sequences from a tissue at each sample point and sequences obtained from that tissue at the seroconversion sample. The average difference between dn and ds, evaluated by t test, was significantly greater than zero for all Q23 tissues (P < 0.001 for all tissue isolates) and Q47 tissues (P < 0.05 for cervical [Cx], P < 0.005 for PBMC [Pb], and P < 0.05 for plasma RNA [Rn] samples) and for Q45 PBMC (P < 0.005) and plasma viral RNA (P < 0.001) sequences.
Phylogenetic and phenetic relationships among tissue variants.
To determine evolutionary relationships among sequences obtained from each individual, a phylogenetic tree was constructed for each subject by neighbor-joining analysis (Fig. 3). Trees representing both Q23 and Q47 sequences showed similar radiating branch patterns, characteristic of rapidly evolving populations (63). The tree representing Q45 sequences clearly showed that two distinct genotypes (4.5% average pairwise difference between the two genotypes) were present at seroconversion and, for PBMC samples, at all subsequent sample points. Only one of these genotypes was detected in Q45 mucosal samples. Sequences from the same date and tissue tended to cluster on trees for all individuals, although there was no bootstrap support greater than 75% for most branches. Therefore, we used a cladistic method described by Slatkin and Maddison (64, 65) to test the hypothesis that sequences obtained from a tissue were more closely related to each other than to sequences from a different tissue. Viral sequences from all three subjects showed significant phylogenetic tissue compartmentalization (Fig. 4). Phylogenetic compartmentalization of tissue variants was statistically significant at all time points sampled for Q47. Statistically significant compartmentalization also occurred for the virus population sampled from Q23 and Q45 but was not evident during the first year of infection. Identical results were obtained if sequences derived only from PBMC and mucosal samples were evaluated (data not shown), with the exception of the 23.5-month PNS sample from Q23 (bootstrap to random tree length ratio = 0.81; standard deviation, 0.22). Thus, we demonstrated that the assemblage of sequences on the phylogenetic trees reflected significant ancestral relationships based on tissue of origin and not chance associations.
FIG. 3.



Phylogenetic trees derived from sequences obtained from Q23, Q47, and Q45. Trees were constructed by the neighbor-joining method. Sequences are represented by diamonds squares, and circles, for clones derived from PBMCs, plasma RNA, and cervical samples, respectively, containing a number that indicates the months PNS that the sample was taken. Branch lengths are drawn to scale. The scale bars represent 0.05 change per average nucleotide position.
FIG. 4.

Phylogenetic and phenetic evaluation of tissue as a character state for Q23, Q47, and Q45. Tree lengths were determined, as described in Materials and Methods, for 100 bootstrap trees and 100 randomly constructed trees. Tree topology for bootstrap trees was based on nucleotide sequence data of the V1, V2, and V3 portions of the envelope gene, but tree lengths were based on character state changes between tissues. The ratio of these tree lengths is plotted for each time point. Error bars indicate 1 standard deviation. Mantel’s test was used to determine phenetic relationships among tissue isolates. Time points at which tissue isolates are phenetically distinct are boxed, and levels of significance are indicated by ∗ (P < 0.05) and ∗∗ (P < 0.01).
Viral sequences are frequently evaluated based on sequence genetic identity (phenetic structure) rather than ancestral affiliation (phylogenetic structure). Although it is often true that samples which exhibit phenetic structure also exhibit phylogenetic structure, the converse is not necessarily true. For example, sequences that cluster on two different branches of a phylogenetic tree may share a most recent common ancestor but not have phenetic structure if the distance separating the branches is great. Time points where tissue sequences from a compartment have significant phenetic structure were determined by using Mantel’s test and are indicated in Fig. 4. Phenetic structure among tissue variants was not apparent at seroconversion in any of the individuals. Significant tissue-specific phenetic structure was detected at all time points after the seroconversion sample from Q47 and transiently for the 9.5-month PNS sample from Q23. Both of the later samples at 23.5 and 29 months PNS from Q23 also showed phenetic structure. In contrast, there was no phenetic tissue affiliation among sequences from Q45 at any time point. Thus, tissue variants developed both phylogenetic and phenetic structure at variable times within the first 2 years of infection in Q23 and Q47. Only phylogenetic, not phenetic, structure was detected for Q45 tissue variants.
Phylogenetic and phenetic relationships of sequences from the same tissue compartment over time.
We applied a variant of the Slatkin and Maddison method (64, 65) to test the hypothesis that within a tissue compartment, sequences obtained at a given time are more closely related to each other than to sequences from that tissue sampled at a different time. This hypothesis of phylogenetic temporal structure would indicate a process of population replacement in each tissue, in which evolutionary lineages that dominate a population at one time are sequentially replaced over time.
Phylogenetic temporal structure arose at different times in viral sequences from PBMCs, plasma, and cervical swabs from all three women (Fig. 5). Throughout the observation period in all three women, mucosal sequences were phylogenetically more closely related to sequences obtained from the same time point than to mucosal sequences derived from an earlier time point. Phylogenetic temporal structure was also observed in sequences obtained from plasma of all individuals, although in Q23 this did not develop until 23.5 months PNS. Similarly, sequences from Q23 and Q47 PBMC demonstrated phylogenetic temporal structure at all time points, but this did not occur in PBMC-derived sequences from Q45 PBMC until 15 months PNS.
FIG. 5.

Phylogenetic evaluation of time as a character state for Q23, Q47, and Q45. Tree lengths were determined, as described in Materials and Methods, for 100 bootstrap trees and 100 randomly constructed trees. Tree topology for bootstrap trees is based on nucleotide sequence data of the V1, V2, and V3 portions of the envelope gene, but tree lengths are based on character state changes between time points for cervical (Cx), plasma RNA (Rn), and PBMC (Pb) samples. The ratio of bootstrap to random tree lengths is plotted for each time point. Error bars indicate 1 standard deviation. Phenetic temporal structure was significant for sequences from all three tissues for Q23 (P < 0.001) and for Q47 and Q45 plasma viral RNA and cervical sequences (P < 0.001 and P < 0.05, respectively).
Phenetic temporal structure was statistically significant for sequences from cervical swabs and plasma from all individuals (P < 0.001 for Q23 and Q47; P < 0.05 for Q45). Only PBMC sequences from Q23 developed detectable phenetic temporal structure (P < 0.001). Thus, in all three women, mucosal variants at each time point were consistently distinct from mucosal samples taken at preceding time points both by phenetic and phylogenetic criteria. The same conclusion applied to sequences obtained from plasma viral RNA, with the exception that there was no support for distinct ancestral lineages (phylogenetic temporal structure) of Q23 plasma virus for the first year of infection. PBMC proviral DNA sequences from the three subjects were the least consistent in showing temporal phylogenetic and phenetic structure.
Qualitative relationships among variants.
The high incidence of nonsynonymous site mutations and the distinct phylogenetic and phenetic relationships of envelope gene sequences from each tissue of these three individuals suggested that the primary structure of the glycoprotein encoded by the gene would change over time. We compared the amino acid sequences of the V1, V2, and V3 regions of sequences obtained at seroconversion to those that subsequently evolved and found that each region had a unique pattern of substitutions (Fig. 6 and 7).
FIG. 6.

Changes in V3 seroconversion amino acid consensus sequences over time for Q23, Q47, and Q45. A reference consensus sequence was generated by determining the most common amino acid present at each position in all tissue isolates obtained at the seroconversion sample from each subject. For each subsequent time, the percentage of all sequences that contained an amino acid that differed from the reference sequence at each position is shown. Open and closed symbols represent years 1 and 2 after seroconversion, respectively. Characters (+, ×) are used if only one tissue compartment was sampled at a time point. The hatched bars represent changes in the final sample evaluated for each individual. Where more than 75% of sequences from the last sample contained a nonconsensus amino acid at a position, the replacement amino acid (single-letter designation) is given above the bar. Pb, PBMCs; Cx, cervical secretions.
FIG. 7.

Changes in predicted amino acid V1 sequence for Q23, Q47, and Q45. A consensus sequence was generated as described in the legend to Fig. 6 for all sequences from the seroconversion (A) and final (B) samples from cervical (Cx), plasma RNA (Rn), and PBMC (Pb) sequences from each subject. ∼, gap introduced to maintain alignment between the two consensus sequences; -, gap introduced to maintain alignment among the sequences from the same time point. Capital letters are used if more than 90% of the amino acids at that position are represented in the consensus, and a lowercase letter indicates that the amino acid is represented more than 50% of the time. Where no consensus occurs, “/” is used.
Amino acid changes from the V3 seroconversion sequence of each individual were restricted to several positions. Dominant changes in Q23 V3 during the observation period involved two positions, G25D and H34Y. Similarly, there was a time-dependent replacement of I2T and D29N in the V3 sequence of Q47. Hence, in both Q23 and Q47, changes in V3 predicted amino acid sequences were restricted to a few sites and were characterized by replacement of a single new amino acid at the position rather than by site heterogeneity.
Consensus sequences were generated for both genotypes found in Q45 tissues. Genotype A was the most prevalent at seroconversion and the only genotype found in cervical samples throughout the sampling period. Variation occurred at several positions in the V3 sequence from genotype A, but none of these resulted in fixation of an amino acid that differed from the seroconversion consensus. Genotype B represented 50% of the sequences recovered at 15 months PNS, and there was a temporal replacement of L13H or L13R and K10R in this sequence.
There was no distinct pattern of amino acid replacements in the V2 region over time in sequences from any of the subjects. Substitutions in this region most frequently involved charged amino acids (data not shown).
Changes in V1 from Q23 and Q47 were characterized by insertions and deletions over time (Fig. 7). At seroconversion, the consensus sequence from Q23 plasma virus differed from that of PBMC and cervical variants, but over the next year, the plasma virus V1 sequence found at seroconversion dominated all tissue variants (data not shown). At all time points after 20.5 months PNS, Q23 variants from mucosa and plasma were lengthened by insertions in V1 that primarily encoded potential N-linked glycosylation sites. This correlated with the development of both phenetic and phylogenetic tissue structure in Q23. Q47 variants were also increased in length, due in part to insertion of a potential N-linked site in V1 of all tissue envelope gene sequences. In the 19-month PNS sample, plasma virus sequences were homogeneous in V1 and were characterized by a V1 that was shortened relative to the seroconversion sequence. Representatives of this sequence were recovered from cervical samples but not those from PBMCs (data not shown). Q45 V1 amino acid sequences did not change over time, but the relative frequency of each genotype was different. Hence, V1 from Q47 evolved by both insertions and deletions, Q23 V1 sequence changes were dominated by insertions, and Q45 V1 sequences were unchanged over time.
DISCUSSION
This is the first report of clade A HIV-1 evolution during the asymptomatic stages of infection in subjects who were infected with, or had developed, a heterogeneous population of virus near the time of seroconversion (57). Evidence from nucleotide substitution analysis indicated that the seroconversion population of plasma virus was under positive selection in all three women. Throughout the observation period, dn was higher than ds, suggesting that the viral population was under consistent positive selection during this early stage of infection. Evolution of sequences from two subjects, Q23 and Q47, resulted in replacement of the viral population found at seroconversion, whereas the diverse seroconversion viral population from the third individual, Q45, was maintained throughout the observation period.
We examined the changes in amino acid composition of the V1, V2, and V3 loops that resulted from envelope evolution in the three subjects. Mutations characterized by insertions and deletions affecting potential glycosylation sites in the V1 region were found in both Q47 and Q23 but not in Q45. Viral evolution in V1 due to insertions or deletions (52, 68) and mutations that alter potential N-linked glycosylation sites have been reported for both HIV (8, 52, 68) and simian immunodeficiency virus (51) infections. Substitutions of charged amino acids were common in V2 sequences from all individuals but were not correlated with sample collection time or tissue type. In all three individuals, mutational events in V3 were limited to specific residues and most of the region remained unchanged over time. Others have reported a similar pattern of nonrandom time-dependent change in V3 (24, 44, 72). Virtually all substitutions in V3 from these individuals were nonsynonymous, as has been reported for cohorts where infection was initiated by a single viral genotype (73). There was a notable absence of substitutions at synonymous sites in V3 in tissues of all individuals until 1.5 to 2 years PNS (data not shown). Thus, these variable domains of gp120 appeared to be under different selective pressures and constraints.
The relationship of viral variants from systemic and mucosal compartments of HIV-1-infected women following seroconversion has not been previously explored. We applied both phylogenetic and phenetic methods to study viral evolutionary dynamics among tissue variants during the first 2 years of asymptomatic infection. The significant compartmental phylogenetic structure in tissue viral variants that was detected in all subjects suggested that sequences sampled from each tissue were the progeny of phylogenetically distinct, free or cell associated, virions that had recently migrated to a tissue and subsequently proliferated. Phylogenetic relatedness among sequences from the same tissue compartment developed rapidly in Q47 but was not observed until the second year of infection for sequences obtained from Q23 and Q45. In these latter two subjects, therefore, viral variants emigrating to different tissue compartments throughout the first year of infection either were monophyletic or had not evolved sufficiently for phylogenetic structure to be detected. Both phylogenetic and phenetic tissue compartmentalization developed over time in the viral populations from Q23 and Q47; however, only phylogenetic, not phenetic, tissue compartmentalization was detected among sequences at the final (1.5-year) sample of Q45. This result is consistent with the greater genetic distance that separated the two distinct variants from the Q45 viral population, which decreased their genetic similarity but did not preclude their genealogical affiliation. It is significant that identical results were obtained when only proviral sequences derived from PBMC and mucosal swab samples were evaluated with the cladistic methods. This finding argues against the premise that tissue compartmentalization was detected because of differential longevity of viral sequences in the plasma and cellular compartments but, rather, reflects actual independent migration and proliferation events in each tissue that gave rise to phylogenetically distinct, tissue-derived variants.
Results from our temporal phylogenetic studies of viral sequences derived from PBMCs, plasma, and cervical secretions provide a unique perspective on viral dynamics. Recent data on virus kinetics suggest that plasma virus is supplied primarily from short-lived, newly infected cells rather than from activation of latently infected cells (15, 55, 69). In this model, each new infection would result in production of related progeny viruses. Depending on sample frequency and mutation rate, subsequent virus populations would be phylogenetically distinct from those sampled earlier. Alternatively, if the majority of plasma virus was produced by continual activation of long-lived, chronically infected cells, then the plasma viral population would not develop distinct phylogenetic affiliations over time. Our data showed that in most samples, plasma viral sequences had both phenetic and phylogenetic temporal structure. Furthermore, if plasma virus was generated from a significant proportion of circulating infected PBMCs or from infected mucosal cells, then at any time point there would be phylogenetic intermixing among sequences in these tissue compartments. The implications from our data are that neither circulating PBMCs nor cells in genital mucosa contribute substantially to the plasma viral pool. Indeed, studies suggest that lymphoid tissues harbor the majority of cells that actively produce virus (20, 54). Thus, our cladistic analyses yield results that are in agreement with findings of investigators obtained by other methods and, additionally, extend the observations to dynamics of clade A HIV-1 infection in women.
It has been reported that the virus transmitted to an individual by sexual contact is a minor component of the virus population in the donor blood (75) and that variants found in the genital secretions of infected women are different from those in peripheral blood (50, 57). A finding that was consistent within our study was that of significant temporal phylogenetic and phenetic structure among mucosal variants in all three women. Temporal structure in the mucosal compartment could result from successive migrations of virions from lineages not well represented in either PBMC or plasma viral sequences. Alternatively, the presence of significant temporal phylogenetic structure could reflect tissue specific evolution of mucosal variants. However, there was no evidence of independent evolution of a mucosal lineage from the phylogenetic trees to support this. Rather, the premise that phylogenetic structure arose by independent migration events and subsequent viral proliferation is consistent with the biology of memory cells homing to specific tissue compartments (38, 39). These cells make a small and transient contribution to the circulating pool of lymphocytes, which would reduce the likelihood that they would be detected in a PBMC sample. Continual seeding of mucosa by a small subset of infected lymphocytes would mean that genital mucosa could be continually challenged with new virus variants, some of which may be better adapted to proliferate in that environment. A mucosal variant that is better fit may be more readily transmitted if it is locally abundant in the donor mucosa and is adapted to infect the cells most likely to be initially encountered in a recipient. A better understanding of features that promote infection and replication of HIV-1 variants in the mucosal environment may help in the development of methods to reduce the risk of sexual and vertical transmission.
It is now well established that the early stages of infection with HIV-1 represent an active, not quiescent, phase of viral infection. In this study, we provide insight into the dynamic events that characterize HIV-1 pathogenesis in women recently infected with subtype A HIV-1. Our study is the first to examine virus evolution in non-subtype-B-infected individuals. Host selection pressure, viral diversity, and tissue specific trafficking of virus-infected cells following transmission of HIV-1 will undoubtedly have a profound effect on disease outcome. It is unclear, however, that the paradigm being established by studies of individuals infected with clade B HIV-1 will apply to diverse populations infected with different HIV-1 subtypes found worldwide. It is important to delineate features which are common to HIV-1 pathogenesis in order to direct strategies for therapies which will be globally effective.
ACKNOWLEDGMENTS
We thank the research staff at Ganjoni Municipal Clinic and Coast Provincial General Hospital, Mombasa, Kenya, for sample collection, P. Lewis for determining plasma viral loads, and J. I. Mullins for critical review of the manuscript.
This work was supported by Public Health Service grants AI38518 (J.O.), AI33873 (J.K.K.), and AI27757 (UW CFAR). M.P. was supported by NIH fellowships AI07140 and K08 AI01290, A.G.R. and G.H.L. were supported by AI32885, and J.J.G. was supported by fellowship CA09229 from the National Cancer Institute.
REFERENCES
- 1.Albert J, Abrahamsson B, Nagy K, Aurelius E, Gaines H, Nystrom G, Fenyo E M. Rapid development of isolate-specific neutralizing antibodies after primary HIV-1 infection and consequent emergence of virus variants which resist neutralization by autologous sera. AIDS. 1990;4:107–112. doi: 10.1097/00002030-199002000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Arendrup M, Nielsen C, Hansen J-E S, Pedersen C, Mathiesen L, Nielsen J O. Autologous HIV-1 neutralizing antibodies: emergence of neutralization-resistant escape virus and subsequent development of escape virus neutralizing antibodies. J Acquired Immune Defic Syndr. 1992;5:303–307. [PubMed] [Google Scholar]
- 3.Balfe P, Simmonds P, Ludlam C A, Bishop J O, Leigh Brown A J. Concurrent evolution of human immunodeficiency virus type 1 in patients infected from the same source: rate of sequence change and low frequency of inactivating mutations. J Virol. 1990;64:6221–6233. doi: 10.1128/jvi.64.12.6221-6233.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Barnett S W, Barboza A, Wilcox C M, Forsmark C E, Levy J A. Characterization of human immunodeficiency virus type 1 strains recovered from the bowel of infected individuals. Virology. 1991;182:802–809. doi: 10.1016/0042-6822(91)90621-h. [DOI] [PubMed] [Google Scholar]
- 5.Bonhoeffer S, Holmes E C, Nowak M A. Causes of HIV diversity. Nature. 1995;376:125. doi: 10.1038/376125a0. . (Letter.) [DOI] [PubMed] [Google Scholar]
- 6.Boom R, Sol C M, Salimans M M, Jansen C L, Wertheim-van Dillen P M, van der Noordaa J. Rapid and simple method for purification of nucleic acids. J Clin Microbiol. 1990;28:493–503. doi: 10.1128/jcm.28.3.495-503.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Borrow P, Lewicki H, Wei X, Horwitz M S, Peffer N, Meyers H, Nelson J A, Gairin J E, Hahn B H, Oldstone M B A, Shaw G M. Antiviral pressure exerted by HIV-1-specific cytotoxic T lymphocytes (CTLs) during primary infection demonstrated by rapid selection of CTL escape virus. Nat Med. 1997;3:205–217. doi: 10.1038/nm0297-205. [DOI] [PubMed] [Google Scholar]
- 8.Bosch M L, Andeweg A C, Schipper R, Kenter M. Insertion of N-linked glycosylation sites in the variable regions of the human immunodeficiency virus type 1 surface glycoprotein through AAT triplet reiteration. J Virol. 1994;68:7566–7569. doi: 10.1128/jvi.68.11.7566-7569.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Boyd M T, Simpson G R, Cann A J, Johnson M A, Weiss R A. A single amino acid substitution in the V1 loop of human immunodeficiency virus type 1 gp120 alters cellular tropism. J Virol. 1993;67:3649–3652. doi: 10.1128/jvi.67.6.3649-3652.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cann A J, Churcher M J, Boyd M, O’Brien W, Zhao J Q, Zack J, Chen I S. The region of the envelope gene of human immunodeficiency virus type 1 responsible for determination of cell tropism. J Virol. 1992;66:305–309. doi: 10.1128/jvi.66.1.305-309.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng-Mayer, C. 1990. Biological and molecular features of HIV-1 related to tissue tropism. AIDS 4(Suppl. 1):s49–s56. [PubMed]
- 12.Cheng-Mayer C, Weiss C, Seto D, Levy J A. Isolates of human immunodeficiency virus type 1 from the brain may constitute a special group of the AIDS virus. Proc Natl Acad Sci USA. 1989;86:8575–8579. doi: 10.1073/pnas.86.21.8575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Coffin J M. Genetic variation in AIDS viruses. Cell. 1986;46:1–4. doi: 10.1016/0092-8674(86)90851-2. [DOI] [PubMed] [Google Scholar]
- 14.Coffin J M. HIV pathogenesis. Lines drawn in epitope wars. Nature. 1995;375:534–535. doi: 10.1038/375534a0. [DOI] [PubMed] [Google Scholar]
- 15.Coffin J M. HIV population dynamics in vivo: implications for genetic variation, pathogenesis, and therapy. Science. 1995;267:483–489. doi: 10.1126/science.7824947. [DOI] [PubMed] [Google Scholar]
- 16.Cordonnier A, Montagnier L, Emerman M. Single amino-acid changes in HIV envelope affect viral tropism and receptor binding. Nature. 1989;340:571–574. doi: 10.1038/340571a0. [DOI] [PubMed] [Google Scholar]
- 17.Delwart E L, Mullins J I, Gupta P, Learn G H, Jr, Holodniy M, Katzenstein D, Walker B D, Singh M K. Human immunodeficiency virus type 1 populations in blood and semen. J Virol. 1998;72:617–623. doi: 10.1128/jvi.72.1.617-623.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Delwart E L, Pan H, Sheppard H W, Wolpert D, Neumann A U, Mullins J I. Slower evolution of human immunodeficiency virus type 1 quasispecies during progression to AIDS. J Virol. 1997;71:7498–7508. doi: 10.1128/jvi.71.10.7498-7508.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Delwart E L, Sheppard H W, Walker B D, Goudsmit J, Mullins J I. Human immunodeficiency virus type 1 evolution in vivo tracked by DNA heteroduplex mobility assays. J Virol. 1994;68:6672–6683. doi: 10.1128/jvi.68.10.6672-6683.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Embretson J, Zupancic M, Ribas J L, Burke A, Racz P, Tenner-Racz K, Haase A T. Massive covert infection of helper T lymphocytes and macrophages by HIV during the incubation period of AIDS. Nature. 1993;362:359–362. doi: 10.1038/362359a0. [DOI] [PubMed] [Google Scholar]
- 21.Epstein L G, Kuiken C, Blumberg B M, Hartman S, Sharer L R, Clement M, Goudsmit J. HIV-1 V3 domain variation in brain and spleen of children with AIDS: tissue-specific evolution within host-determined quasispecies. Virology. 1991;180:583–590. doi: 10.1016/0042-6822(91)90072-j. [DOI] [PubMed] [Google Scholar]
- 22.Felsenstein J. PHYLIP—Phylogeny Inference Package (version 3.2) Cladistics. 1989;5:164–166. [Google Scholar]
- 23.Fisher A G, Ensoli B, Looney D, Rose A, Gallo R C, Saag M S, Shaw G M, Hahn B H, Wong-Staal F. Biologically diverse molecular variants within a single HIV-1 isolate. Nature. 1988;334:444–447. doi: 10.1038/334444a0. [DOI] [PubMed] [Google Scholar]
- 24.Holmes E C, Zhang L Q, Simmonds P, Ludlam C A, Leigh Brown A J. Convergent and divergent sequence evolution in the surface envelope glycoprotein of human immunodeficiency virus type 1 within a single infected patient. Proc Natl Acad Sci USA. 1992;89:4835–4839. doi: 10.1073/pnas.89.11.4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hwang S S, Boyle T J, Lyerly H K, Cullen B R. Identification of the envelope V3 loop as the primary determinant of cell tropism in HIV-1. Science. 1991;253:71–74. doi: 10.1126/science.1905842. [DOI] [PubMed] [Google Scholar]
- 26.Itescu S, Simonelli P F, Winchester R J, Ginsberg H S. Human immunodeficiency virus type 1 strains in the lungs of infected individuals evolve independently from those in peripheral blood and are highly conserved in the C-terminal region of the envelope V3 loop. Proc Natl Acad Sci USA. 1994;91:11378–11382. doi: 10.1073/pnas.91.24.11378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ji J, Hoffmann J S, Loeb L. Mutagenicity and pausing of HIV reverse transcriptase during HIV plus-strand DNA synthesis. Nucleic Acids Res. 1994;22:47–52. doi: 10.1093/nar/22.1.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kampinga G A, Simonon A, Van de Perre P, Karita E, Msellati P, Goudsmit J. Primary infections with HIV-1 of women and their offspring in Rwanda: findings of heterogeneity at seroconversion, coinfection, and recombinants of HIV-1 subtypes A and C. Virology. 1997;227:63–76. doi: 10.1006/viro.1996.8318. [DOI] [PubMed] [Google Scholar]
- 29.Koito K, Harrowe G, Levy J A, Cheng-Meyer C. Functional role of the V1/V2 region of human immunodeficiency virus type 1 envelope glycoprotein gp120 in infection of primary macrophages and soluble CD4 neutralization. J Virol. 1994;68:2253–2259. doi: 10.1128/jvi.68.4.2253-2259.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Korber B T, Kunstman K J, Patterson B K, Furtado M, McEvilly M M, Levy R, Wolinsky S M. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. J Virol. 1994;68:7467–7481. doi: 10.1128/jvi.68.11.7467-7481.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Koup R A. Virus escape from CTL recognition. J Exp Med. 1994;180:779–782. doi: 10.1084/jem.180.3.779. . (Comment.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Koup R A, Safrit J T, Cao Y, Andrews C A, McLeod G, Borkowsky W, Farthing C, Ho D D. Temporal association of cellular immune responses with the initial control of viremia in primary human immunodeficiency virus type 1 syndrome. J Virol. 1994;68:4650–4655. doi: 10.1128/jvi.68.7.4650-4655.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kuiken C L, Zwart G, Baan E, Coutinho R A, van den Hoek J A, Goudsmit J. Increasing antigenic and genetic diversity of the V3 variable domain of the human immunodeficiency virus envelope protein in the course of the AIDS epidemic. Proc Natl Acad Sci USA. 1993;90:9061–9065. doi: 10.1073/pnas.90.19.9061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar S, Tamura K, Nei M. Molecular evolutionary genetics analysis software for microcomputers. Comput Appl Biosci. 1994;10:189–191. doi: 10.1093/bioinformatics/10.2.189. [DOI] [PubMed] [Google Scholar]
- 35.Lewis P, Kreiss J K, John G C, Richardson B A, Mbori Ngacha D, Ndinya Achola J, Overbaugh J. Cell-free human immunodeficiency virus type 1 in breast milk. J Infect Dis. 1998;177:34–39. doi: 10.1086/513816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S-L, Schacker T, Musey L, Shriner D, McElrath M J, Corey L, Mullins J I. Divergent pattern of progression to AIDS after infection from the same source: human immunodeficiency virus type 1 evolution and antiviral responses. J Virol. 1997;71:4284–4295. doi: 10.1128/jvi.71.6.4284-4295.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lukashov V V, Kuiken C L, Goudsmit J. Intrahost human immunodeficiency virus type 1 evolution is related to length of the immunocompetent period. J Virol. 1995;69:6911–6916. doi: 10.1128/jvi.69.11.6911-6916.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Mackay C R, Marston W, Dudler L. Altered patterns of T cell migration through lymph nodes and skin following antigen challenge. Eur J Immunol. 1992;22:2205–2210. doi: 10.1002/eji.1830220904. [DOI] [PubMed] [Google Scholar]
- 39.Mackay C R, Marston W L, Dudler L, Spertini O, Tedder T F, Hein W R. Tissue-specific migration pathways by phenotypically distinct subpopulations of memory T cells. Eur J Immunol. 1992;22:887–895. doi: 10.1002/eji.1830220402. [DOI] [PubMed] [Google Scholar]
- 40.Maddison W P, Maddison D R. MacClade: analysis of phylogeny and character evolution. 3rd ed. Sunderland, Mass: Sinauer Associates; 1992. [Google Scholar]
- 41.Martin H L, Jackson D J, Mandaliya K, Bwayo J, Rakwar J P, Nyange P, Moses S, Ndinya-Achola J O, Holmes K, Plummer F, Ngugi E, Kreiss J. Preparation for AIDS vaccine evaluation in Mombasa, Kenya: establishment of seronegative cohorts of commercial sex workers and trucking company employees. AIDS Res Hum Retroviruses. 1994;10:S235–S237. [PubMed] [Google Scholar]
- 42.McDonald R A, Mayers D L, Chung R C-Y, Wagner K F, Ratto-Kim S, Birx D L, Michael N L. Evolution of human immunodeficiency virus type 1 env sequence variation in patients with diverse rates of disease progression and T-cell function. J Virol. 1997;71:1871–1879. doi: 10.1128/jvi.71.3.1871-1879.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKeating J A, Gow J, Goudsmit J, Pearl L H, Mulder C, Weiss R A. Characterization of HIV-1 neutralization escape mutants. AIDS. 1989;3:777–784. doi: 10.1097/00002030-198912000-00001. [DOI] [PubMed] [Google Scholar]
- 44.McNearney T, Hornickova Z, Markham R, Birdwell A, Arens M, Saah A, Ratner L. Relationship of human immunodeficiency virus type 1 sequence heterogeneity to stage of disease. Proc Natl Acad Sci USA. 1992;89:10247–10251. doi: 10.1073/pnas.89.21.10247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mulder J, McKinney N, Christopherson C, Sninsky J, Greenfield L, Kwok S. Rapid and simple PCR assay for quantitation of human immunodeficiency virus type 1 RNA in plasma: application to acute retroviral infection. J Clin Microbiol. 1994;32:292–300. doi: 10.1128/jcm.32.2.292-300.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nei M, Gojobori T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol Biol Evol. 1986;3:418–426. doi: 10.1093/oxfordjournals.molbev.a040410. [DOI] [PubMed] [Google Scholar]
- 47.Nowak M A, Anderson R M, McLean A R, Wolfs T F, Goudsmit J, May R M. Antigenic diversity thresholds and the development of AIDS. Science. 1991;254:963–969. doi: 10.1126/science.1683006. [DOI] [PubMed] [Google Scholar]
- 48.Nowak M A, May R M, Phillips R E, Rowland-Jones S, Lalloo D G, McAdam S, Klenerman P, Koppe B, Sigmund K, Bangham C R, McMichael A J. Antigenic oscillations and shifting immunodominance in HIV-1 infections. Nature. 1995;375:606–611. doi: 10.1038/375606a0. [DOI] [PubMed] [Google Scholar]
- 49.O’Brien W A, Koyanagi Y, Namazie A, Zhao J-Q, Diagne A, Idler K, Zack J A, Chen I S Y. HIV-1 tropism for mononuclear phagocytes can be determined by regions of gp120 outside the CD4-binding domain. Nature. 1990;348:69–73. doi: 10.1038/348069a0. [DOI] [PubMed] [Google Scholar]
- 50.Overbaugh J, Anderson R J, Ndinya-Achola J O, Kreiss J K. Distinct but related human immunodeficiency virus type 1 variant populations in genital secretions and blood. AIDS Res Hum Retroviruses. 1996;12:107–115. doi: 10.1089/aid.1996.12.107. [DOI] [PubMed] [Google Scholar]
- 51.Overbaugh J, Rudensey L M. Alterations in potential sites for glycosylation predominate during evolution of the simian immunodeficiency virus envelope gene in macaques. J Virol. 1992;66:5937–5948. doi: 10.1128/jvi.66.10.5937-5948.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palmer C, Balfe P, Fox D, May J C, Frederiksson R, Rnyo E-M, McKeating J A. Functional characterization of the V1/V2 region of human immunodeficiency virus type 1. Virology. 1996;220:436–449. doi: 10.1006/viro.1996.0331. [DOI] [PubMed] [Google Scholar]
- 53.Pang S, Shlesinger E S, Daar E S, Moudgil T, Ho D D, Chen I S. Rapid generation of sequence variation during primary HIV-1 infection. AIDS. 1992;6:453–460. doi: 10.1097/00002030-199205000-00003. [DOI] [PubMed] [Google Scholar]
- 54.Pantaleo G, Graziosi C, Demarest J F, Butini L, Montroni M, Fox C H, Orenstein J M, Kotler D P, Fauci A S. HIV infection is active and progressive in lymphoid tissue during the clinically latent stage of disease. Nature. 1993;362:355–358. doi: 10.1038/362355a0. [DOI] [PubMed] [Google Scholar]
- 55.Perelson A S, Neumann A U, Markowitz M, Leonard J M, Ho D D. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271:1582–1586. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 56.Poss M, Gosink J J, Thomas E, Kreiss J K, Ndinya-Achola J, Mandaliya K, Bwayo J, Overbaugh J. Phylogenetic evaluation of Kenyan HIV-1 isolates. AIDS Res Hum Retroviruses. 1997;13:493–499. doi: 10.1089/aid.1997.13.493. [DOI] [PubMed] [Google Scholar]
- 57.Poss M, Martin H, Kreiss J, Nyange P, Mandaliya K, Chohan B, Overbaugh J. Diversity in virus populations from genital mucosa and peripheral blood in women recently infected with human immunodeficiency virus. J Virol. 1995;69:8118–8122. doi: 10.1128/jvi.69.12.8118-8122.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Power C, McArthur J C, Johnson R T, Griffen D E, Glass J D, Perryman S, Chesebro B. Demented and nondemented patients with AIDS differ in brain-derived human immunodeficiency virus type 1 envelope sequences. J Virol. 1994;68:4643–4649. doi: 10.1128/jvi.68.7.4643-4649.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rodrigo A, Mullins J I. Human immunodeficiency virus type 1 molecular evolution and the measure of selection. AIDS Res Hum Retroviruses. 1996;12:1681–1685. doi: 10.1089/aid.1996.12.1681. [DOI] [PubMed] [Google Scholar]
- 60.Saag M S, Hahn B H, Gibbons J, Li Y X, Parks E S, Parks W P, Shaw G M. Extensive variation of human immunodeficiency virus type 1 in vivo. Nature. 1988;334:440–444. doi: 10.1038/334440a0. [DOI] [PubMed] [Google Scholar]
- 61.Sanger F, Nicklen S, Coulson A R. DNA sequencing with chain-terminating inhibitors. Proc Natl Acad Sci USA. 1977;74:5463–5467. doi: 10.1073/pnas.74.12.5463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shioda T, Levy J A, Cheng-Mayer C. Macrophage and T cell-line tropisms of HIV-1 are determined by specific regions of the envelope gp120 gene. Nature. 1991;349:167–169. doi: 10.1038/349167a0. [DOI] [PubMed] [Google Scholar]
- 63.Slatkin M, Hudson R R. Pairwise comparisons of mitochondrial DNA sequences in stable and exponentially growing populations. Genetics. 1991;129:555–562. doi: 10.1093/genetics/129.2.555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Slatkin M, Maddison W P. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics. 1989;123:603–613. doi: 10.1093/genetics/123.3.603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Slatkin M, Maddison W P. Detecting isolation by distance using phylogenies of genes. Genetics. 1990;126:249–260. doi: 10.1093/genetics/126.1.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Smouse P E, Long J C, Sokal R R. Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Syst Zool. 1986;35:627–632. [Google Scholar]
- 67.Waddle D M. Matrix correlation tests support a single origin for modern humans. Nature. 1994;368:452–454. doi: 10.1038/368452a0. [DOI] [PubMed] [Google Scholar]
- 68.Wang N, Zhu T, Ho D D. Sequence diversity of V1 and V2 domains of gp120 from human immunodeficiency virus type 1: lack of correlation with viral phenotype. J Virol. 1995;69:2708–2715. doi: 10.1128/jvi.69.4.2708-2715.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wei X, Ghosh S K, Taylor M E, Johnson V A, Emini E A, Deutsch P, Lifson J D, Bonhoeffer S, Nowak M A, Hahn B, Saag M S, Shaw G M. Viral dynamics in human immunodeficiency virus type 1 infection. Nature. 1995;373:117–126. doi: 10.1038/373117a0. [DOI] [PubMed] [Google Scholar]
- 70.Wolfs T F, Zwart G, Bakker M, Goudsmit J. HIV-1 genomic RNA diversification following sexual and parenteral virus transmission. Virology. 1992;189:103–110. doi: 10.1016/0042-6822(92)90685-i. [DOI] [PubMed] [Google Scholar]
- 71.Wolinsky S M, Korber B T M, Neumann A U, Daniels M, Kunstman K J, Whetsell A J, Furtado M R, Cao Y, Ho D D, Safrit J T, Koup R A. Adaptive evolution of human immunodeficiency virus-type 1 during the natural course of infection. Science. 1996;272:537–542. doi: 10.1126/science.272.5261.537. [DOI] [PubMed] [Google Scholar]
- 72.Yamaguchi Y, Gojobori T. Evolutionary mechanisms and population dynamics of the third variable envelope region of HIV within single hosts. Proc Natl Acad Sci USA. 1997;94:1264–1269. doi: 10.1073/pnas.94.4.1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zhang L, Diaz R S, Ho D D, Mosley J W, Busch M P, Mayer A. Host-specific force in human immunodeficiency virus type 1 evolution in vivo. J Virol. 1997;71:2555–2561. doi: 10.1128/jvi.71.3.2555-2561.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zhang L Q, MacKenzie P, Cleland A, Holmes E C, Leigh Brown A J, Simmonds P. Selection for specific sequences in the external envelope protein of human immunodeficiency virus type 1 upon primary infection. J Virol. 1993;67:3345–3356. doi: 10.1128/jvi.67.6.3345-3356.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Zhu T, Ho H, Wang N, Nam D S, Cao Y, Koup R A, Ho D D. Genotypic and phenotypic charcterization of HIV-1 in patients with primary infection. Science. 1993;261:1179–1181. doi: 10.1126/science.8356453. [DOI] [PubMed] [Google Scholar]
- 76.Zhu T, Wang N, Carr A, Nam D S, Moor-Jankowski R, Cooper D A, Ho D D. Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission. J Virol. 1996;70:3098–3107. doi: 10.1128/jvi.70.5.3098-3107.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zwart G, Langedijk H, van der Hoek L, de Jong J J, Wolfs T F, Ramautarsing C, Bakker M, de Ronde A, Goudsmit J. Immunodominance and antigenic variation of the principal neutralization domain of HIV-1. Virology. 1991;181:481–489. doi: 10.1016/0042-6822(91)90880-k. [DOI] [PubMed] [Google Scholar]