

Abstract
α-Aryl-substituted pyrrolidine moiety is found in many natural alkaloids. Starting from pyrrolidine, we were able to synthesize α-aryl-substituted pyrrolidines in one step using quinone monoacetal as the oxidizing agent and DABCO as the base. We also discovered the reaction condition needed to efficiently remove the N-aryl moiety from the α-arylated product. When the above reaction was carried out without the addition of an aryl nucleophile, the reaction of pyrrolidine and quinone monoacetal in 2,2,2-trifluoroethanol afforded octahydro-dipyrroloquinoline in high yield, which has the same skeleton as that of natural product incargranine B.
Using a quinone monoacetal as the oxidant and DABCO as the base, we report the one-step synthesis of α-aryl-substituted pyrrolidines from pyrrolidine. The reaction of pyrrolidine and quinone monoacetal in 2,2,2-trifluoroethanol afforded octahydro-dipyrroloquinoline in high yield.
Introduction
Pyrrolidine alkaloids are characterized by a five-membered nitrogen-containing ring. Many of the alkaloids in this group have good biological activity and are particularly well known for their effects on the nervous system, with nicotine (also have antioxidant, anti-inflammatory, and antihyperglycemic properties) being the most important pyrrolidine alkaloid. Substituted pyrrolidines are one of the most widely used scaffolds in drug molecules1 and in recent years have been used as catalysts in asymmetric organocatalysis.2
Pyrrolidine alkaloids can have one or two alkyl or aryl substitutions at the two α-positions (Scheme 1). Alkylated pyrrolidine (+)-preussin was an antibacterial and anticancer alkaloid from the fermentation broth of bacterium Aspergillus ochraceus.3 Anisomycin is an antibiotic drug that inhibits bacterial protein and DNA synthesis.4 Simple arylated pyrrolidines such as nicotine have strong bioactivity. Radicamines A and B (isolated from Lobelia chinensis) are glucosidase inhibitors.5 Incargranine B was first reported by Zhang and co-workers6 in 2010, and Lawrence's research group revised its structure by total synthesis.7
Scheme 1. Representative natural alkaloids containing α-substituted pyrrolidine moiety.

Due to the strong bioactivity of substituted pyrrolidines, extensive synthetic efforts have been devoted to the synthesis of these compounds. Among these strategies, a non-traditional and also more environmentally friendly way is the use of redox-neutral intramolecular hydride transfer and subsequent nucleophilic addition process,8,9 as no metal or very strong base is involved as in other methods.10 Seidel and co-workers opened a new avenue for C–H functionalization of pyrrolidine by introducing azomethine ylide as an intermediate.11 Our group previously reported an iterative synthesis of unsymmetrical 2,5-disubstituted pyrrolidines from pyrrolidine by two rounds of redox-neutral α-C–H functionalization, and we synthesized (±)-preussin and its C(3) epimer using this strategy.12 Few studies have been carried out in the literature on the redox-neutral α-C–H arylation of pyrrolidine.13 Recently, we reported a redox-neutral α-C–H arylation of pyrrolidin-3-ol with boronic acid nucleophiles. The hydroxy group at the 3-position of pyrrolidine coordinated with the arylboronic acid and delivered the aryl group to the α-position of pyrrolidine.14 As a continuation of this research project, here we have achieved α-C–H arylation of unsubstituted pyrrolidine.
Results and discussion
We carried out initial investigations using 2,6-di-tert-butyl-1,4-benzoquinone 1 as the oxidizing agent and β-naphthol as the nucleophile. We first tried the reaction in 2,2,2-trifluoroethanol (TFE), which we had used in previous research,12 but it turned out that the reaction gave a complicated mixture of products (entry 1, Table 1). After changing the reaction solvent to isopropanol or less polar solvent toluene, it was also found that only trace amounts of the arylated pyrrolidine were formed (entries 2 and 3). We thought that adding a base to the system might help to abstract the α-C–H of pyrrolidine. When strong base DBU was added to the reaction system, the reaction also gave complicated products (entry 4). The addition of DABCO increased the reaction yield to 30% (entry 5). We found that the use of 4,4-dimethoxycyclohexa-2,5-dien-1-one 2 (p-quinone monoacetal), in which one of the carbonyl groups of the quinone is protected as a ketal, increased the motivation for aromatization and made the reaction easier to proceed. Using quinone monoacetal 2 as the oxidizing agent and adding 0.1 equivalent of DABCO, the reaction gave an increased yield of 59% (entry 6). Increasing the amount of DABCO to 0.2 equivalent resulted in a further increase in yield to 84% (entry 7), but increasing the amount of DABCO further to 0.5 equivalent resulted in a decrease in yield (entry 8). We then carried out further screening of the reaction solvent. The previously used TFE was retested using DABCO as the base (entry 9), but only trace amounts of product were observed. The inapplicability of TFE in this study may be due to the fact that this study requires the base to assist in abstracting the α-C–H of pyrrolidine, the acidic TFE being incompatible with the added base. The reaction gave similar yields in benzene (entry 10) and p-xylene (entry 11). The reaction yields decreased in alcohols and acetonitrile (entries 12–14). Increasing the concentration of the reaction gave α-C–H arylated pyrrolidine in 91% isolated yield (entry 15).
Evaluation of reaction condition for pyrrolidine α-arylationa.
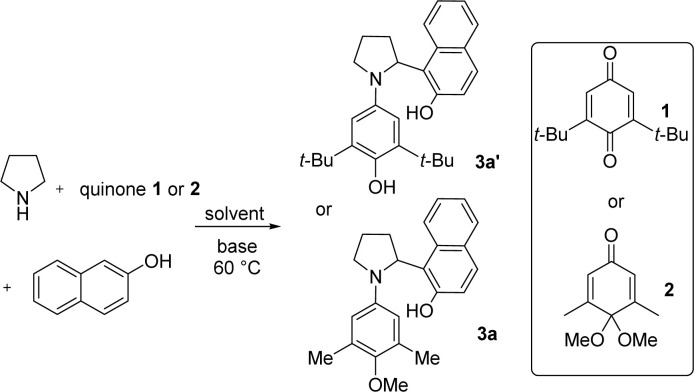
| ||||
|---|---|---|---|---|
| Entry | Quinone | Base (equiv.) | Solvent | Yield |
| 1 | 1 | TFE | 3a′ (trace) | |
| 2 | 1 | i-PrOH | 3a′ (trace) | |
| 3 | 1 | PhCH3 | 3a′ (trace) | |
| 4 | 1 | DBU (0.1) | PhCH3 | 3a′ (trace) |
| 5 | 1 | DABCO (0.1) | PhCH3 | 3a′ (30%) |
| 6 | 2 | DABCO (0.1) | PhCH3 | 3a (59%) |
| 7 | 2 | DABCO (0.2) | PhCH3 | 3a (84%) |
| 8 | 2 | DABCO (0.5) | PhCH3 | 3a (64%) |
| 9 | 2 | DABCO (0.2) | TFE | 3a (trace) |
| 10 | 2 | DABCO (0.2) | Bezene | 3a (84%) |
| 11 | 2 | DABCO (0.2) | p-Xylene | 3a (80%) |
| 12 | 2 | DABCO (0.2) | MeOH | 3a (25%) |
| 13 | 2 | DABCO (0.2) | EtOH | 3a (37%) |
| 14 | 2 | DABCO (0.2) | CH3CN | 3a (30%) |
| 15b | 2 | DABCO (0.2) | PhCH3 | 3a (91%) |
Reactions were performed with pyrrolidine (0.33 mmol), β-naphthol (0.45 mmol), and quinone monoacetal 2 (0.3 mmol) at 60 °C in solvent (conc. = 0.3 M), isolated yield.
Reaction was performed in toluene (conc. = 0.5 M).
Using the optimized reaction condition (Table 1, entry 15), several aromatic nucleophiles were tested and most of them were able to provide α-arylated pyrrolidines in moderate to high yields (Scheme 2). The reactions proceeded with high efficiency when using β- or α-naphthol as well as sesamol (3a–3c). Phenols substituted with methoxyl groups could be attached to the α-position of pyrrolidine with comparable yields (3d–3f). Amine was compatible with the reaction condition, and the desired products 3g and 3h were obtained in good yields. Replacement of naphthol with 8-hydroxyquinoline in 3i led to a decrease in efficiency. Naphthol nucleophiles bearing a halogen atom afforded products 3j and 3k in 66% and 95% yields respectively. With 5,6,7,8-tetrahydro-1-naphthol as the nucleophile, product 3l could be obtained in a moderate yield. As expected, indole derivatives showed good reactivity to give 3m and 3n in 56% and 60% yields respectively. Methylphenylacetylene could also be introduced to form functionalized pyrrolidine 3o in 30% yield.
Scheme 2. Scope of aromatic nucleophiles. [a] Reactions were performed with pyrrolidine (0.33 mmol), quinone monoacetal 2 (0.3 mmol), nucleophile (0.45 mmol), and DABCO (0.06 mmol) at 60 °C in toluene (conc. = 0.5 M). [b] 4 Å molecular sieve (100 mg) was added. [c] Reactions were performed with pyrrolidine (1.5 mmol), quinone monoacetal 2 (0.3 mmol), nucleophile (1.5 mmol), and DABCO (0.06 mmol) at 60 °C in toluene. [d] 15 mol% of CuI was added and the reaction was performed at 80 °C.

Under similar condition, α-substituted pyrrolidines were evaluated (Scheme 3). The α-arylation of α-substituted pyrrolidines, such as those in 4a and 4b, took place only at the less sterically hindered position in 70% and 50% yields, respectively. The protons at the 2- and 5-positions of 4a and 4b do not show correlation in their two-dimensional NOESY spectrum, indicating a trans relationship between the substituents at these two positions. The cis product was not observed in these reactions. We used (S)-prolinol as the substrate in the preparation of 4b, the enantiomeric purity of 4b was determined to be >99%, indicating that no racemization (due to the isomerization of the iminium ion) took place at the α-carbon of (S)-prolinol under the reaction condition. In the literature, the application of the redox-neutral α-C–H functionalization methodology to the functionalization of a piperidine is typically not as good as that of a pyrrolidine, presumably due to the relatively lower reactivity of a piperidine in the formation of the iminium intermediate.11 Tetrahydroisoquinoline was compatible with the reaction conditions to give the corresponding product 4c in 39% yield. This protocol can be applied to piperidine 4d and the 4-phenyl substituted piperidine 4e, but with lower yields. The 2D NOESY spectra of 4e suggest that the phenyl at the 4-position is in trans with the naphthol at the 2-position.
Scheme 3. α-Arylation of substituted pyrrolidines and piperidines. [a] Reactions were performed with cyclic amine (0.33 mmol), quinone monoacetal 2 (0.3 mmol), nucleophile (0.45 mmol), and DABCO (0.06 mmol) at 60 °C in toluene (conc. = 0.5 M).

Redox-neutral pyrrolidine α-functionalization usually gave products with an alkyl or an aryl group attached to the nitrogen atom. To obtain α-substituted pyrrolidines, it is necessary to remove the functional group on the nitrogen atom. Several previous studies have not attempted to remove the functional group on the nitrogen atom.9a,b,d,13 We attempted to remove the aryl group from β-naphthol-substituted pyrrolidine 3a in the presence of oxidizing agent ceric ammonium nitrate (CAN) reported in the literature,15 but we did not isolate the desired product. Our group developed a method to break the carbon–nitrogen bond in the iodine–sodium hydroxide reaction system.12 However, no cleavage product was obtained by direct application of this method. Therefore, in the presence of boron tribromide, we first converted the methoxy group in 3a to the phenolic hydroxyl group in 5a and then used iodine–sodium hydroxide system for the aryl group removal reaction. After optimizing the reaction condition, β-naphthol substituted pyrrolidine 6a was obtained in 83% yield (Scheme 4).
Scheme 4. Removal of the N-aryl moiety from the α-arylated pyrrolidine.

We have also tested the reactions with benzene or naphthalene derivatives that do not have a phenolic hydroxyl group on them, disappointingly, the desired α-arylated products were not formed under the standard condition, suggesting that phenolic hydroxyl group is crucial for the reaction.13b,c However, a more complex product 7 was formed in these experiments. X-ray analysis of 7 showed that it had an octahydro-dipyrroloquinoline skeleton (entry 1, Table 2). Octahydro-dipyrroloquinoline skeleton is a core structure found in several biologically useful alkaloid natural products, such as incargranine B6,7 and seneciobipyrrolidine.16 The azatetracyclic 7 is the epimer of the octahydro-dipyrroloquinoline skeleton in incargranine B.
Rapid assembly of octahydro-dipyrroloquinoline scaffold starting from pyrrolidinea.
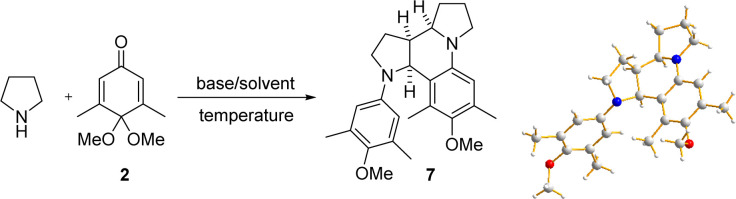
| |||||
|---|---|---|---|---|---|
| Entry | Base (eq.) | Pyrrolidine (eq.) | Solvent | Time (h) | Yield (%) |
| lb | DABCO (0.2) | 5 | PhCH3 | 72 | 16 |
| 2 | DABCO (0.2) | 5 | PhCH3 | 72 | 43 |
| 3 | DABCO (0.2) | 1.1 | PhCH3 | 72 | 50 |
| 4 | DBU (0.2) | 1.1 | PhCH3 | 72 | 24 |
| 5 | DIPEA (0.2) | 1.1 | PhCH3 | 72 | 60 |
| 6 | DIPEA (0.2) | 1.1 | i-PrOH | 8 | 35 |
| 7 | DIPEA (0.2) | 1.1 | TFE | 3 | 63 |
| 8c | DIPEA (0.2) | 1.1 | TFE | 3 | 71 |
| 9c | DIPEA (0.5) | 1.1 | TFE | 3 | 77 |
| 10c | DIPEA (0.1) | 1.1 | TFE | 3 | 82 |
Reactions were performed with pyrrolidine, quinone monoacetal 2 (1.0 mmol), and base in solvent (3 mL) under reflux condition, isolated yield.
Reaction was performed at 60 °C.
TFE (10 mL) was used.
Although fast assembly of the octahydro-dipyrroloquinoline skeleton has been achieved using several pyrrolidine derivatives (most studies produced a mixture of equal amounts of syn and anti dimerization diastereomers),17 there have been no report using pyrrolidine as the starting material. Therefore, we also optimized the reaction condition that can only lead to the syn diastereomer 7 (Table 2). It was found that the yield of 7 was significantly increased when the reaction was carried out at a higher temperature (entry 2, Table 2). Reducing the amount of pyrrolidine resulted in a further increase in yield (entry 3). Using a stronger base such as DBU decreased the yield of the desired product. We therefore chose to add relatively less basic DIPEA to the reaction and obtained 7 in 60% yield, but the reactions took 72 hours to complete under these conditions (entries 1–5). The formation of an iminium ion is more favorable in protic solvents, so we tried the reaction in protic solvents such as isopropanol or TFE and found that the reaction time was considerably shorter in both cases, but the yield of 7 was only 35% in isopropanol (entry 6). The reaction time was reduced to 3 h with TFE as solvent, while the yield of 7 was increased to 63% (entry 7). We speculate that TFE may be able to stabilize the iminium ion intermediate in the reaction.18 We continued to investigate the effect of concentration and amount of the base on the reaction and finally obtained 7 in 82% yield with the addition of 0.1 equivalent of DIPEA (entries 8–10).
Based on the mechanism studies of previously reported redox-neutral α-alkylations of pyrrolidine and substituted pyrrolidines8,9,11 and the preparation of octahydro-dipyrroloquinoline scaffold with pyrrolidine derivatives,17g the mechanism for the formation of the arylated product and the octahydro-dipyrroloquinoline scaffold is shown in Scheme 5. The reaction of pyrrolidine with quinone monoacetal 2 resulted in the formation of iminium ion I, which was transferred to iminium ion II driven by aromatization (the hydrogen atom on the α-carbon of pyrrolidine is abstracted by the applied base). If iminium ion II was captured by an aromatic nucleophile, the reaction produced an α-aryl-substituted pyrrolidine (path A). If the aromatic nucleophile is poorly nucleophilic or the nucleophilic reagent is absent from the reaction system, the cyclic iminium ion II will tautomerize to enamine III. The attack of iminium ion II by enamine III gave intermediate IV, then an intramolecular Pictet–Spengler type reaction of the newly formed iminium ion gave octahydro-dipyrroloquinoline skeleton (path B).
Scheme 5. Proposed mechanism.

Conclusions
In conclusion, we have achieved the direct arylation of pyrrolidine at the α-position in a one-pot method. In the absence of aromatic nucleophile, the same reaction in TFE efficiently constructed an octahydro-dipyrroloquinoline skeleton of the natural product incargranine B in one step. In addition, we could remove the N-aryl group from the α-arylated pyrrolidines in a very high yield. As an alternative to metal-mediated pyrrolidine α-functionalization, our approach is a green and practical method for the synthesis of α-aryl-substituted pyrrolidines.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was financially supported by the NSFC (22171150 and 21772105).
Electronic supplementary information (ESI) available. CCDC 2292799. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d4ra00983e
Notes and references
- (a) Haddad M. Imogaï H. Larchevêque M. The First Enantioselective Synthesis of the cis-2-Carboxy-5-phenylpyrrolidine. J. Org. Chem. 1998;63:5680–5683. [Google Scholar]; (b) Severino E. A. Costenaro E. R. Garcia A. L. L. Correia C. R. D. Probing the Stereoselectivity of the Heck Arylation of Endocyclic Enecarbamates with Diazonium Salts. Concise Syntheses of (2S,5R)-Phenylproline Methyl Ester and Schramm's C-Azanucleoside. Org. Lett. 2003;5:305–308. doi: 10.1021/ol027268a. [DOI] [PubMed] [Google Scholar]; (c) Perni R. B. Farmer L. J. Cottrell K. M. Court J. J. Courtney L. F. Deininger D. D. Gates C. A. Harbeson S. L. Kim J. L. Lin C. Lin K. Luong Y.-P. Maxwell J. P. Murcko M. A. Pitlik J. Rao B. G. Schairer W. C. Tung R. D. Van Drie J. H. Wilson K. Thomson J. A. Inhibitors of Hepatitis C Virus NS3·4A Protease. Part 3: P2 Proline Variants. Bioorg. Med. Chem. Lett. 2004;14:1939–1942. doi: 10.1016/j.bmcl.2004.01.078. [DOI] [PubMed] [Google Scholar]; (d) Hirayama C. Ono H. Tamura Y. Nakamura M. C-Prolinylquercetins from the Yellow Cocoon Shell of the Silkworm, Bombyx Mori. Phytochemistry. 2006;67:579–583. doi: 10.1016/j.phytochem.2005.11.030. [DOI] [PubMed] [Google Scholar]; (e) Larsen A. M. Venskutonyte R. Valadés E. A. Nielsen B. Pickering D. S. Bunch L. Discovery of a New Class of Ionotropic Glutamate Receptor Antagonists by the Rational Design of (2S,3R)-3-(3-Carboxyphenyl)-pyrrolidine-2-carboxylic Acid. ACS Chem. Neurosci. 2011;2:107–114. doi: 10.1021/cn100093f. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dey J. Deckwerth T. L. Kerwin W. S. Casalini J. R. Merrell A. J. Grenley M. O. Burns C. Ditzler S. H. Dixon C. P. Beirne E. Gillespie K. C. Kleinman E. F. Klinghoffer R. A. Voruciclib, a Clinical Stage Oral CDK9 Inhibitor, Represses MCL-1 and Sensitizes High-Risk Diffuse Large B-cell Lymphoma to BCL2 Inhibition. Sci. Rep. 2017;7:18007. doi: 10.1038/s41598-017-18368-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Banik S. M. Levina A. Hyde A. M. Jacobsen E. N. Lewis Acid Enhancement by Hydrogen-Bond Donors for Asymmetric Catalysis. Science. 2017;358:761–764. doi: 10.1126/science.aao5894. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Lu H.-B. Lv J.-M. Zhou C.-H. Zhou M. Fang Y.-X. Dong J.-X. Kato T. Liu Y. Maruoka K. Remarkable Effect of tert-Amine Additives in the Asymmetric Direct Michael Reaction of Ketones with β-Arylnitroethenes Catalyzed by an L-Hydroxyproline-Based Amino Tf-Amide Organocatalyst. Eur. J. Org Chem. 2021:1909–1912. [Google Scholar]; (c) Ren X.-K. Couture B. M. Liu N.-Y. Lall M. S. Kohrt J. T. Fasan R. Enantioselective Single and Dual α-C–H Bond Functionalization of Cyclic Amines via Enzymatic Carbene Transfer. J. Am. Chem. Soc. 2023;145:537–550. doi: 10.1021/jacs.2c10775. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Wang T. Wang W.-B. Fu Y.-M. Zhu C.-F. Cheng L.-J. You Y.-E. Wu X. Li Y.-G. Asymmetric Double Oxidative [3 + 2] Cycloaddition for the Synthesis of CF3-Containing Spiro[pyrrolidin-3,2’-oxindole] Org. Lett. 2023;25:3152–3156. doi: 10.1021/acs.orglett.3c01083. [DOI] [PubMed] [Google Scholar]
- (a) Schwartz R. E. Liesch J. Hensens O. Zitano L. Honeycutt S. Garrity G. Fromtling R. A. Onishi J. Monaghan R. L-657, 398, A Novel Antifungal Agent: Fermentation, Isolation, Structural Elucidation and Biological Properties. J. Antibiot. 1988;41:1774–1779. doi: 10.7164/antibiotics.41.1774. [DOI] [PubMed] [Google Scholar]; (b) Johnson J. H. Phillipson D. W. Kahle A. D. The Relative and Absolute Stereochemistry of the Antifungal Agent Preussin. J. Antibiot. 1989;42:1184–1185. doi: 10.7164/antibiotics.42.1184. [DOI] [PubMed] [Google Scholar]; (c) Buttachon S. Ramos A. A. Inácio Â. Dethoup T. Gales L. Lee M. Costa P. M. Silva A. M. S. Sekeroglu N. Rocha E. Pinto M. M. M. Pereira J. A. Kijjoa A. Bis-Indolyl Benzenoids, Hydroxypyrrolidine Derivatives and Other Constituents from Cultures of the Marine Sponge-Associated Fungus Aspergillus candidus KUFA0062. Mar. Drugs. 2018;16:119. doi: 10.3390/md16040119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Seabra R. Malhão F. Correia A. Costa C. Kijjoa A. Rocha E. Effects and Mechanisms of Action of Preussin, a Marine Fungal Metabolite, against the Triple-Negative Breast Cancer Cell Line, MDA-MB-231, in 2D and 3D Cultures. Mar. Drugs. 2023;21:166. doi: 10.3390/md21030166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grollman A. P. Inhibitors of Protein Biosynthesis II. Mode of Action of Anisomycin. J. Biol. Chem. 1967;242:3226–3233. [PubMed] [Google Scholar]
- Shibano M. Tsukamoto D. Masuda A. Tanaka Y. Kusano G. Two New Pyrrolidine Alkaloids, Radicamines A and B, as Inhibitors of α-Glucosidase from Lobelia chinensis LOUR. Chem. Pharm. Bull. 2001;49:1362–1365. doi: 10.1248/cpb.49.1362. [DOI] [PubMed] [Google Scholar]
- Shen Y.-H. Su Y.-Q. Tian J.-M. Lin S. Li H.-L. Tang J. Zhang W.-D. A Unique Indolo-[1,7]naphthyridine Alkaloid from Incarvillea mairei var. grandiflora (Wehrh.) Grierson. Helv. Chim. Acta. 2010;93:2393–2396. [Google Scholar]
- Brown P. D. Willis A. C. Sherburn M. S. Lawrence A. L. Total Synthesis and Structural Revision of the Alkaloid Incargranine B. Angew. Chem., Int. Ed. 2013;52:13273–13275. doi: 10.1002/anie.201307875. [DOI] [PubMed] [Google Scholar]
- For reviews on amine a-C-H functionalization, see: ; (a) Dutta S. Li B.-W. Rickertsen D. R. L. Valles D. A. Seidel D. C–H Bond Functionalization of Amines: A Graphical Overview of Diverse Methods. SynOpen. 2021;5:173–228. doi: 10.1055/s-0040-1706051. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Chen W.-J. Seidel D. Condensation-Based Methods for the C–H Bond Functionalization of Amines. Synthesis. 2021;53:3869–3908. doi: 10.1055/a-1631-2140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For selected reports of intermolecular redox-neutral α-functionalization of pyrrolidines, see: ; (a) Ma L. Chen W. Seidel D. Redox-Neutral α-Cyanation of Amines. J. Am. Chem. Soc. 2012;134:15305–15308. doi: 10.1021/ja308009g. [DOI] [PubMed] [Google Scholar]; (b) Das D. Seidel D. Redox-Neutral α-C–H Bond Functionalization of Secondary Amines with Concurrent C–P Bond Formation/N-Alkylation. Org. Lett. 2013;15:4358–4361. doi: 10.1021/ol401858k. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Das D. Sun A. X. Seidel D. Redox-Neutral Copper(II) Carboxylate Catalyzed α-Alkynylation of Amines. Angew. Chem., Int. Ed. 2013;52:3765–3769. doi: 10.1002/anie.201300021. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chen W. Seidel D. The Redox-Mannich Reaction. Org. Lett. 2014;16:3158–3161. doi: 10.1021/ol501365j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Li Z.-P. Li C.-J. CuBr-Catalyzed Efficient Alkynylation of sp3 C−H Bonds Adjacent to a Nitrogen Atom. J. Am. Chem. Soc. 2004;126:11810–11811. doi: 10.1021/ja0460763. [DOI] [PubMed] [Google Scholar]; (b) Campos K. R. Klapars A. Waldman J. H. Dormer P. G. Chen C.-Y. Enantioselective, Palladium-Catalyzed α-Arylation of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2006;128:3538–3539. doi: 10.1021/ja0605265. [DOI] [PubMed] [Google Scholar]; (c) Cordier C. J. Lundgren R. J. Fu G. C. Enantioconvergent Cross-Couplings of Racemic Alkylmetal Reagents with Unactivated Secondary Alkyl Electrophiles: Catalytic Asymmetric Negishi α-Alkylations of N-Boc-pyrrolidine. J. Am. Chem. Soc. 2013;135:10946–10949. doi: 10.1021/ja4054114. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Chang Z. Huang J.-L. Wang S. Chen G. Zhao H. Wang R. Zhao D.-P. Copper Catalyzed Late-Stage C(Sp3)-H Functionalization of Nitrogen Heterocycles. Nat. Commun. 2021;12:4342. doi: 10.1038/s41467-021-24671-y. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Yao W. Lu C.-J. Zhan L.-W. Wu Y. Feng J. Liu R.-R. Enantioselective Synthesis of N-N Atropisomers by Palladium-Catalyzed C−H Functionalization of Pyrroles. Angew. Chem., Int. Ed. 2023;62:e202218871. doi: 10.1002/anie.202218871. [DOI] [PubMed] [Google Scholar]
- Seidel D. The Azomethine Ylide Route to Amine C−H Functionalization: Redox-Versions of Classic Reactions and a Pathway to New Transformations. Acc. Chem. Res. 2015;48:317–328. doi: 10.1021/ar5003768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Cheng Y.-F. Rong H.-J. Yi C.-B. Yao J.-J. Qu J. Redox-Triggered α-C–H Functionalization of Pyrrolidines: Synthesis of Unsymmetrically 2,5-Disubstituted Pyrrolidines. Org. Lett. 2015;17:4758–4761. doi: 10.1021/acs.orglett.5b02298. [DOI] [PubMed] [Google Scholar]; (b) Rong H.-J. Yao J.-J. Li J.-K. Qu J. Molecular Iodine-Mediated α-C–H Oxidation of Pyrrolidines to N,O-Acetals: Synthesis of (±)-Preussin by Late-Stage 2,5- Difunctionalizations of Pyrrolidine. J. Org. Chem. 2017;82:5557–5565. doi: 10.1021/acs.joc.7b00361. [DOI] [PubMed] [Google Scholar]
- (a) Chen W.-J. Wilde R. G. Seidel D. Redox-Neutral α-Arylation of Amines. Org. Lett. 2014;16:730–732. doi: 10.1021/ol403431u. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Haldar S. Mahato S. Jana C. K. Metal- and Oxidant-Free Direct sp3 C–H Arylation of Pyrrolidine. Asian J. Org. Chem. 2014;3:44–47. [Google Scholar]; (c) Haldar S. Roy S. K. Maity B. Koley D. Jana C. K. Regio- and Diastereoselective and Enantiospecific Metal-Free C(sp3)-H Arylation: Facile Access to Optically Active 5-Aryl 2,5-Disubstituted Pyrrolidines. Chem.–Eur. J. 2015;21:15290–15298. doi: 10.1002/chem.201502297. [DOI] [PubMed] [Google Scholar]
- Yi C.-B. She Z.-Y. Cheng Y.-F. Qu J. Redox-Neutral α-C−H Functionalization of Pyrrolidin-3-ol. Org. Lett. 2018;20:668–671. doi: 10.1021/acs.orglett.7b03807. [DOI] [PubMed] [Google Scholar]
- (a) Jurberg I. D. Peng B. Wöstefeld E. Wasserloos M. Maulide N. Intramolecular Redox-Triggered C−H Functionalization. Angew. Chem., Int. Ed. 2012;51:1950–1953. doi: 10.1002/anie.201108639. [DOI] [PubMed] [Google Scholar]; (b) Shang M. Chan J. Z. Cao M. Chang Y.-J. Wang Q.-F. Cook B. Torker S. Wasa M. C−H Functionalization of Amines via Alkene-Derived Nucleophiles through Cooperative Action of Chiral and Achiral Lewis Acid Catalysts: Applications in Enantioselective Synthesis. J. Am. Chem. Soc. 2018;140:10593–10601. doi: 10.1021/jacs.8b06699. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Liu Y.-B. Diao H.-L. Hong G.-R. Edward J. Zhang T. Yang G.-Q. Yang B.-M. Zhao Y. Iridium-Catalyzed Enantioconvergent Borrowing Hydrogen Annulation of Racemic 1,4-Diols with Amines. J. Am. Chem. Soc. 2023;145:5007–5016. doi: 10.1021/jacs.2c09958. [DOI] [PubMed] [Google Scholar]
- Tan D.-P. Chou G.-X. Wang Z.-T. Three New Alkaloids from Senecio Scandens. Chem. Nat. Compd. 2014;50:329–332. [Google Scholar]
- (a) Fustero S. Bello P. Miró J. Sánchez-Roselló M. Maestro M. A. González J. del Pozo C. Gold Catalyzed Stereoselective Tandem Hydroamination–Formal Aza-Diels–Alder Reaction of Propargylic Amino Esters. Chem. Commun. 2013;49:1336–1338. doi: 10.1039/c2cc37796a. [DOI] [PubMed] [Google Scholar]; (b) Miró J. Sánchez-Roselló M. González J. del Pozo C. Fustero S. Gold-Catalyzed Tandem Hydroamination/Formal Aza-Diels–Alder Reaction of Homopropargyl Amino Esters: A Combined Computational and Experimental Mechanistic Study. Chem.–Eur. J. 2015;21:5459–5466. doi: 10.1002/chem.201406224. [DOI] [PubMed] [Google Scholar]; (c) Yu X.-L. Kuang L.-P. Chen S. Zhu X.-L. Li Z.-L. Tan B. Liu X.-Y. Counteranion-Controlled Unprecedented Diastereo- and Enantioselective Tandem Formal Povarov Reaction for Construction of Bioactive Octahydro-Dipyrroloquinolines. ACS Catal. 2016;6:6182–6190. [Google Scholar]; (d) Ma C.-L. Li X.-H. Yu X.-L. Zhu X.-L. Hu Y.-Z. Dong X.-W. Tan B. Liu X.-Y. Gold-Catalyzed Tandem Synthesis of Bioactive Spiro-Dipyrroloquinolines and its Application in the One-Step Synthesis of Incargranine B Aglycone and Seneciobipyrrolidine (I) Org. Chem. Front. 2016;3:324–329. [Google Scholar]; (e) Liu L.-Y. Wang C. Liu Q.-Q. Kong Y.-F. Chang W.-X. Li J. Copper(II) Trifluoromethanesulfonate Catalyzed Hydroamination Cyclization–Dimerization Cascade Reaction of Homopropargylic Amines for the Construction of Complex Fused Nitrogen-Containing Tetracycles. Eur. J. Org Chem. 2016:3684–3690. [Google Scholar]; (f) Li S.-S. Zhou L. Wang L. Zhao H.-L. Yu L.-P. Xiao J. Organocatalytic C(sp3)−H Functionalization via Carbocation-Initiated Cascade [1,5]-Hydride Transfer/Cyclization: Synthesis of Dihydrodibenzo[b,e]azepines. Org. Lett. 2018;20:138–141. doi: 10.1021/acs.orglett.7b03492. [DOI] [PubMed] [Google Scholar]; (g) Zhang C. Li J.-B. Wang X. Shen X. Zhu D.-R. Shen R.-W. Metal-Free Synthetic Shortcut to Octahydro-Dipyrroloquinoline Skeletons from 2,5-Cyclohexadienone Derivatives and L-Proline. J. Org. Chem. 2021;86:10397–10406. doi: 10.1021/acs.joc.1c01083. [DOI] [PubMed] [Google Scholar]; (h) Liu C. Tan X.-W. Zhan L.-Z. Jing Y.-R. Wu W.-Q. Ke Z.-F. Jiang H.-F. Palladium-Catalyzed Cascade Cyclization for the Synthesis of Fused Benzo-Aza-Oxa-[5-6-5] Tetracycles. Angew. Chem., Int. Ed. 2022;61:e202215020. doi: 10.1002/anie.202215020. [DOI] [PubMed] [Google Scholar]; (i) Matsumoto K. Nakano R. Yamada K. Hirokane T. Yoshida M. Catalytic and Aerobic Oxidative C−H Annulation Reaction of Saturated Cyclic Amines for Synthesis of Dipyrroloquinolines. Adv. Synth. Catal. 2023;365:323–329. [Google Scholar]
- Tian F.-X. Qu J. Studies on the Origin of the Stabilizing Effects of Fluorinated Alcohols and Weakly Coordinated Fluorine-Containing Anions on Cationic Reaction Intermediates. J. Org. Chem. 2022;87:1814–1829. doi: 10.1021/acs.joc.1c02361. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.