

Abstract
The success of precision medicine relies upon collecting data from many individuals at the population level. Although advancing technologies have made such large-scale studies increasingly feasible in some disciplines such as genomics, the standard workflows currently implemented in untargeted metabolomics were developed for small sample numbers and are limited by the processing of liquid chromatography/mass spectrometry data. Here we present an untargeted metabolomics workflow that is designed to support large-scale projects with thousands of biospecimens. Our strategy is to first evaluate a reference sample created by pooling aliquots of biospecimens from the cohort. The reference sample captures the chemical complexity of the biological matrix in a small number of analytical runs, which can subsequently be processed with conventional software such as XCMS. Although this generates thousands of so-called features, most do not correspond to unique compounds from the samples and can be filtered with established informatics tools. The features remaining represent a comprehensive set of biologically relevant reference chemicals that can then be extracted from the entire cohort’s raw data on the basis of m/z values and retention times by using Skyline. To demonstrate applicability to large cohorts, we evaluated >2000 human plasma samples with our workflow. We focused our analysis on 360 identified compounds, but we also profiled >3000 unknowns from the plasma samples. As part of our workflow, we tested 14 different computational approaches for batch correction and found that a random forest-based approach outperformed the others. The corrected data revealed distinct profiles that were associated with the geographic location of participants.
Keywords: untargeted metabolomics, human plasma, sample preparation, high-resolution mass spectrometry, data analysis, batch correction
Graphical Abstract:
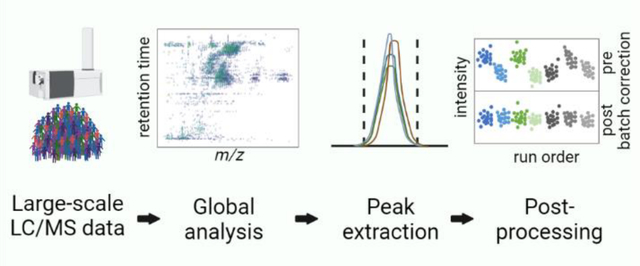
Introduction
The goal of precision medicine is to identify subgroups within the population for whom strategies to prevent, diagnose, and treat disease states can be uniquely tailored.1 It is expected that studies of large, diverse, and longitudinal cohorts will be critical to accelerate progress toward this end over the next decade.2 Indeed, research projects having hundreds of thousands of participants are already emerging with the FinnGen3, UK Biobank,4 and All of Us cohorts5. Given the key role of metabolism in health and diagnostics, the application of untargeted metabolomics to big cohorts promises to inform the practice of precision medicine in ways that are highly complementary to other technologies such as genomics. At this time, however, standard data-processing workflows used in untargeted metabolomics are not amenable to such large sample sizes.
When performing untargeted metabolomics with liquid chromatography/mass spectrometry (LC/MS), a typical biological specimen yields thousands of signals having unique m/z values and retention times, often referred to as “features”. After features are detected from each individual sample, it must be determined which represent the same analyte across all of the LC/MS runs, a process known as alignment or correspondence determination.6 The intensity of every feature from each aligned sample must then be assessed. A complication is that experimental drift is compound specific and introduces nonlinear measurement errors of variable magnitude throughout an experiment. Considering the number of features in a conventional untargeted metabolomics experiment, it is therefore impractical to process and interpret the data by manually inspecting all of the features at the global scale.
Over the last two decades, several software platforms such as mzMine, XCMS, and MS-DIAL have emerged for automated processing of untargeted metabolomics data. These informatics tools are highly effective and widely used for the analysis of small cohorts, but they were not designed to support projects with thousands of samples. Although methods for feature detection can generally be applied to each data file separately, making analysis of large sample numbers cumbersome but feasible, algorithms for correspondence determination are not as readily scaled. Methods such as Obiwarp within XCMS were designed to process all of the data files on one computing workstation at the same time. As the number of samples being evaluated grows, the amount of memory required for data processing increases and can eventually prevent the analysis from being completed. The specific number of samples that can be supported depends upon the user’s available computing power and software, but a recent report indicated that programs for untargeted metabolomics reproducibly crash when processing 250 data files or more.7 Although approaches, such as SLAW7, have developed modified algorithms with a lower computational overhead, user-friendly tools for processing large sample sets remain limited.
The objective of this work was to develop an untargeted metabolomics workflow to support the Long-Life Family Study (LLFS)8, a population-based effort to investigate exceptional longevity and healthy aging. LLFS is a longitudinal, multicenter, multinational, and multigenerational study that requires multi-omics analysis of > 12,000 human plasma samples. Here, as a proof of concept, we present a metabolomics analysis of ~2000 samples. Rather than developing new algorithms for processing large datasets, our strategy was to integrate conventional informatics tools into a workflow that minimizes the data burden of untargeted metabolomics. We first created pooled reference samples by mixing small aliquots of plasma samples from different participant subsets.9,10 We then subjected the pooled reference samples to the standard data-processing pipeline in untargeted metabolomics, thereby generating a table of tens of thousands of features. After filtering features arising from background and degeneracy, we obtained a comprehensive set of unique and biologically relevant metabolites that we could subsequently extract from the data of each individual plasma sample in the cohort by using m/z values and retention times within Skyline. Establishing this workflow enabled us to optimize methods to extract metabolites from plasma and to correct for batch effects and retention-time drifts in population-based studies such as LLFS.
Experimental Section
Samples
Blood samples were collected at participants’ homes in dipotassium ethylenediaminetetraacetic acid (K2- EDTA) collection tubes, which were immediately placed on a frozen gel pack. After shipment to the laboratory via courier or postal service, the samples were then centrifuged to isolate plasma, which was subsequently stored at −80 °C. Pooled samples were prepared from a subset of plasma samples to serve as quality control (QC) samples and for use in peak-list formation and metabolite identification (see Supporting Information). The QC sample was prepared from 58 samples of the first analysis batch. We thereby avoided subjecting samples to an additional freeze-thaw cycle. Generating a pooled sample from all subject specimens in the study was unfeasible because some samples were not available when the analysis started (blood draws were still ongoing). QC aliquots were stored at −80 °C. Additionally, a SPLASH Lipidomix kit (Avanti Polar Lipids), a deuterium labeled lipid mix designed for human plasma analysis, was used as an internal standard in QC samples for lipid metabolite analysis. The QC sample was injected after every 12th research sample.
Metabolite Extraction, LC/MS, and LC/MS/MS Analysis
To maximize coverage while minimizing sample-preparation time, we performed a solid-phase extraction (SPE) to isolate polar and lipid metabolites. One batch of plasma samples at a time was thawed on ice and vortexed. Each batch typically consisted of 92 research samples, 2 QC samples, and 2 blanks. An aliquot of each sample was then transferred into a 96-well SPE plate. A two-step extraction with either acetonitrile/methanol and methyl tert-butyl ether/methanol was used to obtain polar and lipid metabolites in separate fractions (Figure S1). Using SPE eliminates the need for a centrifugation to remove the protein fraction of the samples. The lipid extract was dried under a nitrogen flow and reconstituted prior to analysis via reversed-phase (RP) chromatography coupled to high-resolution mass spectrometry (HRMS) in positive mode. Polar metabolite extracts were directly analyzed (without any drying step) via hydrophilic interaction liquid chromatography (HILIC) coupled to HRMS in negative mode. For both LC/MS analyses, samples were randomized. Additionally, LC/MS/MS data were acquired to aid metabolite identification. Even though we did not collect positive-mode and negative-mode data for both lipid and polar metabolite extracts because of time constraints, doing so would be beneficial when resources permit. Blank samples were injected at the beginning and end of each worklist and used for background peak detection/removal. Representative total ion chromatograms for blank, study, and QC samples for both lipid and polar metabolite extracts are shown in Figure S2. We note that, although substantial signal is observed for the blank samples, features whose intensities were not at least three times higher in study samples than the blank were removed from downstream analysis. A detailed description of the metabolite extraction and LC/MS analysis can be found in the Supporting Information.
Generating Peak Lists
A peak list for polar metabolites was generated by combining the results of centwave11 peak detection (within XCMS12), background subtraction, and adduct selection (CAMERA13) from six pooled samples composed of distinct sample subsets. Additionally, a similar workflow was performed within the AcquireX14 software on three additional pooled samples, and the unfiltered features from this analysis were combined with the XCMS results. The R and Python scripts used to perform the peak detection analysis are available on GitHub (https://github.com/pattilab/metabolomics_workflow) and includes the values of all parameters utilized. A peak list for the lipid metabolites was directly generated based on identifications from Lipid Annotator (Agilent Technologies). We note that any workflow or software can be used to generate peaks lists.
Metabolite Identification
Identification of polar metabolites was supported by matching the accurate mass and MS/MS fragmentation data to our in-house MS/MS library created from authentic reference standards and online MS/MS libraries with our DecoID15 software. For online database searching, the top hit for each feature with a dot-product similarity of greater than 80 was considered as the putative identification. These results were further filtered by using in-house retention times, predicted retention times from a method similar to ReTip16, and manual curation to remove noise peaks, interfered peaks, and incorrect identifications. MSI identification levels17 are given in Tables S1 and S2 for polar and lipid metabolites, respectively. Code and scripts used to perform the automated portion of the metabolite identification workflow are available on GitHub. Lipid iterative MS/MS data were annotated with the Lipid Annotator, and lipid identifications were provided as sum compositions because insufficient information was available to deduce specific fatty-acid compositions. Lipid identifications were subject to the same manual curation as applied to the polar metabolite data. We note that any workflow or software can be used for compound identification.
Extracting Peak Areas
Following the generation of a peak list and metabolite identification, all data files were analyzed in Skyline18 (version 20.1.0.155) batch per batch to obtain peak areas. The m/z values of the metabolite target lists were used to extract peak areas under consideration of retention times or indexed retention times (iRT) (see Supporting Information). Because the data were being acquired over several months, 14 different batch correction approaches were tested for peak-area normalization (see Supporting Information).
Results and Discussion
The presented strategy to analyze large cohorts with untargeted metabolomics is based on the observation that most of the features in an experiment do not correspond to unique metabolites of biological relevance.19 Rather than attempting to evaluate each of these features in every sample, we use a small number of pooled reference samples to annotate features of interest. The process reduces the data burden of untargeted metabolomics such that informatics tools typically applied to targeted studies can be leveraged to profile identified and unidentified features efficiently and rapidly, without the need to subject each research sample to computationally intensive analyses (e.g., peak detection, correspondence determination, peak grouping, and metabolite identification). A schematic of the workflow is shown in Figure 1. As a demonstration, we applied our workflow to analyze a subset of ~2000 human plasma samples from LLFS. A description of each step of the workflow is provided below, with further details in the Experimental Section and Supporting Information.
Figure 1. Pipeline for generating and handling metabolomics data.
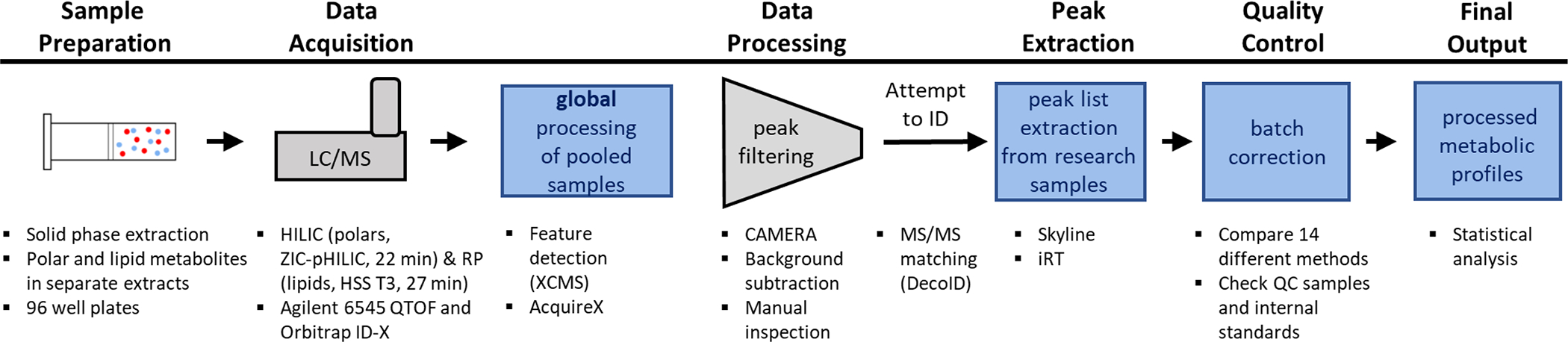
Polar and lipid metabolites are extracted from plasma samples into 96-well plates for LC/MS analysis. A pooled sample is prepared for feature detection, MS/MS acquisition, and use as a QC sample. Untargeted metabolomics analysis is performed on all samples. After detecting features from the pooled sample, background features and degeneracies are filtered. The remaining features are subjected to metabolite identification with DecoID and Lipid Annotator, and the returned putative identifications are manually curated. The peak areas for these metabolites (as well as any unknowns of interest) are extracted from the research samples by using Skyline. Retention-time shifts are manually corrected per batch for polar metabolites and automatically adjusted by using indexed retention times for lipid compounds. The peak areas are imputed and normalized to remove missing values and batch effects from the data. The final output contains the metabolite information (name, m/z, retention time) and normalized metabolite intensities for each research and QC sample.
Sample Preparation and Data Acquisition
Before LC/MS data were acquired for the LLFS sample set, we organized the 2005 plasma samples into 22 batches (mostly 92 samples per batch). Longitudinal samples from the same participant were included within the same batch. Polar and lipid metabolites were extracted from plasma samples into 96-well plates by using SPE. A pooled sample was prepared from the first batch of the LLFS sample set to serve as a QC sample. All samples were analyzed via LC/MS. Additional pooled samples (see Supporting Information) were prepared and analyzed via LC/MS/MS for metabolite identification.
Data Processing for Pooled Samples and Subsequent Data Extraction
The data collected from pooled samples were subjected to a standard processing workflow for untargeted metabolomics. Namely, we applied feature detection, grouping, filtering of background and degeneracy, and MS/MS-based compound identification to form a peak list of putatively identified metabolites that are suitable for multi-omics integration. The peak list of identified polar and lipid metabolites can be found in Tables S1 and S2, respectively. To demonstrate that this workflow is compatible with analysis of unknowns at the scale typically seen in untargeted metabolomics studies, we also added more than 3000 unidentified features to the peak list.
After generating the peak list, peak areas were extracted from the research and QC samples in a batch-by-batch fashion by using Skyline18. The Skyline command-line interface can be used to automatically generate a document for each batch. For polar metabolites separated with HILIC, retention times were stable for all samples within a batch (see Figure S3). Thus, retention-time bounds were set by inspection of the QC samples within each batch, and these bounds were applied to all samples by importing peak boundaries for each sample. Although we used a Python script to generate the peak boundary import file in the current work, this is no longer necessary when using the “Synchronize Integration” function in Skyline 21.2, which was released after we performed our data processing.
Retention-time values were generally stable across all experiments. Even though lipid metabolites tended to show more drift than polar metabolites, only five samples from all of our lipid runs had retention-time deviations greater than 0.25 min. Among those compounds that showed retention-time drifts, lysophosphatidylcholines and lysophosphatidylethanolamines were most pronounced in specific samples within each batch, which may be attributed to matrix effects (Figure S4). Accordingly, we applied the concept of iRT, which was initially established for proteomics20 and recently applied to lipids21. In brief, two to three compounds per lipid class that are of high intensity and easily distinguished from nearby peaks were chosen as indexing compounds. When importing all data files into Skyline in centroid mode, the peaks for the indexing compounds were consistently picked correctly. Incorrect peak assignments were corrected by inspection of the retention time replicate comparison plots and manual adjustment of peak boundaries to ensure that the apex of all indexing peaks was within the integration boundaries. The retention times for the indexing compounds were then used in a Python script to adjust and generate peak boundaries for all compounds in all samples (further details are provided in the Supporting Information). Upon importing those boundaries, correct integration was manually verified before peak areas were exported. We found that this strategy can efficiently correct retention time shifts of over 1 min. In total, after peak area extraction in Skyline, 172 identified polar metabolites, 188 identified lipid metabolites, and 3421 unidentified features from the polar data were profiled across 2001 research samples and 197 QC samples in the LLFS sample set. Of note, four research samples were excluded from downstream analysis because of uncharacteristically low signal abundance. Extracting peak areas with the process described above takes an experienced researcher 20–30 min per batch of samples analyzed. The major time investment is defining and curating the peak list that is formed from analysis of the pooled sample. In the case of LLFS, defining a peak list took several weeks and included acquisition of extensive MS/MS data, manual removal of artifact peaks and interferences, and review of metabolite identifications. The time required to form this peak list will depend on the study and the number of nonbiological and redundant signals filtered.
Comparing Metabolite Coverage from a Pooled Sample to Individually Measured Samples
Limiting data processing to pooled samples greatly reduces the computational burden of untargeted metabolomics, but a potential drawback is that low-abundance compounds only present in a small number of research samples will be missed. To determine the number of features missed by only performing data processing on pooled samples, we evaluated a subset of 58 research samples from LLFS. For this analysis, nine replicate injections of a pooled sample were created by mixing small aliquots of each of the 58 research samples. Then, feature detection, background subtraction, and selection for M +/− H ions on each of the 58 research samples, the 9 pooled samples, and 4 blank samples were performed. Peaks were detected with centwave,11 and features with peak areas less than 10,000 in a particular sample were classified as undetected in that sample. Background subtraction was accomplished by removing features having an intensity in the blank sample that was lower than one-third the intensity of the research or pooled sample.14 M +/– H ions were selected by applying the CAMERA software package.13
After applying our data-processing workflow, we made a list compiling all of the features that were detected from each of the 58 research samples. The list, which was composed of 5894 total features, included features that were only detected from a single research sample. A total of 3241 features (both identified and unknowns) from the list were detected in at least one replicate of the pooled sample (see Figure S5a). To assess the biological relevance of the features not detected in the pooled sample, we searched the m/z values of all features against endogenous metabolites in the Human Metabolome Database22 and the Kyoto Encyclopedia of Genes and Genomes23. In total, 40.4% of the features detected in the pooled sample had at least one hit in the databases. Of the features missed in the pooled sample, only 32.2% had a database hit, suggesting that these missed features were more likely to be exogenous metabolites (e.g., drugs, environmental toxins, and cosmetics). Additionally, features not detected in the pooled sample were found on average in less than 10% of the individual samples (see Figure S5b). In contrast, features detected in the pooled samples were present in the majority of the individual samples (Figure S5c). Features not detected in the pooled samples were also an order of magnitude lower in abundance (Figure S5d,e).
These data reveal that, although there is a reduction in the number of detected features when using a pooled sample, the missing features are above the limit of detection in only a small subset of research samples. It has been suggested previously that features not detected in at least 70% of the samples in an untargeted metabolomics study be excluded from the analysis, irrespective of the data-processing workflow.10 With this threshold, the number of features missed by only subjecting a pooled sample to data processing would be reduced to just 52 (<1% of total features). Another major complication of pursuing features that cannot be detected in the pooled samples is that they are challenging to normalize with respect to batch effects and technical variation. Thus, even though features detected in a small number of samples might be of biological interest, more sensitive methods will need to be developed to evaluate them. At the present time, no matter the data-processing workflow applied, these features are not well suited for large-scale studies.
Data Postprocessing
After extracting peak areas, missing values must be removed from the data to facilitate downstream processing. In our workflow using pooled samples, because of the targeted extraction of peak areas, missing values were infrequent (< 0.03% of all measurements) and most likely arose from metabolites at concentrations below the limit of detection of the instrument rather than random metabolite dropout during peak detection. Thus, we imputed missing values with the half minimum approach24,25. When comparing the frequency of missing values produced with targeted extraction and conventional XCMS-based processing, we found that 9.2% of all peak areas for a single batch of the LLFS samples were missing values when XCMS was employed. In contrast, less than 0.001% of all measurements were missing values when targeted extraction was applied to the same sample set (Figure S6a,b). Moreover, the technical variation in the output peak areas was 5x lower with targeted extraction of peak intensities compared to XCMS (Figure S6c).
Given the size of the LLFS study, the raw data had to be acquired over several months. When combining the extracted data from each group of 92 research samples, we observed strong batch effects as can be seen in Figure 2a and Figure S7a for identified lipid and polar metabolites, respectively. Unfortunately, samples from human subjects for large studies cannot always be randomized into analysis batches because of practical limitations such as longitudinal sample collection or funding timeline constraints. As a result, differences between batches may be due to technical or biological variation (Figure S8). For example, in the case of the LLFS sample set, the last six batches consisted of samples primarily collected in Denmark, whereas the other batches consisted largely of samples collected in the United States.
Figure 2. Correcting for batch effects in metabolomics data.
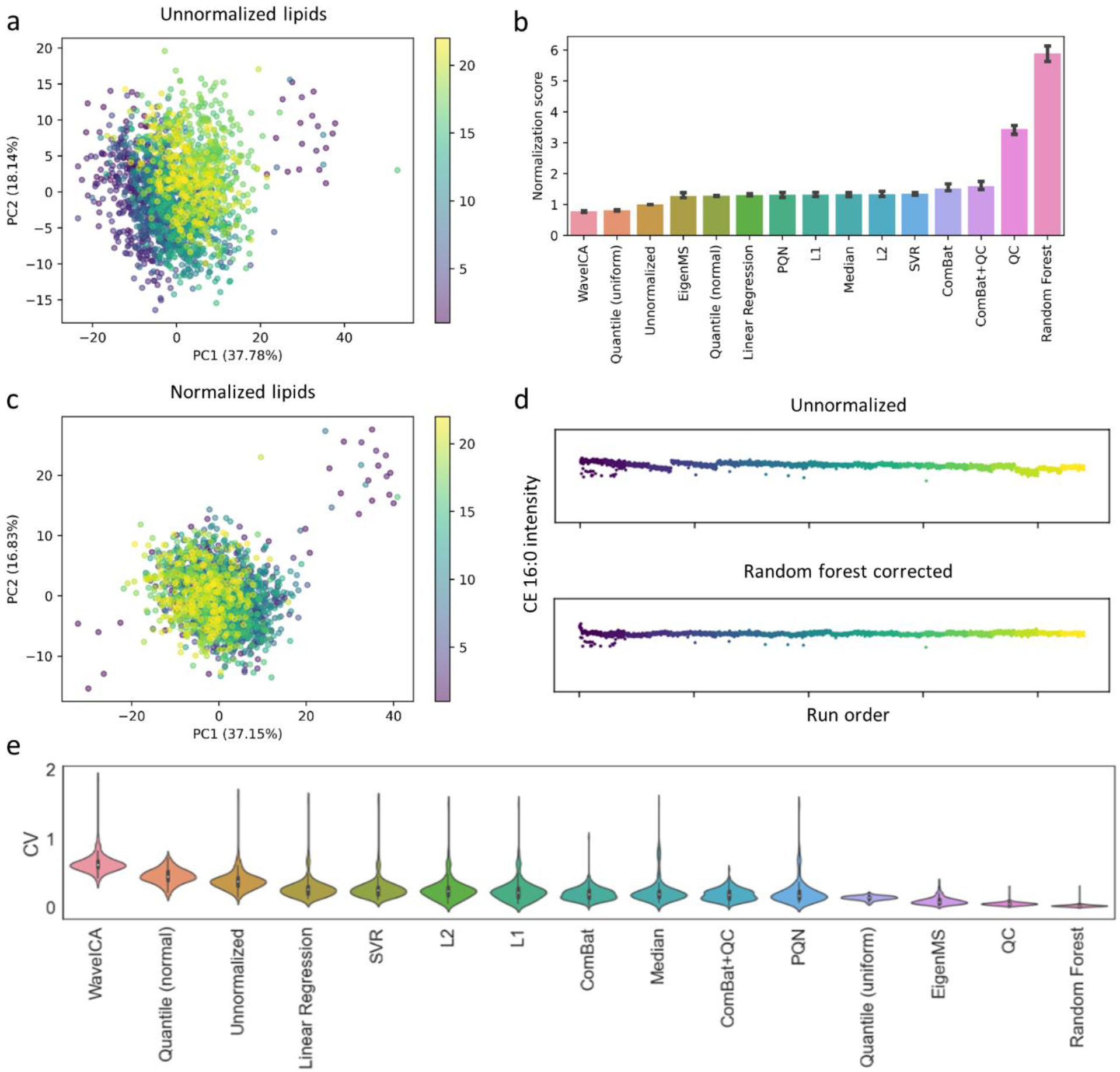
(a) Principal components analysis (PCA) of unnormalized lipid metabolic profiles shows strong batch effects. Each dot represents a unique sample. Dots are colored according to their corresponding batch number. (b) Comparison of 14 different batch-correction algorithms on the lipid metabolic profiles. The normalization score is the change in coefficient of variation (CV) for the research samples (relative to the unnormalized data) divided by the change in CV for the QC samples. A higher score indicates a reduction of technical variation. (c) PCA plot of random forest normalized lipid metabolic profiles shows reduced clustering by batch. (d) Intensity of CE 16:0 as a function of run order for both unnormalized (top) and random forest corrected data (bottom). (e) Violin plots showing the CV distribution of all compounds in the QC samples for each evaluated batch-correction algorithm. The polar metabolite counterpart to these data is shown in Figure S7.
To discriminate between biological and technical variation, identical QC samples across all batches are critical to evaluating and guiding batch-correction methods. Here, we tested 14 different normalization algorithms24,26–30 for their capability to minimize technical variation while keeping biological variance intact (details of this comparison are provided in the Supporting Information). The results of the analysis revealed that the performance of many of the methods is dataset dependent (Figure 2b and Figure S7b), but a random forest based batch-correction algorithm27 outperformed the other evaluated approaches in both the lipid and polar metabolite data. As a secondary validation, we also compared the internal standard variability in the polar metabolite data. We again saw that the random forest based correction reduced both intrabatch and inter-batch variability (Figure S9a–d). When comparing the performance of random forest to the other evaluated methods, we saw that QC, ComBat, and QC+ComBat performed similar to random forest in reducing internal standard CV values, with ComBat+QC achieving the lowest variability (Figure S9e). Overall, although random forest performed well on our data, we recommend that different batch-correction methods be tested before selecting the algorithm to apply to a dataset. Such an evaluation can be performed by adapting the code written for our analysis, which is available on GitHub. After correction, previously batch-affected metabolites no longer showed batch-dependent intensity drifts, and technical variation within the data was reduced (Figure 2c–e, Figure S7c–e). Although some batch-associated clustering remained in the polar metabolic profiles, when examining only the QC samples (Figure S10), no such clustering occurred. These findings indicate that biological differences between batches are driving the observed patterns, rather than technical variability. After batch correction, the metabolic profiles can then be subjected to downstream statistical analysis to identify interesting biological patterns.
Metabolic Profiles Cluster by Geographic Location
Collection of untargeted metabolomics data for the entire LLFS cohort is still underway, but we wished to demonstrate that the described workflow results in metabolic profiles containing biologically relevant information. As such, we investigated differences in the profiles of polar and lipid metabolites that could be attributed to the geographic origin of the samples. The LLFS samples were collected in four distinct field sites: Boston, MA; Pittsburg, PA; New York City, NY; and Odense, Denmark. Given the differences in dietary habits between the United States and Denmark, we surmised that the plasma metabolic profiles would be different between samples collected in the United States and Denmark. In fact, when analyzing the unnormalized metabolic profiles of the identified metabolites profiled in these data, 72 metabolites had maximum absolute fold changes greater than 2 between the field sites and p values of less than 0.05 (one-way ANOVA). Given that samples from the field sites were not uniformly distributed across the sample batches, however, technical variation may artificially introduce separation to their metabolic profiles. Indeed, after performing random forest batch correction, only 45 metabolites met the same statistical cutoffs, demonstrating the importance of batch correction to interpreting metabolomics data. These 45 metabolites led to pronounced clustering of the United States and Denmark samples (Figure 3a). Several of the differentiating lipid metabolites contain multiple unsaturations (e.g., CE 22:5, DG 36:4, LPC 20:5, PC 37:5, PC 38:6, PC 40:7, TG 56:7, TG 58:7, TG 58:8, TG 60:11, and TG 60:12) that may reflect differential abundance of omega-3 and omega-6 polyunsaturated fatty acids (DHA, EPA, linoleic acid, etc.) derived from the diet (Figure 3b). Scandinavian countries have been shown to have elevated levels of circulating omega-3 fatty acids when compared to the United States and other countries with Westernized food habits31, which are potentially due to higher per capita consumption of fish and shellfish. Of the polar metabolites, the most striking difference was inosine (Figure 3c). Inosine is a known dietary metabolite with high concentrations in cow milk32,33. This result indicates a difference in the amount of dairy consumption between individuals in the United States and Denmark, as has been suggested before.34
Figure 3. Metabolic profiles are reflective of geographic location.
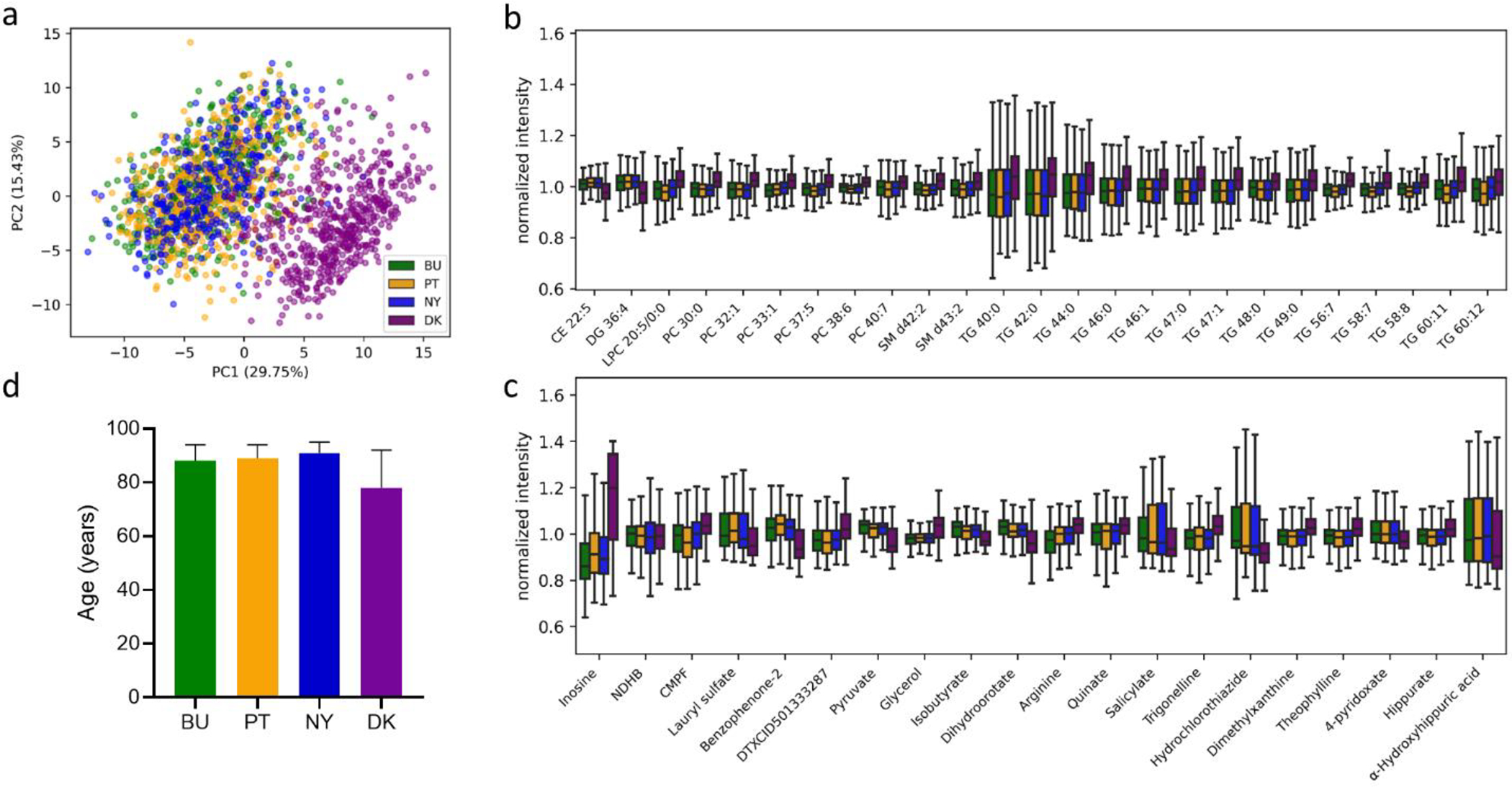
(a) Principal components analysis (PCA) of normalized metabolic profiles (polar and lipid metabolites) shows clustering based on United States (BU = Boston, NY = New York City, PT = Pittsburg) and Denmark (DK, Odense) field sites. Each dot represents a unique sample. Dots are colored according to geographic location. (b) Lipid and (c) polar metabolites associated with geographic location (|FC| > 2, p < 0.05, one-way ANOVA). (d) Age distribution for samples from the different field sites. Data shown are median ± interquartile range, NDHB, N,N-diethyl-4-hydroxybenzamide; CMPF, 3-carboxy-4-methyl-5-propyl-2-furanpropanoic acid.
Although the identified metabolites were the main focus of our analysis for LLFS, where metabolomics data will be linked to corresponding gene and protein measurements, we also wished to determine whether there were differences in unknowns between the sample groups. After filtering (see Supporting Information) and performing statistical analysis of >3000 unknowns from the polar metabolite extracts profiled in this study, we found 29 unique unknowns that were associated with field site location (|FC| > 2, p < 0.05, one-way ANOVA), as shown in Figure S11. The differences between fields sites were dramatic, but participants from Denmark were also on average younger than those from the United States (p <0.0001) and this could have contributed to the observed metabolic profiles (Figure 3d). Notably, however, a principal component analysis of those samples colored by age did not show an age-dependent pattern within United States or Denmark samples (Figure S12), suggesting that the differences cannot solely be attributed to age. An additional potential confounding factor is that the time between blood collection and centrifugation varied between samples and field sites. In a previous study, we showed that this delay can cause alterations in metabolite levels.35 Further statistical analysis of this dataset will be performed that considers covariates and genetic relationships between LLFS participants once data collection for LLFS is complete.
Conclusions
The trend in the omic sciences is to evaluate increasingly large sample sizes approaching tens of thousands to hundreds of thousands of specimens. Larger sample groups increase statistical power and may enable a study to distinguish between subpopulations within a group that have a unique treatment effect, which is the vision of precision medicine. The challenge of using exceptionally large sample cohorts, on the other hand, is the burden of collecting and processing high volumes of data. Applications of untargeted metabolomics to large sample cohorts have been limited because standard software programs are not compatible with population-based studies. In this work, we describe a workflow designed to perform untargeted metabolomics on >12,000 human plasma samples from the LLFS cohort.
To facilitate experimental throughput, we used SPE kits for isolating water-soluble and lipid metabolites. HRMS data can then be collected for each sample by using multiple analytical runs. There are a few potential drawbacks of our experimental approaches. First, because we only dedicated one instrument to data acquisition, polar metabolite extracts were stored while lipid metabolites were being analyzed. It is possible that the storage of the extracts led to the loss of signal for some compounds. Second, although we utilized two chromatographic methods to capture lipid and polar metabolites, some metabolite classes cannot be successfully retained or separated with either method, leading to incomplete coverage. Our metabolite coverage was also limited by using only a single instrument polarity for each fraction. When time and funding permit, acquiring data in both positive and negative mode would extend coverage. Third, given that we identified lipids from positive mode MS/MS spectra, only sum compositions could be determined. Acquiring additional MS/MS spectra in negative mode would allow for the elucidation of fatty-acid composition, and chromatographically resolved isomers could be included in our peak list as distinct species.
To reduce the computational burden of performing global processing of all HRMS data files within the cohort, we focused on processing data from a small number of pooled samples that are created by mixing aliquots of individual samples from our study. By initially limiting data processing to only the pooled samples, we can leverage standard informatics tools in untargeted metabolomics that have been optimized for small sample sizes. This not only facilitates feature detection, but also leads to a considerable data reduction from the removal of adducts, background signals, and other degeneracies. Analysis of the pooled sample does require a significant time investment, but it can be completed in parallel with data acquisition for the research samples and only needs to be performed once. Further, because peak areas for metabolites detected in the pooled sample can be extracted from the research samples as data are generated, the rate-limiting factor in the described workflow is data acquisition rather than data processing. This contrasts with conventional processing workflows that require all data to be acquired prior to analysis and curation.
In the preparation of the reference sample, care should be taken to ensure that pooled samples cover different sample groups and represent the biological diversity present in the study, such as age and gender. The pooled samples are intended to capture the totality of unique compounds across the entire study cohort but, in practice, unique compounds from individual samples are missed. An analysis of our data shows that unique compounds are most often missed when they are only present in a few participant samples at low concentrations, which causes them to be diluted below the limit of detection during pooling. Our results indicate that signals missing from the analysis of pooled samples are more likely to originate from rare exogenous compounds (e.g., chemicals from unique hygiene products or specific environments). While rare exogenous compounds may certainly be biologically interesting, their analysis will require the development of new peak detection, alignment, and annotation algorithms that can be scaled to thousands of samples without loss of functionality or accuracy. We point out that, even when conventional informatics workflows are used for untargeted metabolomics, features only detected in a low percentage of samples are usually discarded because of the difficulties in correcting for technical drift.
When processing untargeted metabolomics data with conventional workflows, retention times are aligned by using correspondence algorithms such as Obiwarp36. Here, we corrected for sample specific retention-time drifts with the application of iRT. This procedure requires some manual intervention from the user to ensure correct peak detection of indexing compounds and to adjust batch-to-batch variations in retention time. We demonstrated that the iRT approach corrected for retention-time drifts in lipid metabolites, but it was not necessary for polar metabolites because we did not observe intrabatch retention-time drifts in these data. Should retention-time drifts be observed for polar metabolites, the iRT approach could easily be extended by determining groups of compounds that have correlated retention-time drifts across samples and using them for indexing.
In large-scale longitudinal studies such as LLFS, it may not be feasible to fully randomize the order in which specimens are analyzed by LC/MS. Correcting for the technical variation between batches of samples is therefore required to measure biological differences accurately. Here, we evaluated 14 different approaches for batch correction and found that accounting for batch effects by using a random forest normalization to estimate drift in each reference chemical was the most effective. Given that normalization relies on QC samples, a drawback to the approach is that drift for chemicals absent from the QC sample cannot be estimated. Further, the QC samples may not reflect batch effects in research samples as the QC sample is repeatedly injected from the sample vial, potentially leading to degradation of chemicals sensitive to oxidation. This study used only a limited number of internal standards, but incorporating additional compounds that cover a broader range of chemical classes would be beneficial for assessing the possibility of metabolite degradation and for evaluating the overall quality of batch-correction results. We also only used a single QC sample in this work, but we recommend implementing a second QC sample that is not used for normalization to identify spurious batch corrections.
Although our approach is not without limitations, we have shown that it enables high-throughput applications of untargeted metabolomics at the population scale, such as LLFS. We also wish to highlight that our workflow does collect HRMS data for every research sample in the cohort. Thus, as new computational resources become available, the data generated can readily be re-evaluated with new tools.
Supplementary Material
Acknowledgments
The authors would like to thank all of the LLFS investigators for their efforts in the collection, coordination, and shipment of the samples analyzed in this study. We would also like to thank Nick Terzich and Thomas White for their contributions to sample preparation and analysis. Furthermore, the authors thank Mary Furlan Feitosa, Joseph H. Lee, Yujing Yao, and Zhezhen Jin for their valuable input during the preparation of this manuscript. LLFS is supported by the National Institutes of Health (NIH) grants U01 AG023749 and U19AG063893. Support to establish a large-scale metabolomics workflow was also provided by NIH grants U01CA235482 and R35ES028365. The TOC was created with BioRender.com.
Footnotes
Competing Interest Statement
The authors declare the following competing financial interests: The Patti laboratory has a research collaboration agreement with Thermo Fisher Scientific and receives financial support from Agilent Technologies. G.J.P is a scientific advisor for Cambridge Isotope Laboratories.
Data and Code Availability
Raw and processed LC/MS data will be made publicly available upon completion of the LLFS study. All code used to perform the analyses presented in this study are available on GitHub (https://github.com/pattilab/metabolomics_workflow). Google Colab notebooks to perform ComBat, QC, and random forest batch correction are linked at the GitHub page.
Supporting Information
Experimental details on sample extraction, LC/MS analysis of polar metabolites, LC/MS analysis of lipid metabolites, preparation of pooled samples, LC/MS/MS analysis, indexed retention times, batch-correction evaluation, analysis of unknown metabolites, and associated references (PDF)
References
- (1).Ashley EA Towards Precision Medicine. Nat. Rev. Genet. 2016, 17 (9), 507–522. 10.1038/nrg.2016.86. [DOI] [PubMed] [Google Scholar]
- (2).Denny JC; Collins FS Precision Medicine in 2030—Seven Ways to Transform Healthcare. Cell 2021, 184 (6), 1415–1419. 10.1016/j.cell.2021.01.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).FinnGen, a Global Research Project Focusing on Genome Data of 500,000 Finns, Launched. In EurekAlert! American Association for the Advancement of Science; EurekAlert! American Association for the Advancement of Science, 2017. [Google Scholar]
- (4).Bycroft C; Freeman C; Petkova D; Band G; Elliott LT; Sharp K; Motyer A; Vukcevic D; Delaneau O; O’Connell J; Cortes A; Welsh S; Young A; Effingham M; McVean G; Leslie S; Allen N; Donnelly P; Marchini J The UK Biobank Resource with Deep Phenotyping and Genomic Data. Nature 2018, 562 (7726), 203–209. 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).The “All of Us” Research Program. N. Engl. J. Med. 2019, 381 (7), 668–676. 10.1056/NEJMsr1809937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Mahieu NG; Genenbacher JL; Patti GJ A Roadmap for the XCMS Family of Software Solutions in Metabolomics. Curr. Opin. Chem. Biol. 2016, 30, 87–93. 10.1016/j.cbpa.2015.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Delabriere A; Warmer P; Brennsteiner V; Zamboni N SLAW: A Scalable and Self-Optimizing Processing Workflow for Untargeted LC-MS. Anal. Chem. 2021. 10.1021/acs.analchem.1c02687. [DOI] [PubMed] [Google Scholar]
- (8).Wojczynski MK; Jiuan Lin S; Sebastiani P; Perls TT; Lee J; Kulminski A; Newman A; Zmuda JM; Christensen K; Province MA; on behalf of the Long Life Family Study. NIA Long Life Family Study: Objectives, Design, and Heritability of Cross-Sectional and Longitudinal Phenotypes. J. Gerontol. Ser. A 2021, glab333. 10.1093/gerona/glab333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Sangster T; Major H; Plumb R; Wilson AJ; Wilson ID A Pragmatic and Readily Implemented Quality Control Strategy for HPLC-MS and GC-MS-Based Metabonomic Analysis. Analyst 2006, 131 (10), 1075–1078. 10.1039/B604498K. [DOI] [PubMed] [Google Scholar]
- (10).Broadhurst D; Goodacre R; Reinke SN; Kuligowski J; Wilson ID; Lewis MR; Dunn WB Guidelines and Considerations for the Use of System Suitability and Quality Control Samples in Mass Spectrometry Assays Applied in Untargeted Clinical Metabolomic Studies. Metabolomics 2018, 14 (6), 72. 10.1007/s11306-018-1367-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Tautenhahn R; Böttcher C; Neumann S Highly Sensitive Feature Detection for High Resolution LC/MS. BMC Bioinformatics 2008, 9 (1), 504. 10.1186/1471-2105-9-504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G XCMS: Processing Mass Spectrometry Data for Metabolite Profiling Using Nonlinear Peak Alignment, Matching, and Identification. Anal. Chem. 2006, 78 (3), 779–787. 10.1021/ac051437y. [DOI] [PubMed] [Google Scholar]
- (13).Kuhl C; Tautenhahn R; Böttcher C; Larson TR; Neumann S CAMERA: An Integrated Strategy for Compound Spectra Extraction and Annotation of Liquid Chromatography/Mass Spectrometry Data Sets. Anal. Chem. 2012, 84 (1), 283–289. 10.1021/ac202450g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Cho K; Schwaiger-Haber M; Naser FJ; Stancliffe E; Sindelar M; Patti GJ Targeting Unique Biological Signals on the Fly to Improve MS/MS Coverage and Identification Efficiency in Metabolomics. Anal. Chim. Acta 2021, 1149, 338210. 10.1016/j.aca.2021.338210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Stancliffe E; Schwaiger-Haber M; Sindelar M; Patti GJ DecoID Improves Identification Rates in Metabolomics through Database-Assisted MS/MS Deconvolution. Nat. Methods 2021, 18 (7), 779–787. 10.1038/s41592-021-01195-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Bonini P; Kind T; Tsugawa H; Barupal DK; Fiehn O Retip: Retention Time Prediction for Compound Annotation in Untargeted Metabolomics. Anal. Chem. 2020, 92 (11), 7515–7522. 10.1021/acs.analchem.9b05765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Sumner LW; Amberg A; Barrett D; Beale MH; Beger R; Daykin CA; Fan TW-M; Fiehn O; Goodacre R; Griffin JL; Hankemeier T; Hardy N; Harnly J; Higashi R; Kopka J; Lane AN; Lindon JC; Marriott P; Nicholls AW; Reily MD; Thaden JJ; Viant MR Proposed Minimum Reporting Standards for Chemical Analysis Chemical Analysis Working Group (CAWG) Metabolomics Standards Initiative (MSI). Metabolomics Off. J. Metabolomic Soc. 2007, 3 (3), 211–221. 10.1007/s11306-007-0082-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Adams KJ; Pratt B; Bose N; Dubois LG; St. John-Williams.; Perrott KM.; Ky.; Kapahi.; Sharma.; MacCoss.; Moseley.; Colton.; MacLean.; Schilling.; Thompson. Skyline for Small Molecules: A Unifying Software Package for Quantitative Metabolomics. J. Proteome Res. 2020, 19 (4), 1447–1458. 10.1021/acs.jproteome.9b00640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Mahieu NG; Patti GJ Systems-Level Annotation of a Metabolomics Data Set Reduces 25 000 Features to Fewer than 1000 Unique Metabolites. Anal. Chem. 2017, 89 (19), 10397–10406. 10.1021/acs.analchem.7b02380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Escher C; Reiter L; MacLean B; Ossola R; Herzog F; Chilton J; MacCoss MJ; Rinner O Using IRT, a Normalized Retention Time for More Targeted Measurement of Peptides. PROTEOMICS 2012, 12 (8), 1111–1121. 10.1002/pmic.201100463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Kirkwood KI; Christopher MW; Burgess JL; Littau SR; Foster K; Richey K; Pratt BS; Shulman N; Tamura K; MacCoss MJ; MacLean BX; Baker ES Development and Application of Multidimensional Lipid Libraries to Investigate Lipidomic Dysregulation Related to Smoke Inhalation Injury Severity. J. Proteome Res. 2022, 21 (1), 232–242. 10.1021/acs.jproteome.1c00820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wishart DS; Feunang YD; Marcu A; Guo AC; Liang K; Vázquez-Fresno R; Sajed T; Johnson D; Li C; Karu N; Sayeeda Z; Lo E; Assempour N; Berjanskii M; Singhal S; Arndt D; Liang Y; Badran H; Grant J; Serra-Cayuela A; Liu Y; Mandal R; Neveu V; Pon A; Knox C; Wilson M; Manach C; Scalbert A HMDB 4.0: The Human Metabolome Database for 2018. Nucleic Acids Res. 2018, 46 (D1), D608–D617. 10.1093/nar/gkx1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kanehisa M; Goto S KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28 (1), 27–30. 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Di Guida R; Engel J; Allwood JW; Weber RJM; Jones MR; Sommer U; Viant MR; Dunn WB Non-Targeted UHPLC-MS Metabolomic Data Processing Methods: A Comparative Investigation of Normalisation, Missing Value Imputation, Transformation and Scaling. Metabolomics 2016, 12 (5). 10.1007/s11306-016-1030-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Wei R; Wang J; Su M; Jia E; Chen S; Chen T; Ni Y Missing Value Imputation Approach for Mass Spectrometry-Based Metabolomics Data. Sci. Rep. 2018, 8 (1), 663. 10.1038/s41598-017-19120-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Johnson WE; Li C; Rabinovic A Adjusting Batch Effects in Microarray Expression Data Using Empirical Bayes Methods. Biostatistics 2007, 8 (1), 118–127. 10.1093/biostatistics/kxj037. [DOI] [PubMed] [Google Scholar]
- (27).Fan S; Kind T; Cajka T; Hazen SL; Tang WHW; Kaddurah-Daouk R; Irvin MR; Arnett DK; Barupal DK; Fiehn O Systematic Error Removal Using Random Forest for Normalizing Large-Scale Untargeted Lipidomics Data. Anal. Chem. 2019, 91 (5), 3590–3596. 10.1021/acs.analchem.8b05592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Deng K; Zhao F; Rong Z; Cao L; Zhang L; Li K; Hou Y; Zhu Z-J WaveICA 2.0: A Novel Batch Effect Removal Method for Untargeted Metabolomics Data without Using Batch Information. Metabolomics 2021, 17 (10), 87. 10.1007/s11306-021-01839-7. [DOI] [PubMed] [Google Scholar]
- (29).Karpievitch YV; Nikolic SB; Wilson R; Sharman JE; Edwards LM Metabolomics Data Normalization with EigenMS. PLOS ONE 2014, 9 (12), e116221. 10.1371/journal.pone.0116221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dieterle F; Ross A; Schlotterbeck G; Senn H Probabilistic Quotient Normalization as Robust Method to Account for Dilution of Complex Biological Mixtures. Application in 1H NMR Metabonomics. Anal. Chem. 2006, 78 (13), 4281–4290. 10.1021/ac051632c. [DOI] [PubMed] [Google Scholar]
- (31).Stark KD; Van Elswyk ME; Higgins MR; Weatherford CA; Salem N Global Survey of the Omega-3 Fatty Acids, Docosahexaenoic Acid and Eicosapentaenoic Acid in the Blood Stream of Healthy Adults. Prog. Lipid Res. 2016, 63, 132–152. 10.1016/j.plipres.2016.05.001. [DOI] [PubMed] [Google Scholar]
- (32).Schlimme E; Raezke KP; Ott FG Ribonucleosides as Minor Milk Constituents. Z. Ernahrungswiss. 1991, 30 (2), 138–152. 10.1007/BF01610069. [DOI] [PubMed] [Google Scholar]
- (33).Schlimme E; Martin D; Meisel H Nucleosides and Nucleotides: Natural Bioactive Substances in Milk and Colostrum. Br. J. Nutr. 2000, 84 Suppl 1, S59–68. 10.1017/s0007114500002269. [DOI] [PubMed] [Google Scholar]
- (34).Vargas-Bello-Pérez E; Faber I; Osorio JS; Stergiadis S Consumer Knowledge and Perceptions of Milk Fat in Denmark, the United Kingdom, and the United States. J. Dairy Sci. 2020, 103 (5), 4151–4163. 10.3168/jds.2019-17549. [DOI] [PubMed] [Google Scholar]
- (35).Schwaiger-Haber M; Stancliffe E; Arends V; Thyagarajan B; Sindelar M; Patti GJ A Workflow to Perform Targeted Metabolomics at the Untargeted Scale on a Triple Quadrupole Mass Spectrometer. ACS Meas. Sci. Au 2021, 1 (1), 35–45. 10.1021/acsmeasuresciau.1c00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Prince JT; Marcotte EM Chromatographic Alignment of ESI-LC-MS Proteomics Data Sets by Ordered Bijective Interpolated Warping. Anal. Chem. 2006, 78 (17), 6140–6152. 10.1021/ac0605344. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.