

Abstract
Introduction:
Congenital anomalies of the kidney and urinary tract (CAKUT) are the most common cause of chronic kidney disease in the first 3 decades of life. Over 40 genes have been identified as causative for isolated human CAKUT. However, many genes remain unknown, and the prioritization of potential CAKUT candidate genes is challenging. To develop an independent approach to prioritize CAKUT candidate genes, we hypothesized that monogenic CAKUT genes are most likely co-expressed along a temporal axis during kidney development and that genes with coinciding high expression may represent strong novel CAKUT candidate genes.
Methods:
We analyzed single-cell mRNA (scmRNA) transcriptomics data of human fetal kidney for temporal sc-mRNA co-expression of 40 known CAKUT genes. A maximum of high expression in consecutive timepoints of kidney development was found for four of the 40 genes (EYA1, SIX1, SIX2, and ITGA8) in nephron progenitor cells a, b, c, d (NPCa–d). We concluded that NPCa–d are relevant for CAKUT pathogenesis and intersected two lists of CAKUT candidate genes resulting from unbiased whole-exome sequencing (WES) with the 100 highest expressed genes in NPCa–d.
Results:
Intersection of the 100 highest expressed genes in NPCa–d with WES-derived CAKUT candidate genes identified an overlap with the candidate genes KIF19, TRIM36, USP35, CHTF18, in each of which a biallelic variant was detected in different families with CAKUT.
Conclusion:
Sc-mRNA expression data of human fetal kidney can be utilized to prioritize WES-derived CAKUT candidate genes. KIF19, TRIM36, USP35, and CHTF18 may represent strong novel candidate genes for CAKUT.
Keywords: Congenital anomalies of the kidney and urinary tract, Single-cell mRNA sequencing, Human fetal kidney
Introduction
Congenital anomalies of the kidney and urinary tract (CAKUT) comprise a large spectrum of congenital malformations that range from severe manifestations, such as renal agenesis, to milder conditions, such as vesicoureteral reflux (VUR). CAKUT represents one of the most frequent birth defects and causes approximately 40% of end-stage renal disease manifesting within the first 3 decades of life [1].
Several lines of evidence in humans and mouse models indicate that CAKUT is often caused by recessive or dominant variants in single genes [2]. CAKUT can occur isolated or as part of a syndromic disorder with extrarenal features [3]. Its pathogenesis includes various genetic reasons like monogenic or polygenic single nucleotide variants, copy number variations, or chromosomal imbalances but also epigenetic and gestational environmental risk factors are discussed to be influential [4]. In this study, we focused on the analysis of potential disease-causing variants for monogenic CAKUT. Whole-exome sequencing (WES) is a powerful tool that helped identify causative genes for CAKUT. To date, 40 genes are known to cause isolated CAKUT if mutated, explaining monogenic causes in 5–20% of CAKUT patients. Over 153 genes have been reported to cause syndromic CAKUT [5]. However, probably hundreds of monogenic CAKUT genes remain unknown [3]. The discovery of novel CAKUT genes remains challenging for many reasons: (i) its genetic heterogeneity, reflected by the existence of more than 200 genes associated with murine CAKUT; (ii) there is strong pleiotropy, i.e., many monogenic CAKUT genes are associated with defects in organ systems other than the urinary tract; (iii) genetic phenomena that loosen genotype-phenotype correlations such as variable expressivity and incomplete penetrance; (iv) WES analysis yields many more heterozygous variants than biallelic variants, leading to difficulties in variant prioritization and identification of candidate genes with a dominant mode of inheritance. Therefore, to prioritize large amounts of, especially heterozygous, variants in potential CAKUT candidate genes, an independent orthogonal layer of evidence is needed.
In this paper, we are utilizing recently published single-cell mRNA (sc-mRNA) sequencing data of 6,602 human fetal kidney cells by Hochane et al. [6] to prioritize candidate genes resulting from WES data analysis of 731 families with CAKUT. We hypothesized that genes causing monogenic CAKUT are most likely co-expressed in peaks along a temporal axis during kidney development. Our approach resulted in the prioritization of KIF19, TRIM36, USP35, and CHTF18 as likely novel CAKUT candidate genes.
Materials and Methods
WES in 731 Families with CAKUT
The study was approved by the Institutional Review Boards of the Boston Children’s Hospital and the Institutional Review Boards of institutions where families have been recruited. The here used cohort was built from 2005 to 2019. After obtaining informed consent, WES was performed for a total of 731 unrelated families with at least one affected relative (in total, 1,362 individuals including 822 affected individuals and 540 unaffected individuals), as described before [7, 8].
WES was performed as previously reported [7, 9]. In brief, genomic DNA was isolated from blood lymphocyte or saliva samples and subjected to WES. After alignment to the human reference genome (NCBI build 38/hg19), detected variants were filtered as previously described [7, 9]. Variants with minor allele frequencies >1% in dbSNP (version 151), synonymous, and intronic variants outside splice site regions were excluded as likely benign. The included variants were analyzed as described in the next step.
Variant Calling and Generation of Candidate Genes
The evaluation of WES data was performed following a standardized a priori filtering process according to the guidelines of the American College of Medical Genetics as previously described [7, 9]. To summarize, we evaluated WES data for potential causative variants in genes reported to cause isolated and syndromic CAKUT in humans, as well as candidate genes reported to cause CAKUT in mice. Then, the remaining variants were prioritized for their potential candidate status considering evolutionary conservation, in silico prediction of deleteriousness, and population frequency. Sanger sequencing was used to confirm variants in the original patient’s DNA and for segregation analysis in familial DNA (parents or siblings), if available. Phenotype and functional aspects of each variant/gene were discussed in a nephron-genetic panel for each of the 731 families before final candidate decisions were made. Candidate genes were assigned to different statuses: (A) novel single CAKUT candidate genes in individuals whose WES analysis revealed a possibly disease-causing variant in only one candidate gene, and (B) novel multiple CAKUT candidate genes in individuals whose WES analysis revealed possibly disease-causing variants in more than one candidate gene.
CNV analysis was performed for one of the families (A1808) as part of another project without results. For this approach, CNV analysis was not considered as the patients have isolated CAKUT, and CNVs possibly affecting multiple genes are not likely to cause the patients’ phenotype without extrarenal findings.
Temporal Co-Expression of 40 Known Isolated Human CAKUT Genes Using Hochane’s Human Fetal Kidney sc-mRNA Transcriptomics Dataset
To develop an independent approach to prioritize WES-derived CAKUT candidate genes, first, we analyzed 40 genes known to cause isolated CAKUT, if mutated (shown in online suppl. Table 1; for all online suppl. material, see https://doi.org/10.1159/000531770), for temporal co-expression of sc-mRNA in human fetal kidney cells as provided by Hochane et al. [6, 7]. Hochane et al. [6] studied gene expression dynamics in the human fetal kidney at week 16 of gestation using sc-mRNA sequencing. Based on the literature on marker genes derived from mouse kidney development, they identified 22 developmental cell types and concluded their temporal relationship. To provide an independent approach to supporting the developmental progression of the identified cell types, the researchers used Monocle 2. This algorithm clusters and classifies single cells and performs pseudotime analysis based on machine learning [10]. By analyzing the gene expression patterns of individual cells and inferring the temporal progression of gene expression changes, Monocle 2 enables the reconstruction of developmental trajectories for heterogenous cell populations [11]. Sc-mRNA expression data for each gene are publicly available on the Semrau lab’s “Human Fetal Kidney Atlas” website (https://home.physics.leidenuniv.nl/~semrau/humanfetalkidneyatlas/). Furthermore, their raw data with missense z-scores for all detected genes are provided for download.
We assessed the expression data deposited in the GEO database (GSM3143601) and used the assigned cell type labels generated by Hochane et al. [6] after t-distributed stochastic neighbor embedding map clustering. To generate a heatmap to display the results, we then calculated z-scores for each gene based on this gene’s fraction of expression-positive cells per cluster.
We extracted sc-mRNA expression data for 40 known isolated human CAKUT genes and arranged them in a heatmap with the cell types arranged by their developmental order as determined by Hochane et al. [6] (shown in Fig. 1). Then, we arranged the order of genes to point out clusters of genes that share a maximum of consecutive co-expression over a maximum of timepoints.
Fig. 1.
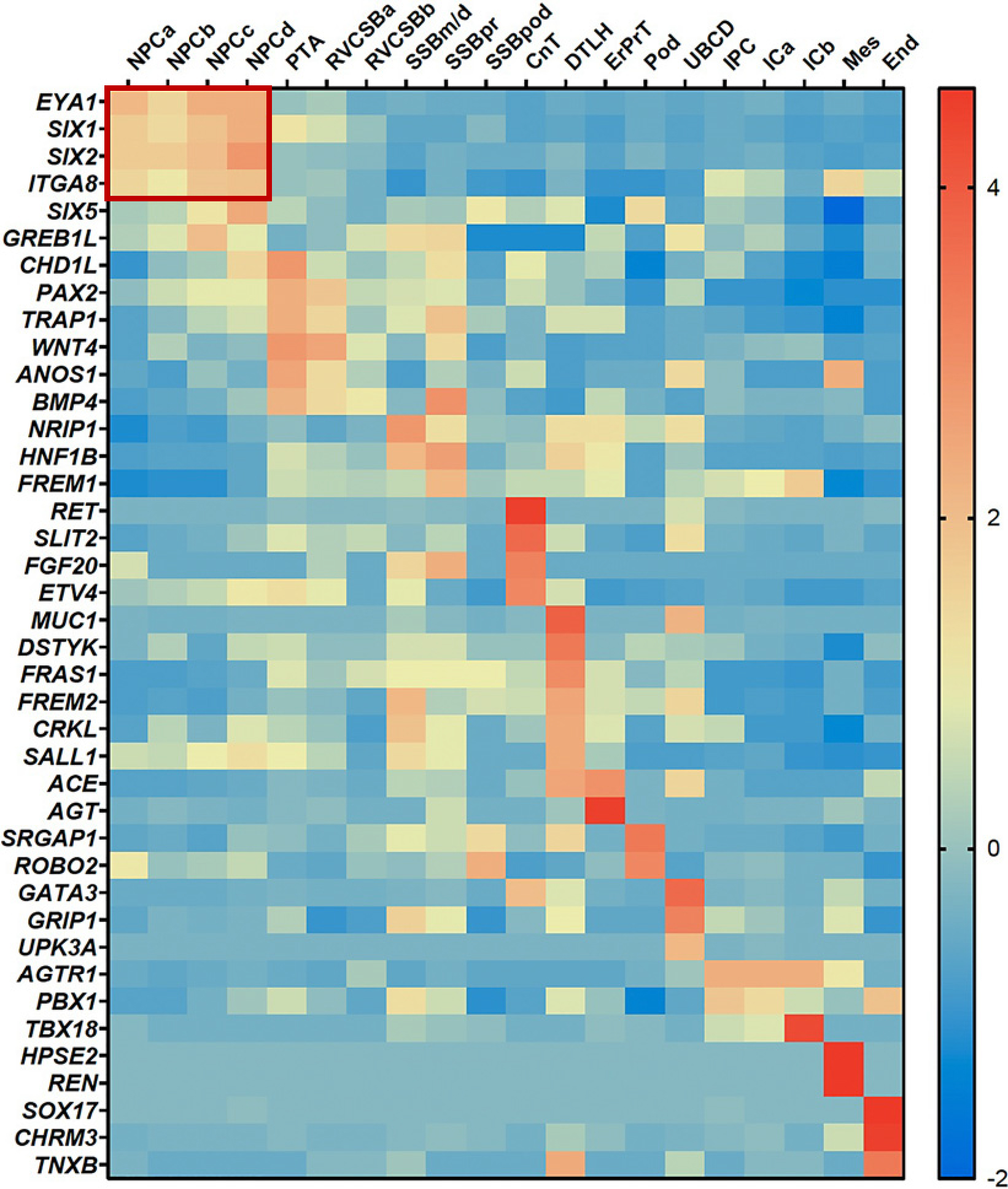
Temporal co-expression of 40 known isolated human CAKUT genes in sc-mRNA expression data of human fetal kidney cells. Four of 40 known isolated human CAKUT genes show temporal co-expression in nephron progenitor cells a–d (NPCa–d): EYA1, SIX1, SIX2, and ITGA8. Each of the rows corresponds to the expression data of one gene and each column from left to right corresponds to one specific cell type in their order from early to late development of the kidney. The order of genes is arranged in a way that a maximum of genes shows consecutive co-expression in a maximum of timepoints. The expression data and developmental order of cell types are based on sc-mRNA sequencing data of week 16 human fetal kidney cells by Hochane et al. [6]. CAKUT, congenital anomalies of the kidney and urinary tract; sc-mRNA, single-cell mRNA; NPCa–d, nephron progenitor cells a–d; PTA, pretubular aggregate; RVCSBa–b, renal vesicle/comma-shaped body a–b; SSB m/d, pr, pod, S-shaped body medial/distal, proximal precursor cells, podocyte precursor cells; CnT, connecting tubule; DTLH, distal tubule/loop of Henle; ErPrT, early proximal tubule; Pod, podocyte; UBCD, ureteric bud/collecting duct; IPC, interstitial progenitor cells; ICa–b, interstitial cells a–b; Mes, mesangial cells; End, endothelial cells.
Intersection of 369 WES-Derived CAKUT Candidate Genes with the Highest Expressed Genes in NPCa–d according to Hochane’s sc-mRNA Database
Second, based on the outcome of the first step, and to prioritize CAKUT candidate genes, we analyzed two lists of WES-derived CAKUT candidate genes for intersection with the 100 highest expressed genes in nephron progenitor cells a, b, c, d (NPCa–d) (shown in Fig. 2). To determine the 100 highest expressed genes in NPCa–d, we assessed the expression data and calculated the z-score as described above. Then, we ranked the genes by the calculated z-score of each NPCa–d (shown in online suppl. Table 2). These 100 highest expressed genes for each cell type NPCa, NPCb, NPCc, and NPCd were then analyzed for overlap within each other and then for intersection with the following lists:
Fig. 2.
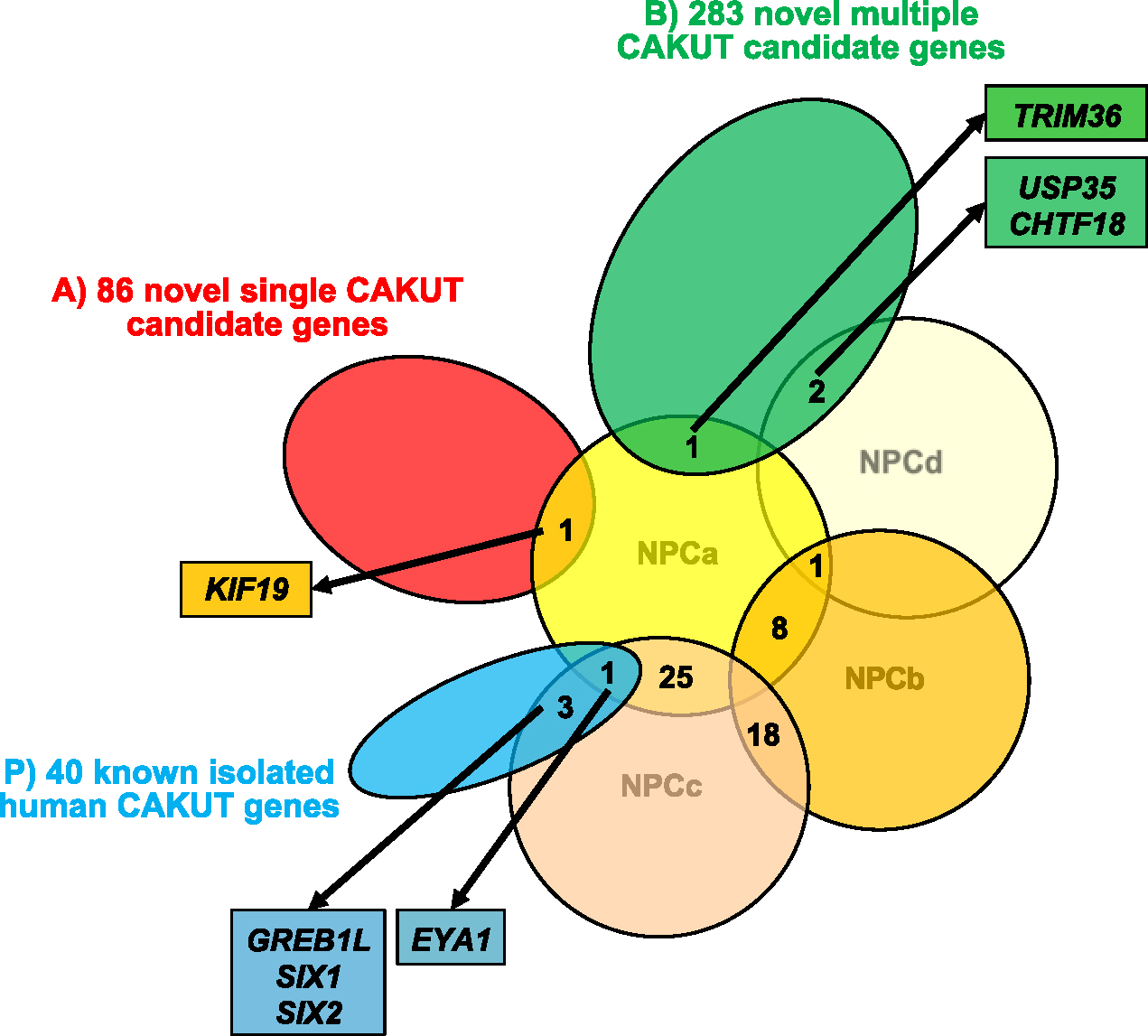
Intersection of WES-derived CAKUT candidate genes with the highest expressed genes in NPCa–d according to the sc-mRNA database of Hochane et al. [6]. Four of 40 known isolated human CAKUT genes (EYA1, SIX1, SIX2, ITGA8) show temporal co-expression in NPCa–d (shown in Fig. 1). Thus, the expression pattern of NPCa–d might reveal further human CAKUT candidate genes. We analyzed the 100 highest expressed genes in each cell type NPCa, NPCb, NPCc, and NPCd for overlap within each other and then for intersection with the following lists: As a pretest, 40 well-established genes known to cause isolated CAKUT in humans (P; indicated in blue color); and then to prioritize CAKUT candidate, 86 novel single CAKUT candidate genes (A; indicated in red color) in individuals whose WES analysis revealed a possibly disease-causing variant in only one candidate gene; and 283 novel multiple CAKUT candidate genes (B; indicated in green color) in individuals whose WES analysis revealed possibly disease-causing variants in more than one candidate gene. Note that in total 52 of the 100 highest expressed genes in each NPC type overlap with each other, rendering a total list of 348 instead of 400 highest expressed genes in NPCa–d: 25 of the 100 highest expressed genes in NPCa overlap with NPCc, 8 of the 100 highest expressed genes in NPCa overlap with NPCb, 18 of the 100 highest expressed genes in NPCb overlap with NPCc, one gene overlaps with three lists of 100 highest expressed genes in NPCa, NPCb, and NPCd. The pretest (P) showed that four of the 40 well-established CAKUT genes (EYA1, SIX1, SIX2, GREB1L) are intersecting with the 100 highest expressed genes in NPCa–d. The intersection of the highest expressed genes in NPCa–d with list (A) identified overlap between KIF19 and NPCa. The intersection with list (B) identified overlap between TRIM36 and NPCa, as well as overlap between USP35 and CHTF18 both with NPCd. WES, whole-exome sequencing; CAKUT, congenital anomalies of the kidney and urinary tract; NPCa–d, nephron progenitor cell type a–d; sc-mRNA, single-cell mRNA.
As a pretest (P), we overlapped the 40 well-established genes known to cause isolated CAKUT in humans with the 100 highest expressed genes in NPCa–d (shown in Fig. 2). To prioritize CAKUT candidate genes, overlap was analyzed for (A) 86 novel single CAKUT candidate genes in individuals whose WES analysis revealed a possibly disease-causing variant in only one candidate gene, and (B) 283 novel multiple CAKUT candidate genes in individuals whose WES analysis revealed possibly disease-causing variants in more than one candidate gene, both resulting from unbiased WES in 731 families with CAKUT (shown in Fig. 2).
As a negative control (NC), we first evaluated the sc-mRNA expression data of the 59 known monogenic causes of human nephrotic syndrome. As described above, we arranged the cell types according to their temporal order and their expression pattern (shown in online suppl. Fig. 1a). Second, we analyzed the 100 highest expressed genes in NPCa–d for intersection with 59 known human genes for nephrotic syndrome (shown in online suppl. Fig. 1b) [12]. It is suitable as a NC as all genes are well-established disease-causing genes for nephrotic syndrome (shown in online suppl. Table 3) [12].
Results
Four of 40 Known Isolated Human CAKUT Genes Show Temporal Co-Expression in NPCa–d
Our evaluation of 40 known isolated human CAKUT genes showed a maximum of high expression in consecutive timepoints of kidney development for four of the 40 genes (EYA1, SIX1, SIX2, and ITGA8) in NPC a, b, c, and d (shown in Fig. 1). NPCa–d are the four earliest cell types of 22 developmental and mature renal cell types identified in week 16 human fetal kidney cells by Hochane et al. [6]. Using pseudotime analysis, they showed that NPCb and NPCc derive simultaneously from NPCa. NPCb and NPCc give rise to NPCd which are followed by the pretubular aggregate and further developmental kidney cell types. We therefore concluded that NPCa–d are relevant for CAKUT pathogenesis.
KIF19, TRIM36, USP35, and CHTF18 Are WES-Derived Candidate Genes that Are Also Highly Expressed in NPC
We then used the above-described findings as an independent approach to prioritize WES-derived candidate genes and analyzed two lists of WES-derived monogenic CAKUT candidate genes for intersection with the 100 highest expressed genes in NPCa–d. For overlap analysis, we used the 100 highest expressed genes in each NPCa, NPCb, NPCc, and NPCd. Note that in total 52 of the 100 highest expressed genes in each NPC type overlap with each other, rendering a total list of 348 instead of 400 highest expressed genes in NPCa–d: 25 of the 100 highest expressed genes in NPCa overlap with NPCc, 8 of the 100 highest expressed genes in NPCa overlap with NPCb, 18 of the 100 highest expressed genes in NPCb overlap with NPCc, one gene overlaps with three lists of 100 highest expressed genes in NPCa, NPCb, and NPCd (shown in Fig. 2).
The pretest showed that four of the 40 well-established CAKUT genes (EYA1, SIX1, SIX2, GREB1L) are intersecting with the 100 highest expressed genes in NPCa–d. We confirmed that CAKUT candidate genes intersecting with the 100 highest expressed genes in NPC might therefore be good candidates to be disease-causing in CAKUT like EYA1, SIX1, SIX2, and GREB1L (shown in Fig. 2).
By intersecting the highest expressed genes in NPCa–d with list (A) of novel single CAKUT candidate genes, we identified an overlap between kinesin family member 19 (KIF19) and NPCa (shown in Fig. 2). By WES, a homozygous missense variant (c.664G>A, p.(Ala222Thr)) in KIF19 was discovered in an individual with consanguineous parents and bilateral VUR. KIF19 encodes a key regulator of ciliary in the ciliary tip, the very top of the primary cilia [13].
By intersection of the highest expressed genes in NPCa–d with (B) novel multiple CAKUT candidate genes, we identified an overlap between TRIM36 and NPCa, and an overlap between USP35 and CHTF18 with NPCd (shown in Fig. 2). Variants in all three genes were identified by WES in individuals with different manifestation of CAKUT (shown in online suppl. Table 4). In tripartite motif containing 36 (TRIM36), a homozygous missense variant (c.361G>C, p.(Asp121His)) was identified in an individual with non-consanguineous parents and right dysplastic kidney with VUR and left kidney rotational anomaly. In ubiquitin specific peptidase 35 (USP35), two heterozygous variants, one missense and one nonsense (c.103C>T, p.(Arg35Cys); c.1711G>T, p.(Gly571*)) were identified in an individual with non-consanguineous parents and right cystic dysplastic kidney and urachus remnant. In chromosome transmission fidelity factor 18a (CHTF18), a homozygous missense variant (c.2564G>A, p.(Arg855Gln)) was identified in an individual with non-consanguineous parents and left renal agenesis.
For all four candidate genes, respective knockout mouse models (Kif19a, Trim36, Usp35, and Chtf18) do not present with renal developmental defects. However, in all cases, there are only a few (n = 1–3) alleles and genetic backgrounds reported in the Mouse Genome Informatics (MGI) database. As the CAKUT phenotype is not strictly lethal, targeted analysis of renal phenotypes could yield results.
All four candidate genes were submitted to GeneMatcher. However, the GeneMatcher submission did not yield further families with variants in the four candidate genes.
The NC showed that of the 59 known genes for human nephrotic syndrome 8 are highly expressed in S-shaped body podocyte precursor cells and 15 are highly expressed in podocytes (shown in online suppl. Fig. 1a). The intersection of the highest expressed genes in NPCa–d with the 59 known human genes for nephrotic syndrome (NS) showed no intersection of genes (shown in online suppl. Fig. 1b).
Discussion
To develop an independent approach to prioritize WES-derived CAKUT candidate genes, we evaluated 40 genes known to cause isolated monogenic CAKUT in humans for consecutive temporal co-expression in developmental kidney cells by interrogating single-cell transcriptomics data of the human fetal kidney at week 16 [6]. Four of the 40 known CAKUT genes (EYA1, SIX1, SIX2, and ITGA8) showed consecutive temporal clustering in the four earliest cell types of kidney development: NPCa, NPCb, NPCc, and NPCd (shown in Fig. 1). Based on these findings, we intersected two lists of independent WES-derived CAKUT candidate genes with the 100 highest expressed genes in NPCa–d. The two lists comprise (A) 86 novel single CAKUT candidate genes from individuals whose WES analysis revealed a possibly disease-causing variant in only one candidate gene, and (B) 283 novel multiple CAKUT candidate genes from individuals whose WES analysis revealed possibly disease-causing variants in more than one candidate gene (shown in Fig. 2).
The intersection analysis with (A) identified KIF19, a novel single CAKUT candidate gene overlapping with NPCa; interestingly, the most promising candidate gene, KIF19, is also the highest expressed gene in NPCa. The variant was identified in a patient with VUR. Defects of the lower urinary tract are a part of the broad phenotype of CAKUT. Variable expressivity and pleiotropy contribute to the wide spectrum of phenotypes ranging from renal agenesis to VUR from the same gene [3]. This explains why a candidate gene found in a patient expressing VUR could cause a renal phenotype and show high expression in developmental kidney cells like NPC. Intersection with (B) identified TRIM36, a novel multiple CAKUT candidate gene overlapping with NPCa, and USP35 and CHTF18, both novel multiple CAKUT candidate genes overlapping with NPCd. Therefore, we consider these genes to be strong candidate genes for human CAKUT. For further evaluation of the role of the here discussed CAKUT candidate genes KIF19, TRIM36, USP35, and CHTF18 in CAKUT pathogenesis, additional families with variants in the respective genes, as well as functional analysis in cell culture or model organisms, are warranted.
In the first step of the analysis, only four of the 40 known isolated human CAKUT genes showed consecutive high co-expression in fetal kidney cells raising the question of the involvement of other cell types in CAKUT pathogenesis. CAKUT represents a complex disease of the developing kidney suggesting a variety of cell types involved in the pathogenesis of CAKUT. However, in this approach, we focused on genes with consecutive high expression in succeeding cell types based on the hypothesis that monogenic CAKUT genes are most likely co-expressed along a temporal axis during kidney development. We are aware that this only represents one strategy to approach the challenge of a complex and heterogeneous disease like CAKUT. Furthermore, some of the 40 known genes for isolated human CAKUT were considered as marker genes in the clustering of human fetal kidney developmental cell types. Therefore, it is not surprising that we found the strongest evidence for genes as candidate genes for CAKUT in NPCs. Thus, the role of other kidney and non-kidney cell types in CAKUT development remains to be determined.
Gene expression in kidney development has been approached from different aspects. The temporospatial expression of single CAKUT candidate genes has been determined by antibody staining and RT-PCR in human fetal kidney tissue. Analysis of the expression data allowed the conclusion of each candidate gene’s role in renal development [14, 15]. Moreover, multiple studies evaluated sc-mRNA expression in kidneys. Park et al. [16] performed sc-mRNA analysis in adult mouse kidney. Menon et al. [17] report on sc-mRNA data of five human fetal kidneys between weeks 12 and 19 of gestation. They detected 11 different cell clusters with further subclustering. We also compared the sc-mRNA transcriptomics data of human fetal kidney of Hochane et al. [6] with the sc-mRNA transcriptomics data of Lindström et al. [18]. In both publications, single-cell sequencing of the human fetal kidney with subsequent pseudotime analysis was performed. Hochane et al. [6] examined the human fetal kidney at gestational week 16 and Lindström et al. [18] examined the nephrogenic niche at gestational week 17. Both studies identified 22 developmental cell types, however, Lindström et al. [18] combined their data into 17 cell types for their online data display. EYA1, SIX1, SIX2, and ITGA8 showed high expression in NPCs of the Lindström database as well (shown in online suppl. Fig. 2). However, we continued our analysis with the scRNA transcriptomics data from Hochane et al. [6], as the published data of human fetal kidney cells are more recent and have slightly more granularity.
Many studies evaluating fetal kidney sc-mRNA expression utilize organs from only one timepoint of embryonal development. For example, Hochane et al. [6] used week 16 human fetal kidneys and Lindström et al. [18] used week 17 human fetal kidneys, respectively. As the development of the kidney occurs in parallel through consecutive formation of the renal vesicle and the recruitment of NPCs from proximal to distal depending on time, sc-mRNA data of the human fetal kidney at one timepoint provides comprehensive data of the different states of kidney development [18].
To the best of our knowledge, we here provide the first systematic analysis of temporo-spatial expression of known human CAKUT genes and potential novel CAKUT candidate genes using sc-mRNA expression data in human fetal kidney cells. This could offer an orthogonal layer for the analysis of large WES analysis datasets.
To conclude, we were able to identify a peak of temporal co-expression in four known isolated human CAKUT genes in NPCa–d, which led to the prioritization of four WES-derived candidate genes with coinciding high expression in NPC as strong novel candidate genes for CAKUT. These genes are KIF19, TRIM36, USP35, and CHTF18. Our study represents a new approach of combining two orthogonal criteria for the identification of novel CAKUT candidate genes: first, the results from WES data analysis, and second, the sc-mRNA sequencing data. This approach provides new opportunities to prioritize WES-derived CAKUT candidate genes and to improve the evaluation process.
Supplementary Material
Funding Sources
C.-H.W.W. was supported by funding from the American College of Medical Genetics and Genomics Foundation (ACMG/Takeda Next-Generation Biochemical Genetics Award) and the National Institutes of Health (Grant No. T32-GM007748). D.M.C. is funded by the Eugen Drewlo Chair for Kidney Research and Innovation at the Schulich School of Medicine and Dentistry at Western University, London, ON, Canada, and the Academic Medical Organization of Southwestern Ontario (AMOSO) Innovation Fund. Financial support is provided by the department of medicine, Schulich School of Medicine and Dentistry, University of Western Ontario.
F.H. is the William E. Harmon Professor of Pediatrics at Harvard Medical School. His research was supported by grants from the National Institutes of Health (DK076683). Sequencing and data processing were performed by the Yale Centers for Mendelian Genomics, funded by the National Human Genome Research Institute (U54 HG006504).
F.H. and S.Sh. are supported by grants from the Beggs Family Foundation. This research was also supported by the Isabella Forrest Julian Research Fund for Pediatric Post Kidney Transplant Research. S.Se. was supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation; 442070894).
N.M. is supported by funding from the National Institutes of Health (K08-DK127011). L.M.S. and A.A.H. were supported by the Biomedical Education Program.
Footnotes
Conflict of Interest Statement
The authors declare no conflict of interest to declare.
Statement of Ethics
This study was conducted ethically in accordance with the World Medical Association Declaration of Helsinki. This study protocol was reviewed and approved by Boston Children’s Hospital and with approval number IRB-P00006200. Prior to inclusion, informed consent of each individual or its legal guardians, respectively, was obtained.
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.
References
- 1.Chesnaye N, Bonthuis M, Schaefer F, Groothoff JW, Verrina E, Heaf JG, et al. Demographics of paediatric renal replacement therapy in Europe: a report of the ESPN/ERA-EDTA registry. Pediatr Nephrol. 2014;29(12):2403–10. [DOI] [PubMed] [Google Scholar]
- 2.Vivante A, Kohl S, Hwang DY, Dworschak GC, Hildebrandt F. Single-gene causes of congenital anomalies of the kidney and urinary tract (CAKUT) in humans. Pediatr Nephrol. 2014;29(4):695–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Ven AT, Vivante A, Hildebrandt F. Novel insights into the pathogenesis of monogenic congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29(1):36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nicolaou N, Renkema KY, Bongers EMHF, Giles RH, Knoers NVAM. Genetic, environmental, and epigenetic factors involved in CAKUT. Nat Rev Nephrol. 2015;11(12):720–31. [DOI] [PubMed] [Google Scholar]
- 5.Connaughton DM, Hildebrandt F. Personalized medicine in chronic kidney disease by detection of monogenic mutations. Nephrol Dial Transplant. 2020;35(3):390–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hochane M, van den Berg PR, Fan X, Bérenger-Currias N, Adegeest E, Bialecka M, et al. Single-cell transcriptomics reveals gene expression dynamics of human fetal kidney development. Plos Biol. 2019;17(2):e3000152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.van der Ven AT, Connaughton DM, Ityel H, Mann N, Nakayama M, Chen J, et al. Whole-exome sequencing identifies causative mutations in families with congenital anomalies of the kidney and urinary tract. J Am Soc Nephrol. 2018;29(9):2348–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Seltzsam S, Wang C, Zheng B, Mann N, Connaughton DM, Wu CHW, et al. Reverse phenotyping facilitates disease allele calling in exome sequencing of patients with CAKUT. Genet Med. 2022;24(2):307–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Connaughton DM, Bukhari S, Conlon P, Cassidy E, O’Toole M, Mohamad M, et al. The Irish kidney gene project--prevalence of family history in patients with kidney disease in Ireland. Nephron. 2015;130(4):293–301. [DOI] [PubMed] [Google Scholar]
- 10.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14(10):979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Song D, Li JJ. PseudotimeDE: inference of differential gene expression along cell pseudotime with well-calibrated p-values from single-cell RNA sequencing data. Genome Biol. 2021;22(1):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Connaughton DM, Kennedy C, Shril S, Mann N, Murray SL, Williams PA, et al. Monogenic causes of chronic kidney disease in adults. Kidney Int. 2019;95(4):914–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Niwa S, Nakajima K, Miki H, Minato Y, Wang D, Hirokawa N. KIF19A is a microtubule-depolymerizing kinesin for ciliary length control. Dev Cell. 2012;23(6):1167–75. [DOI] [PubMed] [Google Scholar]
- 14.Lozic M, Minarik L, Racetin A, Filipovic N, Saraga Babic M, Vukojevic K, et al. CRKL, AIFM3, AIF, BCL2, and UBASH3A during human kidney development. Int J Mol Sci. 2021;22(17):9183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veljačić Visković D, Lozić M, Vukoja M, Šoljić V, Vukojević K, Glavina Durdov M, et al. Spatio-temporal expression pattern of CAKUT candidate genes DLG1 and KIF12 during human kidney development. Biomolecules. 2023;13(2):340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Park J, Shrestha R, Qiu C, Kondo A, Huang S, Werth M, et al. Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science. 2018; 360(6390):758–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Menon R, Otto EA, Kokoruda A, Zhou J, Zhang Z, Yoon E, et al. Single-cell analysis of progenitor cell dynamics and lineage specification in the human fetal kidney. Development. 2018;145(16):dev164038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lindström NO, De Sena Brandine G, Tran T, Ransick A, Suh G, Guo J, et al. Progressive recruitment of mesenchymal progenitors reveals a time-dependent process of cell fate acquisition in mouse and human nephrogenesis. Dev Cell. 2018;45(5):651–60.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the article and in its online supplementary material.