

Abstract
Purpose:
To achieve eradication of solid tumors, we examined how many neoantigens need to be targeted with how many TCRs by which type of T cells.
Experimental Design:
Unmanipulated, naturally expressed (autochthonous) neoantigens were targeted with adoptively transferred TCR-engineered autologous T cells (TCR-therapy). TCR-therapy used CD8+ T cell subsets engineered with TCRs isolated from CD8+ T cells (CD8+TCR-therapy), CD4+ T cell subsets engineered with TCRs isolated from CD4+ T cells (CD4+TCR-therapy) or combinations of both. The targeted tumors were established for at least 3 weeks and derived from primary autochthonous cancer cell cultures, resembling natural solid tumors and their heterogeneity as found in humans.
Results:
Relapse was common with CD8+TCR-therapy even when targeting multiple different autochthonous neoantigens on heterogeneous solid tumors. CD8+TCR-therapy was only effective against homogenous tumors artificially derived from a cancer cell clone. By contrast, a combination of CD8+TCR-therapy with CD4+TCR-therapy, each targeting one neoantigen, eradicated large and established solid tumors of natural heterogeneity. CD4+TCR-therapy targeted a mutant neoantigen on tumor stroma while direct cancer cell recognition by CD8+TCR-therapy was essential for cure. In vitro data were consistent with elimination of cancer cells requiring a four-cell cluster composed of TCR-engineered CD4+ and CD8+ T cells together with antigen-presenting cells (APCs) and cancer cells.
Conclusion:
Two cancer-specific TCRs can be essential and sufficient to eradicate heterogeneous solid tumors expressing unmanipulated, autochthonous targets. We demonstrate that simplifications to adoptive TCR-therapy are possible without compromising efficacy.
Keywords: Neoantigen, adoptive T cell transfer, cancer immunotherapy, TCR gene therapy, Four-cell cluster
Introduction
Despite advances in immunotherapy with checkpoint inhibitors or adoptive transfer of tumor-infiltrating lymphocytes (TILs), achieving curative treatment of solid malignancies remains challenging in cancer therapy. Immunotherapies can achieve long-term survival, but unfortunately only in a fraction of patients and only in certain types of cancers; relapse is still common (1,2). However, when treatments are effective, mutant neoantigens appear to be the relevant immunological targets (3,4). These antigens had already been recognized as major targets for cancer rejection in experimental models and had been called “individually specific” or “unique” antigens. It was then discovered that most of these antigens were mutant neoantigens encoded by cancer-specific, nonsynonymous single nucleotide variants (nsSNVs) (5,6). The extreme diversity of nsSNVs explains the individual specificity of most mutant neoantigens which can now be easily identified by sequencing of the cancer genome (7). These antigens differ between cancers of individuals even when they are of the same histologic type or from the same site of origin (8). Mutant neoantigens in immunotherapy are important because they are truly cancer-specific; targeting them spares normal cells, tissues and organs. Additionally, they are found in virtually all types of cancers (9).
For current immunotherapies, neoantigen-specific T cells must be activated by checkpoint blockade or TILs must be successfully expanded before adoptively transferred. However, the specific T cells may be irreversibly programmed not to be activated and not to proliferate sufficiently in response to their cognate antigen. (10). This problem may be overcome when TCRs are isolated from failing T cells and transferred into functional peripheral T cells that lack cognate TCRs for mutant neoantigens (11,12). Experimentally, such a TCR-therapy using a single TCR recognizing a mutant neoantigen with very high affinity to MHC I eradicated large, long-established solid tumors (13). However, eradication could only be achieved when cancer cells expressed the mutant neoantigen at artificially high levels. Unmanipulated, autochthonous tumors regularly escaped as antigen loss variants (13).
Previous studies in vitro have shown that the probability of immune escape from T cell therapy can be greatly reduced by targeting simultaneously several independent neoantigens on the same cancer cell (14). In the present study, we examined this principle in vivo by targeting solid tumors that were established for at least 3 weeks. Escape was common even when TCR-therapy combined three TCRs isolated from CD8+ T cells (CD8+TCRs) for three different autochthonous antigens in the same cancer. Eradication was only achieved when the tumors had been artificially re-derived from a cancer cell clone to obtain homogeneity. However, eradication without relapse of the naturally heterogeneous tumors was only possible when a population of CD8+ T cells engineered with one CD8+TCR was combined with a population of CD4+ T cells engineered with one CD4+TCR. The CD4+TCR targeted an MHC II-restricted neoantigen and did not recognize cancer cells but only tumor stroma. For eradication, direct cancer cell recognition by the CD8+TCR was essential. Effective cancer cell elimination in vitro was consistent with a need of a four-cell type interaction between CD4+ and CD8+ T cells together with professional antigen presenting cells (APCs) and cancer cells. This study provides evidence that a single pair of cancer-specific TCRs can be sufficient to eradicate large and long-established heterogeneous tumors expressing unmanipulated amounts of the targeted neoantigens.
Material and Methods
Mice
Both female and male mice were used in this study and were between 3 to 8 months old. Mice were euthanized when tumor sizes reached more than 2 cm3 or mice appeared hunched and weak. Littermates of the same sex were randomly assigned to experimental groups on the day of adoptive T cell transfer. C3H/HeN mice (RRID:MGI:2160972) were obtained from Envigo (Huntingdon, Cambridgeshire, United Kingdom). C3H Rag2−/− (C3H.129S6-Rag2tm1Fwa) mice were obtained from Douglas Hanahan (University of California, San Francisco, CA, USA). C3H CD8−/− (C3H.129S2-Cd8atm1Mak) mice were generated in-house by crossing C3H/HeN mice with C57BL/6 CD8−/− (B6.129S2-Cd8atm1Mak, RRID:MGI:3789587) mice for 20 generations. C3H CD4−/− (C3H.129S2-Cd4atm1Mak) mice were generated in house by crossing C3H/HeN mice with C57BL/6 CD4−/− (B6.129S2-Cd4atm1Mak, RRID:IMSR_JAX:002663) mice for more than 20 generations. C57BL/6, C57BL/6 CD8−/− (B6.129S2-Cd8atm1Mak), C57BL/6 CD4−/− (B6.129S2-Cd4atm1Mak) and C57BL/6 Rag1−/− (B6.129S7-Rag1tm1Mom, RRID:IMSR_JAX:002216) mice were purchased from the Jackson Laboratory (Bar Harbor, ME, USA). OT-I Rag1−/− (B6.129S7-Rag1tm1Mom Tg(TcraTcrb)1100Mjb, RRID:IMSR_TAC:4175) mice have been described previously (13). Spleen of C3H CD8−/−, C3H CD4−/−, OT-I Rag−/− and C57BL/6 mice were used as T cell sources for TCR gene transfer.
Generation and isolation of TCR genes
T cell clones specific for 6132A (5,15), 6139B (15,16) and 8101 (17) have been characterized before. TCR sequences from T cell clones were obtained by 5’-RACE-PCR (TaKaRa, Kusatsu, Japan) following manufactures protocol, codon optimized (GeneArt, Thermo Fisher Scientific, Waltham, MA, USA) and integrated into the pMP71 vector using NotI and EcoRI flanked restriction sides as described (13). The CD8+TCR anti-mDDX5 has been described before (13). Generation of mCherry and GFP linked TCR vectors anti-6132A-A1-mCherry (CD8+TCR), anti-mRPL9-GFP and anti-mRPL26-GFP (both CD4+TCRs) have been described previously (18). For CDR3 sequences of isolated CD8+TCRs and CD4+TCRs see Table S2 and S3. The anti-mRPL26 CD4+TCR recognizes the 6139B-cancer-specific H96Y substitution in the ribosomal protein L26 (referred to as mRPL26H96Y84–108 and presented on I-Ek, (16)). The anti-6139B-A CD8+TCR specifically recognizes antigen “A” on the syngeneic UV-induced cancer cell line 6139B (15).
TCR-engineering of T cells
TCR gene transfer was conducted as previously described (13). Plat-E packaging cells were transfected with pMP71-anti-mDDX5, -anti-mNav3, anti-8101-C, -anti-6132A-A1, -anti-6132A-A4, -anti-6139B-A, -anti-mRPL9 or -anti-mRPL26 vectors by calcium phosphate precipitation. 42 h after transfection, virus containing supernatant was harvested and filtrated through a 0.45 μm syringe filter (VWR, Radnor, PA, USA) before used. Spleens were isolated and erythrocytes were lysed for 3 min with 0.017 M TRIS, 0.14 M ammonium chloride (both Sigma-Aldrich, St. Louis, MO, USA). Cells were cultured in complete medium containing Roswell Park Memorial Institute medium (RPMI 1640, Corning, Corning, NY, USA) 10 % FBS (Gemini, Sacramento, CA, USA), 2 mM l-glutamine, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/mL penicillin, 100 μg/mL streptomycin (all purchased from Life Technologies, Carlsbad, CA, USA), 50 μM 2-mercaptoethanol (Thermo Fisher Scientific, Waltham, MA, USA), 50 μg/mL gentamicin (VWR, Radnor, PA, USA) and were supplemented with 40 U/mL recombinant human IL-2 (Peprotech, Rocky Hill, NJ, USA) and cell suspension was transferred into a 24-Well plate (Greiner Bio-One, Kremsmuenster, Austria) coated with 1.4 μg/mL αCD3 (University of Chicago, Frank W. Fitch Monoclonal Antibody Facility, Clone 145-2C11.1) and 0.2 μg/mL αCD28 (Clone 37.51, Biolegend, San Diego, CA, USA) at a concentration of 3 × 106 cells/mL. On the first day after spleen removal, 0.5 mL of corresponding virus containing supernatant containing 8 μg/mL protamine sulfate (Sigma-Aldrich, St. Louis, MO, USA) was added per well and cells were spinoculated (800 × g, 90 min, 32 ° C). Overnight, a 12-Well plate (Greiner Bio-One, Kremsmuenster, Austria) was coated with RetroNectin [12.5 μg/mL (TaKaRa, Kusatsu, Japan)] and centrifuged with 1.5 mL virus containing supernatant (3000 × g, 90 min, 4 ° C) on the next day. Afterwards supernatants were removed and 5 × 106 of CD8+ T cells in complete medium containing 50 ng/mL recombinant human IL-15 (Peprotech, Rocky Hill, NJ, USA) or same amounts of CD4+ T cells in complete medium with 40 U/mL IL-2, were transferred to the virus coated 12-Well plate and followed by spinoculation (800 × g, 90 min, 32 ° C). Transduction rate was confirmed by flow cytometry using LSR II (BD Bioscience, Franklin Lakes, NJ, USA) and T cells were used 3 days after transduction for adoptive transfer. TCR-engineered CD8+ T cells were maintained in complete medium with 17 ng/mL IL-15 and used after 12 days for in vitro assays, while TCR-engineered CD4+ T cells were maintained in complete medium with 40 U/mL IL-2 and used after 4 days for in vitro analysis respectively.
Cell lines
6132A, 6132B, 6139B cancer cell lines originated from UV-treated C3H/HeN (15) and were used as specificity controls for anti-6132A-CD8+TCRs. 6132B is a second cancer cell line from the 6132 mouse and 6132-HLF is used as autologous normal heart-lung fibroblasts control. 6139B represents another UV-induced syngeneic cancer from a different mouse. 8101 cancer cell lines originated from UV-treated C57BL/6 mice and were generated in our laboratory together with heart-lung fibroblasts as normal tissue control (15,17). All of these tumors were only minimally cultured in vitro and never passaged in vivo. This is important because immunoselection may occur even after a single transplantation into a naïve immunocompetent host (18,19). From the original 8101 cancer cell line, two different clone series were generated (Series A and B). Series A was described before (17) and series B was generated accordingly (20). Furthermore, 8101 clone 12 was recloned accordingly after outgrowth in B6 Rag1−/− mice. The 6132A-cerulean cancer cell line was described before (18). The 6132A B2m−/− cell line was generated using CRISPR-Cas9. Single guide (sg) RNAs targeting exon 1 of the murine B2m gene were designed using the sg RNA design tool from the Broad Institute (21). The corresponding sense and antisense DNA oligomers (IDT, Coralville, IA, USA) were compared to earlier publications (22), annealed and cloned over an BbsI side into PX458 as described (23). The sg RNA 5’ – CATGGCTCGCTCGGTGACCC – 3’ was successfully used to generate 6132A B2m−/− cancer cells which were sorted (FACSAriaII, BD Bioscience, Franklin Lakes, NJ, USA) for MHC class I negative populations after transient calcium phosphate transfection and cloned afterwards to establish a pure MHC class I loss cell line. All cell lines were maintained in Dulbecco’s Modified Eagle Medium (DMEM, Corning, Corning, NY, USA) supplemented with 10 % FBS (Gemini, Sacramento, CA, USA) and 2 mM l-glutamine (Life Technologies, Carlsbad, CA, USA) and cultured at 10 % CO2 in a 37 °C dry incubator. Plat-E (RRID:CVCL_B488) packaging cells (24) used for TCR gene transfer were maintained in DMEM supplemented with 10 % FBS and cultured at 5 % CO2 in a 37 °C dry incubator. To identify whether antigens are presented by either H-2Kb or H-2Db, 58−/− cells expressing either one of the H-2b alleles were generated by retroviral transduction as described (13). EL4 (RRID:CVCL_0255) and 58−/− cells were maintained in Roswell Park Memorial Institute medium (RPMI 1640, Corning, Corning, NY, USA) containing 10 % FBS (Gemini, Sacramento, CA, USA) at 5 % CO2 in a 37 °C dry incubator. Before use, tumor cell lines were authenticated by sequencing and/or co-culture with antigen-specific T cells and by morphology. All cell lines were shortly passaged after thawing of the initial frozen stock to generate master cell banks. Working batches were passaged no longer than 4 weeks.
Treatment of tumor-bearing mice
Cancer cells were injected s.c. into the shaved back of either C57BL/6 Rag1−/− or C3H Rag2−/− mice (1 × 107 8101 or 6132A). Tumor volumes were measured along 3 orthogonal axes, every 2 – 3 days and were calculated as (a × b × c) ÷ 2. T cells expressing either the CD8+TCRs (anti-6132A-A1, anti-6132A-A4 or anti-6139B-A) or the CD4+TCRs (anti-mRPL9 or anti-mRPL26) were injected i.p. Spleen cells from either C57BL/6 or OT-1 Rag−/− mice were used as TCR recipient when 8101 tumors were treated. Spleen cells from C3H CD4−/− or C3H CD8−/− were used as TCR recipient when 6132A tumors were treated. T cells expressing the CD8+TCRs anti-mDDX5, anti-mNav3 or anti-8101-C were injected i.p. or i.v.. The number of TCR+ T cells was always calculated based on transduction rate on the day of treatment prior to T cell transfer. Per recipient, 2 × 106 TCR+ CD8+ or CD4+ T cells were injected when given alone. When multiple TCR+ CD8+ T cells were combined or TCR+ CD8+ T cells were combined with TCR+ CD4+ T cells, the number of TCR+ T cells of each population injected i.p. per recipient was 1 × 106. Mice were randomized into different treatment groups on the day of adoptive T cell transfer. Mice were euthanized when tumor sizes reached more than 2 cm3 or mice appeared hunched and weak.
Generation of 8101-immune T cells
C57BL/6, C57BL/6 CD8−/− and C57BL/6 CD4−/− mice were injected s.c. with 1 × 107 8101 cancer cells derived from the autochthonous primary cancer cell cultures. These cancer cells regularly form tumors in mice lacking T cells, but rarely grow in young fully immunocompetent mice (25,26). Therefore, no conditioning of cancer cells was required for immunization of B6 CD4−/−, B6 CD8−/− or B6 WT mice. Mice were boosted s.c. after 6 weeks with another dose of 1 × 107 8101 cancer cells. Additional 6 weeks later, spleens were removed, red blood cells were lysed with 0.017 M TRIS, 0.14 M ammonium chloride (both Sigma-Aldrich, St. Louis, MO, USA) solution and one full spleen was used for i.p. T cell transfer per recipient. Spleen from C57BL/6 CD8−/− mice were used for transfer of 8101-immune CD4+ T cells while spleen from C57BL/6 CD4−/− mice were used for transfer of 8101-immune CD8+ T cells.
Isolation of CD4+ T cells, CD11b+ and F4/80+ cells
CD4+ T cells were isolated from 8101-immune C57BL/6 spleen cells by magnetic cell sorting (Miltenyi, Bergisch Gladbach, Germany) following manufacturer’s protocol. Purity was confirmed by flow cytometry and CD4+ T cells were used immediately for adoptive T cell transfer. For isolation of CD11b+ and F4/80+ cells, 6132A tumors grown in C3H Rag2−/− mice were removed and single cell suspensions were generated by enzymatic digestion. Tumors were minced, 2 mg/mL Collagenase D and 100 U/mL DNAse I (both Roche, Indianapolis, IN, USA) were added and the suspension was incubated for 20 min at 37 °C in RPMI on a horizontal shaker, following addition of trypsin in Hanks’ Balance Salt Solution (HBSS, MP Biomedicals LLC, Solon, OH, USA) to a final concentration of 0.025% and the cell suspension was incubated for another 15 min at 37 °C on a horizontal shaker. The tumor cell suspension was filtered over a 40 μm cell strainer (Thermo Fisher Scientific, Waltham, MA, USA) and CD11b+ or F4/80+ cells were enriched by magnetic cell sorting (Miltenyi, Bergisch Gladbach, Germany) following manufacturer’s protocol. Successful isolation was confirmed by flow cytometry before used for T cell stimulation.
Antigen presentation and T cell stimulation
Analyzing antigen presentation by indicated cancer and stromal cells, co-cultures were performed for 24 h with CD8+ T cells engineered with CD8+TCRs or with CD4+ T cells engineered with CD4+TCRs. Co-cultures with TCR-engineered CD8+ T cells have been described before (27). In brief, 5 × 104 CD8+TCR-engineered CD8+ T cells or 5 × 104 CD4+TCR-engineered CD4+ T cells were added to 1 × 105 cancer cells or stromal cells. For TCR independent stimulation, 8 μg/mL αCD3 (University of Chicago, Frank W. Fitch Monoclonal Antibody Facility, Clone 145-2C11.1) and 2 μg/mL αCD28 (Clone 37.51, Biolegend, San Diego, CA, USA) was used. In addition, antigen in form of cancer cell lysate loaded on CD11b+ cells isolated from the spleens of C3H/HeN mice were performed as previously described (5). 6132A or 6139B cancer cells were adjusted to 1 × 107 cells/mL in RPMI 1640 (Corning, Corning, NY, USA) before three cycles of freezing in liquid nitrogen and thawing at 37 °C were conducted. Cancer cell lysate was immediately used for CD11b+ cell loading with 1 × 105 cancer cell equivalents. Always after 24 h, supernatants were removed and tested for IFN-γ by enzyme—linked immunosorbent assay (ELISA, Ready-SET-Go!, eBioscience, San Diego, CA, USA), following the manufacturer’s protocol. Light absorbance at 450 nm was read with the microplate reader VERSAmax (Molecular Devices LLC, San Jose, CA, USA). No stimulation and TCR-independent stimulation using CD3- and CD28-specific antibodies or ionomycin (1 μM, Sigma-Alderich, St. Louis, MO, USA) and phorbol myristate acetate (MAX, 5ng/mL, Sigma-Alderich, St. Louis, MO, USA) were used.
Flow cytometry and antibodies
1 μg Fc receptor block (anti-mouse 2.4G2, RRID:AB_626927)) was added to samples and cells were incubated with 50 μL phosphate buffer saline (PBS) containing 0.2 μg of indicated anti-mouse antibodies: CD3+ [145-2C11, Fluorescein isothiocyanate (FITC), RRID:AB_2536119)], CD4+ [GK1.5, Allophycocyanin (APC), RRID:AB_2535396)], CD8+ [53–6.7, Allophycocyanin (APC), RRID:AB_2536105)], CD11b+ [M1/70, Allophycocyanin (APC) or Allophycocyanin/Cyanine 7 (APC/Cy7), RRID:AB_2857951)], CD11c [N418, Allophycocyanin (APC), RRID:AB_2621557)], F4/80 [BM8, PerCP/Cyanine 5.5 (PerCP/Cy5.5) or BV421, RRID:AB_2534315)], Gr1 [RB6-8C5, R-Phycoerythrin (PE), BD Bioscience, RRID:AB_2621803)], Ly6G [1A8, Fluorescein isothiocyanate (FITC), RRID:AB_2877150)], Ly6C [AL-21, PerCP/Cyanine 5.5 (PerCP/Cy5.5), BD Bioscience, RRID:AB_1727558)], CD274 (PD-L1, 10F.9G2, R-Phycoerythrin (PE), RRID:AB_2535471)], IDO (mIDO-48, R-Phycoerythrin (PE), Invitrogen, RRID:AB_2688156)], TCRvb2 [B20.6, R-Phycoerythrin (PE), RRID:AB_2535326)], TCRvb4 [KT4, R-Phycoerythrin (PE), BD Bioscience, RRID:AB_394812)] TCRvb6 [RR4-7, R-Phycoerythrin (PE), RRID:AB_394701)], TCRvb8.1,8.2 [KJ16-133.18, R-Phycoerythrin (PE), RRID:AB_1134109)], TCRvb8.3 [8C1, R-Phycoerythrin (PE), RRID:AB_1937250)], H-2Kk [36-7-5, Fluorescein isothiocyanate (FITC) or R-Phycoerythrin (PE), RRID:AB_313612)], H-2Dk [15-5-5, R-Phycoerythrin (PE), RRID:AB_313480)], H-2Kb [AF6-88.5, Allophycocyanin (APC)], H-2Db [KH95, R-Phycoerythrin (PE), RRID:AB_313512)], I-Ek [14-4-4S, Alexa Fluor 647 (AF647), RRID:AB_493214)], I-Ab [AF6-120.1, Fluorescein isothiocyanate (FITC), RRID:AB_313725)]. If not indicated otherwise, antibodies were purchased from Biolegend (San Diego, CA, USA). BD Horizon 510 (BD Biosciences, Franklin Lakes, NJ, USA) was used for Life/Dead stain. Samples were washed in PBS and analyzed using LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA). Data analysis was performed using FlowJo software (RRID:SCR_008520, TreeStar, Ashland, OR, USA).
CFSE labelling of T cells
TCR-engineered CD4+ or CD8+ T cells were stained with 1 μm CFSE (Biolegend, San Diego, CA, USA) at 37 °C on a horizontal shaker protected from light for 20 min. Afterwards, cells were cocultured for 72 h with CD11b+ cells isolated from 6132A tumors before analyzed for the CFSE signal by flow cytometry using LSR II flow cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
IFN-γ exposure in vitro
Cancer cells were cultured for 48 h either with or without 25 ng/mL IFN-γ (28) (Genentech, San Francisco, CA, USA) before MHC class I and II surface expression was analyzed by flow cytometry.
Blood serum cytokine analysis
Blood was taken by buccal bleeding with a 5 mm animal lancet (Medipoint Inc, Mineola, NY, USA) at day of adoptive T cell transfer and every following 3 days for a total of 21 days. Blood (100 μL) was collected in tubes containing 50 μL heparin (80 U/mL, Pfizer, New York, NY, USA). Cells were spun down at 9,000 × g for 0.5 min and supernatants were harvested and analyzed for IFN-γ with Legendplex according to manufacture protocol (Biolegend, San Diego, CA, USA). Cell pellets were used for T cell detection and quantification was performed with AccuCount Rainbow beads (Spherotech, Lake Forest, IL, USA) according to manufactures specifications by flow cytometry.
Whole-exome and RNA sequencing
Both genomic DNA and total RNA were extracted from the 8101 and 6132A cell lines, using AllPrep DNA/RNA mini kit (Qiagen, Venlo, The Netherlands). For whole-exome sequencing, 3 μg of genomic DNA was subjected to library construction, using SureSelectXT Mouse All Exon V1 (Agilent Technologies, Santa Clara, CA, USA). RNAseq libraries were prepared from 1 μg of total RNA using TruSeq Stranded Total RNA Library Prep kit (Illumina, San Diego, CA, USA). The prepared whole-exome and RNAseq libraries were quantified by 2200 Tape Station (Agilent Technologies, Santa Clara, CA, USA), and then sequenced by 150 bp paired-end reads on NextSeq 500 Sequencer (Illumina, San Diego, CA, USA).
In vitro model and MTT assay
6132A cancer cells were cultured in 96-well plates (Greiner Bio-One, Kremsmuenster, Austria) over night with or without CD11b+ cells isolated either from the spleen of tumor-free C3H/HeN mice or from 6132A tumors grown in C3H Rag−/− mice in complete RPMI 1640 (Corning, Corning, NY, USA) medium with 10 % FBS (Gemini Bio-Products, West Sacramento, CA, USA), 2 mM l-glutamine, 50 μM 2—mercaptoethanol, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/mL penicillin, 100 μg/mL streptomycin and 50 μg/mL gentamicin (all purchased from Invitrogen, Carlsbad, CA, USA). TCR-egnineered CD8+ and CD4+ T cells were added one day later and plates were cultured for 7 days at 10 % CO2. Then, the medium containing non-attached cells was removed, attached cells were carefully washed once with PBS and replaced with fresh complete medium. After culturing for additional 5 days, live cells were analyzed by MTT (Sigma-Alderich, St. Louis, MO, USA) as described (5).
Time-lapse imaging
6132A-cerulean cancer cells and DiD-labelled (Thermo Fisher Scientific, Waltham, MA, USA) CD11b+ cells isolated from spleens of tumor-free C3H/HeN mice were cultured overnight in a 35 mm glass-bottom dish (MatTek, Ashland, MA, USA), containing 2.5 mL culture medium, consisting of RPMI 1640 w/o riboflavin (US Biological, Salem, MA, USA) with 10 % FBS (Gemini Bio-Products, West Sacramento, CA, USA), 2 mM l-glutamine, 50 μM 2—mercaptoethanol, 1 mM sodium pyruvate, 0.1 mM non-essential amino acids, 100 U/mL penicillin, 100 μg/mL streptomycin and 50 μg/mL gentamicin (all purchased from Invitrogen, Carlsbad, CA, USA). Imaging was performed the next day with an Olympus VivaView incubator-based, epifluorescence microscope (Olympus Corporation of the Americas, Center Valley, PA, USA) and run by MetaMorph software (Molecular Devices LLC, Sunnyvale, CA, USA). Incubator settings were kept at 37 °C and 5 % CO2. Positions with appropriate numbers of cancer cells (between 3 and 10) were recorded, and anti-6132A-A1-mCherry-CD8+ and either anti-mRPL9-GFP- or anti-mRPL26-GFP-CD4+ T cells resuspended in the same culture medium were added afterwards. The montage was set up to proceed from left to right, top to bottom for eight rows and eight columns to set an 8 × 8 field of view. This sequence of images corresponds to Fiji’s built-in montage feature (29). Each 8 × 8 montage time-point took 25 min to image and was followed up for 72 h to evaluate experimental success of cancer cell disappearance or outgrowth. In addition, four 2 × 2 montages were chosen, equally spaced from one another, from within the 8 × 8 montage. The 2 × 2 montages (Position 1, 2, 3 and 4 in Figure 5C) were imaged one at a time every 90 s for 5.5 h, with the 8 × 8 montage recorded in between the four positions. An infrared differential interference contrast (DIC) channel and 4 color channels were used (CFP, Cy5, GFP, RFP). The CFP channel used excitation at 436/20 (center/bandpass; nm) with a 480/40 filter to capture the Cerulean emission at 458 nm. The Cy5 channel used a 700/75 filter, with excitation at 620/60, to capture the DiD, which emits at 665 nm. The GFP channel used excitation at 470–495 nm with a 515/25 filter to capture the GFP emission at 510 nm. The RFP channel used excitation at 530–550 nm with a 593/46 filter to capture the mCherry emission at 610 nm.
Four cell-type interaction analysis
Images were analyzed using the Fiji (RRID:SCR_002285) distribution of ImageJ (RRID:SCR_003070) (29), and were gathered in sets of adjacent acquisition tiles, at multiple “z” planes (initially), where z position relates to the focus of the microscope, and sorted by color channel, x-y position, z-position and time point in separated files and analyzed using Fiji software. Separate images were merged together into montages to display the whole imaged area. Then, the raw image data were processed to decrease background noise and to remove bright outliers (e.g., large debris) in two ways. The first method (18), generates understandable videos and images. The second method functions through a high-precision edge-finding method that operates on the DIC channel, where an automated macro draws outlines around objects and then searches each color channel for an intensity above a user-defined threshold. It then colors in each outline accordingly. This method produces accurate counts of cancer cells (through a watershed routine) and their interactions but is more visually abstract. Processed images were analyzed to search for cell interactions using the cluster-finding macro previously described (18), where overlapping colors that correspond to different cell types were used to classify clusters of cells into varying types of two-, three- and four-cell interactions. To account for optical inconsistencies between single frames, the macro was modified to perform a z-projection of the current frame and the next four frames, where it took the maximum intensity at each x and y value of each frame and superimposed it on a single image. The macro then re-split the cancer cells in the z-projection to account for blurred boundaries. Two-cell interaction counts were found by subtracting the number of three- and four-cell interaction counts from the counts of pairwise interactions given by the macro. Graphical depiction was done using Spline fits with RStudio’s (RStudio Inc. Boston, MA, USA, RRID:SCR_000432) ggplot2 package (30,31). The default settings for geom_spline were used for all plots: mapping = NULL, data = NULL, stat = “spline”, position = “identity”, na.rm = FALSE, show.legend = NA, inherit.aes = TRUE, weight = NULL, df = NULL, spar = NULL, cv = FALSE, all.knots = FALSE, nknots = stats::.nknots.smspl, df.offset = 0, penalty = 1, control.spar = list(), tol = NULL.
Statistics
All statistical analyses were performed using GraphPad Prism software (GraphPad, San Diego, CA, USA, RRID:SCR_002798)). Data points indicate either means of biological duplicates of a representative experiment or are experimental replicates summarized as mean ± standard deviation. The method used to present the data is always indicated in the figure legend. Statistical analysis of the four cell-type interaction model was performed with RStudio (RStudio Inc. Boston, MA, USA, RRID:SCR_000432). Students t-tests were used to determine significant differences between means of cell-to-cell interaction counts over 220 time points per position. P-values were then determined from the test statistic and summarized in Supplementary Table S1. The test used data from four positions, with three degrees of freedom.
Study approval
Mice were bred and maintained in a specific pathogen-free barrier facility at The University of Chicago according to Institutional Animal Care and Use Committee (IACUC) guidelines. All animal experiments were approved by the IACUC of The University of Chicago.
Data availability statement
The raw sequencing data generated in this study are publicly available in Sequence Read Archive (SRA) BioProject ID PRJNA1054787 at accession numbers SAMN38933246 (6132A), SAMN38933247 (6132-HLF), SAMN38933248 (8101), SAMN38933249 (8101-HLF), SAMN38933250 (8101 Clone 4), SAMN38933251 (8101 Clone 6), SAMN38933252 (8101 Clone 12), SAMN38933253 (8101 Clone 13), SAMN38933254 (8101 tumor fragment 1), SAMN38933255 (8101 tumor fragment 2), SAMN38933256 (8101 tumor fragment 3), SAMN38933257 (8101 tumor fragment 4), SAMN38933258 (8101 tumor fragment 6), SAMN38933259 (8101 tumor fragment 7), SAMN38933260 (8101 tumor fragment 16), SAMN38933268 (8101 tumor fragment 18), SAMN38933261 (8101 tumor fragment 20), SAMN38933269 (8101 tumor fragment 21), SAMN38933270 (8101 tumor fragment 22), SAMN38933271 (8101 tumor fragment 23), SAMN38933272 (8101 tumor fragment 24), SAMN38933273 (8101 tumor fragment 27), SAMN38933262 (8101 tumor fragment 26), SAMN38933263 (8101 tumor fragment 30), SAMN38933264 (8101 tumor fragment 36), SAMN38933265 (8101 tumor fragment 37), SAMN38933266 (8101 tumor fragment 39), SAMN38933267 (8101 tumor fragment 53). All other data and materials generated in this study are available upon request from the corresponding author.
Results
Heterogeneous cancers escape from a combination of CD8+TCRs targeting multiple independent, cancer-specific antigens
Using the 8101 tumor model, we previously identified a cancer-specific S551F substitution in the DEAD box helicase 5 protein causing the immunodominant neoantigen DDX5S551F547–554 (mDDX5) (17), which binds with sub-nM affinity to H-2Kb (25). The DDX5 protein, also known as RNA helicase p68, can be an important driver in tumor development (32) and thus an ideal target for adoptive TCR-therapy. Nevertheless, and consistent with earlier studies (13), anti-mDDX5 CD8+TCR-T cells failed to eradicate large solid tumors that arose from s.c. injected cancer cells derived from the autochthonous primary 8101 tumor expressing mDDX5 (Fig. 1A, upper left). Some tumors regressed after treatment, but rapidly relapsed. Cloning of 8101 cancer cells identified either mDDX5-positive or -negative phenotypes (17). Genetic differences between the two phenotypes were analyzed by whole-exome sequencing of two mDDX5-positive (A4 and A12) and two mDDX5-negative clones (A6 and A13) (Fig. 1B). While clones of the same phenotype were similar, the mutational signature between the two phenotypes was remarkably distinct. Only 266 mutations were shared by both phenotypes, while unique mutations for the mDDX5-positive (3,080) and mDDX5-negative (1,777) phenotypes were considered to be specific gene-signatures. These gene-signatures were used to determine whether cells of both phenotypes were present in spatially distinct tumor fragments of the original, autochthonous 8101 tumor (Fig. 1B). While all but two fragments contained 100% of the mDDX5-positive gene-signatures, there were large differences between the individual fragments in the percentage of the mDDX5-negative gene-signatures, indicating a significant heterogeneity of the autochthonous 8101 tumor.
Figure 1. 8101 tumors escape from a combination of multiple CD8+TCRs targeting independent cancer-specific antigens.
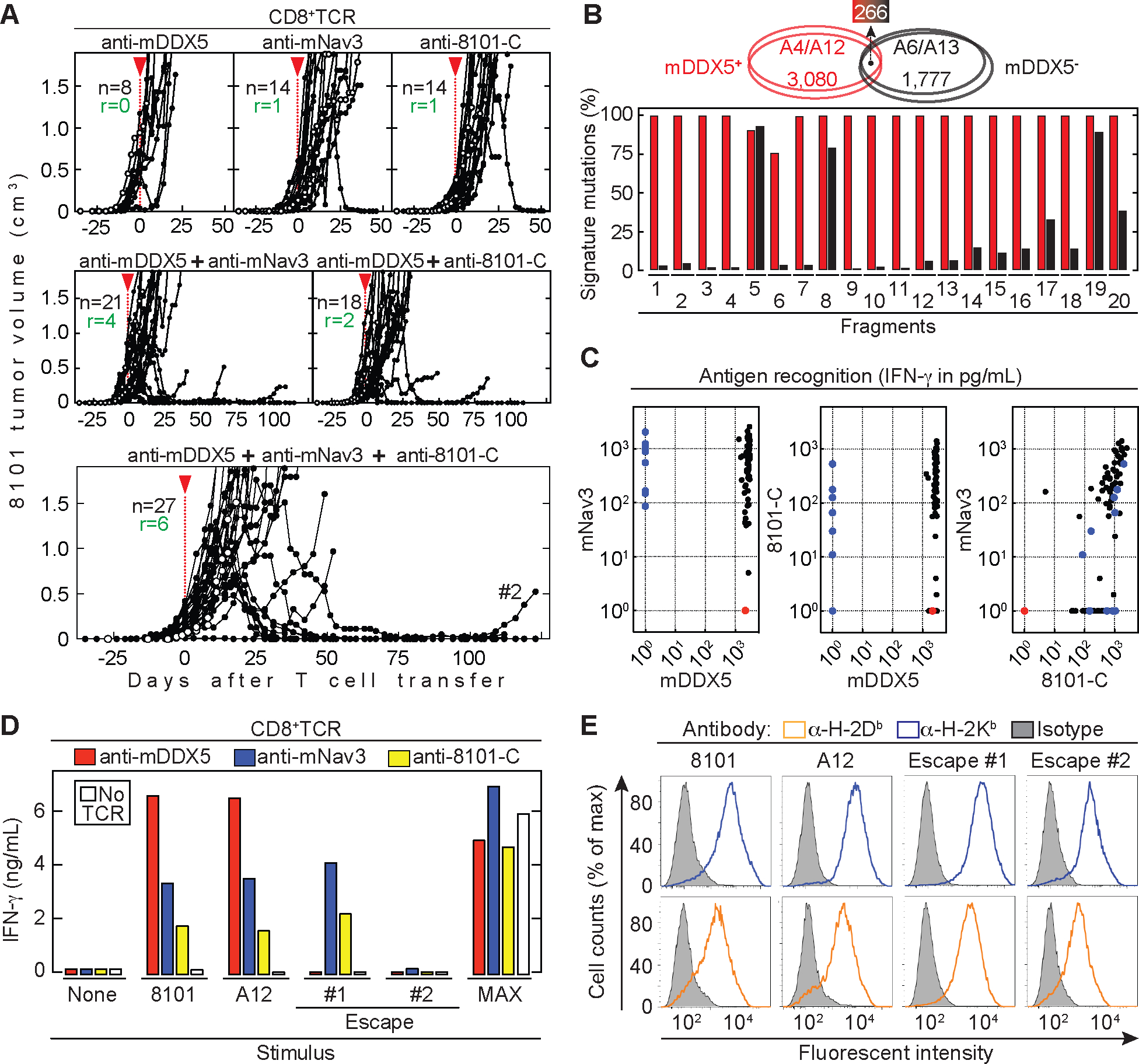
(A) 8101 tumor-bearing C57BL/6 Rag−/− mice were treated around 30 days after cancer cell inoculation as indicated by the red arrow head. Indicated are total numbers of mice (n) and the number of mice that rejected the tumor (r). Average tumor size: 290 ± 277 mm3. Data are summarized from at least three independent experiments. Open circles indicate untreated mice as controls. CD8+TCR therapy Upper panels: Either anti-mDDX5 (n = 8, left), or anti-mNav3 (n = 14, middle) or anti-8101-C (n = 14, right). Middle panels: A combination of the anti-mDDX5 and anti-mNav3 CD8+TCRs (n = 21, left) or anti-mDDX5 and anti-8101-C CD8+TCRs (n = 18, right). Lower panel: Combination using anti-mDDX5, anti-mNav3 and anti-8101-C (n = 27). (B) Signature mutations in mDDX5-positive and -negative 8101 clonotypes. Upper panel: Comparison of signature mutations between mDDX5-positive (A4 and A12 (red ellipses)) and mDDX5-negative (A6 and A13 (black ellipses)) 8101 clones. Total number of unique and shared mutations is indicated. Lower panel: Frequency of signature mutations of either mDDX5-positive (red) and -negative (black) clonotypes in fragments of the original 8101 tumor. (C) Recognition patterns of 83 cancer cell clones of the 8101 “B series”, were used for stimulation of T cells engineered with the anti-mDDX5, anti-mNav3 or anti-8101-C CD8+TCR. Shown is one representative out of two independent experiments. Clones that are only mNav3 (blue) or only mDDX5 positive (red) are indicated. (D) 8101 cancer cells, 8101 clone A12 and 8101 variants that escaped either anti-mDDX5-TCR (#1) or therapy with all three CD8+TCRs (#2) were used for T cell stimulation or (E) for MHC class I stain. Data are means of duplicates from one out of two independent experiments.
In order to overcome relapse from heterogeneity, we isolated another CD8+TCR from a T cell clone that recognized both phenotypes (17). Tandem minigenes (Supplementary Fig. 1) were used to identify the target epitope which is derived from the mutated neuron navigator 3 gene (mNav3S1490F1484–1492) and is presented by H-2Db. The mNav3 neoantigen, a mutant tumor suppressor important for cancerous growth (33), was one of the 266 mutations shared between both phenotypes and detected in all of the 20 obtained 8101 tumor fragments. Thus, the mNav3 neoantigen should be useful targeting every spatial part of the autochthonous 8101 tumor. However, with only one exception in 14 mice, 8101 tumors regularly escaped therapy with anti-mNav3 CD8+TCR-T cells (Fig. 1A, upper middle panel).
The analysis of antigenic heterogeneity using cancer cell clones has practical limitations, as the escape from targeting a single tumor antigen has a probability of 10−4 (one variant cell in 10,000 cancer cells (34). Therefore, targeting two independent antigens on a single cancer cell would reduce the chance of escape to 10−8 (14). Nevertheless, tumors also escaped even when both mDDX5 and mNav3 CD8+TCRs, which had similarly high avidity for their respective targets (Supplementary Fig. 2), were combined (Fig. 1A, left middle panel). Following this hypothesis, tumor escape might be nearly impossible when three antigens are targeted because the theoretical frequency of escape would be 10−12 (one variant cell in 1012 cancer cells). Therefore, we isolated a third CD8+TCR (anti-8101-C) from 8101-immunized C57BL/6 mice that recognized another independent 8101 cancer-specific antigen. This is supported by the different recognition patterns of previously generated 8101 cancer cell clones (20) (Supplementary Fig. 3A). All of these clones could be targeted with the combination of the CD8+TCRs, with most clones being recognized by all three (Fig. 1C) and others by two or at least one of the three CD8+TCRs (Supplementary Fig. 3B). Most 8101 tumors escaped when anti-8101-C CD8+TCR-T cells were used alone (Fig. 1A, upper right). Similar results were observed when anti-mDDX5 were combined with anti-8101-C T cells (Fig. 1A, middle right). Surprisingly however, even combination therapy using all three CD8+TCRs mostly failed (Fig. 1A, lower panel). In our previous work, we isolated an 8101 variant (Fig. 1D, Escape #1) which escaped from anti-mDDX5 treatment and showed loss of mDDX5 expression (13). This variant was no longer recognized by anti-mDDX5 T cells but was still recognized by anti-mNav3 and anti-8101-C T cells. Therefore, we determined whether escape from all three CD8+TCRs was also due to loss of expression of the targeted antigens. An 8101 tumor that relapsed 100 days after transfer of all three CD8+TCR-T cell populations was no longer recognized by any of them (Fig. 1D, Escape #2), even though the MHC class I molecules were still expressed (Fig. 1E).
Eradication by CD8+TCRs alone requires targeting of three independent, cancer-specific antigens on an artificially homogeneous tumor
The experiments shown in Fig. 1 model the realistic setting of a heterogeneous cancer as it arises in an individual. However, the 8101 tumor model may be exceptionally heterogeneous and failures of TCR-therapy might have been avoided using a less heterogeneous 8101 cancer cell population. Thus, we used clone A12 (17) which is recognized by all three CD8+TCRs (Fig. 1D) and is representative for the majority of 8101 cancer cell clones (Fig. 1C and Supplementary Fig. 3B). Furthermore, s.c. injected A12 cancer cells were regularly rejected by mice harboring only T cells expressing the anti-mDDX5 CD8+TCR, while the same mice failed to reject the uncloned original heterogeneous 8101 tumor (20). Preventing outgrowth of inoculated cancer cells is easier than eradicating established solid tumors that are actively growing and continue to diversify. Therefore, we generated clones from an established A12 tumor and examined them for mDDX5 expression. Our results (Supplementary Fig. 3C) suggest that A12-derived tumors were about 10-fold more homogeneous than the original 8101 tumor. Nevertheless, all A12-derived tumors escaped therapy using only anti-mDDX5 CD8+TCR-T cells (Fig. 2A). Escape variants induced weaker or no anti-mDDX5 CD8+TCR-T cell activation, indicating reduced amounts of antigen expression (Fig. 2B). Similar results were demonstrated using the anti-mNav3 CD8+TCR (Fig. 2C, left). Even targeting mDDX5 and mNav3 together led to tumor eradication in only 50% of the mice (Fig. 2C, right). Eradication of established A12 tumors was only achieved when using the combination of all three CD8+TCRs (anti-mDDX5, anti-mNav3 and anti-8101-C) (Fig. 2D). This result shows that the chosen CD8+TCRs can be effective in achieving tumor rejection. However, the difficulty of finding multiple CD8+TCRs that recognize different cancer-specific antigens against a patient’s tumor with little or no heterogeneity led us to search for other strategies to overcome therapeutic failure.
Figure 2. Homogeneous tumors developed from a cancer cell clone are eradicated by combination of three CD8+TCRs.
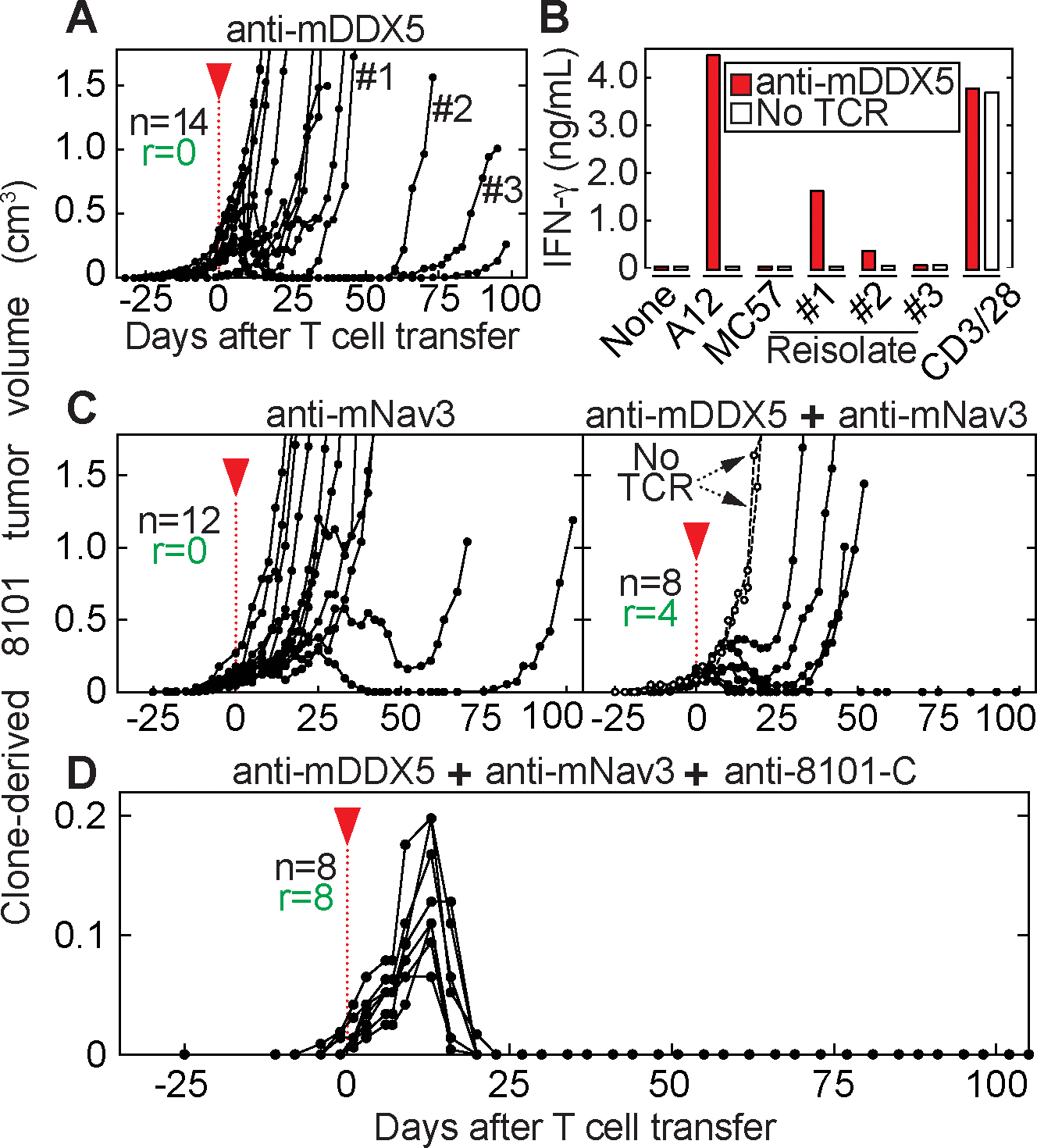
(A – D) B6 Rag−/− mice bearing 8101 clone A12 tumors were treated 25 or 30 days after cancer cell inoculation as indicated by the red arrow head. Average tumor sizes were 80 ± 82 mm3 at day of T cell transfer. Indicated are total mice (n) and the number of mice that rejected the tumor (r). Shown are data from at least two independent experiments. (A) anti-mDDX5 CD8+TCR-T cells were used for therapy (n = 14). Relapsed tumors were isolated (#1, #2 and #3). (B) 8101 cancer cell variants (see A), in vitro-cultured A12 and MC57 cancer cells were used for anti-mDDX5 T cell stimulation. Data are means of duplicates. (C) Anti-mNav3 CD8+TCR T cells were used for T cell transfer (n = 12, left). In addition, a combination of anti-mDDX5 together with anti-mNav3 CD8+TCR was used (n = 8, right). Mice without T cell treatment are shown as controls. (D) Combination therapy using anti-mDDX5, anti-mNav3 and anti-8101-C CD8+TCRs (n = 8).
Cotransfer of cancer-specific CD4+ T cells together with a single CD8+TCR eradicates a heterogeneous tumor
Our previous work showed that CD4+ T cells can cooperate with CD8+ T cells during the effector phase and prevent outgrowth of transplanted cancer cells (35). We therefore examined whether naturally heterogeneous, uncloned 8101 tumors can be eradicated when CD4+ T cells from 8101-immunized mice are co-transferred with anti-mDDX5 CD8+TCR-T cells. Fig. 3A shows that large, long-established 8101 tumors were eradicated when anti-mDDX5 CD8+TCR-T cells were combined with polyclonal 8101-immune CD4+ T cells. The combination of CD4+ T cells with a single CD8+TCR was as effective as combining polyclonal CD4+ and polyclonal CD8+ T cells from 8101-immunized mice (Fig. 3B). Polyclonal 8101-immune CD8+ T cells, generated in C57BL/6 CD4−/− mice, were ineffective alone (Fig. 3C, left), while, interestingly, polyclonal 8101-immune CD4+ T cells, generated in C57BL/6 CD8−/− mice, caused significant destruction of the tumors in 5 out of 6 mice followed by a temporary to long-term arrest of tumor growth (Fig. 3C, right), even though all mice eventually died of progressing cancers.
Figure 3. One CD8+TCR and one CD4+TCR are essential and sufficient for tumor eradication.
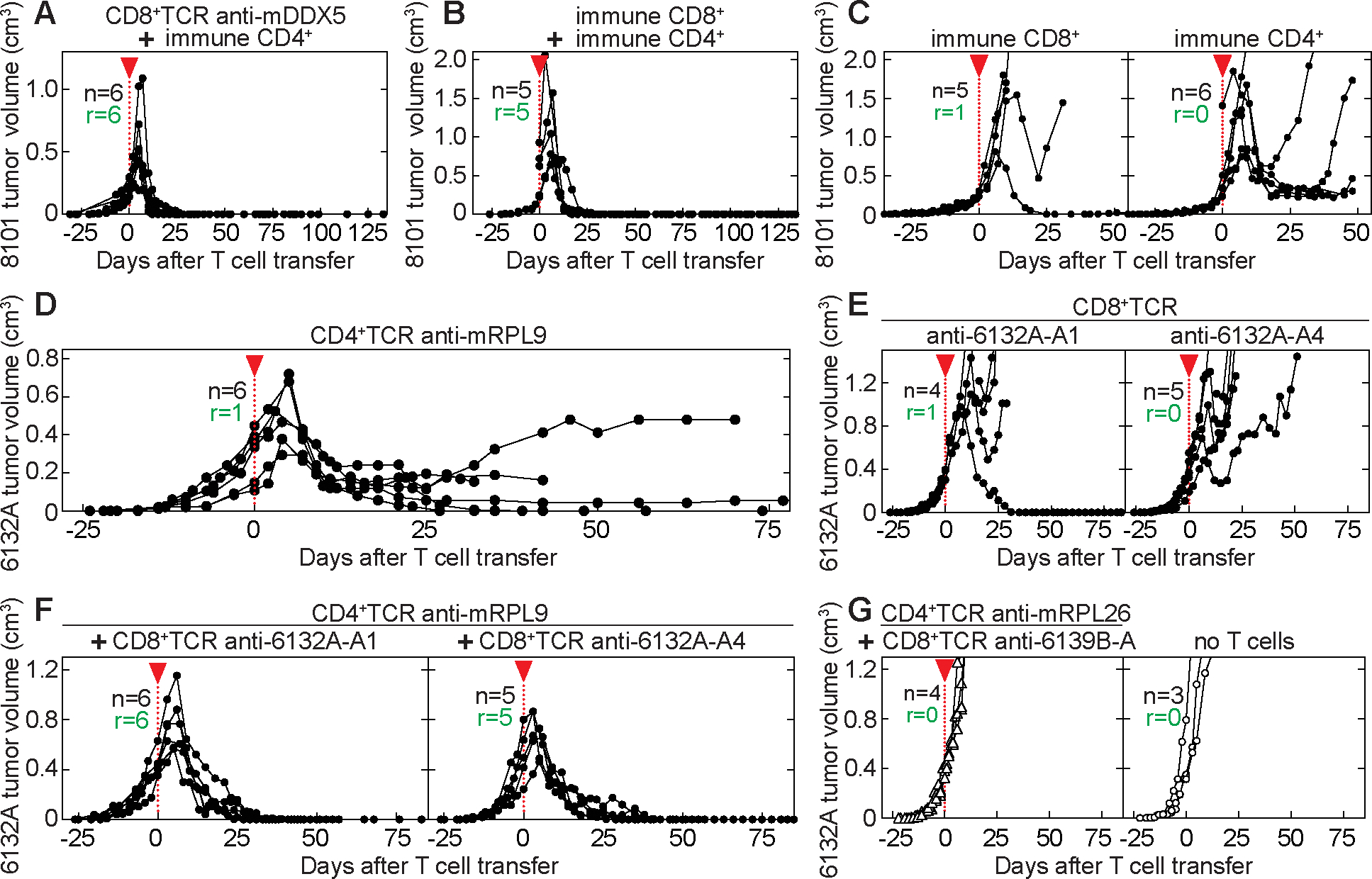
(A – G) Indicated are total mice (n) and the number of mice that rejected the tumor (r). Start of T cell transfer is indicated by the red arrow head. (A – B) 8101 tumor-bearing B6 Rag−/− mice were treated 35 days after cancer cell inoculation. Average tumor sizes were 429 ± 297 mm3 at day of T cell transfer and are summarized from three independent experiments. Therapy used: (A) anti-mDDX5 CD8+TCR together with 8101-immune CD4+ T cells (n = 6). (B) 8101-immune CD4+ and CD8+ T cells (n = 5). (C) 8101-immune CD8+ T cells (n = 5, left) or 8101-immune CD4+ T cells (n = 6, right). (D – G) 6132A tumor-bearing C3H Rag−/− mice were treated 25 days after cancer cell injection. Average tumor sizes were 448 ± 149 mm3 at day of T cell transfer and are summarized from three independent experiments. Therapy used: (D) anti-mRPL9 CD4+ T cells (n = 6), (E) anti-6132A-A1 (left, n = 4) or anti-6132A-A4 CD8+ T cells (right, n = 5) or (F) a combination of CD8+ T cells (anti-6132A-A1 (left, n = 6) or anti-6132A-A4 (right, n = 5)) with anti-mRPL9 CD4+ T cells, and (G) a combination of CD8+ and CD4+ T cells expressing TCRs of unrelated specificity (CD8+TCR: anti-6139B-A, CD4+TCR: anti-mRPL26, left, n = 4). Untreated mice are shown as control (right, n = 3).
One CD8+TCR and one CD4+TCR are essential and sufficient for tumor eradication
We used the UV-induced 6132A tumor model to be able to analyze the efficacy of a single cancer-specific CD4+TCR, a single cancer-specific CD8+TCR, or a combination of the two. First, we isolated the CD4+TCR from a CD4+ T cell hybridoma (5). This hybridoma recognizes the 6132A-cancer-specific L47H substitution in the ribosomal protein L9 (5) resulting in the MHC class II I-Ek-restricted mutant neoantigen mRPL9L47H41–53 expressed by 6132A cancer cells that are known to be negative for MHC class II (36). Our previous work had shown that mRPL9-specific CD4+ T cells are able to prevent outgrowth of 6132A cancer cells by recognizing stroma only (5). Next, we investigated the efficacy of this CD4+TCR against established tumors. Interestingly, treatment with anti-mRPL9 CD4+TCR-T cells destroyed large, established 6132A tumors (Fig. 3D) similar to the destruction of 8101 tumors with polyclonal 8101-immune CD4+ T cells (Fig. 3C, right). In both models, a much smaller tumor persisted following their destruction. Only 1 out of 6 established 6132A tumors were completely eradicated (Fig. 3D). However, we also observed one relapse from anti-mRPL9 CD4+TCR-T cells, which retained the mRPL9 antigen (Supplementary Fig. 4).
Subsequently, we investigated the effects of CD8+TCR-T cells alone against established 6132A tumors. We isolated two different CD8+TCRs (anti-6132A-A1 and anti-6132A-A4) from T cell clones generated from 6132A-immunized C3H/HeN mice (15). Anti-6132A-A1 or anti-6132A-A4 CD8+TCR-T cells recognized “unique” cancer-specific antigens on 6132A cancer cells, but no other cells from the exact same host (Supplementary Fig. 5A). As observed in the 8101-tumor model, neither anti-6132A-A1 nor anti-6132A-A4 CD8+TCR-T cells were effective in eradicating 6132A tumors (Fig. 3E). The two TCRs recognized two independent 6132A cancer-specific antigens, since the variant that had escaped anti-6132A-A4 CD8+TCR-therapy (Fig. 3E, right) was no longer recognized by the anti-6132A-A4 CD8+TCR, but remained sensitive to the anti-6132A-A1 CD8+TCR (Supplementary Fig. 5B and 5C).
Due to the synergy observed in the 8101-tumor model, we tested whether co-transfer of one CD4+TCR with one CD8+TCR would suffice for tumor eradication. Indeed, complete eradication of large, long-established 6132A tumors was achieved by using a combination of either anti-6132A-A1 or anti-6132A-A4 CD8+TCR-T cells with anti-mRPL9 CD4+TCR-T cells in all of the 11 treated mice (Fig. 3F). To prove that the polyclonal population of TCR-engineered T cells had no effect on their own in tumor eradication, we isolated a CD4+TCR (anti-mRPL26) and a CD8+TCR (anti-6139B-A) specific for the syngeneic 6139B cancer model (15,16). The combination of these CD4+TCR- and CD8+TCR-T cells was as ineffective in eradicating established 6132A tumors as transferring no T cells (Fig. 3G). To further exclude that direct recognition of cancer cells by CD4+TCR-engineered T cells contributed to tumor destruction in vivo, we incubated 6132A cancer cells with high amounts of IFN-γ to determine possible upregulation of MHC class II. However, I-Ek expression remained undetectable, while levels of MHC class I increased significantly on 6132A cancer cells (Supplementary Fig. 6). Similar results were obtained for 8101 cancer cells. In contrast, CD11b+ cells isolated from both mouse strains were positive for MHC class I and II.
CD8+TCRs require direct cancer cell recognition for tumor eradication and need help from CD4+TCRs for expansion
Blood samples from treated mice showed high IFN-γ values about 6 days after TCR-therapy, but only when CD4+TCR-T cells were used either alone or in combination. The contribution of CD8+TCR-T cells was minor (Supplementary Fig. 7). It seems as CD4+ T cells contribute mainly by cytokine secretion. We had observed previously that only indirect (stromal) recognition of cancer antigens by CD8+ and CD4+ T cells was sufficient in preventing outgrowth of cancer cells (35). Thus, we determined the stromal composition of 6132A tumors by staining for CD11b, CD11c, F4/80, Gr1, Ly6G and Ly6C (Supplementary Fig. 8). The majority of the stroma is characterized by CD11b+ cells which consist mainly, ~80 %, of tumor-associated macrophages (F4/80+ TAMs). These TAMs express IDO and PD-L1 consistent with an immunosuppressive environment. Both anti-6132A CD8+ T cells and anti-mRPL9 CD4+ T cells recognized bulk CD11b+ stromal cells isolated from 6132A tumors (Supplementary Fig. 9A and 9B). However, conventional methods of loading CD11b+ cells isolated from the spleen with 6132A cancer cell lysates were only successful for stimulation of CD4+TCR-T cells. Intriguingly, F4/80+ TAMs were as good in stimulating both T cell populations as bulk CD11b+ cells. Yet, stromal cells only induced proliferation of CD4+TCR- and not of CD8+TCR-T cells. Nonetheless, CD8+ T cells might have the advantage over CD4+ T cells to recognize cancer cells directly. Therefore, we investigated whether stromal recognition alone by CD8+TCR-T cells when combined with CD4+TCR-T cells was also sufficient for solid tumor elimination or whether direct recognition of 6132A cancer cells was required for eradication. Thus, we knocked out the murine beta-2 microglobulin (B2m) in 6132A cancer cells. These cancer cells were negative for staining of H-2Kk and H-2Dk (Fig. 4A) and were no longer recognized by the anti-6132A-A1 CD8+TCR (Fig. 4B). As expected, 6132A-specific CD8+TCR-T cells could still recognize cognate antigen on stromal cells in the 6132A tumor (Fig. 4C). However, the combination of anti-mRPL9 CD4+TCR- and anti-6132A-A1 CD8+TCR-T cells no longer eradicated the solid tumor, but only achieved cancer regression followed by growth arrest (Fig. 4D).
Figure 4. The CD8+TCRs require direct cancer cell recognition for tumor eradication and help from CD4+ T cells for expansion.
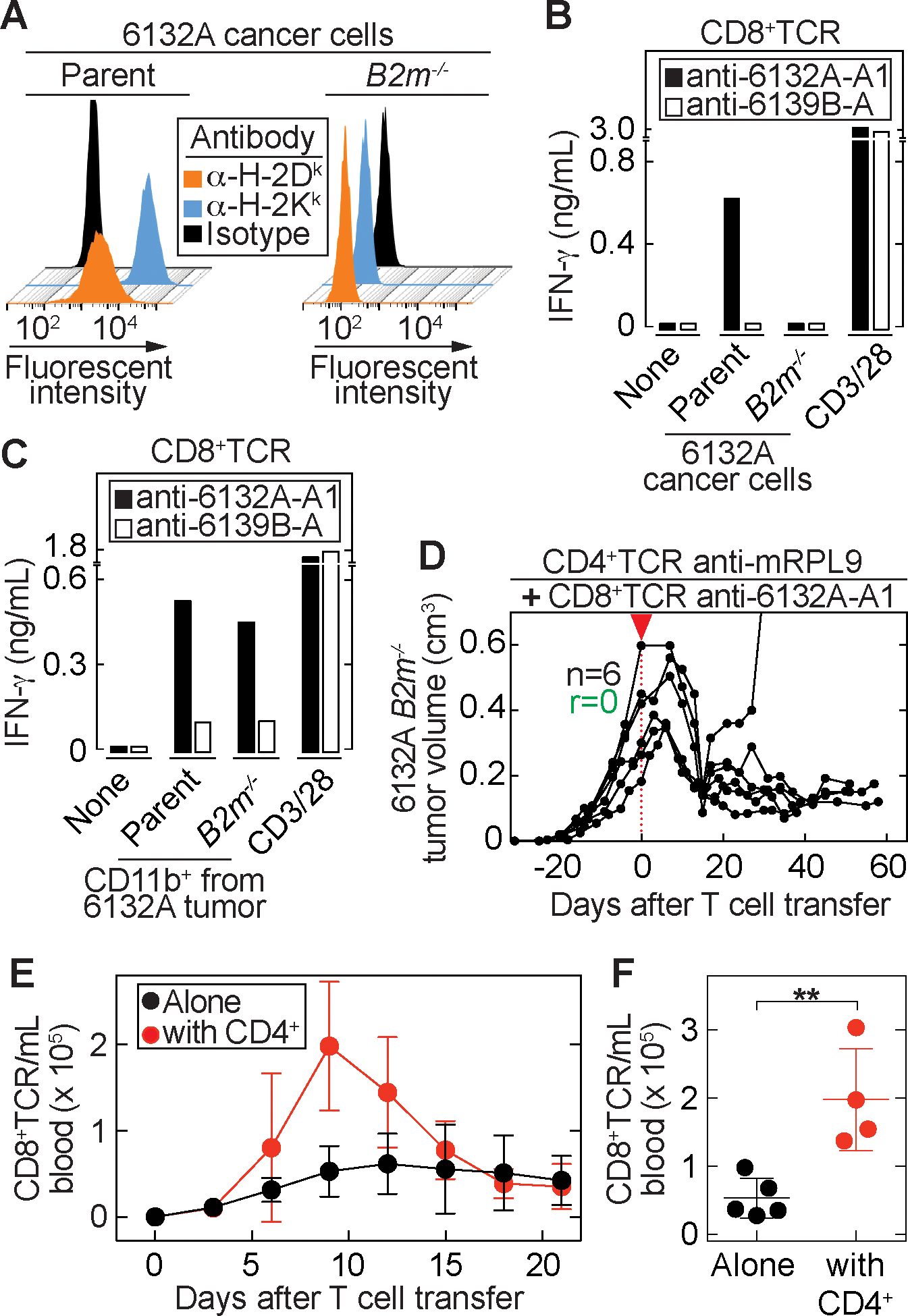
(A) MHC class I surface expression of parental 6132A and its beta-2-microglobulin knockout variant (B2m−/−). (B - C) anti-6132A-A1 or anti-6139B-A CD8+TCR T cells were stimulated with (B) Parental and B2m−/− cancer cells or (C) CD11b+ cells, isolated from parental or B2m−/− tumors. Data are means of duplicates from one representative out of three independent experiments. (D) B2m−/− tumor bearing C3H Rag−/− mice were treated around 30 days after tumor injection as indicated by the red arrow head. Average tumor sizes: 369 ± 150 mm3. anti-6132A-A1 CD8+TCR-T cells together with anti-mRPL9 CD4+TCR-T cells were used for treatment (n = 6). Indicated are the total numbers of mice (n) and the number of mice that rejected the tumor (r). Results are from two independent experiments. (E – F) 6132A tumor-bearing C3H Rag−/− mice were treated with anti-6132A-A1 (n = 2) or anti-6132A-A4 (n = 3) CD8+TCR-T cells alone or in combination with anti-mRPL9 CD4+TCR-T cells (anti-6132A-A1 combination (n = 2), anti-6132A-A4 combination (n = 2)). (E) Peripheral blood was analyzed at indicated time points. Total numbers of CD8+ and Vβ6+ (anti-6132A-A1) or Vβ8.3+ (anti-6132A-A4) T cells were determined. (F) Cell numbers were compared at day 9 after T cell transfer between CD8+ T cells transferred alone (n = 5) or in combination with CD4+ T cells (n = 4). Significance between groups was determined by a two-tailed Student’s t-test with **P ≤ 0.01.
To determine whether CD4+TCR-T cells provide help for CD8+TCR-T cells for proliferation in vivo, we analyzed the expansion of T cells after adoptive transfer. When CD8+TCR-T cells were used alone, only a small increase in numbers was observed while the combination with anti-mRPL9 CD4+TCR-T cells led to strong expansion (Fig. 4E). About 4x more CD8+TCR-T cells were detected at day 9 after transfer when used together with CD4+TCR-T cells compared to when used alone (Fig. 4F), showing that CD8+ T cells receive help from CD4+ T cells for improved expansion.
A four cell-type interaction is essential for cancer cell elimination.
To model the interaction of CD4+TCR- and CD8+TCR-T cells, stromal cells, and cancer cells in vitro, we developed a culture assay showing the required interaction partners needed for successful cancer cell elimination. 6132A cancer cells were cultured with CD11b+ cells, CD4+TCR- and CD8+TCR-T cells for 7 days. Then T cells were removed and the cancer cells were cultured for additional 5 days before analyzed for viability (Fig. 5A). Almost no live cells were detected when cancer cells were cultured with CD11b+, anti-mRPL9 CD4+TCR- and anti-6132A-A1 CD8+TCR-T cells. When CD11b+ cells were missing or mRPL26-specific CD4+TCR-T cells were used as non-specific CD4+ T cell help, 6132A cancer cells were viable and abundant. To investigate whether all four cell types interact with each other to achieve effective cancer cell elimination, we established a time-lapse imaging method that can determine the occurrence of four cell-type interactions (18). Cerulean-labeled 6132A cancer cells were cultured with DiD-labelled CD11b+ cells, anti-6132A-A1 mCherry+ CD8+TCR-T cells and either anti-mRPL9 (specific help) or anti-mRPL26 (non-specific help) GFP+ CD4+TCR-T cells (Fig. 5A). In the “specific” situation, cancer cells disappeared within 72 h while they proliferated in the “non-specific” situation (Fig. 5B and Supplementary Video S1 and S2). When analyzing cell-to-cell interactions, no differences were found for cancer cells that interacted either with CD8+ T cells alone (Fig. 5C, upper panel) or together with CD8+ T cells and CD11b+ cells (Fig. 5C, middle panel). However, when analyzing interactions between all four cell-types (cancer cells with CD11b+, CD8+ and CD4+ T cells, Fig. 5C, lower panel) we found a higher percentage of cancer cells being in a four cell-type interaction over the whole time in the “specific” situation. Furthermore, quantification of these interactions showed a significant difference (Supplementary Table S1) for the four cell-type interaction of cancer cells when comparing the “specific” with the “non-specific” situation.
Figure 5. A four cell-type interaction is essential for cancer cell elimination.
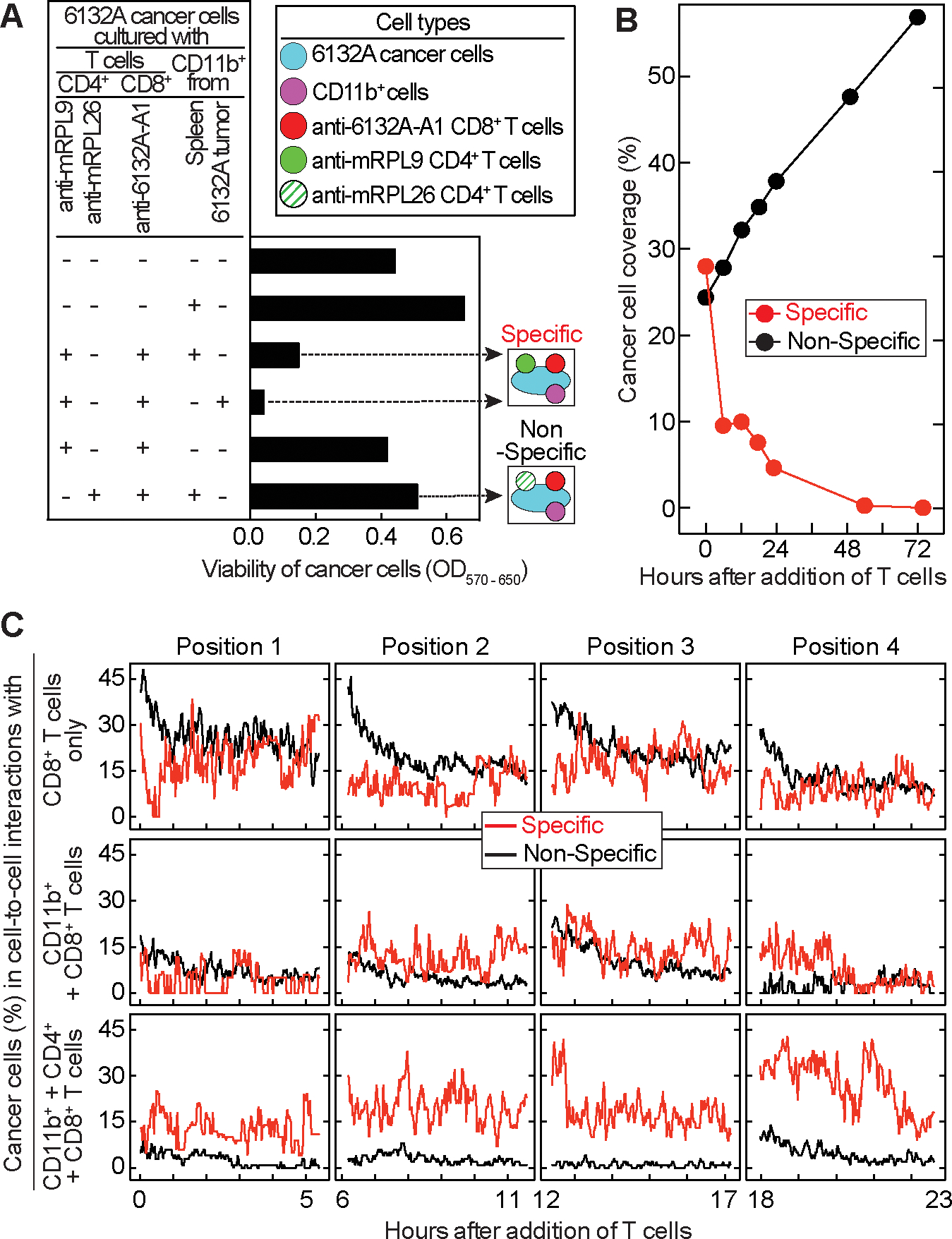
(A) 6132A cancer cells were cultured with or without CD11b+ cells isolated either from spleens of C3H/HeN mice or from 6132A tumors. Ten days after incubation with CD8+ and CD4+ T cells, living cells in culture were analyzed using MTT. Cell types and the two experimental settings (specific and non-specific) analyzed in (B – C) are color-coded. (B) Cerulean-labelled 6132A cancer cells were cultured over night with DiD-labeled CD11b+ cells isolated from spleen of C3H/HeN mice. mCherry-labeled anti-6132A-A1 CD8+TCR-T cells were added together with either GFP-labeled anti-mRPL9 (specific, red) or anti-mRPL26 CD4+TCR-T cells (non-specific, black). Coverage of cancer cells was determined for 72 h. (C) Percentage of cancer cells that are in cell-to-cell contacts with either CD8+ T cells alone (top), CD8+ T cells and CD11b+ cells (middle) or CD8+, CD4+ T cells and CD11b+ cells (bottom) over 24 h.
Discussion
Here we investigated how adoptive transfer of autologous neoantigen-specific TCR-engineered T cells (TCR-therapy) can be effective in rejecting long-established and naturally heterogeneous tumors. Mimicking solid tumors as they occur in human patients, TCR-therapy was initiated three weeks after cancer cell inoculation to exclude the participation of the early acute inflammatory innate immune response (37). Furthermore, the 8101 and 6132A cell lines were derived from primary cancer cell cultures, only minimally expanded in vitro and never passaged in vivo. Our tumor models therefore reflected important characteristics of human cancer: (i) natural heterogeneity and (ii) unmanipulated expression amounts of autochthonous neoantigens which is in contrast to homogeneous models using non-syngeneic antigen artificially overexpressed for successful TCR-therapy (38). Our autochthonous primary cancer cell cultures regularly form tumors in immunodeficient mice, but rarely grow in young fully immunocompetent mice (25,26). Using mice without endogenous T cells was essential to exclude the participations of such T cells (39,40) and to prove that a single pair of cancer-specific TCRs - i.e., one CD4+TCR targeting the tumor stroma and one CD8+TCR recognizing the cancer cells directly - can be sufficient to eradicate established heterogeneous solid tumors. Neither external addition of IL-2 nor immune checkpoint blockade was required. However, limitations of our reductionist approach include (i) how targeting of additional antigens by endogenous T cells and/or (ii) how upregulation of antigen presentation following pre-conditioning to deplete endogenous T cells (41) might affect heterogeneous tumors escaping TCR-therapy. Nevertheless, targeting MHC class I and II antigens was still required even when mice with an endogenous T cell repertoire received TCR-therapy (39,40).
Most tumors showed an intrinsic resistance to adoptive CD8+TCR-therapy even though a combination of three CD8+TCRs was used. Even when artificially re-cloned for greater homogeneity, most tumors still seemed similarly unaffected by transfer of CD8+TCR-T cells alone and all three CD8+TCRs were needed in combination to reject artificially homogeneous tumors. Since paracrine availability of IFN-γ produced by CD8+ T cells can increase their local motility and cytotoxicity and is seemingly necessary at the site of rejection (42), insufficient local levels of IFN-γ may have contributed to the failure when a single type of CD8+TCR-T cell was used. In the naturally heterogeneous tumors, about 10 % of cancer cells lacked at least one of the targeted neoantigens and a mosaic antigen expression pattern in tumors after outgrowth has been observed before (35). Therefore, even when the combination of three CD8+TCRs was used, T cells that enter tumor areas with low antigen abundance may not always receive sufficient antigenic stimulation to release enough IFN-γ for paracrine stimulation and effective activation; thus most heterogeneous solid tumors escape. Even when occasionally CD8+TCR combinations were initially effective, an MHC I positive relapse variant that had lost expression of all three targeted neoantigens was observed. Thus, and in agreement with earlier studies (13), CD8+TCR-therapy on its own selects for antigen-loss variants. However, we cannot exclude the participation of T cell exhaustion in treatment failures.
Our results suggest that IFN-γ production by CD4+ T cells is key to rejection of solid tumors when combined with only a single type of CD8+TCR. Spatial gradients of cytokines result in a variable degree of cytokine exposure which is important for cell-to-cell communication and activation (43,44). Cytokines such as IFN-γ, therefore, act most efficiently in diffusion-limited spaces. In addition, we found that cotransfer of CD4+TCR-T cells greatly helped CD8+TCR-T cells to proliferate, which is consistent with classical DC-dependent help in lymph nodes (45). Those two effects, increased IFN-γ levels and increased numbers of CD8+TCR-T cells, combined could lead to more efficient killing of cancer cells since multiple CD8+ T cells may be needed to kill one cancer cell in vivo (46).
In accordance with our (36) and other (39) tumor models, most human solid cancers do not express MHC class II and do not allow for direct recognition by CD4+ T cells, even though melanoma represent a notable exception (47,48). However, even the success of TIL-therapy against melanoma might be attributed to a combination of CD8+ and CD4+ T cells (49). In our studies, eradication of established solid tumors required direct cancer cell recognition by CD8+ T cells when combined with CD4+ T cells.
There is an increasing realization that CD4+ and CD8+ T cells are both essential to prevent the outgrowth of inoculated cancer cells (35,50,51). Engineering CD8+ and CD4+ T cells with only MHC-I-restricted TCRs might lead to similar therapeutic efficiency. Yet, such an approach would only target the cross-presented MHC-I antigen on the tumor stroma which would exclude seemingly required MHC-II presented antigens from therapy.
Effective elimination of cancer cells may rely on the collaboration of CD4+ and CD8+ T cells at the effector phase (35,52). In our model, the efficacy of cancer cell destruction in vitro by cancer cell-recognizing CD8+ T cells and stromal cell-recognizing CD4+ T cells depended on a four-cell interaction between CD4+ and CD8+ T cells together with antigen presenting cells (APCs) and cancer cells. While the formation of four-cell clusters have yet to be confirmed in vivo, it can be concluded that the recognition of stromal cells by CD4+ T cells induces the secretion of IFN-γ, which can upregulate the expression of MHC class I and tumor antigen (53) in the cancer cell, thus enabling more effective recognition by CD8+ T cells.
Together, we achieved curative effects of naturally heterogeneous tumors with just two TCRs: a stromal cell-recognizing CD4+TCR and a cancer cell-recognizing CD8+TCR. Our finding that these two types of TCRs can be essential and sufficient for eradication of solid tumors shows that simplification of TCR gene therapy may be possible without sacrificing efficacy.
Supplementary Material
Translational relevance.
Targeting mutant neoantigens might be required for eradication of established solid tumors. Some cancers harbor ~10, other cancers hundreds of somatic mutations. These mutations can result in cancer-specific antigens each of which may be presented by multiple patient-specific MHC alleles. Recognition of such antigens is usually mediated by T cells of multiple different T cell receptor (TCR) clonotypes. To achieve curative therapy with TCR-engineered autologous T cells (TCR-therapy), information is needed on how many TCRs are required targeting which types of neoantigens by which type of T cells. Previous studies revealed that targeting an unmanipulated autochthonous neoantigen using one TCR in one type of T cells cannot eradicate established tumors. Here we show that two neoantigen-specific TCRs, one recognizing tumor stroma and one cancer cells directly, can be essential and sufficient for eradication of solid tumors. This demonstrates that simplifications to neoantigen-specific TCR-therapy may be possible without sacrificing efficacy.
Acknowledgements
This work was supported by National Institute of Health grants [R01-CA022677 and R01-CA037156 to H.Schreiber]; Harriet and Allan Wulfstat, and the Gerald O. Mann Foundation [to H. Schreiber]; the Cancer Research Foundation [to H. Schreiber]; and a gift of Janet D. Rowley [to K. Schreiber and H. Schreiber]. Deutsche Forschungsgemeinschaft [409512914 to M. Leisegang]; Berliner Krebsgesellschaft e.V. [LEFF201901 to M. Leisegang]; German Cancer Consortium [to M. Leisegang]; and Einstein Foundation Berlin [A-2019-559 to M. Leisegang]. The David and Etta Jonas Center for Cellular Therapy, The University of Chicago [to H. Schreiber and M. Leisegang]; and The University of Chicago Cancer Center. We also thank Michael Bishop, Vytas Bindokas, Madeline Steiner and Yanran He for technical support, critical comments and/or valuable suggestions.
Footnotes
Conflict of interest: The authors declare no potential conflict of interest.
References
- 1.Haslam A, Prasad V. Estimation of the Percentage of US Patients With Cancer Who Are Eligible for and Respond to Checkpoint Inhibitor Immunotherapy Drugs. JAMA Netw Open 2019;2(5):e192535 doi 10.1001/jamanetworkopen.2019.2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rosenberg SA, Restifo NP. Adoptive cell transfer as personalized immunotherapy for human cancer. Science 2015;348(6230):62–8 doi 10.1126/science.aaa4967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tran E, Robbins PF, Rosenberg SA. ‘Final common pathway’ of human cancer immunotherapy: targeting random somatic mutations. Nat Immunol 2017;18(3):255–62 doi 10.1038/ni.3682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gubin MM, Zhang X, Schuster H, Caron E, Ward JP, Noguchi T, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature 2014;515(7528):577–81 doi 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Monach PA, Meredith SC, Siegel CT, Schreiber H. A unique tumor antigen produced by a single amino acid substitution. Immunity 1995;2(1):45–59. [DOI] [PubMed] [Google Scholar]
- 6.Wölfel T, Hauer M, Schneider J, Serrano M, Wölfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995;269:1281–4. [DOI] [PubMed] [Google Scholar]
- 7.Robbins PF, Lu YC, El-Gamil M, Li YF, Gross C, Gartner J, et al. Mining exomic sequencing data to identify mutated antigens recognized by adoptively transferred tumor-reactive T cells. Nat Med 2013;19(6):747–52 doi 10.1038/nm.3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Segal NH, Parsons DW, Peggs KS, Velculescu V, Kinzler KW, Vogelstein B, et al. Epitope landscape in breast and colorectal cancer. Cancer Res 2008;68(3):889–92 doi 10.1158/0008-5472.Can-07-3095. [DOI] [PubMed] [Google Scholar]
- 9.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA, Jr., Kinzler KW. Cancer genome landscapes. Science 2013;339(6127):1546–58 doi 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Philip M, Fairchild L, Sun L, Horste EL, Camara S, Shakiba M, et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017;545(7655):452–6 doi 10.1038/nature22367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Leidner R, Sanjuan Silva N, Huang H, Sprott D, Zheng C, Shih YP, et al. Neoantigen T-Cell Receptor Gene Therapy in Pancreatic Cancer. N Engl J Med 2022;386(22):2112–9 doi 10.1056/NEJMoa2119662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim SP, Vale NR, Zacharakis N, Krishna S, Yu Z, Gasmi B, et al. Adoptive Cellular Therapy with Autologous Tumor-Infiltrating Lymphocytes and T-cell Receptor-Engineered T Cells Targeting Common p53 Neoantigens in Human Solid Tumors. Cancer Immunol Res 2022:OF1–OF15 doi 10.1158/2326-6066.Cir-22-0040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Leisegang M, Engels B, Schreiber K, Yew PY, Kiyotani K, Idel C, et al. Eradication of Large Solid Tumors by Gene Therapy with a T-Cell Receptor Targeting a Single Cancer-Specific Point Mutation. Clin Cancer Res 2016;22(11):2734–43 doi 10.1158/1078-0432.Ccr-15-2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wortzel RD, Philipps C, Schreiber H. Multiple tumour-specific antigens expressed on a single tumour cell. Nature 1983;304:165–7. [DOI] [PubMed] [Google Scholar]
- 15.Ward PL, Koeppen H, Hurteau T, Schreiber H. Tumor antigens defined by cloned immunological probes are highly polymorphic and are not detected on autologous normal cells. J Exp Med 1989;170(1):217–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Beck-Engeser GB, Monach PA, Mumberg D, Yang F, Wanderling S, Schreiber K, et al. Point mutation in essential genes with loss or mutation of the second allele: relevance to the retention of tumor-specific antigens. J Exp Med 2001;194(3):285–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dubey P, Hendrickson RC, Meredith SC, Siegel CT, Shabanowitz J, Skipper JC, et al. The immunodominant antigen of an ultraviolet-induced regressor tumor is generated by a somatic point mutation in the DEAD box helicase p68. J Exp Med 185(4):695–705, 1997. Feb 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Diehl MI, Wolf SP, Bindokas VP, Schreiber H. Automated cell cluster analysis provides insight into multi-cell-type interactions between immune cells and their targets. Exp Cell Res 2020:112014 doi 10.1016/j.yexcr.2020.112014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matsushita H, Vesely MD, Koboldt DC, Rickert CG, Uppaluri R, Magrini VJ, et al. Cancer exome analysis reveals a T-cell-dependent mechanism of cancer immunoediting. Nature 2012;482(7385):400–4 doi 10.1038/nature10755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schreiber K, Karrison TG, Wolf SP, Kiyotani K, Steiner M, Littmann ER, et al. Impact of TCR Diversity on the Development of Transplanted or Chemically Induced Tumors. Cancer Immunol Res 2020;8(2):192–202 doi 10.1158/2326-6066.Cir-19-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, et al. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol 2016;34(2):184–91 doi 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Das K, Eisel D, Lenkl C, Goyal A, Diederichs S, Dickes E, et al. Generation of murine tumor cell lines deficient in MHC molecule surface expression using the CRISPR/Cas9 system. PLoS One 2017;12(3):e0174077 doi 10.1371/journal.pone.0174077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 2013;8(11):2281–308 doi 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Morita S, Kojima T, Kitamura T. Plat-E: an efficient and stable system for transient packaging of retroviruses. Gene Ther 2000;7(12):1063–6 doi 10.1038/sj.gt.3301206. [DOI] [PubMed] [Google Scholar]
- 25.Schreiber K, Arina A, Engels B, Spiotto MT, Sidney J, Sette A, et al. Spleen cells from young but not old immunized mice eradicate large established cancers. Clin Cancer Res 2012;18(9):2526–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kripke ML. Latency, histology, and antigenicity of tumors induced by ultraviolet light in three inbred mouse strains. Cancer Res 1977;37(5):1395–400. [PubMed] [Google Scholar]
- 27.Engels B, Engelhard VH, Sidney J, Sette A, Binder DC, Liu RB, et al. Relapse or eradication of cancer is predicted by peptide-major histocompatibility complex affinity. Cancer Cell 2013;23(4):516–26 doi 10.1016/j.ccr.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gray PW, Goeddel DV. Cloning and expression of murine immune interferon cDNA. Proc Natl Acad Sci U S A 1983;80(19):5842–6 doi 10.1073/pnas.80.19.5842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, et al. Fiji: an open-source platform for biological-image analysis. Nat Methods 2012;9(7):676–82 doi 10.1038/nmeth.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wickham H ggplot2: Elegant Graphics for Data Analysis. Springer-Verlag New York; 2016. [Google Scholar]
- 31.R Core Team. R: A language and environment for statistical computing. 2018. [Google Scholar]
- 32.Hashemi V, Masjedi A, Hazhir-Karzar B, Tanomand A, Shotorbani SS, Hojjat-Farsangi M, et al. The role of DEAD-box RNA helicase p68 (DDX5) in the development and treatment of breast cancer. J Cell Physiol 2019;234(5):5478–87 doi 10.1002/jcp.26912. [DOI] [PubMed] [Google Scholar]
- 33.Cohen-Dvashi H, Ben-Chetrit N, Russell R, Carvalho S, Lauriola M, Nisani S, et al. Navigator-3, a modulator of cell migration, may act as a suppressor of breast cancer progression. EMBO Mol Med 2015;7(3):299–314 doi 10.15252/emmm.201404134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tomlinson IP, Novelli MR, Bodmer WF. The mutation rate and cancer. Proc Natl Acad Sci U S A 1996;93(25):14800–3 doi 10.1073/pnas.93.25.14800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schietinger A, Philip M, Liu RB, Schreiber K, Schreiber H. Bystander killing of cancer requires the cooperation of CD4(+) and CD8(+) T cells during the effector phase. J Exp Med 2010;207(11):2469–77 doi 10.1084/jem.20092450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mumberg D, Monach PA, Wanderling S, Philip M, Toledano AY, Schreiber RD, et al. CD4(+) T cells eliminate MHC class II-negative cancer cells in vivo by indirect effects of IFN-gamma. Proc Natl Acad Sci U S A 1999;96(15):8633–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schreiber K, Rowley DA, Riethmuller G, Schreiber H. Cancer immunotherapy and preclinical studies: why we are not wasting our time with animal experiments. Hematol Oncol Clin North Am 2006;20(3):567–84. [DOI] [PubMed] [Google Scholar]
- 38.Poncette L, Chen X, Lorenz FK, Blankenstein T. Effective NY-ESO-1-specific MHC II-restricted T cell receptors from antigen-negative hosts enhance tumor regression. J Clin Invest 2019;129(1):324–35 doi 10.1172/JCI120391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brightman SE, Becker A, Thota RR, Naradikian MS, Chihab L, Zavala KS, et al. Neoantigen-specific stem cell memory-like CD4(+) T cells mediate CD8(+) T cell-dependent immunotherapy of MHC class II-negative solid tumors. Nat Immunol 2023;24(8):1345–57 doi 10.1038/s41590-023-01543-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schaettler MO, Desai R, Wang AZ, Livingstone AJ, Kobayashi DK, Coxon AT, et al. TCR-engineered adoptive cell therapy effectively treats intracranial murine glioblastoma. J Immunother Cancer 2023;11(2) doi 10.1136/jitc-2022-006121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klebanoff CA, Khong HT, Antony PA, Palmer DC, Restifo NP. Sinks, suppressors and antigen presenters: how lymphodepletion enhances T cell-mediated tumor immunotherapy. Trends Immunol 2005;26(2):111–7 doi 10.1016/j.it.2004.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bhat P, Leggatt G, Waterhouse N, Frazer IH. Interferone-y derived from cytotoxic lymphocytes directly enhances their motility and cytotoxicity. Cell Death and Disease 2017. doi 10.1038/cddis.2017.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Oyler-Yaniv A, Oyler-Yaniv J, Whitlock BM, Liu Z, Germain RN, Huse M, et al. A Tunable Diffusion-Consumption Mechanism of Cytokine Propagation Enables Plasticity in Cell-to-Cell Communication in the Immune System. Immunity 2017(46):609–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Thurley K, Gerecht D, Friedmann E, Höfer T. Three-Dimensional Gradients of Cytokine Signaling between T Cells. PLOS Computational Biology 2015. doi 10.1371/journal.pcbi.1004206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ahrends T, Spanjaard A, Pilzecker B, Babala N, Bovens A, Xiao Y, et al. CD4(+) T Cell Help Confers a Cytotoxic T Cell Effector Program Including Coinhibitory Receptor Downregulation and Increased Tissue Invasiveness. Immunity 2017;47(5):848–61 e5 doi 10.1016/j.immuni.2017.10.009. [DOI] [PubMed] [Google Scholar]
- 46.Halle S, Halle O, Forster R. Mechanisms and Dynamics of T Cell-Mediated Cytotoxicity In Vivo. Trends Immunol 2017;38(6):432–43 doi 10.1016/j.it.2017.04.002. [DOI] [PubMed] [Google Scholar]
- 47.Muranski P, Boni A, Antony PA, Cassard L, Irvine KR, Kaiser A, et al. Tumor-specific Th17-polarized cells eradicate large established melanoma. Blood 2008;112(2):362–73 doi blood-2007-11-120998 [pii] 10.1182/blood-2007-11-120998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hunder NN, Wallen H, Cao J, Hendricks DW, Reilly JZ, Rodmyre R, et al. Treatment of metastatic melanoma with autologous CD4+ T cells against NY-ESO-1. N Engl J Med 2008;358(25):2698–703 doi 10.1056/NEJMoa0800251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hall MS, Teer JK, Yu X, Branthoover H, Snedal S, Rodriguez-Valentin M, et al. Neoantigen-specific CD4(+) tumor-infiltrating lymphocytes are potent effectors identified within adoptive cell therapy products for metastatic melanoma patients. J Immunother Cancer 2023;11(10) doi 10.1136/jitc-2023-007288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kreiter S, Vormehr M, van de Roemer N, Diken M, Lower M, Diekmann J, et al. Mutant MHC class II epitopes drive therapeutic immune responses to cancer. Nature 2015;520(7549):692–6 doi 10.1038/nature14426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Alspach E, Lussier DM, Miceli AP, Kizhvatov I, DuPage M, Luoma AM, et al. MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 2019;574(7780):696–701 doi 10.1038/s41586-019-1671-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 2010;70(21):8368–77 doi 10.1158/0008-5472.Can-10-1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Decker T, Kovarik P, Meinke A. GAS elements: a few nucleotides with a major impact on cytokine-induced gene expression. J Interferon Cytokine Res 1997;17(3):121–34 doi 10.1089/jir.1997.17.121. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw sequencing data generated in this study are publicly available in Sequence Read Archive (SRA) BioProject ID PRJNA1054787 at accession numbers SAMN38933246 (6132A), SAMN38933247 (6132-HLF), SAMN38933248 (8101), SAMN38933249 (8101-HLF), SAMN38933250 (8101 Clone 4), SAMN38933251 (8101 Clone 6), SAMN38933252 (8101 Clone 12), SAMN38933253 (8101 Clone 13), SAMN38933254 (8101 tumor fragment 1), SAMN38933255 (8101 tumor fragment 2), SAMN38933256 (8101 tumor fragment 3), SAMN38933257 (8101 tumor fragment 4), SAMN38933258 (8101 tumor fragment 6), SAMN38933259 (8101 tumor fragment 7), SAMN38933260 (8101 tumor fragment 16), SAMN38933268 (8101 tumor fragment 18), SAMN38933261 (8101 tumor fragment 20), SAMN38933269 (8101 tumor fragment 21), SAMN38933270 (8101 tumor fragment 22), SAMN38933271 (8101 tumor fragment 23), SAMN38933272 (8101 tumor fragment 24), SAMN38933273 (8101 tumor fragment 27), SAMN38933262 (8101 tumor fragment 26), SAMN38933263 (8101 tumor fragment 30), SAMN38933264 (8101 tumor fragment 36), SAMN38933265 (8101 tumor fragment 37), SAMN38933266 (8101 tumor fragment 39), SAMN38933267 (8101 tumor fragment 53). All other data and materials generated in this study are available upon request from the corresponding author.