

Abstract
Purpose
Pneumococcal meningitis is a major cause of hearing loss and permanent neurological impairment despite widely available antimicrobial therapies to control infection. Methods to improve hearing outcomes for those who survive bacterial meningitis remains elusive. We used a mouse model of pneumococcal meningitis to evaluate the impact of mononuclear phagocytes on hearing outcomes and cochlear ossification by altering the expression of CX3CR1 and CCR2 in these infected mice.
Methods
We induced pneumococcal meningitis in approximately 500 C57Bl6 adult mice using live Streptococcus pneumoniae (serotype 3, 1 × 105 colony forming units (cfu) in 10 µl) injected directly into the cisterna magna of anesthetized mice and treated these mice with ceftriaxone daily until recovered. We evaluated hearing thresholds over time, characterized the cochlear inflammatory response, and quantified the amount of new bone formation during meningitis recovery. We used microcomputed tomography (microCT) scans to quantify cochlear volume loss caused by neo-ossification. We also performed perilymph sampling in live mice to assess the integrity of the blood-perilymph barrier during various time intervals after meningitis. We then evaluated the effect of CX3CR1 or CCR2 deletion in meningitis symptoms, hearing loss, macrophage/monocyte recruitment, neo-ossification, and blood labyrinth barrier function.
Results
Sixty percent of mice with pneumococcal meningitis developed hearing loss. Cochlear fibrosis could be detected within 4 days of infection, and neo-ossification by 14 days. Loss of spiral ganglion neurons was common, and inner ear anatomy was distorted by scarring caused by new soft tissue and bone deposited within the scalae. The blood-perilymph barrier was disrupted at 3 days post infection (DPI) and was restored by seven DPI. Both CCR2 and CX3CR1 monocytes and macrophages were present in the cochlea in large numbers after infection. Neither chemokine receptor was necessary for the induction of hearing loss, cochlear fibrosis, ossification, or disruption of the blood-perilymph barrier. CCR2 knockout (KO) mice suffered the most severe hearing loss. CX3CR1 KO mice demonstrated an intermediate phenotype with greater susceptibility to hearing loss compared to control mice. Elimination of CX3CR1 mononuclear phagocytes during the first 2 weeks after meningitis in CX3CR1-DTR transgenic mice did not protect mice from any of the systemic or hearing sequelae of pneumococcal meningitis.
Conclusions
Pneumococcal meningitis can have devastating effects on cochlear structure and function, although not all mice experienced hearing loss or cochlear damage. Meningitis can result in rapid progression of hearing loss with fibrosis starting at four DPI and ossification within 2 weeks of infection detectable by light microscopy. The inflammatory response to bacterial meningitis is robust and can affect all three scalae. Our results suggest that CCR2 may assist in controlling infection and maintaining cochlear patency, as CCR2 knockout mice experienced more severe disease, more rapid hearing loss, and more advanced cochlear ossification after pneumococcal meningitis. CX3CR1 also may play an important role in the maintenance of cochlear patency.
Supplementary Information
The online version contains supplementary material available at 10.1007/s10162-024-00935-4.
Keywords: Meningitis, Labyrinthitis ossificans, Cochlea, Inflammation
Introduction
Infections of the central nervous system (CNS) are life-threatening illnesses that can result in loss of life, severe brain damage, and profound hearing loss. Bacterial meningitis represents one of the most common forms of CNS infection, particularly in young children, and these infections are a major target of childhood vaccination [1]. While the incidence of bacterial meningitis decreased after development of effective vaccines, sequelae of bacterial meningitis remain severe and confer substantial morbidity. Early treatment with antibiotics is necessary for survival, and late effects of meningitis, such as deafness, still occur in 27% of survivors [2]. The specific mechanisms of hearing loss and cochlear damage after meningitis are not well understood. We propose that both the infection and the immune response to infection contribute to permanent cochlear injury. We adopted an experimental bacterial meningitis model in adult mice to study the process that leads to hearing disability and to evaluate the contributions of two specific populations of immune cells, CCR2 inflammatory monocytes, and CX3CR1 monocytes and macrophages. The function of these two chemokine receptors has been implicated in infectious and inflammatory conditions affecting the nervous system and has been shown to impact disease outcome [3–12].
Prior animal studies in bacterial meningitis have demonstrated many aspects of cochlear pathology after meningitis. Animal models and postmortem human temporal bone studies after meningitis have shown rampant infection and inflammation within the labyrinth characterized by abundant leukocytes and bacteria in the scalae [13–18]. Fibrosis and ossification of the inner ear fluid spaces often follow the inflammatory response [13, 19]. Ossification can occur within weeks of infection, and cochlear fibrosis and ossification are accompanied by exceptionally poor hearing outcomes. Studies using systemic immunosuppressive agents such as corticosteroids demonstrate improved hearing outcomes in patients receiving corticosteroids along with antibiotics for bacterial meningitis, supporting the idea that the inflammatory response to infection may contribute to long-term hearing deficits after meningitis [20–23]. These findings are also supported by animal studies that demonstrate improved hearing and spiral ganglion survival associated with steroid administration at the onset of infection [20, 22, 24]. However, adverse effects associated with high-dose steroids prevent their consistent use. Expanding treatment options to preserve hearing in meningitis could have a major health impact for those who survive this life-threatening infection.
In these experiments, we evaluated the time course of hearing loss by testing mice from 4 days post infection to 9 months post infection, characterized the tissue response to infection, and analyzed anatomic and histologic changes associated with meningitis. We performed a quantitative assessment of neo-ossification using microCT scanning, evaluated the effect of altering chemokine receptor expression in structural and functional changes of the cochlea, and characterized the transient loss of the blood-perilymph barrier associated with meningitis using perilymph sampling from live mice. Through these experiments, we have detailed the existing challenges to improving hearing outcomes and altering the downstream sequelae after the massive inflammatory response that accompanies pneumococcal meningitis.
Materials and Methods
Animals
C57Bl6 mice with the following genetic modifications were used for this study. CX3CR1 and CCR2 reporter mice (CX3CR1+/GFPCCR2+/RFP), thy 1 and NK1.1 reporter mice (thy-1+/YFP and NK1.1+/tdTom), CX3CR1 knockout mice (CX3CR1GFP/GFP CCR2+/RFP), CCR2 knockout mice (CX3CR1+/GFP CCR2RFP/RFP), and CX3CR1-DTR knock in mice (CX3CR1+/DTR) were raised according to recommendations by the Institutional Animal Care and Use Committee at Washington University School of Medicine. At 7–8 weeks of age, WT mice and transgenic mice were randomly assigned to control or meningitis experimental groups. A total of approximately 500 mice, both male and female mice, were used for all experiments described here.
Depletion of CX3CR1 Mononuclear Phagocytes
CX3CR1-DTR knock in mice received intraperitoneal diphtheria toxin (200 ng/20 gm mouse weight) at days 1, 3, 5, and 7 after being infected with S. pneumoniae on day 1. CX3CR1-DTR mice were tested by ABR and killed 10 days post infection. Depletion of monocytes, macrophages, and microglia was confirmed by histology of the spleen, brain, and cochlea. Mice were housed in a BSL2 animal care facility with access to water and food ad libitum with daily checks by our research team.
Mouse Meningitis
Streptococcus pneumoniae bacteria (ATCC 6303, serotype 3) were grown from a frozen stock on soy agar; colonies were selected and suspended in saline. The density was adjusted to 3 × 108 CFU/ml and then diluted to the working solution. Mice were anesthetized with ketamine (100 mg/kg) and xylazine (20 mg/kg) and received 1 × 105 CFU/10 uL of Streptococcus pneumoniae by injection into the cisterna magna through the posterior neck skin (see Supplemental Fig. 1). Control mice received an identical volume of saline into the intrathecal space. Mice were administered ceftriaxone (100–200 mg/kg day) to treat bacterial meningitis until they achieved their baseline body weight and resumed normal activity. A severity scale was used as follows: 0: normal, 1: intermittent slowing of movement, 2: consistent slow movement and ambulatory, 3: not consistently ambulating, resting for periods of time lasting longer than 1 min, 4: not ambulating, matted fur, and moving head and limbs, 5: hunched and movement slow and rare. When mice reached a score of 5, they were euthanized. Ceftriaxone (100 mg/kg) was administered on the day of infection and for up to 2 weeks post infection. For those mice in the steroid and antibiotic treatment group, dexamethasone was given (0.5 mg/kg) IP each time ceftriaxone was given throughout the course of treatment. Mice were euthanized if they were persistently lethargic for over 24 h or if they lost more than 15% of their body weight. Survival times varied from 2, 5, 7, 10, 14, 28, 100, 135, 180, and 270 DPI. Mice were housed in a BSL2 animal care facility with access to water and food ad libitum with daily checks by our research team for the first 30 days of infection, and weekly thereafter, and by our veterinarian from the Department of Comparative Medicine.
Auditory Brainstem Response (ABR)
ABRs were elicited by tone pips played in a sound tube inserted into the mouse ear canal, and responses were recorded via subcutaneous electrodes placed in the postauricular region, vertex, and ground placed near the tail. The stimuli were 5-ms tone pips (0.5-ms rise-fall with a cos2 onset envelope, delivered at 41/s). The response was amplified (×10,000), filtered (100 Hz–3 kHz), and averaged using BioSig computer software (System 3, Tucker-Davis Technologies, Alachua, FL). The sound level was raised in 5 dB steps from 5 dB SPL up to 95 dB SPL. At each sound level, 512 responses were averaged with stimulus polarity alternated. Response waveforms were discarded as artifacts if the peak-to-peak voltage exceeded 15 µV. Stimulus frequencies ranged from 5.6 to 64 kHz. Thresholds were tested at 5.6, 8, 11, 16, 22, 32, 45, and 64 kHz. The threshold was determined by a single observer who noted the lowest sound level at which a recognizable waveform was seen on a screen of tracings stacked from lowest to highest sound level. Waveforms were confirmed as auditory evoked responses by observing amplitude and latency changes with increasing stimulus intensity.
Cochlear Histology and Immunohistochemistry
Histologic Preparation
Anesthetized mice were perfused with 4% paraformaldehyde by thoracotomy and intracardiac puncture for 60 s. Temporal bones were removed, and intralabyrinthine perfusion of fixative was performed through the opened oval and round windows. After fixation for 45 min, cochleas were decalcified in 1M EDTA for 3 days at 4 °C.
For plastic embedding, after decalcification, cochleas were osmicated (1% OsO4 in dH2O) for 60 min, dehydrated in serial ethanols and propylene oxide, embedded in Araldite resin, and allowed to cure for 3 days at 60 °C. The cochleas were serially sectioned with a carbide steel knife in 40 µm sections.
For immunohistochemistry, cochleas were then immersed in cryoprotection solution overnight, frozen in 30% sucrose on dry ice, and cut in 30 µm sections from a round window to an oval window on a horizontal sliding microtome. The spleen was harvested to use as a positive control for immunohistochemistry of leukocyte markers and in CX3CR1-DTR mice to assess monocyte/macrophage depletion. Serial sections were stored in cryostorage solution at −20 °C. Cochlear sections were washed in phosphate-buffered saline (PBS, pH 7.4) in glass wells. Sections were incubated in 3% serum for 1 h, then incubated overnight in primary antibody (see Table 1 for all antibody reagents) in a humidified chamber on a nutator at room temperature. Sections were then incubated in secondary antibody for 1 h, coverslipped, and examined under DIC and confocal microscopy.
Table 1.
List of reagents for immunohistochemistry
| 1st Ab | 2nd Ab | Diluted Ratio | Vendors | Cat. # |
|---|---|---|---|---|
| Rat anti-CD45 IgG1 | 1:5000 | AbD Serotec | MCA1388 | |
| Alexa Fluor® 594 goat anti-rat IgG | 1:500 | Fisher-Invitrogen | A11007 | |
| Rabbit anti-MPO IgG | 1:250 | Abcam | ab9535 | |
| Alexa Fluor® 594 goat anti-rabbit IgG | 1:500 | Fisher-Invitrogen | A11012 | |
| Rabbit anti-CD3 IgG1κ | 1:1000 | Agilent (DAKO) | M7254 (A0452) | |
| Alexa Fluor® 594 goat anti-rabbit IgG | 1:500 | Fisher-Invitrogen | A11012 | |
| Alexa Fluor® 647 goat anti-rabbit IgG | 1:500 | Fisher-Invitrogen | A21245 | |
| Rabbit anti-CD19 | 1:100 | Cell Signaling | 3574 | |
| Alexa Fluor® 594 goat anti-rabbit IgG | 1:500 | Fisher-Invitrogen | A11012 | |
| Rat anti-CD68 IgG2a | 1:200 | AbD Serotec | MCA1957 | |
| Alexa Fluor® 594 goat anti-rat IgG | 1:500 | Fisher-Invitrogen | A11007 | |
| Rat anti-CD4 IgG2b | 1:200 | Fisher-Invitrogen | 14-0041-82 | |
| Alexa Fluor® 647 goat anti-rat IgG | 1:500 | Fisher-Invitrogen | A21247 | |
| Rat anti-CD8a IgG2a | 1:500 | Fisher-Invitrogen | 14-0081-82 | |
| Alexa Fluor® 647 goat anti-rat IgG | 1:500 | Fisher-Invitrogen | A21247 | |
| Rabbit anti-Iba1 | 1:500 | Fujifilm | 019-19741 | |
| Alexa Fluor® 488 goat anti-rabbit IgG | 1:500 | Fisher-Invitrogen | A11008 |
Reporter mice: CX3CR1-GFP (Daniel Littman, NYU); CCR2-RFP (Israel Charo, Gladstone Institute UCSF); Thy1-YFP (Joshua Sanes, Harvard Medical School); NK1.1-Td Tomato (Wayne Yokoyama, Washington University)
Antibody Characterization
Negative controls were performed for all experiments in which secondary antibody was used alone without primary antibody to detect background levels of staining. Concentrations of primary and secondary antibody were adjusted to reduce background signal. Positive control tissue for leukocyte markers was located in cochlear sections that contained bone marrow of the temporal bone where leukocytes of all lineages were detected. Spleen sections were also used as positive controls. Specificity was confirmed by colocalization with other expected markers.
Microcomputed Tomography (microCT)
Mice recovered from acute meningitis were selected for ABR testing and CT imaging at 135 and 270 days after infection. Mice were anesthestized with ketamine and xylazine and placed in a microCT scanner (VivaCT40, ScancoMedical, Switzerland). The mouse head was positioned in a head holder within the scanner, and regions of interest were captured using a 10-µm voxel size. Images of the entire inner ear were captured in 400 coronal sections. Reconstructions were performed in the parasagittal and axial planes, and 3D reconstructions were performed using AMIRA software. The inner ear fluid volumes for the scala tympani were calculated, and regions of new bone formation within the scalae were identified and quantified.
Perilymph Sampling
Mice recovering from meningitis (3, 7, and 100 DPI) were anesthetized and injected with 0.3 ml of sodium fluorescein IP (10 mg/ml). A control group of uninfected mice was also injected with sodium fluorescein. Tracheostomy was performed. The mouse was placed in a head holder in the right lateral decubitus position. An incision was made behind the left pinna, and the posterior semicircular canal was exposed. A retractor was placed, the otic capsule was dried, and cyanoacrylate (CA) glue was applied, followed by layers of two-part silicone (Kwik-Cast, WPI, Sarasota, FL). The silicone was shaped to form a cup around the entry site in the posterior semicircular canal to ensure that the collected fluid did not contact surrounding tissue or blood [25]. The bone covered in CA in the center of the silicone cup overlying the canal was curetted with a small pick. Entry into the labyrinth was achieved by removal of bone over the dome of the posterior semicircular canal. Fluid sampling was initiated 30 min after the initial injection of fluorescein. Perilymph was collected in a calibrated glass capillary tube (approximately 0.5 μl volume per sample). Five samples were collected sequentially; each sample required 15–30 s to collect. Care was taken to ensure no contact occurred between the inner ear fluid and surrounding blood, serum, or tissues. Two separate samples of 10 µl of blood were taken from a neck vein to measure serum fluorescein concentration.
Blood-Perilymph Barrier Assessment
Perilymph and blood samples were diluted in 100 μl of phosphate-buffered saline. Blood was centrifuged, and the serum supernatant was collected. Fluorescence intensity of the perilymph and serum samples was measured on a Spectramax i3 microplate reader (Molecular Devices, Sunnyvale, CA). The samples of known concentration from the dilution series were used to calculate a standard curve, and the concentrations of fluorescein from the original samples were interpolated. Fluorescein concentrations of perilymph were compared with serum concentration and expressed as a percentage reflecting the fraction of fluorescein concentration in the perilymph with respect to the serum concentration.
Statistical Analysis
A mixed model approach using SAS 9.4 was used for statistical analysis of the data. This allowed for random effects and exploring differences between test conditions through repeated measures. The Kenward-Roger approximation was used to adjust for unequal variance. Akaike’s information criteria (AIC) and Bayesian information criteria (BIC) were used to select covariance matrices for the best-fit model.
Study Approval
All described experiments were reviewed and approved by the Institutional Animal Care Use Committee (IACUC) at Washington University.
Data Availability
Underlying data and supporting work will be available through PubMed through this published article and through direct inquiry.
Results
Pneumococcal Meningitis Induces Hearing Loss and Weight Loss and Is Associated with 50% Mortality
Hearing loss was observed in most surviving mice after pneumococcal meningitis. ABR thresholds 7 days post infection (DPI) are shown in Fig. 1A. Mice demonstrated a broad range of hearing outcomes after pneumococcal meningitis with most mice experiencing profound hearing loss and a minority of mice retaining near normal hearing thresholds. The average of these ABR thresholds was 70–90 dB SPL across frequencies, and the standard deviation was large (shaded light red area and red icons, Fig. 1A). Control uninfected littermates demonstrated hearing thresholds within an expected range, (Fig. 1A, black icons). Hearing loss after meningitis progressed rapidly. Some mice had achieved their final hearing outcome by 14 DPI.
Fig. 1.
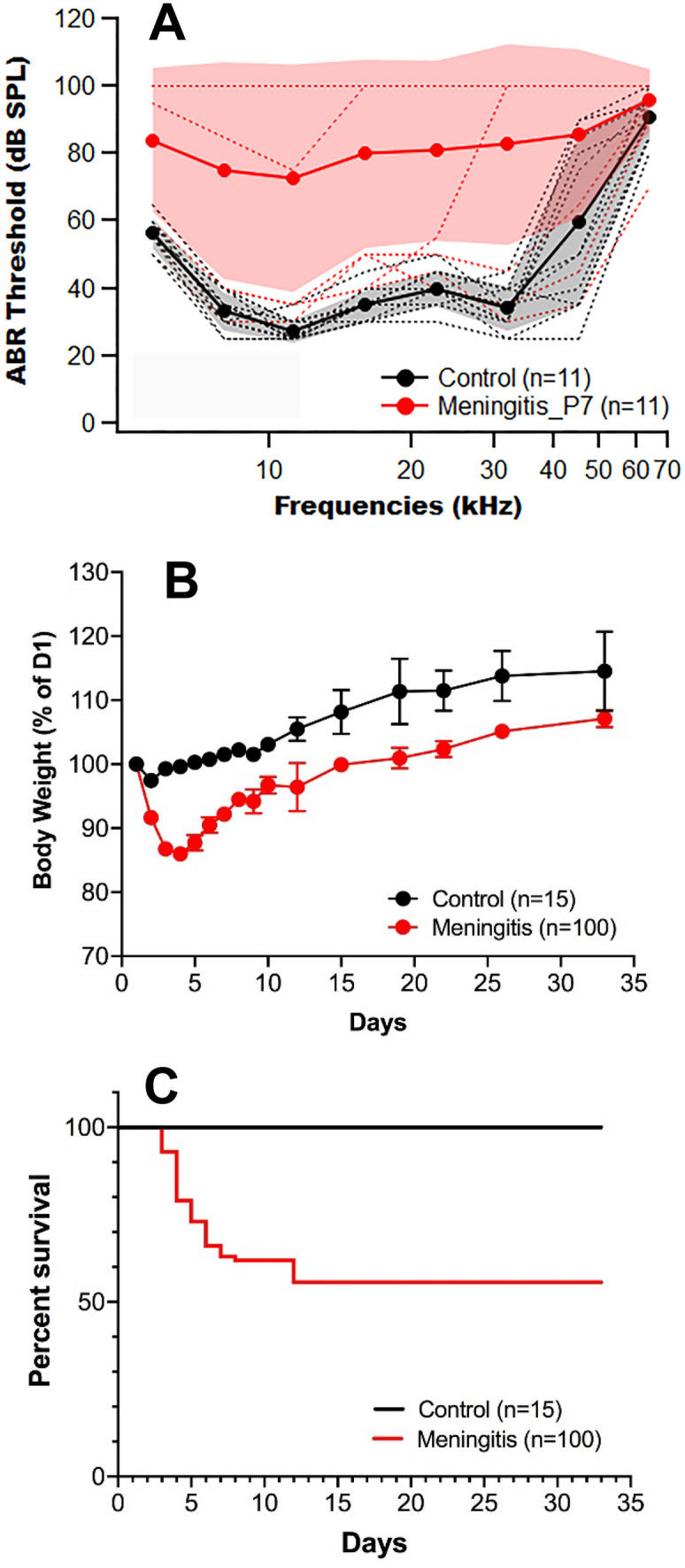
Pneumococcal meningitis caused hearing loss, weight loss, and 50% mortality in WT mice. A ABR thresholds showed variable hearing loss in mice with pneumococcal meningitis at 7 DPI. ABR thresholds of uninfected (black) and meningitic mice (red). Shaded area: standard deviation. B Weight loss after meningitis was reversed by 5 DPI. Meningitic mice remained smaller than control mice. C Survival after pneumococcal meningitis was 50% at 35 DPI
Weight loss was noted in infected mice during the first five DPI. While mice began to regain weight by day 5, meningitic mice were consistently smaller than control mice throughout the recovery period (Fig. 1B). At their nadir, most mice had lost approximately 15% of their body weight. Survival from pneumococcal meningitis was 50–60%. If mice survived the first 2 weeks of infection, they typically survived long term (Fig. 1C). Generally, mice that experienced the greatest weight loss demonstrated the poorest clinical scores in functional activity, although a significant number of poorly performing mice survived with antibiotic therapy. Mice with the most severe weight loss experienced higher rates of mortality.
Cochlear Histopathology: Inflammatory Infiltrates, Fibrosis, and Hemorrhage After Bacterial Meningitis
A wide range of histologic changes were observed in the inner ear after pneumococcal meningitis. The final endpoints were diverse, although there were some common findings among some animals that experienced hearing loss. Figure 2 demonstrates low magnification images of representative cochlear sections imaged with differential interference contrast (DIC) at 30 DPI. Cochleas at this time frequently demonstrated inflammatory cell infiltration of the scalae, and in some cases, fibrosis and ossification, that were typically concentrated in the basal turn. Ossification was more commonly observed in scala tympani than in scala vestibuli. In mice that exhibited minimal or no hearing loss, cochlear changes were subtle, and the scalae remained fluid-filled and contained few inflammatory cells. In some cases of meningitis, we observed minimal changes under light microscopy (Fig. 2A, B). In cases of severe to profound hearing loss, fibrosis was often observed in the scala tympani of the basal turn (Fig. 2C, E). Fibrosis occurred rapidly starting at four DPI and advanced through 30 DPI. Among those mice with severe to profound hearing loss and advanced fibrosis, encroachment of soft tissue in scala tympani advanced into the apical turn. Scala tympani obliteration was irregular and discontinuous along the cochlear duct in some ears (Fig. 2D). In some cases of fulminant meningitis, hemorrhage was observed within the scalae where perilymph was replaced with whole blood (Fig. 2F). In these rare cases, we observed erythrocytes filling the perilymphatic and endolymphatic spaces. In the cochlea depicted in Fig. 2F, the only area where a small region of endolymph remained was in the scala media in the apical turn. In cases of overt hemorrhage such as this, we surmised that frank rupture of cochlear vessels resulted in the scalae filling with blood.
Fig. 2.

Mouse cochleas 1 month after meningitis display a range of scalar patency. A No fibrosis or cellular infiltrates were observed in the scalae of some mice. Normal hearing, normal histology. B No obvious cochlear histopathology at low magnification with moderate threshold shift. C–F Profound hearing loss with variable degrees of inflammation, fibrosis, and ossification. Arrows: inflammatory infiltrates and fibrosis in scala tympani (×4 objective). Scale bar: 200 µm
Histologic Changes in the Cochlea: Sensory Cell Loss
Serial sections of plastic-embedded cochleas were used to examine histopathology after meningitis. There were diverse findings in mice that suffered profound hearing loss. In some cases, mice with profound hearing loss had relatively normal appearing cochleas on light microscopy. Other cochleas demonstrated loss of sensory cells during the first week of infection as demonstrated in Fig. 3B (compare with control Fig. 3A). We observed loss of outer hair cells and swollen, distorted inner hair cells, accompanied by scattered inflammatory cells and cellular debris in the scala tympani. In this cochlea, the inflammatory cells were confined to the scala tympani and did not migrate into the scala media, and the reticular lamina appeared intact. Three to four months after infection, loss of spiral ganglion neurons was extensive (Fig. 3D, 100 DPI compared to control (Fig. 3C)). Reduction in neurons was frequently associated with fibrosis and ossification of the scalae, and extensive loss of hair cells. Rosenthal’s canal was nearly vacant in some cases with sparse afferent fibers, leaving large open spaces. The dark myelinated fibers were missing in cochleas with depletion of neurons (arrow, Fig. 3D).
Fig. 3.
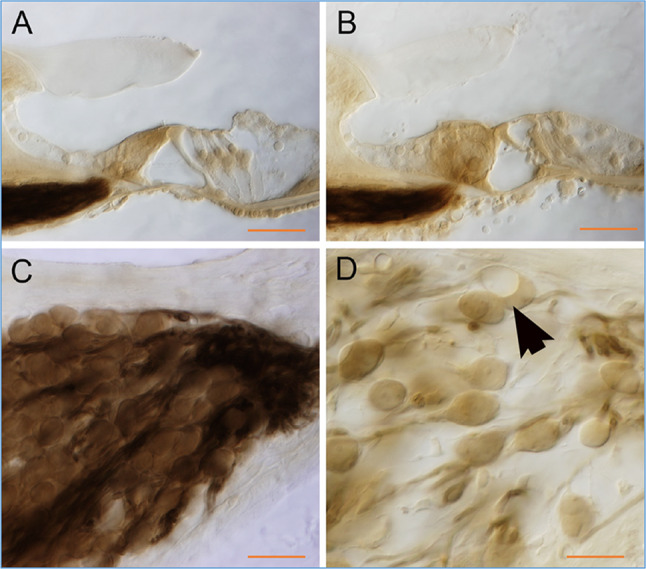
Hair cells are damaged, and spiral ganglion neurons are partially depleted after pneumococcal meningitis. A Control organ of Corti. B Four DPI, damaged sensory epithelium, and cellular debris in scala tympani and tunnel of Corti. C Control spiral ganglion. D 100 DPI, SGNs depleted with ghosts of prior SGNs (arrow). Scale bars: A, B 75 µm, C 30 µm, and D 20 µm
Late-stage findings in severely affected post-meningitic cochleas were detectable at low magnification. Fibrosis and scarring in all three scalae (Fig. 4A), distortion of Reissner’s membrane due to scar formation (Fig. 4B), extensive new bone deposition, and loss of spiral ganglion neurons and afferent fibers (Fig. 4C) were observed in surviving mice after bacterial meningitis. Rosenthal’s canal appeared nearly depleted of neurons (Fig. 4A–C) and newly formed blood vessels developed in the fibrotic scar of the scalae where, previously, there was only perilymph (Fig. 4C). There were unique forms of scarring from the sensory epithelium to Reissner’s membrane (Fig. 4A) and scar in the scala vestibuli and tenting of Reissner’s membrane mimicking the appearance of endolymphatic hydrops (Fig. 4B). The stria vascularis was atrophic with loss of membrane surface area of intermediate and marginal cells, and the spiral ligament was frequently depleted of fibrocytes (Fig. 4D).
Fig. 4.

Chronic changes after meningitis: labyrinthitis ossificans, scarring of Reissner’s membrane, loss of spiral ganglion neurons, and strial atrophy. A Low magnification: apical turn with ossification of scala tympani (arrows). B Fibrosis of the scala vestibuli, tethering Reissner’s membrane (arrow) inducing the appearance of endolymphatic hydrops, depletion of spiral ligament fibrocytes, loss of sensory cells, thickening of the basilar membrane, and ossification of the scala tympani. C Loss of spiral ganglion neurons (long arrow) and areas of dense fibrosis in scala tympani. Lacunae around vessels (arrow heads). D Severe strial atrophy. Scale bars: A 200 µm, B 150 µm, C 100 µm, and D 50 µm
Cochlear Inflammatory Response to Pneumococcal Meningitis
Histologic changes in the mouse cochlea after meningitis were further evaluated using immunohistochemistry to assess the inflammatory response to infection. Figure 5A shows the spiral ganglion of a control mouse cochlea with healthy spiral ganglion neurons (magenta) and sparse cochlear macrophages (green) around the ganglion, in the osseus spiral lamina, and the spiral limbus (inset shows orientation of the spiral limbus and spiral ganglion). Two days post infection, many CCR2 monocytes were present in the scala media and scala tympani (Fig. 5B). The presence of leukocytes in scala media was a rare event, even in severely inflamed ears. Five days post infection, CCR2 monocytes nearly filled the scala tympani. CCR2 and CX3CR1 monocytes and macrophages were observed in the ganglion and among the afferent fibers (Fig. 5C). Cellular debris and CCR2 monocytes were seen in scala media while large ramified macrophages occupied the spiral ligament (green). CCR2 monocytes were present in the lower spiral ligament and the scala tympani (Fig. 5D). At 2 weeks post infection, fibrosis was detected in the scala tympani, accompanied by a reduction in spiral ganglion neurons (magenta), and dying natural killer (NK) cells (red) ingested by CX3CR1 macrophages (green) in the scala tympani and spiral ligament (Fig. 5E). At 2 weeks after infection, the basal turn of the cochlea had developed dense fibrosis with a thick rind of CX3CR1 macrophages incorporated in scar (Fig. 5F).
Fig. 5.

Robust cochlear inflammation was observed during pneumococcal meningitis. A Control mouse with healthy spiral ganglion neurons (magenta) and sparse CX3CR1 cochlear macrophages (green). Arrowheads indicate resident macrophages, and single arrow indicates afferent fibers of the spiral ganglion. B Two DPI: CCR2 monocytes (magenta) were present in the scala media and scala tympani. C Five DPI: CCR2 monocytes nearly filled the scala tympani. D Five DPI: cellular debris and CCR2 monocytes in scala media, scala tympani, and spiral ligament. E Fourteen DPI: fibrosis in scala tympani, loss of spiral ganglion neurons, and dying NK cells ingested by CX3CR1 macrophages (arrows). F Fourteen DPI: fibrosis with thick rind in scala tympani incorporating CX3CR1 macrophages in scar. A, E thy1YFP: magenta; CX3CR1GFP: green; NK1.1 Td−Tomato: red. B–D, F CCR2RFP: magenta; CX3CR1GFP: green. The insets demonstrate regions of the inner ear where images were captured. Representative samples from over 50 mouse cochleas. Scale bars: A, C, E 25 µm. B, D 50 µm. F 100 µm
In most meningitic mice, large numbers of inflammatory cells and bacteria were observed in the perilymph-filled spaces during the first week of infection. Many leukocytes lysed or were phagocytosed within the first few days after infection. Antibody labeling of neutrophils, lymphocytes, and macrophages showed a large variation of immune cells within the cochlear duct although many of these cells did not appear viable. Figure 6 shows representative cochlear sections of the basal turn seven DPI using CD45 immunohistochemistry and CX3CR1GFP reporter mice. CD45 labels all white blood cells, and CX3CR1 is expressed in monocytes, macrophages, NK cells, microglia, and some T cells [26]. In some infected mice, we observed preservation of hearing and little cochlear inflammation. The number and distribution of inflammatory cells in these cochleas were comparable to those in healthy control animals (Fig. 6A, D, G), and most of these leukocytes were CX3CR1+ monocytes and macrophages. In other mice with moderate hearing loss after meningitis, a greater number of leukocytes (CD45+) were observed in the spiral ligament and the scala tympani (Fig. 6B, E, H). In these ears, the spiral ligament contained a large number of inflammatory cells, and the basilar membrane was thick and populated with mostly monocytes and macrophages. In mice with profound hearing loss, infiltration of leukocytes was extensive (Fig. 6C, F, I). The immediate migration of inflammatory cells into the scalae filled the lumen with inflammatory cells and was followed by the rapid formation of dense fibrous tissue and, subsequently, further consolidated with labyrinthitis ossificans.
Fig. 6.

Cochlear inflammation was observed after pneumococcal meningitis at varying levels. Minor inflammation and a normal distribution of cochlear macrophages were observed in some post-meningitic ears (A, D, G). Those mice that demonstrated preserved hearing thresholds appeared minimally affected by inflammatory changes. Others demonstrated leukocytes throughout the spiral ligament and scattered in the scala tympani (B, E, H). This mouse demonstrated thresholds in the moderate to severe hearing loss range. Rampant cochlear inflammation was sometimes observed where scala tympani was filled with leukocytes and the spiral ligament appeared replaced by inflammatory cells (C, F, I). This appearance of extensive leukocyte infiltration of the ligament and the scala tympani was frequently seen in mice with the worst hearing thresholds. Magenta: CD45; green: CX3CR1. Representative images were taken from a selection of over 50 mice. Scale bar: 100 µm for all figures
CX3CR1 and CCR2 Protect Hearing in Pneumococcal Meningitis
Both CCR2 and CX3CR1 leukocytes were recruited in large numbers to the cochlea during pneumococcal meningitis, and CX3CR1-expressing cochlear macrophages remained abundant in the scar that formed in the scalae months after the initial infection. Prior reports have established a role for both of these chemokine receptors in hearing outcomes after noise and ototoxicity [5, 9, 10, 27, 28]. Therefore, we investigated the roles of CCR2 and CX3CR1 expression on hearing outcomes after bacterial meningitis using knockout mice and DTR-mediated-macrophage depleted mice (CX3CR1-DTR). Single knockout/double reporter mice (CX3CR1GFP/GFP CCR2+/RFP and CX3CR1+/GFP CCR2RFP/RFP) were experimental subjects, and double heterozygous (CX3CR1+/GFP CCR2+/RFP) reporter mice are shown as controls. ABR thresholds after meningitis shown in Fig. 7A–C were performed at 7, 14, 30, 60, 90, 120, and 270 days post infection (DPI).
Fig. 7.

CX3CR1 KO mice have poorer hearing than heterozygotes, and CCR2 KO mice have the worst hearing after pneumococcal meningitis. CCR2KO mice also experienced greater weight loss and more severe clinical scores. A ABR thresholds in CX3CR1 KO mice at 0.5, 1, 2, 3, 4, and 9 months after meningitis. B ABR thresholds of reporter mice at 0.5, 1, 2, 3, 4, and 9 months after meningitis (control group). C ABR thresholds in CCR2 KO mice at 2, 3, and 4 months after meningitis. ABRs were not repeated after 4 months due to profound HL in all mice in this group. Statistical analysis demonstrated significant differences in ABR thresholds in control vs CX3CR1 KO mice and differences between control and CCR2KO mice at all matched time points after meningitis. D Weight loss after meningitis was worse in CCR2 KO mice (red) compared with the other two genotypes (green, black). E Clinical scores after meningitis in CX3CR1 KO, reporter, and CCR2 KO mice. Clinical scores showed worse outcomes in CCR2 KO mice. In Fig. 8D, E, genotype abbreviations are HET/HET (CX3CR1+/GFP CCR2+/RFP), KO/HET (CX3CR1GFP/GFP CCR2+/RFP), and HET/KO (CX3CR1+/GFP CCR2RFP/RFP)
Hearing was worse in CX3CR1 null mice after pneumococcal meningitis compared to heterozygotes (Fig. 7A, B). CCR2 null mice demonstrated the greatest susceptibility to hearing loss in meningitis of all three groups (Fig. 7A–C). We observed a monotonic increase in average ABR threshold over time in all groups. Figure 7B shows the time course of post-meningitis hearing loss in reporter mice. At the lower frequencies (5.6, 8, 11, 16 kHz) at all time points, there was improved hearing when compared to CX3CR1 or CCR2 null mice. Figure 7C shows that hearing loss in CCR2 knockout mice was the most severe and rapid of all groups after meningitis. Profound hearing loss was observed by 2 months post infection, and by 4 months, there were no mice with residual low frequency hearing. Note that there were differences in baseline hearing thresholds between the two knockout strains (Fig. 7A, C), although the cause of high frequency hearing loss in CCR2 KO mice is not known. In Supplemental Fig. 2, we show ABR thresholds of CCR2 KO mice demonstrating elevated high frequency thresholds in healthy aged mice (4 months of age). Nevertheless, there remains a large difference between thresholds in post-meningitic CCR2 KO mice with no responses across all frequencies and control CCR2 KO mice that demonstrate isolated high frequency hearing loss.
In Fig. 7D, body weight after meningitis is shown for each genotype plotted over time, and in Fig. 7E, clinical score is shown. CCR2 KO mice are shown in red, CX3CR1 KO mice are shown in green, and control mice are shown in black. Control mice demonstrated the most rapid recovery in body weight and clinical score, while the CCR2 KO mice experienced the slowest recovery. CCR2 deletion resulted in more severe and more rapid hearing loss and slower recovery from weight loss and motor deficits assessed by clinical score. CX3CR1 KO mice were comparable to control mice with respect to weight loss and clinical score. Both demonstrated near complete recovery from meningitis by 12 DPI. CCR2 KO mice had not yet completely recovered at 28 DPI.
Cochlear Monocytes and Macrophages Are More Abundant in CX3CR1 Knockout Mice in Pneumococcal Meningitis
We observed large numbers of CX3CR1 expressing monocytes and macrophages in the cochlea after meningitis. Figure 8 shows representative cochlear sections at nine DPI in CX3CR1 heterozygous and CX3CR1 null mice. Most of the macrophages and monocytes observed after meningitis were concentrated in the lateral wall and the spiral ganglion. In some ears, large numbers of inflammatory cells were present in the scalae. Cochlear monocytes/macrophages were quantified in CX3CR1 heterozygous and knockout mice, and these data are shown in Fig. 8E. CX3CR1 knockout mice recruited more cochlear myeloid cells during meningitis and developed worse hearing loss compared to control mice (Fig. 7A, B).
Fig. 8.

CX3CR1 deletion increases monocyte and macrophage migration into the cochlea during bacterial meningitis. Representative figures of CX3CR1+/GFP mice (HET) (A, B). CX3CR1GFP/GFP (KO) (C, D). On the left are uninfected mice, and the right shows 9 days post meningitis infection (DPI). CX3CR1 monocytes and macrophages accumulated in large numbers in the spiral ligament after meningitis. CX3CR1 KO mice showed no difference in resting macrophage number in the cochlea (A, C) but demonstrated enhanced accumulation of monocytes and macrophages after meningitis (C, D). E Numbers of cochlear macrophages in HET (blue) and KO (green) mice, and average macrophages per 30-µm section (black) demonstrating a statistically significant difference in cochlear myeloid cells in infected CX3CR1 knockout mice when compared with heterozygous mice (n = 6 for each genotype, error bars reflect standard deviation, p = 0.03)
Depletion of CX3CR1 Myeloid Cells Did Not Ameliorate Hearing Loss Resulting from Pneumococcal Meningitis
Diphtheria toxin administered to CX3CR1+/DTR mice successfully eliminated CX3CR1 monocytes, macrophages, and microglia 1 week prior to infection with meningitis and for 10 days after infection (Supplemental Fig. 3). CX3CR1-depleted mice survived the challenge of bacterial meningitis to 10 DPI despite impaired immunity. CX3CR1-depleted mice demonstrated worse hearing thresholds compared to wild-type mice although this difference was not statistically significant due to wide variations in threshold (Fig. 9). CX3CR1-depleted mice did not experience better outcomes than CX3CR1 knockout mice. CX3CR1+ mice demonstrated the most favorable outcome with respect to survival, weight gain, and hearing loss. Neither CX3CR1 knockout (CX3CR1−/−) nor CX3CR1 monocyte and macrophage depletion (DT administered to CX3CR1+/DTR mice) improved hearing outcomes in pneumococcal meningitis.
Fig. 9.
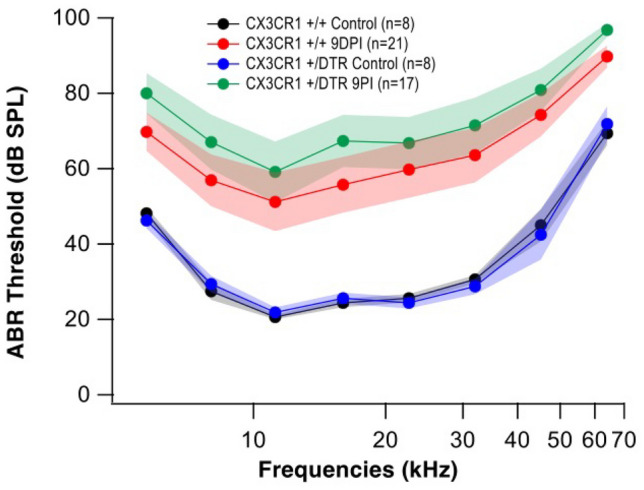
Elimination of CX3CR1 cells did not protect hearing in pneumococcal meningitis. Hearing thresholds were slightly worse in mice after elimination of mononuclear phagocytes prior to infection with meningitis, but this difference was not statistically significant (p = 0.2445). Mice were tested at 9 DPI
Cochlear Ossification and Remodeling
Mice underwent imaging studies using micro-computerized tomography scans (microCT) during the recovery period after meningitis. These research CT scans provide high-resolution images (10-µm3 voxel size) and permit computation of the cochlear luminal volume using 3D reconstruction in living mice. Examples of the microCT images are shown in Fig. 10A, B. Serial CT images were reconstructed in three dimensions using AMIRA software, and these three-dimensional renderings were used to calculate the volumes of the fluid space within the scalae. Examples of the three-dimensional reconstructions are displayed in Fig. 10C with profiles of the labyrinth shown from different angles. New bone growth in the scala tympani is shown in purple. Histologic sections of the mouse cochlea that correlate to these CT scans are shown in Fig. 10D, E where new bone is present amidst cochlear fibrosis in the basal turn of the scala tympani. Cochlear fluid volumes in the basal turn of the scala tympani were compared across three genotypes: CX3CR1 null, CCR2 null, and wild-type mice at three time points: 3, 4.5, and 9 months. The effect of dexamethasone treatment on ossification/fluid volume was also assessed.
Fig. 10.

Labyrinthitis ossificans: imaged by microCT scan and correlated to histology. High-resolution microcomputed tomography was performed in mice recovering from pneumococcal meningitis to identify new bone formation in the inner ear. A Control mouse cochlea. B Post-meningitic cochlea revealed bone deposition in the basal turn (arrows in ST compare control to ossified ST). C Three-dimensional reconstructed images of the mouse cochlea demonstrate regions of neo-ossification in purple. D Low magnification image of mouse cochlea at 5 months post infection shows fibrosis and ossification of the basal turn in scala tympani with ascending scar in scala tympani of the upper basal and lower apical turns (arrows) as reflected in microCT. E Scala tympani in the basal turn replaced with bone and soft tissue
Figure 11A shows the calculated fluid volume in the basal turn of scala tympani in wild-type (WT) healthy control mice compared with WT mice 3 months after pneumococcal meningitis. While there was considerable variation in fluid volumes of post-meningitic mice, we observed an average reduction of scala tympani fluid of approximately 20% attributable to neo-ossification after meningitis. Figure 11B shows scala tympani volumes at 4.5 months after meningitis in wild-type, CCR2 KO, and CX3CR1 KO mice. A large range of variation was observed within each group. However, wild-type mice demonstrated the best preservation of scalar fluid volume. CCR2 KO mice experienced the greatest reduction in scalar volume and the most advanced ossification of the scala tympani.
Fig. 11.

Labyrinthitis ossificans caused reduction in scala tympani volume after pneumococcal meningitis. Ossification was more severe in CCR2 knockout mice. Dexamethasone reduced but did not eliminate neo-ossification. A Fluid volumes of the scala tympani (ST) in the basal turn: uninfected control ears compared to meningitic ears in wild-type mice at 3 months post infection. Scala tympani volume was reduced after meningitis due to neo-ossification (p = 0.022). B Fluid volumes of ST at 5 months after meningitis: comparison of 3 genotypes: control (double heterozygous CX3CR1+/GFPCCR2+/RFP), CCR2 knockout (CCR2RFP/RFP), and CX3CR1 knockout (CX3CR1GFP/GFP) mice. Cochlear fluid volume after meningitis was smaller in CCR2KO mice compared to control (p = 0.010, WT vs CCR2KO). Cochlear volumes were not significantly different after meningitis in CX3CR1 null mice (CX3CR1 KO mice vs. WT mice, p = 0.159). C Dexamethasone was given IP with ceftriaxone on the day of infection. Dexamethasone attenuated ossification and maintained higher fluid volumes in scala tympani at 3 months post infection (p = 0.028)
WT mice treated with dexamethasone demonstrated less cochlear ossification and better preservation of fluid volume compared to WT meningitic mice without dexamethasone (Fig. 11C). Hearing remained severely compromised after meningitis despite corticosteroid administration early after onset of pneumococcal meningitis (data not shown).
The Blood-Perilymph Barrier Was Transiently Disrupted in Some Mice During Pneumococcal Meningitis
Blood-perilymph barrier function was evaluated in live, anesthetized mice with meningitis. These studies were performed by sampling perilymph and blood simultaneously and measuring the concentration of perilymph fluorescein and reporting it as a fraction of the fluorescein measured in serum (see Hirose et al. [28] for detailed methods). Figure 12 shows the relative concentration of fluorescein in perilymph from control mice and meningitic mice at three time points (3, 7, and 100 DPI) after onset of infection. Each data point represents the perilymph concentration of fluorescein (re: serum) in a single mouse. The value of each data point reflects the percent entry of fluorescein into the perilymph with respect to the serum concentration of fluorescein. A low percentage indicates a robust, minimally permeable barrier, and 100% indicates the absence of a barrier, where fluorescein concentration is the same in perilymph and in serum. In control mice, the relative concentration of fluorescein in perilymph ranged from 0.2 to 8% of serum fluorescein concentration, consistent with a well-functioning barrier. Three days post meningitis, the blood-perilymph barrier was dysregulated and demonstrated a wide range of leakiness. The concentration of fluorescein in perilymph reached 95% in some mice. We observed a large degree of variability in barrier function during early meningitis. By day 7 post infection, the blood-perilymph barrier was completely closed in all animals, despite the large numbers of inflammatory cells in the cochlea demonstrated by histopathology at this time point. At seven DPI, the measured fluorescein concentration was lower than in control ears, suggesting less fluorescein entry into perilymph from the circulation than baseline. One hundred days after infection, perilymph sampling was not successful in most mice. Only 1 out of 15 mice produced perilymph through surgical entry at the posterior semicircular canal, and the relative fluorescein concentration in perilymph was normal at that time. Inability to extract perilymph was not surprising, as we observed fibrosis and ossification of the cochlear aqueduct and the scala tympani in many mice by 100 days after meningitis.
Fig. 12.
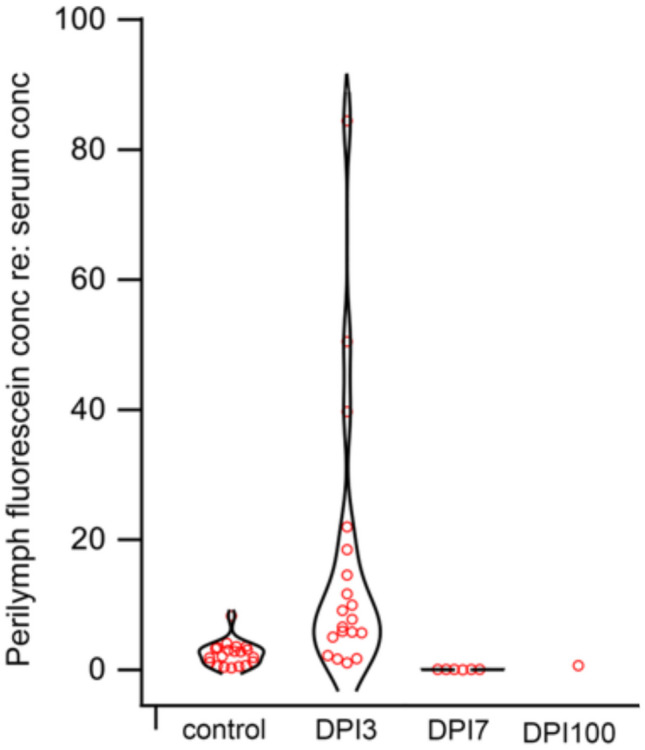
Severe transient disruption of the blood-perilymph barrier (BPB) occurs during pneumococcal meningitis. Control ears showed perilymph fluorescein concentration of 2–8% of fluorescein concentration in circulation. At 3 DPI, the BPB was severely disrupted (range: 5–85%). At 7 DPI, the BPB was closed with lower permeability than baseline (1–2%). At 100 DPI, perilymph sampling was difficult due to ossification of the labyrinth. Each data point represents an average of three sequential samples from the same mouse
We evaluated CCR2 null mice and CX3CR1 null mice to determine if elimination of either chemokine receptor influenced blood-perilymph barrier properties in meningitis. We discovered that at three DPI, when we observed a highly leaky barrier, mice of all genotypes experienced similarly leaky BPB properties, and genotype did not result in any change in this finding among meningitic mice (data not shown).
Summary of Cochlear Changes and Timeline After Pneumococcal Meningitis
Direct inoculation of Streptococcus pneumoniae in cerebrospinal fluid resulted in approximately 50% mortality within 14 days. Weight loss and lethargy were observed in most recovering mice with weight loss reversing at 5 DPI. During the first 5 days, some mice experienced abundant inflammatory cell entry into the CSF and perilymph, profound blood-perilymph barrier, and blood brain barrier dysfunction, and cochlear apoplexy was observed in a small number of mice. Initiation of fibrosis was seen as early as 4 DPI, and acute loss of sensory cells was observed in cases of severe inflammation and tissue destruction. In some cases, inflammatory cells entered the endolymph as well as the perilymph. We did not observe vestibular deficits (circling, head tilt, difficulty ambulating) after pneumococcal meningitis.
Between 2 and 4 weeks post infection, we observed progressive cochlear fibrosis and neo-ossification. Macrophages were present in the cochlear fibrotic scar, and rapid progression of hearing loss ensued while acute meningitis symptoms resolved. Weight gain resumed, and the blood-perilymph barrier was reestablished. Neither elimination of CCR2 nor deletion of CX3CR1 attenuated cochlear neo-ossification.
From week 4 to week 8, neo-ossification advanced, spiral ganglion cells were severely reduced in number, hearing loss progressed, and fluid volume of the scalae due was reduced due to new bone deposition.
Two to nine months post infection, mice experienced progressive labyrinthitis ossificans, profound hearing loss in 60–70% of surviving mice, fibrosis of the cochlear aqueduct with obstruction of CSF flow into the perilymph.
Discussion
Pneumococcal meningitis is often associated with rapid onset, profound hearing loss accompanied by severe distortions of cochlear anatomy, including obliteration of fluid spaces with soft tissue and new bone. Pneumococcal meningitis resulting in labyrinthitis ossificans provides limited options for hearing rehabilitation. This mouse model of meningitis demonstrates the short window between onset of infection and development of hearing loss and subsequent fibrosis and ossification. Hearing loss progressed rapidly after infection, detected as early as 3 days post infection (3 DPI). Within 2 weeks of infection, hearing was severely impaired, and hearing thresholds continued to worsen through 9 months post infection. Approximately 60–70% of mice developed hearing loss after pneumococcal meningitis, and many of these animals had little to no residual hearing.
Pneumococcal meningitis induced by direct inoculation of bacteria into the cerebrospinal fluid (CSF) causes inflammation in the central nervous system and in the inner ear due to direct communication between the perilymph and the CSF [29]. The cochlear aqueduct connects the subarachnoid space to the labyrinth in the hook region of the scala tympani (basal end of the cochlea). Thus, it is not surprising that inflammatory cells and fibrosis were first observed in the scala tympani near the cochlea aqueduct. Inflammatory cells were sometimes observed in scala vestibuli and, rarely, in scala media. Inflammatory cells were observed within the spiral ligament, spiral limbus, and spiral ganglion where cochlear inflammatory cells are commonly seen after noise or ototoxicity. After 2 weeks, cochlear monocytes and macrophages were embedded in scar that occupied the previously fluid-filled scalae. We observed initial signs of fibrosis in the scala tympani as early as 4 days post infection.
Leukocytes recruited to the cochlea during the first week of infection included neutrophils, lymphocytes, CCR2+ monocytes, CX3CR1+ monocytes and macrophages, and NK cells during the first week of infection. These inflammatory cells were present in the spiral ligament, the scala tympani, at times in the scala vestibuli, and on rare occasion, the scala media. Leukocytes are accumulated in the spaces containing perilymph, whereas the endolymph-filled compartments usually excluded them. The inflammatory response was largely resolved after 3 weeks, and fibrous tissue and scar progressed after resolution of the initial inflammatory response. Three months after infection, CX3CR1 monocytes and macrophages remained abundant in mouse cochleas recovered from meningitis. These mononuclear phagocytes were concentrated in the spiral ligament and in the scar tissue that replaced the perilymph in the scala tympani.
The etiology of hearing loss after pneumococcal meningitis could be attributed to numerous factors. In some cases, cochlear structural damage was extensive, and fluid spaces were filled with soft tissue and bone such that sound could no longer be transmitted through displacement of the basilar membrane. Scarring and ossification were observed frequently and ranged from focal areas of fibrosis in the basal turn of scala tympani to replacement of the entire cochlear duct with soft tissue and bone. Any amount of neo-ossification within the cochlear lumen was associated with profound hearing loss. In some mice, spiral ganglion cells were severely reduced in number. Some mice had widespread loss of hair cells. The cause of hearing loss was easily identified in these cases, although there were numerous mice with profound or severe hearing loss without clear histologic findings to indicate the etiology.
In rare cases, we observed blood in all scalar compartments. We were not able to determine the exact source of bleeding, and there were likely multiple vascular beds that were compromised to result in blood in both the endolymph and perilymph. Intracochlear hemorrhage has been reported in postmortem human temporal bones after acute pneumococcal meningitis [14]. The histologic appearance of these cochleas filled with blood implies a major vascular event that resulted in irreversible loss of hearing and could be described as cochlear apoplexy.
In many ears, histologic changes were more subtle, and the etiology of hearing loss could not be determined by light microscopy. Pneumococcal meningitis was found to induce profound hearing loss in some cases without fibrosis, ossification, or hemorrhage. In these ears where anatomy was largely preserved and hair cells and spiral ganglion cells were intact, the etiology of hearing loss is not determined.
We considered the possibility that modulating the inflammatory response to pneumococcal meningitis could mitigate cochlear damage or ossification resulting from pneumococcal infection. This concept has been previously explored in experimental animal models and in human subjects with the use of anti-inflammatory and immunosuppressive agents during bacterial meningitis. Bacterial meningitis is known to induce a robust inflammatory response in the meninges and CSF that results in an influx of inflammatory cells and molecules into the CSF and causes dysregulation of CSF resorption [30–33]. The inability to resorb CSF can result in elevated intracranial pressure and hydrocephalus. With hopes to attenuate the inflammatory response, improve CSF resorption, protect the CNS, and improve hearing outcomes, various anti-inflammatory or immune-suppressive interventions have been trialed in bacterial meningitis. In these studies, hearing loss is one of many sequelae that have been assessed. The use of systemic dexamethasone has been evaluated extensively, and hearing has been shown to be protected by dexamethasone given at the time of diagnosis along with antibiotics in pneumococcal meningitis [34]. Other investigators have explored the use of other immunosuppressive or other specific immune-activating therapies to alter the sequelae of meningitis [23, 34–38]. Overall, most studies demonstrated that reducing the impact of acute inflammation during early infection provided partial protection against hearing loss. Furthermore, circulating monocytes recruited to the cochlea may serve as precursors to differentiated osteoclasts [39–42]. Monocyte infiltration may initiate pathologic bone resorption resulting in concomitant bone deposition leading to scalar ossification. Thus, modulating or reprogramming monocyte differentiation after entry into the cochlea may attenuate labyrinthitis ossificans.
Here, we examined the effects of deleting chemokine receptors CCR2 or CX3CR1 and evaluated ABR thresholds and cochlear histology/patency in these mice after pneumococcal meningitis. Neither knockout mouse was protected from weight loss, hearing loss, or ossification of the cochlea. We also depleted monocytes and macrophages using CX3CR1-DTR transgenic mice. Administering diphtheria toxin in these mice successfully depleted monocytes, macrophages, and microglia for the duration of DT administration, and mice survived pneumococcal meningitis after CX3CR1 depletion. However, their hearing outcomes were not improved by eliminating these mononuclear phagocytes. We did not deplete CX3CR1 cells in the later stages of meningitis; thus, the contribution of macrophages and monocytes in long-term cochlear remodeling is not known. The only intervention that demonstrated a significant improvement in outcome was systemic dexamethasone in combination with antibiotic at the onset of infection. Mice receiving dexamethasone demonstrated a higher average scala tympani fluid volume, a modest improvement in low frequency hearing, and no reduction in survival.
In other forms of inner ear injury, we have found that the innate immune response can influence hearing outcomes. In the case of aminoglycoside antibiotic ototoxicity, CCR2 expression in inflammatory monocytes exacerbated hearing loss when animals were exposed to low-dose lipopolysaccharide (LPS) prior to ototoxic antibiotics [28]. Systemic pretreatment with LPS resulted in worse hearing outcomes in ototoxicity by 20–30 dB across all frequencies. Large numbers of CCR2+ inflammatory monocytes were noted in the inner ear after LPS priming which were not present after aminoglycoside treatment alone. With LPS priming, CCR2 deletion attenuated the effects of ototoxicity. Not only were CCR2 KO mice protected from the effects of LPS on ototoxicity, but they were also resistant to ototoxic agents in general [28].
Therefore, CCR2 was a plausible target to modify outcomes in post-infectious remodeling of the inner ear. However, CCR2 deletion did not prove to be effective in protecting against hearing loss or labyrinthine ossification after pneumococcal meningitis. CCR2 null mice suffered worse weight loss, a longer duration of illness exhibited by lethargy and poor weight gain, and more severe cochlear ossification by quantitative measurements using microCT. While CCR2 monocytes contributed to ototoxic hearing loss, CCR2 was protective in pneumococcal meningitis. We postulate that the beneficial antimicrobial effect of CCR2 outweighed the bystander effects of cochlear inflammation. Eliminating CCR2 ultimately resulted in prolonged infection and a worse outcome for the inner ear after meningitis.
Hearing loss was worse in CX3CR1 knockout mice compared to controls in pneumococcal meningitis. The local effects of CX3CR1 in the cochlea suggest a protective role of cochlear macrophages that may be important for clearance of infection and local control of disease in bacterial meningitis. Systemic symptoms, such as weight loss, gross motor activity, and antibiotic needs, were similar in CX3CR1 null mice compared to wild-type mice. Prior work has demonstrated the impact of CX3CR1 expression on the inner ear. CX3CR1 is an important regulator of monocyte/macrophage entry from the vasculature into the inner ear [27, 43]. In other conditions such as noise and ototoxicity, CX3CR1 null mice recruit an excess of monocytes and macrophages to the inner ear compared to wild-type ears, suggesting that CX3CR1 is involved in regulation of myeloid cell entry into the labyrinth [27]. In addition, we have observed that CX3CR1-deleted monocytes and macrophages demonstrate a more destructive phenotype, poorly regulated as a result of the loss of CX3CR1 signaling [6, 9, 10].
Production of inflammatory chemokines and cytokines has been proposed as an important source of cochlear injury in bacterial meningitis [44, 45]. However, other than systemic corticosteroids, no specific agents have been recommended for clinical use. A study of pneumococcal meningitis in rabbits evaluated the effect of granulocyte colony stimulatory factor (GCSF) [46]. GCSF is used during cancer chemotherapy to increase the availability of white blood cells by stimulating the bone marrow to accelerate leukocyte release into circulation and enhance the immune response. These experiments did not show improved survival in pneumococcal meningitis after GCSF. GCSF did not reduce bacterial counts in the CSF, although there was reduced bacteria in the blood [46]. In another study investigating the effects of GCSF in bacterial meningitis in rats, hearing loss was accelerated, and cochlear structural damage was worse with GCSF [35]. Both animal experiments and clinical trials in humans have demonstrated that dexamethasone, if initiated early during infection, provides protection against hearing loss in pneumococcal meningitis [20–22, 24, 35, 43]. Most believe that the beneficial effects of dexamethasone are the result of potent suppression of the inflammatory response in the CSF and perilymph in bacterial meningitis. In our experiments, dexamethasone did attenuate cochlear ossification at 3 months post infection; however, preservation of hearing was modest.
Neurotrophins, such as NT-3, offer another approach to hearing protection in meningitis. Animal experiments in pneumococcal meningitis proved NT-3 to be effective in preserving spiral ganglion neurons and improving hearing outcomes by ABR in mice [36]. The hearing benefit provided by NT-3 was comparable to that observed by dexamethasone; however, it did not improve patency of the scala tympani. Other studies using antioxidants such as N-acetyl cysteine have been tested in mice and do not appear to provide protection [47]. Free radical scavengers have been shown to be protective of CNS neuronal damage associated with group B streptococcal meningitis although we do not have evidence of hearing protection [48]. Metformin has been tested as a neuroprotective agent and holds promise. The mechanism of metformin’s protective effect is not known. Early studies in pneumococcal meningitis show that metformin protects against hearing loss and neuronal injury in meningitic rats [49]. Interferon gamma (IFNg), the cardinal inflammatory cytokine associated with viral and bacterial infections, was found to be a critical element leading to breakdown of the blood brain barrier and to induce neuronal damage in the hippocampus and cortex in pneumococcal meningitis that was abrogated in IFNg knockout mice [44, 45, 50, 51]. While IFNg is known for its potent antiviral and antimicrobial function, how to reduce collateral damage associated with IFNg production has not been solved.
An important and disabling outcome of pneumococcal meningitis is profound hearing loss, which is accompanied by cochlear remodeling, ossification of the cochlear duct, and spiral ganglion cell loss. Labyrinthitis ossificans is a final common pathway for various forms of inner ear injury, rendering the cochlea ineffective. Furthermore, the loss of spiral ganglion neurons in many cases of meningitis eliminates the critical element necessary for successful rehabilitation with cochlear implantation. As our understanding of this disease process advances, interventions that successfully retain an open cochlear duct by preventing labyrinthine ossification and preserving spiral ganglion neurons will result in major advances for people with hearing loss after recovery from bacterial meningitis. Ultimately, preservation of cochlear patency and preservation of native acoustic hearing would be our goal, and more effective therapies to address this long-term complication of meningitis are needed.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
We thank Dr. Dorina Kallogjeri for her help with the statistical analysis of the quantitative data and Dr. Celeste Morley for sharing live Streptococcus pneumoniae from her laboratory.
Abbreviations
- CFU
Colony forming units
- CNS
Central nervous system
- CSF
Cerebrospinal fluid
- DPI
Days post infection
- DT
Diphtheria toxin
- GCSF
Granulocyte colony-stimulating factor
- GFP
Green fluorescent protein
- KO
Knockout
- LPS
Lipopolysaccharide
- NK cell
Natural killer cell
- RFP
Red fluorescent protein
- SPL
Sound pressure level
- WT
Wild type
Author Contribution
Keiko Hirose conceived the project, designed the experiments, performed the mouse surgery, reviewed the data, performed the statistical analyses, created the figures, and wrote the manuscript. Song Zhe Li performed the induction of meningitis, collected the ABR data, harvested the organs, performed the immunohistochemistry, performed confocal microscopy, and obtained the microCT scan data. Ruth Gill performed segmentation of the CT scan data and performed measurements of the cochlear volumes. Jared Hartsock assisted with mouse surgery for perilymph sampling, measurement and analysis of blood, perilymph, and CSF fluorescence.
Funding
This work was supported by a grant from the National Institute of Deafness and Communication Disorders (DC011315) to KH.
Declarations
Competing Interests
The authors declare no competing interests.
Footnotes
Brief Summary
CCR2 and CX3CR1 are expressed by monocytes and macrophages that are recruited to the cochlea during acute bacterial meningitis and are protective against hearing loss and ossification induced by bacterial meningitis.
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Brouwer MC, Tunkel AR, van de Beek D. Epidemiology, diagnosis, and antimicrobial treatment of acute bacterial meningitis. Clin Microbiol Rev. 2010;23(3):467–492. doi: 10.1128/CMR.00070-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kloek AT, Brouwer MC, Schmand B, Tanck MWT, van de Beek D. Long-term neurologic and cognitive outcome and quality of life in adults after pneumococcal meningitis. Clin Microbiol Infect. 2020;26(10):1361–1367. doi: 10.1016/j.cmi.2020.01.020. [DOI] [PubMed] [Google Scholar]
- 3.Lee S, et al. CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer’s disease mouse models. Am J Pathol. 2010;177(5):2549–2562. doi: 10.2353/ajpath.2010.100265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hirose K, Li S-Z. The role of monocytes and macrophages in the dynamic permeability of the blood-perilymph barrier. Hear Res. 2019;374:49–57. doi: 10.1016/j.heares.2019.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sautter NB, Shick EH, Ransohoff RM, Charo IF, Hirose K. CC chemokine receptor 2 is protective against noise-induced hair cell death: studies in CX3CR1(+/GFP) mice. J Assoc Res Otolaryngol. 2006;7(4):361–372. doi: 10.1007/s10162-006-0051-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cardona AE, et al. Control of microglial neurotoxicity by the fractalkine receptor. Nat Neurosci. 2006;9(7):917–924. doi: 10.1038/nn1715. [DOI] [PubMed] [Google Scholar]
- 7.Hughes PM, Botham MS, Frentzel S, Mir A, Perry VH. Expression of fractalkine (CX3CL1) and its receptor, CX3CR1, during acute and chronic inflammation in the rodent CNS. Glia. 2002;37(4):314–327. doi: 10.1002/glia.10037. [DOI] [PubMed] [Google Scholar]
- 8.Saederup N, et al. Selective chemokine receptor usage by central nervous system myeloid cells in CCR2-red fluorescent protein knock-in mice. PLoS ONE. 2010;5(10):e13693. doi: 10.1371/journal.pone.0013693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kaur T, et al. Fractalkine signaling regulates macrophage recruitment into the cochlea and promotes the survival of spiral ganglion neurons after selective hair cell lesion. J Neurosci Off J Soc Neurosci. 2015;35(45):15050–15061. doi: 10.1523/JNEUROSCI.2325-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sato E, Shick EH, Ransohoff R, Hirose K. CX3CR1 expression in cochlear macrophages down-regulates kanamycin ototoxicity. Assoc Res Otolaryngol Abstr. 2008;31:247. [Google Scholar]
- 11.Stojković L, et al. The association of V249I and T280M fractalkine receptor haplotypes with disease course of multiple sclerosis. J Neuroimmunol. 2012;245(1–2):87–92. doi: 10.1016/j.jneuroim.2011.12.028. [DOI] [PubMed] [Google Scholar]
- 12.Blauth K, Zhang X, Chopra M, Rogan S, Markovic-Plese S. The role of fractalkine (CX3CL1) in regulation of CD4(+) cell migration to the central nervous system in patients with relapsing-remitting multiple sclerosis. Clin Immunol Orlando Fla. 2015;157(2):121–132. doi: 10.1016/j.clim.2015.01.001. [DOI] [PubMed] [Google Scholar]
- 13.Brodie HA, Thompson TC, Vassilian L, Lee BN. Induction of labyrinthitis ossificans after pneumococcal meningitis: an animal model. Otolaryngol Head Neck Surg. 1998;118(1):15–21. doi: 10.1016/S0194-5998(98)70369-9. [DOI] [PubMed] [Google Scholar]
- 14.Igarashi M, Saito R, Alford BR, Filippone MV, Smith JA. Temporal bone findings in pneumococcal meningitis. Arch Otolaryngol. 1974;99(2):79–83. doi: 10.1001/archotol.1974.00780030085001. [DOI] [PubMed] [Google Scholar]
- 15.Kesser BW, Hashisaki GT, Spindel JH, Ruth RA, Scheld WM. Time course of hearing loss in an animal model of pneumococcal meningitis. Otolaryngol Neck Surg. 1999;120(5):628–637. doi: 10.1053/hn.1999.v120.a92772. [DOI] [PubMed] [Google Scholar]
- 16.Klein M, Koedel U, Pfister H-W, Kastenbauer S. Morphological correlates of acute and permanent hearing loss during experimental pneumococcal meningitis. Brain Pathol. 2003;13(2):123–132. doi: 10.1111/j.1750-3639.2003.tb00012.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Merchant SN, Gopen Q. A human temporal bone study of acute bacterial meningogenic labyrinthitis. Am J Otol. 1996;17(3):375–385. [PubMed] [Google Scholar]
- 18.Møller MN, Brandt C, Østergaard C, Caye-Thomasen P. Bacterial invasion of the inner ear in association with pneumococcal meningitis. Otol Neurotol. 2014;35(5):e178. doi: 10.1097/MAO.0000000000000305. [DOI] [PubMed] [Google Scholar]
- 19.Nabili V, Brodie HA, Neverov NI, Tinling SP. Chronology of labyrinthitis ossificans induced by streptococcus pneumoniae meningitis. Laryngoscope. 1999;109(6):931–935. doi: 10.1097/00005537-199906000-00017. [DOI] [PubMed] [Google Scholar]
- 20.Bhatt SM, et al. The impact of dexamethasone on hearing loss in experimental pneumococcal meningitis. Pediatr Infect Dis J. 1995;14(2):93–96. doi: 10.1097/00006454-199502000-00002. [DOI] [PubMed] [Google Scholar]
- 21.Brouwer MC, McIntyre P, Prasad K, van de Beek D. Corticosteroids for acute bacterial meningitis. Cochrane Database Syst Rev. 2015;9:4405. doi: 10.1002/14651858.CD004405.pub5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kim HH, Addison J, Suh E, Trune DR, Richter C-P. Otoprotective effects of dexamethasone in the management of pneumococcal meningitis: an animal study. Laryngoscope. 2007;117(7):1209–1215. doi: 10.1097/MLG.0b013e318058195f. [DOI] [PubMed] [Google Scholar]
- 23.Worsøe L, Brandt CT, Lund SP, Østergaard C, Thomsen J, Cayé-Thomasen P. Intratympanic steroid prevents long-term spiral ganglion neuron loss in experimental meningitis. Otol Neurotol. 2010;31(3):394–403. doi: 10.1097/MAO.0b013e3181d2796c. [DOI] [PubMed] [Google Scholar]
- 24.Liechti FD, Grandgirard D, Leib SL. Bacterial meningitis: insights into pathogenesis and evaluation of new treatment options: a perspective from experimental studies. Future Microbiol. 2015;10(7):1195–1213. doi: 10.2217/fmb.15.43. [DOI] [PubMed] [Google Scholar]
- 25.Mynatt R, Hale SA, Gill RM, Plontke SK, Salt AN. Demonstration of a longitudinal concentration gradient along scala tympani by sequential sampling of perilymph from the cochlear apex. J Assoc Res Otolaryngol. 2006;7(2):182–193. doi: 10.1007/s10162-006-0034-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hirose K, Discolo CM, Keasler JR, Ransohoff R. Mononuclear phagocytes migrate into the murine cochlea after acoustic trauma. J Comp Neurol. 2005;489(2):180–194. doi: 10.1002/cne.20619. [DOI] [PubMed] [Google Scholar]
- 27.Sato E, Shick HE, Ransohoff RM, Hirose K. Expression of fractalkine receptor CX3CR1 on cochlear macrophages influences survival of hair cells following ototoxic injury. J Assoc Res Otolaryngol. 2010;11(2):223–234. doi: 10.1007/s10162-009-0198-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hirose K, Li S-Z, Ohlemiller KK, Ransohoff RM. Systemic lipopolysaccharide induces cochlear inflammation and exacerbates the synergistic ototoxicity of kanamycin and furosemide. J Assoc Res Otolaryngol. 2014;15(4):555–570. doi: 10.1007/s10162-014-0458-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Salt AN, Hirose K. Communication pathways to and from the inner ear and their contributions to drug delivery. Hear Res. 2018;362:25–37. doi: 10.1016/j.heares.2017.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Scheld WM, Dacey RG, Winn HR, Welsh JE, Jane JA, Sande MA. Cerebrospinal fluid outflow resistance in rabbits with experimental meningitis. Alterations with penicillin and methylprednisolone. J Clin Invest. 1980;66(2):243–253. doi: 10.1172/JCI109850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lahrtz F, Piali L, Spanaus K-S, Seebach J, Fontana A. Chemokines and chemotaxis of leukocytes in infectious meningitis. J Neuroimmunol. 1998;85(1):33–43. doi: 10.1016/S0165-5728(97)00267-1. [DOI] [PubMed] [Google Scholar]
- 32.Klein M, et al. MyD88-dependent immune response contributes to hearing loss in experimental pneumococcal meningitis. J Infect Dis. 2007;195(8):1189–1193. doi: 10.1086/512859. [DOI] [PubMed] [Google Scholar]
- 33.Perny M, Solyga M, Grandgirard D, Roccio M, Leib SL, Senn P. Streptococcus pneumoniae-induced ototoxicity in organ of Corti explant cultures. Hear Res. 2017;350:100–109. doi: 10.1016/j.heares.2017.04.012. [DOI] [PubMed] [Google Scholar]
- 34.Brower MC, McIntyre P, de Gans J, Prasad K, van Beek D. Corticosteroids for acute bacterial meningitis (review) Cochrane Database Syst Rev. 2010 doi: 10.1002/14651858.CD004405.pub3. [DOI] [PubMed] [Google Scholar]
- 35.Brandt CT, et al. Hearing loss and cochlear damage in experimental pneumococcal meningitis, with special reference to the role of neutrophil granulocytes. Neurobiol Dis. 2006;23(2):300–311. doi: 10.1016/j.nbd.2006.03.006. [DOI] [PubMed] [Google Scholar]
- 36.Demel C, et al. Reduced spiral ganglion neuronal loss by adjunctive neurotrophin-3 in experimental pneumococcal meningitis. J Neuroinflammation. 2011;8(1):7. doi: 10.1186/1742-2094-8-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Barichello T, et al. Targets for adjunctive therapy in pneumococcal meningitis. J Neuroimmunol. 2015;278:262–270. doi: 10.1016/j.jneuroim.2014.11.015. [DOI] [PubMed] [Google Scholar]
- 38.Bewersdorf JP, Grandgirard D, Koedel U, Leib SL. Novel and preclinical treatment strategies in pneumococcal meningitis. Curr Opin Infect Dis. 2018;31(1):85–92. doi: 10.1097/QCO.0000000000000416. [DOI] [PubMed] [Google Scholar]
- 39.Teitelbaum SL. Glucocorticoids and the osteoclast. Clin Exp Rheumatol. 2015;33(4 Suppl 92):S37–39. [PubMed] [Google Scholar]
- 40.Fujikawa Y, Quinn JM, Sabokbar A, McGee JO, Athanasou NA. The human osteoclast precursor circulates in the monocyte fraction. Endocrinology. 1996;137(9):4058–4060. doi: 10.1210/endo.137.9.8756585. [DOI] [PubMed] [Google Scholar]
- 41.Madel M-B, et al. Immune function and diversity of osteoclasts in normal and pathological conditions. Front Immunol. 2019;10:1408. doi: 10.3389/fimmu.2019.01408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jacome-Galarza CE, et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nature. 2019;568(7753):541–545. doi: 10.1038/s41586-019-1105-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sato E, Shick HE, Ransohoff RM, Hirose K. Repopulation of cochlear macrophages in murine hematopoietic progenitor cell chimeras: the role of CX3CR1. J Comp Neurol. 2008;506(6):930–942. doi: 10.1002/cne.21583. [DOI] [PubMed] [Google Scholar]
- 44.Too LK, Ball HJ, McGregor IS, Hunt NH. The pro-inflammatory cytokine interferon-gamma is an important driver of neuropathology and behavioural sequelae in experimental pneumococcal meningitis. Brain Behav Immun. 2014;40:252–268. doi: 10.1016/j.bbi.2014.02.020. [DOI] [PubMed] [Google Scholar]
- 45.Yau B, Mitchell AJ, Too LK, Ball HJ, Hunt NH. Interferon-γ-induced nitric oxide synthase-2 contributes to blood/brain barrier dysfunction and acute mortality in experimental Streptococcus pneumoniae meningitis. J Interferon Cytokine Res. 2016;36(2):86–99. doi: 10.1089/jir.2015.0078. [DOI] [PubMed] [Google Scholar]
- 46.Ostergaard C, et al. Pretreatment with granulocyte colony-stimulating factor attenuates the inflammatory response but not the bacterial load in cerebrospinal fluid during experimental pneumococcal meningitis in rabbits. Infect Immun. 1999;67(7):3430–3436. doi: 10.1128/IAI.67.7.3430-3436.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Högen T, et al. Adjunctive N-acetyl-l-cysteine in treatment of murine pneumococcal meningitis. Antimicrob Agents Chemother. 2013;57(10):4825–4830. doi: 10.1128/AAC.00148-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Leib SL, Kim YS, Chow LL, Sheldon RA, Täuber MG. Reactive oxygen intermediates contribute to necrotic and apoptotic neuronal injury in an infant rat model of bacterial meningitis due to group B streptococci. J Clin Invest. 1996;98(11):2632–2639. doi: 10.1172/JCI119084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Muri L, Le ND, Zemp J, Grandgirard D, Leib SL. Metformin mediates neuroprotection and attenuates hearing loss in experimental pneumococcal meningitis. J Neuroinflammation. 2019;16(1):156. doi: 10.1186/s12974-019-1549-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Mitchell AJ, et al. Inflammasome-dependent IFN-γ drives pathogenesis in Streptococcus pneumoniae meningitis. J Immunol. 2012;189(10):4970–4980. doi: 10.4049/jimmunol.1201687. [DOI] [PubMed] [Google Scholar]
- 51.Yau B, Too LK, Ball HJ, Hunt NH. TIGR4 strain causes more severe disease than WU2 strain in a mouse model of Streptococcus pneumoniae meningitis: a common pathogenic role for interferon-γ. Microbes Infect. 2017;19(7–8):413–421. doi: 10.1016/j.micinf.2017.04.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Underlying data and supporting work will be available through PubMed through this published article and through direct inquiry.