

Abstract
Innate immunity must be tightly regulated to enable sensitive pathogen detection while averting autoimmunity triggered by pathogen-like host molecules. A hallmark of viral infection, double-stranded RNAs (dsRNAs) are also abundantly encoded in mammalian genomes, necessitating surveillance mechanisms to distinguish “self” from “nonself.” ADAR1, an RNA editing enzyme, has emerged as an essential safeguard against dsRNA-induced autoimmunity. By converting adenosines to inosines (A-to-I) in long dsRNAs, ADAR1 covalently marks endogenous dsRNAs, thereby blocking the activation of the cytoplasmic dsRNA sensor MDA5. Moreover, beyond its editing function, ADAR1 binding to dsRNA impedes the activation of innate immune sensors PKR and ZBP1. Recent landmark studies underscore the utility of silencing ADAR1 for cancer immunotherapy, by exploiting the ADAR1-dependence developed by certain tumors to unleash an antitumor immune response. In this perspective, we summarize the genetic and mechanistic evidence for ADAR1's multipronged role in suppressing dsRNA-mediated autoimmunity and explore the evolving roles of ADAR1 as an immuno-oncology target.
Keywords: ADAR1, RNA editing, MDA5, dsRNA, PKR, ZBP1
INTRODUCTION
Innate immune pathways that sense double-stranded RNA (dsRNA) form an essential layer of antiviral defense in mammals (Fig. 1; Hur 2019). However, besides signaling viral infection, dsRNAs can also form within endogenous transcripts, predominantly between the transposable repeat sequences that occupy nearly half of the human genome. Thus, the protective role of dsRNA sensors needs to be constantly balanced with the risk of erroneously launching an immune response against cellular RNAs (Chen and Hur 2022).
FIGURE 1.

Major cytoplasmic nucleic sensing pathways in mammals. The RIG-I pathway (activated by 5′-triphosphorylated, blunt-ended dsRNA), MDA5 pathway (activated by long dsRNAs), and cGAS-STING pathway (activated by dsDNA) converge to induce interferon signaling. Meanwhile, PKR dimerization on dsRNA primarily leads to translational arrest and cell death. ADAR1 blocks the recognition of endogenous dsRNAs by MDA5 and PKR to prevent constitutive interferon activation and apoptosis.
Adenosine-to-inosine (A-to-I) RNA editing constitutes an essential mechanism for marking cellular dsRNAs as “self” and repressing autoimmunity (Quin et al. 2021). In mammals, A-to-I RNA editing is carried out by two catalytically active ADAR proteins, ADAR1 and ADAR2. A third homolog, ADAR3, lacks deaminase activity and instead functions as a negative regulator of editing in the brain (Fig. 2A; Nishikura 2016; Oakes et al. 2017; Tan et al. 2017). The ubiquitously expressed ADAR1 protein carries out most of the cellular editing, predominantly targeting long dsRNAs that form between the complementary sequences of transposable repeat elements (Tan et al. 2017). Meanwhile, ADAR2 primarily functions in the brain and is responsible for most A-to-I editing events in coding regions, including an essential recoding event in the neurotransmitter receptor GRIA2 (Sommer et al. 1991; Higuchi et al. 2000; Tan et al. 2017). ADAR1 comes in two isoforms that exhibit significant differences in molecular structure, subcellular localization, and function. The shorter p110 isoform is expressed from a constitutively active promoter and resides in the nucleus, whereas the longer p150 isoform, featuring an additional nucleic acid binding domain, initiates from an upstream interferon-inducible promoter and shuttles between the nucleus and cytoplasm (George and Samuel 1999; Sun et al. 2021). Unlike the site-specific editing events in coding sequences, typically confined to short and imperfect duplex regions, the long transposable repeat-derived dsRNAs tend to be edited by ADAR1 promiscuously, producing clusters of inosines that are referred to as hyper-editing (Porath et al. 2014). These clusters of editing events can markedly alter the structure of cellular dsRNAs by replacing canonical A-U base pairs with the less stable I-U wobbles and can transform dsRNAs–protein interactions, including the recognition by dsRNA sensors (Fig. 2B; Bass and Weintraub 1988; Strobel et al. 1994; Serra et al. 2004; Wright et al. 2007; Ahmad et al. 2018).
FIGURE 2.
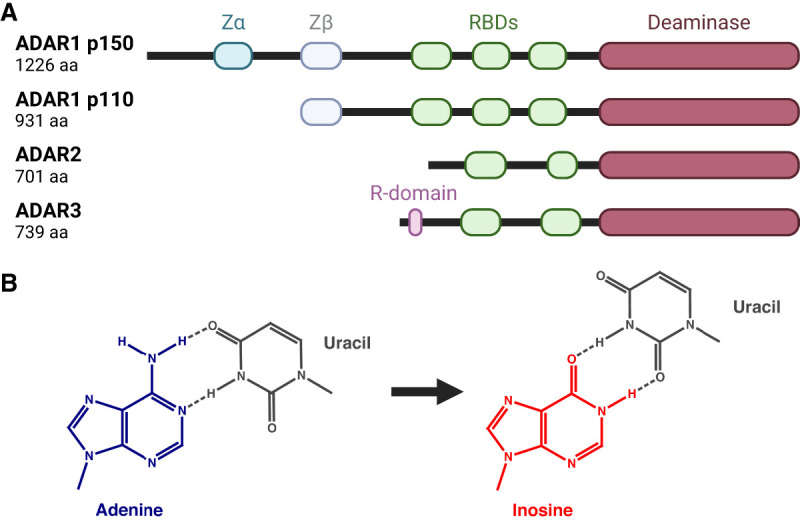
ADAR proteins and RNA editing. (A) Human ADAR proteins. (RBD) RNA-binding domain. (B) Deamination of adenine to inosine changes the base pair geometry and lowers the stability of base-pairing.
Our understanding of the physiological roles of ADAR1 has undergone a significant shift since the original discovery that ADAR1 was essential for survival. Initially centered on the biological functions of individual editing events, more recent research has established a global role of ADAR1 in regulating the cellular “dsRNAome.” Genetic studies of mouse models and human disease have identified ADAR1 as a pivotal regulator of dsRNA innate immunity, bearing implications for autoinflammatory diseases and cancer. Among the pathways responsible for sensing dsRNA, the predominant in vivo function of ADAR1 appears to be the antagonism of MDA5 through A-to-I editing. Nonetheless, recent findings highlight editing-independent mechanisms that also play a crucial role in maintaining cellular homeostasis. These mechanisms involve the competition for dsRNA binding between ADAR1 and PKR, and between ADAR1 and ZBP1. In the following sections, we present the evidence supporting ADAR1's role as a suppressor of dsRNA immunity, with a primary focus on the ADAR1–dsRNA–MDA5 axis. We conclude with a discussion of the emergent promise of targeting ADAR1 in cancer immunotherapy.
THE ADAR1–dsRNA–MDA5 AXIS OF LONG dsRNA-MEDIATED IMMUNITY
Genetic evidence for the ADAR1–dsRNA–MDA5 interplay in mouse
Genetic studies in mice were instrumental in establishing ADAR1's functions in dsRNA immunity. Both ADAR1 and ADAR2 are essential for survival in mice (Higuchi et al. 2000; Hartner et al. 2004; Wang et al. 2004). In the case of ADAR2, a single recoding event in the neurotransmitter receptor GRIA2 accounts for the lethal deletion phenotype (Higuchi et al. 2000). In contrast, the mechanism behind the lethal phenotype of ADAR1 deletion proved more complex and more challenging to delineate.
The knockout of ADAR1 in mice leads to embryonic lethality due to extensive liver damage (Hartner et al. 2004; Wang et al. 2004). At the molecular level, ADAR1-deficient mouse embryos display hallmarks of an activated innate immune response, characterized by excessive production of interferon and the induction of interferon-stimulated genes (ISGs) (Hartner et al. 2009). These observations strongly hinted at ADAR1's involvement in controlling innate immunity.
Nevertheless, the early attempts to identify the responsible molecular pathways were unsuccessful. Knockout of PKR, a dsRNA sensor that induces translational arrest and apoptosis, failed to rescue the lethal Adar1 null phenotype (Wang et al. 2004). The OAS-RNase L pathway, which likewise is triggered by cytoplasmic dsRNAs, also appeared to be unaffected in vivo (Wang et al. 2004). It was not until a decade later that the question of ADAR1's role in innate immunity was revisited with new mouse models, including mice lacking components of the long dsRNA-sensing MDA5–MAVS pathway (Mannion et al. 2014; Liddicoat et al. 2015, 2016; Pestal et al. 2015; Heraud-Farlow et al. 2017; Li et al. 2017; Costa Cruz et al. 2020). MDA5 had not yet been linked to dsRNA innate immunity at the time of the initial rescue studies (Andrejeva et al. 2004; Yoneyama et al. 2004). Yet, of all tested cytoplasmic nucleic acid sensors and interferon pathway components, only the knockout of the dsRNA sensor MDA5 or its downstream effector MAVS rescued embryonic death in mice lacking functional ADAR1 (Mannion et al. 2014; Liddicoat et al. 2015; Pestal et al. 2015; Heraud-Farlow et al. 2017; Li et al. 2017; Costa Cruz et al. 2020). Removal of the dsRNA sensor RIG-I, DNA sensing adaptor STING, interferon receptors (IFNαR, IFNγR), or the transcriptional activator of interferon signaling STAT1 provided no rescue (Mannion et al. 2014; Pestal et al. 2015; Liddicoat et al. 2016).
Mechanistically, it was the A-to-I editing activity of ADAR1 that obstructed MDA5 activation and the induction of ISGs. This was evidenced by the fact that a catalytically inactive ADAR1 point mutant produced an embryonically lethal phenotype similar to that observed in full ADAR1 knockout (Liddicoat et al. 2015). Notably, mice lacking the A-to-I editing function of ADAR1 were able to reach their full life span when MDA5 was eliminated, indicating tight coupling between ADAR1-mediated editing and dsRNA sensing by MDA5 (Liddicoat et al. 2015; Heraud-Farlow et al. 2017). Consistent with a role in suppressing cytoplasmic dsRNA sensing, the removal of ADAR1's cytoplasmic, interferon-inducible p150 isoform alone was sufficient to replicate the Adar1 null phenotype (Ward et al. 2011; Pestal et al. 2015). Collectively, these genetic studies pinpointed MDA5–MAVS as the primary pathway that is negatively regulated by ADAR1-mediated editing in vivo. This conclusion was later corroborated by a mouse model completely devoid of A-to-I editing by ADAR1 and ADAR2 (Chalk et al. 2019). A quadruple mutant mouse was generated by breeding ADAR1 editing-deficient mice that were rescued by MDA5 knockout with ADAR2-deficient mice that were rescued by an A-to-G substitution (mimicking A-to-I) in ADAR2's essential target GRIA2. The completely editing-deficient mice have normal life spans without apparent phenotypes, indicating that editing is only vitally important within MDA5's dsRNA targets and the GRIA2 mRNA. Together, the genetic studies in mouse models have established an essential function of ADAR1-mediated editing in suppressing spontaneous MDA5 activation by endogenous dsRNAs.
Clinical evidence for the ADAR1–dsRNA–MDA5 interplay in humans
The ADAR1–dsRNA–MDA5 axis is conserved from mouse to human, as revealed by genetic studies of human immune disorders. ADAR1 loss-of-function (LOF) and MDA5 gain-of-function (GOF) mutations give rise to an array of rare autoinflammatory diseases (i.e., conditions characterized by an overactive innate immune system) (Rice et al. 2012, 2014, 2020; Hayashi and Suzuki 2013; Oda et al. 2014; Crow et al. 2015; Crow and Stetson 2022). These so-called type-I interferonopathies range from relatively mild skin conditions in individuals with one LOF ADAR1 allele to lethal neurodevelopmental disorders in the presence of MDA5 GOF mutations or two LOF ADAR1 alleles (Crow et al. 2015; Rice et al. 2020). Despite differences in symptoms, all tested patients exhibit up-regulation of type-I interferon activity, mirroring the constitutive interferon signaling observed in ADAR1-deficient mice.
Both ADAR1 and MDA5 mutations have been linked to the rare neurodevelopmental disease Aicardi–Goutières syndrome (AGS), which can also be caused by mutations in other genes involved in nucleic acid sensing (Crow et al. 2015). AGS typically starts in childhood, manifesting with brain and skin abnormalities and often severe intellectual and physical disability (Crow and Manel 2015). Most AGS patients with ADAR1 mutations contain two mutated alleles, with at least one mutation in the deaminase domain, whereas the most common compound mutation is located in the p150 isoform-specific Zα domain (Rice et al. 2012). All disease-linked ADAR1 mutations are predicted to decrease the deaminase activity or weaken dsRNA binding (Rice et al. 2012; Matthews et al. 2016; Karki et al. 2024). In contrast, the MDA5 mutations behind AGS and other autoinflammatory disorders confer gain of function by stabilizing MDA5 filament formation on dsRNA (Rice et al. 2014, 2020; Yu et al. 2021). These pathogenic mutations typically cluster around the ATPase active site, disrupting the ATPase activity and consequently slowing the dissociation of MDA5 monomers from dsRNA (Peisley et al. 2012; Rice et al. 2014; Yu et al. 2018). The resulting stabilization of dsRNA binding makes MDA5 less sensitive to imperfections in dsRNA structure and A-to-I editing, leading to constitutive MDA5 activation by endogenous dsRNAs (Ahmad et al. 2018; Yu et al. 2018). Conversely, MDA5 LOF variants can be protective against certain autoimmune diseases, including type-I diabetes, psoriasis, psoriatic arthritis, vitiligo, and coronary artery disease, although excessive MDA5 LOF increases the susceptibility to certain viral infections (Nejentsev et al. 2009; Shigemoto et al. 2009; Li et al. 2010; Asgari et al. 2017; Budu-Aggrey et al. 2017; Dand et al. 2017; Jin et al. 2017; Lamborn et al. 2017; Zaki et al. 2017; Emdin et al. 2018; Cananzi et al. 2021; Chen et al. 2021a).
The clinical manifestation of ADAR1 and MDA5 variants, when considered alongside mouse genetic studies, suggests a precisely tuned balance between MDA5's sensing of endogenous dsRNAs and the counteracting editing activity of ADAR1 (Fig. 3). Disruption of this delicate balance through perturbations in any component—be it ADAR1 loss, heightened expression of endogenous dsRNA, or MDA5 gain of function—can escalate dsRNA sensing by MDA5, leading to autoinflammation. Adjusting each of the three components of the ADAR1–dsRNA–MDA5 axis holds the potential not only to address autoinflammatory diseases but also transform treatments that benefit from transient interferon induction, as epitomized by the emerging applications in cancer immunotherapy.
FIGURE 3.

The ADAR1–dsRNA–MDA5 axis of innate immunity. In their unedited form, long cytoplasmic dsRNAs serve as substrates for MDA5 filament formation, leading to the induction of interferon response. By catalyzing A-to-I editing in endogenous dsRNAs, ADAR1 prevents MDA5 oligomerization and constitutive interferon activation. Boxes indicate the physiological consequences of genetic perturbations of ADAR1 and MDA5 activity in mice and humans.
Mapping immunogenic dsRNAs
The immunogenic “self” dsRNAs represent the most enigmatic and likely most complex component of the ADAR1–dsRNA–MDA5 axis. Initiating the MDA5–MAVS dsRNA-sensing pathway requires the formation of stable MDA5 filaments on long dsRNAs, typically comprising at least several hundred base pairs (Fig. 1; Cadena and Hur 2019). Interactions between adjacent dsRNA-bound MDA5 monomers stabilize dsRNA binding, counteracting the rapid ATP-dependent dissociation of individual monomers (Cadena and Hur 2019). The MDA5 filaments activate the downstream effector MAVS by bringing together MDA5's amino-terminal caspase activation and recruitment domains to form oligomers, which in turn nucleate filaments of MAVS CARD domains (Fig. 1; Wu and Hur 2015). Once in its active filamentous state, MAVS recruits TRAF molecules, triggering the interferon signaling cascade (Wu and Hur 2015).
MDA5 filaments are best sustained by long perfect dsRNAs, such as those arising from viral replication (Peisley et al. 2012). Nevertheless, the genetic studies discussed above demonstrated that cellular dsRNAs can sufficiently activate MDA5–MAVS to trigger lethal autoinflammatory responses, unless these dsRNAs are “neutralized” by ADAR1-mediated editing (Fig. 3). The vast majority of the endogenous dsRNAs are presumably formed by inverted repeats where most of ADAR1-mediated hyper-editing takes place (Tan et al. 2017; Reich and Bass 2019; Levanon et al. 2023). Most of these repeats are located in introns and intergenic regions, making them unlikely to be exposed to cytoplasmic dsRNA sensors like MDA5. Nevertheless, a small fraction of tandem inverted repeats occur in mature mRNA, particularly in the untranslated regions (UTRs), and are exported into the cytosol, where they become potential MDA5 ligands capable of inducing an innate immune response. Through A-to-I editing, the cytoplasmic ADAR1 p150 isoform destabilizes and reshapes the dsRNA structure, interfering with MDA5 filament formation and thereby mitigating the risk of inflammation (Fig. 3; Strobel et al. 1994; Serra et al. 2004; Wright et al. 2007; Ahmad et al. 2018).
The precise identity of cellular dsRNAs that require A-to-I editing to avoid MDA5 sensing has been a topic of extensive investigation and debate (Chen and Hur 2022; Levanon et al. 2023; Cottrell et al. 2024). One strategy for identifying potentially immunogenic cytoplasmic dsRNAs involves isolating the editing sites that are selectively introduced by the cytosolic, interferon-inducible ADAR1 p150 isoform, in contrast to nuclear ADARs (Kim et al. 2021; Sun et al. 2021, 2022; Kleinova et al. 2023). Studies using this approach have consistently highlighted an enrichment for UTR-localized inverted Alu repeats (IR Alus) among p150-selective sites and suggest that only a minuscule fraction of IR Alus may be immunogenic (Kim et al. 2021; Sun et al. 2022; Levanon et al. 2023). Intriguingly, some of the most potent immunogenic dsRNAs may not derive from Alu repeats at all but instead originate from cis-natural antisense transcripts (cis-NATs)—that is, complementary RNAs that are transcribed in opposite directions (Faghihi and Wahlestedt 2009; Li et al. 2022; Sun et al. 2022). Unlike IR Alus, which form relatively short, ∼300-bp imperfect duplexes, cis-NATs constitute perfectly complementary dsRNAs spanning hundreds to over 1000 bp, making them much more suitable ligands for stable MDA5 multimerization (Sun et al. 2022; Levanon et al. 2023; Cottrell et al. 2024). Although cis-NATs are rare compared to IR Alus, their ideal properties as MDA5 substrates suggest that they may carry disproportionate weight in the pool of immunogenic dsRNAs. Indeed, cis-NAT editing was implicated in a number of autoinflammatory and immune-related diseases based on their enrichment at the genomic loci defined by genome-wide association studies (Li et al. 2022). Moreover, cis-NATs but not the primate-specific IR Alus tend to be conserved from mice to humans and may have played a pivotal role in the conservation of the ADAR1–dsRNA–MDA5 axis (Sun et al. 2022).
Immunogenic dsRNAs comprise a dynamic category of cellular dsRNAs that is shaped by tissue-specific transcriptional programs, among other factors. For instance, neural progenitor cells (NPCs) stand out for their elevated burden of immunogenic dsRNA compared to other cell types (Chung et al. 2018; Sun et al. 2022; Dorrity et al. 2023). This increased burden results both from the distinct identities of RNAs expressed in NPCs and from cell-type specific variations in RNA expression levels. Moreover, the global lengthening of the 3′ UTRs of neuronal mRNAs contributes to the heightened dsRNA load in neurons (Dorrity et al. 2023). The overabundance of immunogenic dsRNAs in neuronal cells may explain why in the ADAR1-linked genetic disorder AGS, inflammation is largely confined to the brain. The cell-type specific immunogenic dsRNA burden may also account for inflammation patterns in other diseases, such as type-I diabetes (Knebel et al. 2024). This subject warrants further exploration across the spectrum of autoimmune and related conditions.
BEYOND A-TO-I EDITING: THE INVOLVEMENT OF ADAR1 IN PKR AND ZBP1 PATHWAYS OF dsRNA IMMUNITY
Competition between ADAR1 and PKR for dsRNA binding
The essential role of ADAR1-mediated dsRNA editing in mitigating MDA5-induced dsRNA autoimmunity is well-established. However, mouse and in vitro studies have also unveiled that ADAR1 juggles several functions across dsRNA-sensing pathways, some of which may not require the deaminase function at all.
In the landmark genetic studies in mice that elucidated the ADAR1–dsRNA–MDA5 axis, an initially puzzling observation arose from rescue experiments of mice with full ADAR1 knockout versus those with a deaminase-inactivating mutation. Concurrent MDA5 knockout provided full rescue only in mice expressing editing-deficient ADAR1 but not in those entirely lacking ADAR1 (Pestal et al. 2015). The latter were still rescued from embryonic lethality but only survived for two days postbirth. Knockout of MDA5's downstream adaptor MAVS yielded analogous partial rescue of Adar1 null mice (Mannion et al. 2014; Pestal et al. 2015). This implied that other, editing-independent functions of ADAR1 become important during later development (Mannion et al. 2014; Pestal et al. 2015). Two studies published in 2023 finally identified the dsRNA sensor PKR as the missing link, by demonstrating that knocking out PKR in addition to MDA5 (or its downstream adaptor MAVS) rescued ADAR1 knockout mice to adulthood (Hu et al. 2023; Sinigaglia et al. 2023). Previously, PKR knockout alone was known to be ineffective in rescuing the embryonic lethal phenotype of ADAR1 knockout, despite abundant evidence for ADAR1 suppressing PKR activation in vitro (Wang et al. 2004; Toth et al. 2009; Li et al. 2017; Chung et al. 2018; Corbet et al. 2021). The new in vivo studies reconcile these seemingly conflicting observations. The accompanying mechanistic studies revealed that ADAR1 operates through two distinct mechanisms: it prevents MDA5 activation through A-to-I editing of immunogenic dsRNAs, and it competes with PKR for dsRNA binding, thereby preventing PKR-induced translational arrest (Hu et al. 2023). Both pathways must be suppressed by ADAR1 to prevent fatal inflammation in mice, explaining why disabling MDA5/MAVS and PKR individually was insufficient to fully compensate for ADAR1 loss in earlier mouse studies.
From structural and biochemical perspectives, PKR and MDA5 form distinct interactions with dsRNA, contributing to the different modes of ADAR1 intervention. Whereas MDA5 requires long dsRNAs spanning hundreds of base pairs to form stable filaments, PKR's active form is a dsRNA-induced dimer, requiring as few as 33 bp of dsRNA (Hull and Bevilacqua 2016). Once dimerized, PKR undergoes autophosphorylation, generating the active pPKR form that phosphorylates the translation initiation factor eIF2α, leading to global translational shutdown and ultimately apoptosis (Hull and Bevilacqua 2016). Besides differences in the dsRNA footprint and downstream steps, PKR and MDA5 utilize different domains for dsRNA binding. Whereas MDA5 engages dsRNA through its helicase domains, PKR contains two dsRNA-binding domains (dsRBDs). dsRBDs represent a conserved class of RNA-binding motifs and are also present in ADAR proteins (Fig. 2A). In the case of ADAR1, three dsRBDs are located upstream of its deaminase domain. The shared cytoplasmic localization and similar RNA-binding domain composition can explain the ability of ADAR1 p150 to efficiently compete with PKR for binding cytosolic dsRNAs (Cottrell et al. 2024). Indeed, overexpression of ADAR1's dsRBD domains alone was sufficient to suppress PKR activation, as was cytoplasmic expression of dsRBDs from other dsRNA-binding proteins (Hu et al. 2023).
Although PKR's dsRNA targets may still be edited by ADAR1, the mismatches potentially introduced by A-to-I editing seem to have a much smaller role in preventing the binding of PKR compared to blocking MDA5 filaments. This observation can be rationalized by PKR only requiring short continuous dsRNA regions, which will still be present in most edited dsRNAs. On the other hand, even sparse mismatches introduced by ADAR1 may interfere with stable multimerization of MDA5 on long dsRNA duplexes. The distinct dsRNA length requirements of PKR and MDA5 also imply that the sets of dsRNA recognized by each sensor are not identical, even though for both IR Alus comprise a major subset of targets (Ahmad et al. 2018; Kim et al. 2018; Sun et al. 2022).
Competition between ADAR1 and ZBP1 and the roles of ADAR1's Zα domain in innate immunity
ADAR1 p150 stands out among mammalian ADARs in several ways: its cytoplasmic localization, expression from an IFN-inducible promoter, and the possession of a unique amino-terminal Zα domain (Fig. 2A). As discussed earlier, the former two features clearly position ADAR1 p150 to interact with cytosolic dsRNA-sensing pathways. Recent investigations now show that the third distinguishing feature, a Z-form RNA and DNA binding domain, also contributes to the role of ADAR1 p150 in curbing dsRNA immunity.
Although the majority of AGS-causing ADAR1 mutations reside in the deaminase domain, surprisingly, the most common ADAR1 compound mutation in AGS patients, Pro193Ala, is located in the Zα domain (Crow and Manel 2015). Found in only one other human protein, ZBP1, this domain recognizes the Z-conformation of dsDNA and dsRNA, which can be adopted by purine–pyrimidine repeat sequences (Herbert et al. 1997; Schwartz et al. 1999; Brown et al. 2000; Placido et al. 2007). Consistent with Z-RNA binding by ADAR1, purine–pyrimidine tracts can accelerate the editing of RNA substrates in vitro and may enhance the editing of certain substrates in vivo (Koeris et al. 2005). In a crystal structure of the ADAR1 Zα domain complexed with Z-form DNA, Pro193 (the residue mutated in AGS) contacts the Z-DNA backbone, suggesting that the alanine substitution may weaken the binding of certain dsRNAs; alternatively, the structural role of Pro193 may result in misfolding of the Zα domain upon mutation (Schwartz et al. 1999; de Reuver et al. 2021). The importance of Pro193 for RNA binding and editing is further supported by the decreased editing of a representative substrate in HEK293 cells expressing the Pro193Ala mutant (Mannion et al. 2014).
Several studies recently zeroed in on the role of the Zα domain in innate immune regulation in vivo (de Reuver et al. 2021, 2022; Maurano et al. 2021; Nakahama et al. 2021; Tang et al. 2021; Jiao et al. 2022). Mice with mutations in the Zα domain typically exhibit mild or no defects and show subtle changes in editing (de Reuver et al. 2021; Maurano et al. 2021; Nakahama et al. 2021; Tang et al. 2021; Liang et al. 2023). Severe detrimental effects are observed only when the AGS-linked Pro193Ala mutation is introduced in the absence of a second Adar1 p150 allele, resulting in a shortened life span for a subset of animals (Maurano et al. 2021; Liang et al. 2023). In all mouse models, the observed interferon up-regulation was MDA5–MAVS dependent, with additional involvement of the dsRNA sensors PKR and LGP2. Thus, the Zα domain exerts a modest effect on ADAR1 p150's ability to modulate dsRNA immunity, but this role may be exacerbated in certain contexts.
ADAR1 p150 shares the Zα domain with only one other mammalian protein, ZBP1, which is likewise implicated in innate immunity through as yet incompletely understood mechanisms (de Reuver and Maelfait 2023). The potential for competition between the two proteins for binding the same Z-form prone RNA targets spawned a collection of recent studies focusing on ZBP1 (de Reuver et al. 2022; Hubbard et al. 2022; Jiao et al. 2022). These investigations uncovered the involvement of ZBP1 in triggering the interferon response in ADAR1-deficient mice and found that ZBP1 knockout could extend the survival of ADAR1 knockout mice, although to much shorter life spans than seen in the complete rescue by MDA5 and PKR knockout (de Reuver et al. 2022; Hubbard et al. 2022; Jiao et al. 2022; Hu et al. 2023; Sinigaglia et al. 2023). These studies paint a nuanced picture of the importance of ADAR1's Zα domain while providing support for a model of competition between ADAR1 p150 and ZBP1 (de Reuver and Maelfait 2023).
Taken together, the interactions of ADAR1 with the MDA5, PKR, and ZBP1 dsRNA-sensing pathways demonstrate how ADAR1 utilizes its versatile domain architecture to safeguard the cell from dsRNA autoimmunity on multiple fronts. The A-to-I editing activity of the deaminase domain is responsible for preventing MDA5 multimerization, the dsRBDs compete with PKR for dsRNA binding; and the Zα domain confers the unique ability to compete with ZBP1 for binding of select dsRNAs. This spectrum of activities by ADAR1 epitomizes the intricate competitive landscape of endogenous dsRNAs and their interaction partners (Cottrell et al. 2024). With the key players and pathways now identified, a deeper mechanistic understanding of the involved pathways will be a key priority, along with therapeutic development guided by such understanding.
ADAR1 IN CANCER
Evidence for ADAR1's role as a checkpoint of anticancer immunity
The neutralizing role of ADAR1 in long dsRNA-mediated immunity can be exploited by cancer cells to promote tumor growth and evade immune recognition (Bhate et al. 2019). Here, we briefly discuss the potential for targeting ADAR1 in cancer immunotherapy and highlight the promise of activating the MDA5, PKR, and/or ZBP1 dsRNA-sensing pathways through ADAR1 inhibition.
Targeting the innate immune system presents a potent antitumor strategy, as has been successfully demonstrated for the cGAS-STING and RIG-I pathways, which run parallel to the MDA5 pathway (Fig. 1; Kasumba and Grandvaux 2019; Reisländer et al. 2020; Cao et al. 2022). In preclinical settings, durable tumor regression can be achieved by treating tumors with synthetic cGAS-STING and RIG-I agonists in combination with immune checkpoint inhibitors or, in some cases, as standalone treatments (Kasumba and Grandvaux 2019; Reisländer et al. 2020). The analogous promise of activating MDA5 to combat cancer is supported by a variety of experimental evidence. Similarly to RIG-I and STING agonists, synthetic MDA5 ligand Poly(IC:LC)—a dsRNA derivative—enhances the potency of a cancer vaccine (Kasumba and Grandvaux 2019). Moreover, the efficacy of epigenetic cancer therapy was attributed to MDA5 activation by endogenous dsRNAs that become up-regulated in cancer cells upon treatment with DNA methyltransferase inhibitors or inhibitors of other epigenetic processes (Chiappinelli et al. 2015; Roulois et al. 2015; Brocks et al. 2017; Cuellar et al. 2017; Sheng et al. 2018; Zhang et al. 2018; Mehdipour et al. 2020; for review, see Chen et al. 2021b). Similarly, treating mouse tumors with spliceosome-targeted therapies leads to cytoplasmic accumulation of misspliced dsRNAs and activation of dsRNA innate immune pathways, including MDA5 (Bowling et al. 2021). Despite the clear therapeutic potential of MDA5 activation in cancer, specific MDA5 agonists without severe toxicities are yet to be identified (Kasumba and Grandvaux 2019). Potentially circumventing this challenge, the ADAR1–MDA5 antagonism provides a unique opportunity to indirectly activate MDA5 through ADAR1 inhibition.
ADAR1 is overexpressed in various cancers, and its pro-oncogenic effects have been compellingly linked to the silencing of endogenous dsRNA-sensing pathways. In the pivotal studies that first identified ADAR1 as an immuno-oncology target, ADAR1 consistently emerged as a top suppressor of anticancer immunity (Manguso et al. 2017; Gannon et al. 2018; Ishizuka et al. 2019; Liu et al. 2019). Deletion of ADAR1 sensitized certain tumors to checkpoint inhibitor therapy, resulting in reduced tumor growth. This effect was associated with the activation of interferon signaling in cancer cells and the inflammation of the tumor microenvironment, dependent on the MDA5–MAVS axis (Gannon et al. 2018; Ishizuka et al. 2019; Liu et al. 2019). Furthermore, the ADAR1 knockout triggered tumor cell growth arrest and apoptosis through the PKR pathway (Gannon et al. 2018; Ishizuka et al. 2019; Liu et al. 2019). Consistent with our current understanding of ADAR1's distinct modes of interference with MDA5 and PKR sensing, ADAR1 impeded both pathways albeit through different mechanisms (Gannon et al. 2018). Although in both cases dependent on the cytoplasmic p150 isoform, MDA5 but not PKR suppression required editing (Gannon et al. 2018). In vivo, the MDA5 and the PKR pathways each were sufficient to prime Adar1 null tumors for immune checkpoint inhibitor (anti-PD1) blockade (Ishizuka et al. 2019). Thus, ADAR1-dependent suppression of the MDA5 and PKR dsRNA-sensing pathways is essential for tumor growth in a subset of cancers, making tumors vulnerable to ADAR1 loss and, prospectively, pharmacological ADAR1 inhibition (Fig. 4). In line with these results, ADAR1 knockdown also enhances the effects of epigenetic therapy, boosting MDA5 activation by DNMTi-induced endogenous dsRNA (Mehdipour et al. 2020). More recently, ADAR1's role in blocking ZBP1-dependent cell death was also implicated in cancer, indicating additional opportunities for intervention in the realm of cancer immunotherapy (Karki et al. 2021; Zhang et al. 2022; de Reuver and Maelfait 2023).
FIGURE 4.

ADAR1 promotes tumor growth by suppressing dsRNA sensing. (Left) Increased ADAR1 expression in tumors (red arrow) leads to efficient editing of endogenous dsRNAs, including abnormal dsRNAs that may accumulate in cancer cells. Through editing dependent and independent mechanisms, ADAR1 inhibits spontaneous activation of MDA5 and PKR, thus promoting tumor growth and preventing immune recognition, even in the presence of checkpoint inhibitors. (Right) ADAR1 deletion unleashes the innate immune response against cellular dsRNAs in cancer cells, leading to inflammation and enhanced sensitivity to checkpoint inhibitors (via the MDA5 pathway) and triggering apoptosis (via the PKR pathway).
Toward ADAR1-based cancer therapies
Taken together, accumulating evidence suggests that perturbations in the core components of the ADAR1–dsRNA–MDA5 axis, along with parallel pathways involving PKR and ZBP1, could be leveraged for therapeutic benefit in cancer. A crucial consideration is establishing tolerance levels for ADAR1 inhibition or activation of MDA5, PKR, and ZBP1 in patients. The severity of certain ADAR1 and MDA5-linked autoinflammatory diseases warrants caution, as does the potential for cell death beyond the tumor. Nonetheless, the elevated ADAR1 expression observed in some cancers suggests a therapeutic window for the specific activation of dsRNA sensors in tumors.
The recent mechanistic insights offer many opportunities for fine-tuning the potency and specificity of ADAR1's antitumor effects. For example, selectively inhibiting the deaminase activity should induce MDA5-dependent interferon up-regulation without triggering PKR-dependent apoptosis or ZBP1-dependent cell death. Meanwhile, targeting the RNA-binding activity of ADAR1 would likely induce several dsRNA-sensing pathways, including PKR. Each of these strategies may be advantageous in different scenarios.
Research on ADAR1's roles in innate immunity and cancer alike underscores the presence of a pool of endogenous dsRNAs in mammalian cells that must be edited by ADAR1 to prevent constitutive MDA5 activation. A better understanding of the nature of these immunogenic dsRNAs may enable their manipulation as specific MDA5 agonists in cancer therapies and beyond. Epigenetic therapy seems to trigger innate immune signaling by increasing the expression of some immunogenic RNAs (Chen et al. 2021b). Alternative strategies to induce the expression of dsRNAs, for example, by inhibiting RNA methylation, or more targeted approaches may also help achieve therapeutically appropriate levels of MDA5 activation (Liu et al. 2020, 2021; Chelmicki et al. 2021).
CONCLUDING REMARKS AND FUTURE DIRECTIONS
Genetic studies in human patients and mouse models have firmly established the central role of ADAR1 in regulating dsRNA-mediated innate immunity. It is now clear that this same role is exploited by cancer cells to promote tumor growth and evade immune detection. Promisingly, targeted inhibition of ADAR1 in tumors, once therapeutically possible, may provide a more specific and safer approach to triggering an interferon response in tumors than, for example, global DNA demethylation in epigenetic therapy. To harness the full potential of ADAR1 and dsRNA sensing as therapeutic targets in cancer, several key questions need to be addressed.
First, specific ADAR1 inhibitors and MDA5 or PKR agonists are yet to be identified. Progress in this area will depend on advances in structural and mechanistic understanding of ADAR1 and the innate immunity pathways it interacts with. In particular, the efforts to inhibit ADAR1 would greatly benefit from high-resolution structural data. At a broader level, determining the most effective types of perturbations (ADAR1 repression, activation of dsRNA sensors, or increasing the tumor's dsRNA burden), or combinations thereof, is essential to achieve potent antitumor effects with minimal toxicity.
Additionally, the design of specific MDA5 agonists would benefit from an improved understanding of the identities and features of cellular immunogenic dsRNAs. Despite recent advances, studies of cellular dsRNAs and their immunogenic potential remain an active research frontier. Intriguingly, rather than comprising a defined set of RNAs, the cellular repertoire of immunogenic dsRNAs appears to be dynamic and vary between tissues, developmental stages, and disease contexts. This dynamic nature underscores the importance of studying immunogenic dsRNAs in physiologically relevant model systems that accurately reproduce in vivo complexity.
The surge in interest in ADAR1 biology promises a wealth of new insights in the coming years. We envision that the greatest advances will arise from the synthesis of continued fundamental studies with new ventures into clinical applications, together paving the way for ADAR1-based therapies in cancer and beyond.
COMPETING INTEREST STATEMENT
J.B.L. is a cofounder of AIRNA and a consultant for Risen Pharma. I.J. is an employee of AIRNA Corporation.
ACKNOWLEDGMENTS
Work in the Li Lab is funded in part by National Institutes of Health grants R35GM144100, R01 GM102484, R01 GM124215, R01 MH115080, and R01AI182437. Figures were created with BioRender.com.
Footnotes
Article is online at http://www.rnajournal.org/cgi/doi/10.1261/rna.079953.124.
Freely available online through the RNA Open Access option.
REFERENCES
- Ahmad S, Mu X, Yang F, Greenwald E, Park JW, Jacob E, Zhang CZ, Hur S. 2018. Breaching self-tolerance to Alu duplex RNA underlies MDA5-mediated inflammation. Cell 172: 797–810.e13. 10.1016/j.cell.2017.12.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrejeva J, Childs KS, Young DF, Carlos TS, Stock N, Goodbourn S, Randall RE. 2004. The V proteins of paramyxoviruses bind the IFN-inducible RNA helicase, mda-5, and inhibit its activation of the IFN-β promoter. Proc Natl Acad Sci 101: 17264–17269. 10.1073/pnas.0407639101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari S, Schlapbach LJ, Anchisi S, Hammer C, Bartha I, Junier T, Mottet-Osman G, Posfay-Barbe KM, Longchamp D, Stocker M, et al. 2017. Severe viral respiratory infections in children with IFIH1 loss-of-function mutations. Proc Natl Acad Sci 114: 8342. 10.1073/pnas.1704259114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bass BL, Weintraub H. 1988. An unwinding activity that covalently modifies its double-stranded RNA substrate. Cell 55: 1089–1098. 10.1016/0092-8674(88)90253-X [DOI] [PubMed] [Google Scholar]
- Bhate A, Sun T, Li JB. 2019. ADAR1: a new target for immuno-oncology therapy. Mol Cell 73: 866–868. 10.1016/j.molcel.2019.02.021 [DOI] [PubMed] [Google Scholar]
- Bowling EA, Wang JH, Gong F, Wu W, Neill NJ, Kim IS, Tyagi S, Orellana M, Kurley SJ, Dominguez-Vidaña R, et al. 2021. Spliceosome-targeted therapies trigger an antiviral immune response in triple-negative breast cancer. Cell 184: 384–403.e21. 10.1016/j.cell.2020.12.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brocks D, Schmidt CR, Daskalakis M, Jang HS, Shah NM, Li D, Li J, Zhang B, Hou Y, Laudato S, et al. 2017. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet 49: 1052–1060. 10.1038/ng.3889 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown BA, Lowenhaupt K, Wilbert CM, Hanlon EB, Rich A. 2000. The Zα domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc Natl Acad Sci 97: 13532–13536. 10.1073/pnas.240464097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budu-Aggrey A, Bowes J, Stuart PE, Zawistowski M, Tsoi LC, Nair R, Jadon DR, McHugh N, Korendowych E, Elder JT, et al. 2017. A rare coding allele in IFIH1 is protective for psoriatic arthritis. Ann Rheum Dis 76: 1321. 10.1136/annrheumdis-2016-210592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadena C, Hur S. 2019. Filament-like assemblies of intracellular nucleic acid sensors: commonalities and differences. Mol Cell 76: 243–254. 10.1016/j.molcel.2019.09.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cananzi M, Wohler E, Marzollo A, Colavito D, You J, Jing H, Bresolin S, Gaio P, Martin R, Mescoli C, et al. 2021. IFIH1 loss-of-function variants contribute to very early-onset inflammatory bowel disease. Hum Genet 140: 1299–1312. 10.1007/s00439-021-02300-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Cordova AF, Li L. 2022. Therapeutic interventions targeting innate immune receptors: a balancing act. Chem Rev 122: 3414–3458. 10.1021/acs.chemrev.1c00716 [DOI] [PubMed] [Google Scholar]
- Chalk AM, Taylor S, Heraud-Farlow JE, Walkley CR. 2019. The majority of A-to-I RNA editing is not required for mammalian homeostasis. Genome Biol 20: 268. 10.1186/s13059-019-1873-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chelmicki T, Roger E, Teissandier A, Dura M, Bonneville L, Rucli S, Dossin F, Fouassier C, Lameiras S, Bourc'his D. 2021. m6A RNA methylation regulates the fate of endogenous retroviruses. Nature 591: 312–316. 10.1038/s41586-020-03135-1 [DOI] [PubMed] [Google Scholar]
- Chen YG, Hur S. 2022. Cellular origins of dsRNA, their recognition and consequences. Nat Rev Mol Cell Biol 23: 286–301. 10.1038/s41580-021-00430-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen J, Jing H, Martin-Nalda A, Bastard P, Rivière JG, Liu Z, Colobran R, Lee D, Tung W, Manry J, et al. 2021a. Inborn errors of TLR3- or MDA5-dependent type I IFN immunity in children with enterovirus rhombencephalitis. J Exp Med 218: e20211349. 10.1084/jem.20211349 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen R, Ishak CA, De Carvalho DD. 2021b. Endogenous retroelements and the viral mimicry response in cancer therapy and cellular homeostasis. Cancer Discov 11: 2707–2725. 10.1158/2159-8290.CD-21-0506 [DOI] [PubMed] [Google Scholar]
- Chiappinelli KB, Strissel PL, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote NS, Cope LM, Snyder A, et al. 2015. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162: 974–986. 10.1016/j.cell.2015.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H, Calis JJA, Wu X, Sun T, Yu Y, Sarbanes SL, Dao Thi VL, Shilvock AR, Hoffmann HH, Rosenberg BR, et al. 2018. Human ADAR1 prevents endogenous RNA from triggering translational shutdown. Cell 172: 811–824.e14. 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbet GA, Burke JM, Parker R. 2021. ADAR1 limits stress granule formation through both translation-dependent and translation-independent mechanisms. J Cell Sci 134: jcs258783. 10.1242/jcs.258783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Cruz PH, Kato Y, Nakahama T, Shibuya T, Kawahara Y. 2020. A comparative analysis of ADAR mutant mice reveals site-specific regulation of RNA editing. RNA 26: 454–469. 10.1261/rna.072728.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell KA, Andrews RJ, Bass BL. 2024. The competitive landscape of the dsRNA world. Mol Cell 84: 107–119. 10.1016/j.molcel.2023.11.033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Manel N. 2015. Aicardi–Goutières syndrome and the type I interferonopathies. Nat Rev Immunol 15: 429–440. 10.1038/nri3850 [DOI] [PubMed] [Google Scholar]
- Crow YJ, Stetson DB. 2022. The type I interferonopathies: 10 years on. Nat Rev Immunol 22: 471–483. 10.1038/s41577-021-00633-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow YJ, Chase DS, Lowenstein Schmidt J, Szynkiewicz M, Forte GMA, Gornall HL, Oojageer A, Anderson B, Pizzino A, Helman G, et al. 2015. Characterization of human disease phenotypes associated with mutations in TREX1, RNASEH2A, RNASEH2B, RNASEH2C, SAMHD1, ADAR, and IFIH1. Am J Med Genet A 167: 296–312. 10.1002/ajmg.a.36887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar TL, Herzner A-M, Zhang X, Goyal Y, Watanabe C, Friedman BA, Janakiraman V, Durinck S, Stinson J, Arnott D, et al. 2017. Silencing of retrotransposons by SETDB1 inhibits the interferon response in acute myeloid leukemia. J Cell Biol 216: 3535–3549. 10.1083/jcb.201612160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dand N, Mucha S, Tsoi LC, Mahil SK, Stuart PE, Arnold A, Baurecht H, Burden AD, Callis Duffin K, Chandran V, et al. 2017. Exome-wide association study reveals novel psoriasis susceptibility locus at TNFSF15 and rare protective alleles in genes contributing to type I IFN signalling. Hum Mol Genet 26: 4301–4313. 10.1093/hmg/ddx328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Reuver R, Maelfait J. 2023. Novel insights into double-stranded RNA-mediated immunopathology. Nat Rev Immunol 10.1038/s41577-023-00940-3 [DOI] [PubMed] [Google Scholar]
- de Reuver R, Dierick E, Wiernicki B, Staes K, Seys L, De Meester E, Muyldermans T, Botzki A, Lambrecht BN, Van Nieuwerburgh F, et al. 2021. ADAR1 interaction with Z-RNA promotes editing of endogenous double-stranded RNA and prevents MDA5-dependent immune activation. Cell Rep 36: 109500. 10.1016/j.celrep.2021.109500 [DOI] [PubMed] [Google Scholar]
- de Reuver R, Verdonck S, Dierick E, Nemegeer J, Hessmann E, Ahmad S, Jans M, Blancke G, Van Nieuwerburgh F, Botzki A, et al. 2022. ADAR1 prevents autoinflammation by suppressing spontaneous ZBP1 activation. Nature 607: 784–789. 10.1038/s41586-022-04974-w [DOI] [PubMed] [Google Scholar]
- Dorrity TJ, Shin H, Wiegand KA, Aruda J, Closser M, Jung E, Gertie JA, Leone A, Polfer R, Culbertson B, et al. 2023. Long 3′UTRs predispose neurons to inflammation by promoting immunostimulatory double-stranded RNA formation. Sci Immunol 8: eadg2979. 10.1126/sciimmunol.adg2979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emdin CA, Khera AV, Chaffin M, Klarin D, Natarajan P, Aragam K, Haas M, Bick A, Zekavat SM, Nomura A, et al. 2018. Analysis of predicted loss-of-function variants in UK Biobank identifies variants protective for disease. Nat Commun 9: 1613. 10.1038/s41467-018-03911-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faghihi MA, Wahlestedt C. 2009. Regulatory roles of natural antisense transcripts. Nat Rev Mol Cell Biol 10: 637–643. 10.1038/nrm2738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon HS, Zou T, Kiessling MK, Gao GF, Cai D, Choi PS, Ivan AP, Buchumenski I, Berger AC, Goldstein JT, et al. 2018. Identification of ADAR1 adenosine deaminase dependency in a subset of cancer cells. Nat Commun 9: 5450. 10.1038/s41467-018-07824-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- George CX, Samuel CE. 1999. Human RNA-specific adenosine deaminase ADAR1 transcripts possess alternative exon 1 structures that initiate from different promoters, one constitutively active and the other interferon inducible. Proc Natl Acad Sci 96: 4621–4626. 10.1073/pnas.96.8.4621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartner JC, Schmittwolf C, Kispert A, Müller AM, Higuchi M, Seeburg PH. 2004. Liver disintegration in the mouse embryo caused by deficiency in the RNA-editing enzyme ADAR1. J Biol Chem 279: 4894–4902. 10.1074/jbc.M311347200 [DOI] [PubMed] [Google Scholar]
- Hartner JC, Walkley CR, Lu J, Orkin SH. 2009. ADAR1 is essential for the maintenance of hematopoiesis and suppression of interferon signaling. Nat Immunol 10: 109–115. 10.1038/ni.1680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Suzuki T. 2013. Dyschromatosis symmetrica hereditaria. J Dermatol 40: 336–343. 10.1111/j.1346-8138.2012.01661.x [DOI] [PubMed] [Google Scholar]
- Heraud-Farlow JE, Chalk AM, Linder SE, Li Q, Taylor S, White JM, Pang L, Liddicoat BJ, Gupte A, Li JB, et al. 2017. Protein recoding by ADAR1-mediated RNA editing is not essential for normal development and homeostasis. Genome Biol 18: 166. 10.1186/s13059-017-1301-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herbert A, Alfken J, Kim Y-G, Mian IS, Nishikura K, Rich A. 1997. A Z-DNA binding domain present in the human editing enzyme, double-stranded RNA adenosine deaminase. Proc Natl Acad Sci 94: 8421–8426. 10.1073/pnas.94.16.8421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higuchi M, Maas S, Single FN, Hartner J, Rozov A, Burnashev N, Feldmeyer D, Sprengel R, Seeburg PH. 2000. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature 406: 78–81. 10.1038/35017558 [DOI] [PubMed] [Google Scholar]
- Hu S-B, Heraud-Farlow J, Sun T, Liang Z, Goradia A, Taylor S, Walkley CR, Li JB. 2023. ADAR1p150 prevents MDA5 and PKR activation via distinct mechanisms to avert fatal autoinflammation. Mol Cell 83: 3869–3884.e7. 10.1016/j.molcel.2023.09.018 [DOI] [PubMed] [Google Scholar]
- Hubbard NW, Ames JM, Maurano M, Chu LH, Somfleth KY, Gokhale NS, Werner M, Snyder JM, Lichauco K, Savan R, et al. 2022. ADAR1 mutation causes ZBP1-dependent immunopathology. Nature 607: 769–775. 10.1038/s41586-022-04896-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hull CM, Bevilacqua PC. 2016. Discriminating self and non-self by RNA: roles for RNA structure, misfolding, and modification in regulating the innate immune sensor PKR. Acc Chem Res 49: 1242–1249. 10.1021/acs.accounts.6b00151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hur S. 2019. Double-stranded RNA sensors and modulators in innate immunity. Annu Rev Immunol 37: 349–375. 10.1146/annurev-immunol-042718-041356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishizuka JJ, Manguso RT, Cheruiyot CK, Bi K, Panda A, Iracheta-Vellve A, Miller BC, Du PP, Yates KB, Dubrot J, et al. 2019. Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565: 43–48. 10.1038/s41586-018-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao H, Wachsmuth L, Wolf S, Lohmann J, Nagata M, Kaya GG, Oikonomou N, Kondylis V, Rogg M, Diebold M, et al. 2022. ADAR1 averts fatal type I interferon induction by ZBP1. Nature 607: 776–783. 10.1038/s41586-022-04878-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Y, Andersen GHL, Santorico SA, Spritz RA. 2017. Multiple functional variants of IFIH1, a gene involved in triggering innate immune responses, protect against vitiligo. J Invest Dermatol 137: 522–524. 10.1016/j.jid.2016.09.021 [DOI] [PubMed] [Google Scholar]
- Karki R, Sundaram B, Sharma BR, Lee S, Malireddi RKS, Nguyen LN, Christgen S, Zheng M, Wang Y, Samir P, et al. 2021. ADAR1 restricts ZBP1-mediated immune response and PANoptosis to promote tumorigenesis. Cell Rep 37: 109858. 10.1016/j.celrep.2021.109858 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karki A, Campbell KB, Mozumder S, Fisher AJ, Beal PA. 2024. Impact of disease-associated mutations on the deaminase activity of ADAR1. Biochemistry 63: 282–293. 10.1021/acs.biochem.3c00405 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasumba DM, Grandvaux N. 2019. Therapeutic targeting of RIG-I and MDA5 might not lead to the same rome. Trends Pharmacol Sci 40: 116–127. 10.1016/j.tips.2018.12.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y, Park J, Kim S, Kim M, Kang M-G, Kwak C, Kang M, Kim B, Rhee H-W, Kim VN. 2018. PKR senses nuclear and mitochondrial signals by interacting with endogenous double-stranded RNAs. Mol Cell 71: 1051–1063.e6. 10.1016/j.molcel.2018.07.029 [DOI] [PubMed] [Google Scholar]
- Kim JI, Nakahama T, Yamasaki R, Costa Cruz PH, Vongpipatana T, Inoue M, Kanou N, Xing Y, Todo H, Shibuya T, et al. 2021. RNA editing at a limited number of sites is sufficient to prevent MDA5 activation in the mouse brain. PLoS Genet 17: e1009516. 10.1371/journal.pgen.1009516 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleinova R, Rajendra V, Leuchtenberger AF, Lo Giudice C, Vesely C, Kapoor U, Tanzer A, Derdak S, Picardi E, Jantsch MF. 2023. The ADAR1 editome reveals drivers of editing-specificity for ADAR1-isoforms. Nucleic Acids Res 51: 4191–4207. 10.1093/nar/gkad265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knebel UE, Peleg S, Dai C, Cohen-Fultheim R, Jonsson S, Poznyak K, Israeli M, Zamashanski L, Glaser B, Levanon EY, et al. 2024. Disrupted RNA editing in beta cells mimics early-stage type 1 diabetes. Cell Metab 36: 48–61.e6. 10.1016/j.cmet.2023.11.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeris M, Funke L, Shrestha J, Rich A, Maas S. 2005. Modulation of ADAR1 editing activity by Z-RNA in vitro. Nucleic Acids Res 33: 5362–5370. 10.1093/nar/gki849 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamborn IT, Jing H, Zhang Y, Drutman SB, Abbott JK, Munir S, Bade S, Murdock HM, Santos CP, Brock LG, et al. 2017. Recurrent rhinovirus infections in a child with inherited MDA5 deficiency. J Exp Med 214: 1949–1972. 10.1084/jem.20161759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levanon EY, Cohen-Fultheim R, Eisenberg E. 2023. In search of critical dsRNA targets of ADAR1. Trends Genet 10.1016/j.tig.2023.12.002 [DOI] [PubMed] [Google Scholar]
- Li Y, Liao W, Cargill M, Chang M, Matsunami N, Feng B-J, Poon A, Callis-Duffin KP, Catanese JJ, Bowcock AM, et al. 2010. Carriers of rare missense variants in IFIH1 are protected from psoriasis. J Invest Dermatol 130: 2768–2772. 10.1038/jid.2010.214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Banerjee S, Goldstein SA, Dong B, Gaughan C, Rath S, Donovan J, Korennykh A, Silverman RH, Weiss SR. 2017. Ribonuclease L mediates the cell-lethal phenotype of double-stranded RNA editing enzyme ADAR1 deficiency in a human cell line. eLife 6: e25687. 10.7554/eLife.25687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Gloudemans MJ, Geisinger JM, Fan B, Aguet F, Sun T, Ramaswami G, Li YI, Ma JB, Pritchard JK, et al. 2022. RNA editing underlies genetic risk of common inflammatory diseases. Nature 608: 569–577. 10.1038/s41586-022-05052-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z, Chalk AM, Taylor S, Goradia A, Heraud-Farlow JE, Walkley CR. 2023. The phenotype of the most common human ADAR1p150 Zα mutation P193A in mice is partially penetrant. EMBO Rep 24: e55835. 10.15252/embr.202255835 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Piskol R, Chalk AM, Ramaswami G, Higuchi M, Hartner JC, Li JB, Seeburg PH, Walkley CR. 2015. RNA editing by ADAR1 prevents MDA5 sensing of endogenous dsRNA as nonself. Science 349: 1115–1120. 10.1126/science.aac7049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liddicoat BJ, Hartner JC, Piskol R, Ramaswami G, Chalk AM, Kingsley PD, Sankaran VG, Wall M, Purton LE, Seeburg PH, et al. 2016. Adenosine-to-inosine RNA editing by ADAR1 is essential for normal murine erythropoiesis. Exp Hematol 44: 947–963. 10.1016/j.exphem.2016.06.250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H, Golji J, Brodeur LK, Chung FS, Chen JT, deBeaumont RS, Bullock CP, Jones MD, Kerr G, Li L, et al. 2019. Tumor-derived IFN triggers chronic pathway agonism and sensitivity to ADAR loss. Nat Med 25: 95–102. 10.1038/s41591-018-0302-5 [DOI] [PubMed] [Google Scholar]
- Liu J, Dou X, Chen C, Chen C, Liu C, Xu MM, Zhao S, Shen B, Gao Y, Han D, et al. 2020. N6-methyladenosine of chromosome-associated regulatory RNA regulates chromatin state and transcription. Science 367: 580–586. 10.1126/science.aay6018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Gao M, He J, Wu K, Lin S, Jin L, Chen Y, Liu H, Shi J, Wang X, et al. 2021. The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature 591: 322–326. 10.1038/s41586-021-03313-9 [DOI] [PubMed] [Google Scholar]
- Manguso RT, Pope HW, Zimmer MD, Brown FD, Yates KB, Miller BC, Collins NB, Bi K, LaFleur MW, Juneja VR, et al. 2017. In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547: 413–418. 10.1038/nature23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mannion NM, Greenwood SM, Young R, Cox S, Brindle J, Read D, Nellåker C, Vesely C, Ponting CP, McLaughlin PJ, et al. 2014. The RNA-editing enzyme ADAR1 controls innate immune responses to RNA. Cell Rep 9: 1482–1494. 10.1016/j.celrep.2014.10.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matthews MM, Thomas JM, Zheng Y, Tran K, Phelps KJ, Scott AI, Havel J, Fisher AJ, Beal PA. 2016. Structures of human ADAR2 bound to dsRNA reveal base-flipping mechanism and basis for site selectivity. Nat Struct Mol Biol 23: 426–433. 10.1038/nsmb.3203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maurano M, Snyder JM, Connelly C, Henao-Mejia J, Sidrauski C, Stetson DB. 2021. Protein kinase R and the integrated stress response drive immunopathology caused by mutations in the RNA deaminase ADAR1. Immunity 54: 1948–1960.e5. 10.1016/j.immuni.2021.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehdipour P, Marhon SA, Ettayebi I, Chakravarthy A, Hosseini A, Wang Y, de Castro FA, Loo Yau H, Ishak C, Abelson S, et al. 2020. Epigenetic therapy induces transcription of inverted SINEs and ADAR1 dependency. Nature 588: 169–173. 10.1038/s41586-020-2844-1 [DOI] [PubMed] [Google Scholar]
- Nakahama T, Kato Y, Shibuya T, Inoue M, Kim JI, Vongpipatana T, Todo H, Xing Y, Kawahara Y. 2021. Mutations in the adenosine deaminase ADAR1 that prevent endogenous Z-RNA binding induce Aicardi-Goutières-syndrome-like encephalopathy. Immunity 54: 1976–1988.e7. 10.1016/j.immuni.2021.08.022 [DOI] [PubMed] [Google Scholar]
- Nejentsev S, Walker N, Riches D, Egholm M, Todd JA. 2009. Rare variants of IFIH1, a gene implicated in antiviral responses, protect against type 1 diabetes. Science 324: 387–389. 10.1126/science.1167728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishikura K. 2016. A-to-I editing of coding and non-coding RNAs by ADARs. Nat Rev Mol Cell Biol 17: 83–96. 10.1038/nrm.2015.4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oakes E, Anderson A, Cohen-Gadol A, Hundley HA. 2017. Adenosine deaminase that acts on RNA 3 (ADAR3) binding to glutamate receptor subunit B pre-mRNA inhibits RNA editing in glioblastoma. J Biol Chem 292: 4326–4335. 10.1074/jbc.M117.779868 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda H, Nakagawa K, Abe J, Awaya T, Funabiki M, Hijikata A, Nishikomori R, Funatsuka M, Ohshima Y, Sugawara Y, et al. 2014. Aicardi-Goutières syndrome is caused by IFIH1 mutations. Am J Hum Genet 95: 121–125. 10.1016/j.ajhg.2014.06.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peisley A, Jo MH, Lin C, Wu B, Orme-Johnson M, Walz T, Hohng S, Hur S. 2012. Kinetic mechanism for viral dsRNA length discrimination by MDA5 filaments. Proc Natl Acad Sci 109: E3340–E3349. 10.1073/pnas.1208618109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pestal K, Funk CC, Snyder JM, Price ND, Treuting PM, Stetson DB. 2015. Isoforms of RNA-editing enzyme ADAR1 independently control nucleic acid sensor MDA5-driven autoimmunity and multi-organ development. Immunity 43: 933–944. 10.1016/j.immuni.2015.11.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Placido D, Brown BA, Lowenhaupt K, Rich A, Athanasiadis A. 2007. A left-handed RNA double helix bound by the Zα domain of the RNA-editing enzyme ADAR1. Structure 15: 395–404. 10.1016/j.str.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porath HT, Carmi S, Levanon EY. 2014. A genome-wide map of hyper-edited RNA reveals numerous new sites. Nat Commun 5: 4726. 10.1038/ncomms5726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quin J, Sedmík J, Vukić D, Khan A, Keegan LP, O'Connell MA. 2021. ADAR RNA modifications, the epitranscriptome and innate immunity. Trends Biochem Sci 46: 758–771. 10.1016/j.tibs.2021.02.002 [DOI] [PubMed] [Google Scholar]
- Reich DP, Bass BL. 2019. Mapping the dsRNA world. Cold Spring Harb Perspect Biol 11: a035352. 10.1101/cshperspect.a035352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reisländer T, Groelly FJ, Tarsounas M. 2020. DNA damage and cancer immunotherapy: a STING in the tale. Mol Cell 80: 21–28. 10.1016/j.molcel.2020.07.026 [DOI] [PubMed] [Google Scholar]
- Rice GI, Kasher PR, Forte GM, Mannion NM, Greenwood SM, Szynkiewicz M, Dickerson JE, Bhaskar SS, Zampini M, Briggs TA, et al. 2012. Mutations in ADAR1 cause Aicardi-Goutieres syndrome associated with a type I interferon signature. Nat Genet 44: 1243–1248. 10.1038/ng.2414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, del Toro Duany Y, Jenkinson EM, Forte GMA, Anderson BH, Ariaudo G, Bader-Meunier B, Baildam EM, Battini R, Beresford MW, et al. 2014. Gain-of-function mutations in IFIH1 cause a spectrum of human disease phenotypes associated with upregulated type I interferon signaling. Nat Genet 46: 503–509. 10.1038/ng.2933 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice GI, Park S, Gavazzi F, Adang LA, Ayuk LA, Van Eyck L, Seabra L, Barrea C, Battini R, Belot A, et al. 2020. Genetic and phenotypic spectrum associated with IFIH1 gain-of-function. Hum Mutat 41: 837–849. 10.1002/humu.23975 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen SY, Han H, Liang G, Jones PA, Pugh TJ, et al. 2015. DNA-demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162: 961–973. 10.1016/j.cell.2015.07.056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz T, Rould MA, Lowenhaupt K, Herbert A, Rich A. 1999. Crystal structure of the Zα domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science 284: 1841–1845. 10.1126/science.284.5421.1841 [DOI] [PubMed] [Google Scholar]
- Serra MJ, Smolter PE, Westhof E. 2004. Pronounced instability of tandem IU base pairs in RNA. Nucleic Acids Res 32: 1824–1828. 10.1093/nar/gkh501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng W, LaFleur MW, Nguyen TH, Chen S, Chakravarthy A, Conway JR, Li Y, Chen H, Yang H, Hsu P-H, et al. 2018. LSD1 ablation stimulates anti-tumor immunity and enables checkpoint blockade. Cell 174: 549–563.e19. 10.1016/j.cell.2018.05.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigemoto T, Kageyama M, Hirai R, Zheng J, Yoneyama M, Fujita T. 2009. Identification of loss of function mutations in human genes encoding RIG-I and MDA5: implications for resistance to type I diabetes. J Biol Chem 284: 13348–13354. 10.1074/jbc.M809449200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinigaglia K, Cherian A, Vukic D, Melicherova J, Linhartova P, Du Q, Zerad L, Stejskal S, Malik R, Prochazka J, et al. 2023. Aberrant activation of the innate immune sensor PKR by self dsRNA is prevented by direct interaction with ADAR1. bioRxiv 10.1101/2023.08.29.555105 [DOI]
- Sommer B, Köhler M, Sprengel R, Seeburg PH. 1991. RNA editing in brain controls a determinant of ion flow in glutamate-gated channels. Cell 67: 11–19. 10.1016/0092-8674(91)90568-J [DOI] [PubMed] [Google Scholar]
- Strobel SA, Cech TR, Usman N, Beigelman L. 1994. The 2,6-diaminopurine riboside.5-methylisocytidine wobble base pair: an isoenergetic substitution for the study of G.U pairs in RNA. Biochemistry 33: 13824–13835. 10.1021/bi00250a037 [DOI] [PubMed] [Google Scholar]
- Sun T, Yu Y, Wu X, Acevedo A, Luo J-D, Wang J, Schneider WM, Hurwitz B, Rosenberg BR, Chung H, et al. 2021. Decoupling expression and editing preferences of ADAR1 p150 and p110 isoforms. Proc Natl Acad Sci 118: e2021757118. 10.1073/pnas.2021757118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun T, Li Q, Geisinger JM, Hu S-B, Fan B, Su S, Tsui W, Guo H, Ma J, Li JB. 2022. A small subset of cytosolic dsRNAs must be edited by ADAR1 to evade MDA5-mediated autoimmunity. bioRxiv 10.1101/2022.08.29.505707 [DOI]
- Tan MH, Li Q, Shanmugam R, Piskol R, Kohler J, Young AN, Liu KI, Zhang R, Ramaswami G, Ariyoshi K, et al. 2017. Dynamic landscape and regulation of RNA editing in mammals. Nature 550: 249–254. 10.1038/nature24041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Q, Rigby RE, Young GR, Hvidt AK, Davis T, Tan TK, Bridgeman A, Townsend AR, Kassiotis G, Rehwinkel J. 2021. Adenosine-to-inosine editing of endogenous Z-form RNA by the deaminase ADAR1 prevents spontaneous MAVS-dependent type I interferon responses. Immunity 54: 1961–1975.e5. 10.1016/j.immuni.2021.08.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toth AM, Li Z, Cattaneo R, Samuel CE. 2009. RNA-specific adenosine deaminase ADAR1 suppresses measles virus-induced apoptosis and activation of protein kinase PKR. J Biol Chem 284: 29350–29356. 10.1074/jbc.M109.045146 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Miyakoda M, Yang W, Khillan J, Stachura DL, Weiss MJ, Nishikura K. 2004. Stress-induced apoptosis associated with null mutation of ADAR1 RNA editing deaminase gene. J Biol Chem 279: 4952–4961. 10.1074/jbc.M310162200 [DOI] [PubMed] [Google Scholar]
- Ward SV, George CX, Welch MJ, Liou L-Y, Hahm B, Lewicki H, de la Torre JC, Samuel CE, Oldstone MB. 2011. RNA editing enzyme adenosine deaminase is a restriction factor for controlling measles virus replication that also is required for embryogenesis. Proc Natl Acad Sci 108: 331–336. 10.1073/pnas.1017241108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DJ, Rice JL, Yanker DM, Znosko BM. 2007. Nearest neighbor parameters for inosine·uridine pairs in RNA duplexes. Biochemistry 46: 4625–4634. 10.1021/bi0616910 [DOI] [PubMed] [Google Scholar]
- Wu B, Hur S. 2015. How RIG-I like receptors activate MAVS. Curr Opin Virol 12: 91–98. 10.1016/j.coviro.2015.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneyama M, Kikuchi M, Natsukawa T, Shinobu N, Imaizumi T, Miyagishi M, Taira K, Akira S, Fujita T. 2004. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat Immunol 5: 730–737. 10.1038/ni1087 [DOI] [PubMed] [Google Scholar]
- Yu Q, Qu K, Modis Y. 2018. Cryo-EM structures of MDA5-dsRNA filaments at different stages of ATP hydrolysis. Mol Cell 72: 999–1012.e6. 10.1016/j.molcel.2018.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Herrero del Valle A, Singh R, Modis Y. 2021. MDA5 disease variant M854K prevents ATP-dependent structural discrimination of viral and cellular RNA. Nat Commun 12: 6668. 10.1038/s41467-021-27062-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaki M, Thoenes M, Kawalia A, Nürnberg P, Kaiser R, Heller R, Bolz HJ. 2017. Recurrent and prolonged infections in a child with a homozygous IFIH1 nonsense mutation. Front Genet 8: 130. 10.3389/fgene.2017.00130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Pandey S, Travers M, Sun H, Morton G, Madzo J, Chung W, Khowsathit J, Perez-Leal O, Barrero CA, et al. 2018. Targeting CDK9 reactivates epigenetically silenced genes in cancer. Cell 175: 1244–1258.e26. 10.1016/j.cell.2018.09.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Yin C, Fedorov A, Qiao L, Bao H, Beknazarov N, Wang S, Gautam A, Williams RM, Crawford JC, et al. 2022. ADAR1 masks the cancer immunotherapeutic promise of ZBP1-driven necroptosis. Nature 606: 594–602. 10.1038/s41586-022-04753-7 [DOI] [PMC free article] [PubMed] [Google Scholar]