

ABSTRACT
The transmembrane serine protease 2 (TMPRSS2) activates the outer structural proteins of a number of respiratory viruses including influenza A virus (IAV), parainfluenza viruses, and various coronaviruses for membrane fusion. Previous studies showed that TMPRSS2 interacts with the carboxypeptidase angiotensin-converting enzyme 2 (ACE2), a cell surface protein that serves as an entry receptor for some coronaviruses. Here, by using protease activity assays, we determine that ACE2 increases the enzymatic activity of TMPRSS2 in a non-catalytic manner. Furthermore, we demonstrate that ACE2 knockdown inhibits TMPRSS2-mediated cleavage of IAV hemagglutinin (HA) in Calu-3 human airway cells and suppresses virus titers 100- to 1.000-fold. Transient expression of ACE2 in ACE2-deficient cells increased TMPRSS2-mediated HA cleavage and IAV replication. ACE2 knockdown also reduced titers of MERS-CoV and prevented S cleavage by TMPRSS2 in Calu-3 cells. By contrast, proteolytic activation and multicycle replication of IAV with multibasic HA cleavage site typically cleaved by furin were not affected by ACE2 knockdown. Co-immunoprecipitation analysis revealed that ACE2-TMPRSS2 interaction requires the enzymatic activity of TMPRSS2 and the carboxypeptidase domain of ACE2. Together, our data identify ACE2 as a new co-factor or stabilizer of TMPRSS2 activity and as a novel host cell factor involved in proteolytic activation and spread of IAV in human airway cells. Furthermore, our data indicate that ACE2 is involved in the TMPRSS2-catalyzed activation of additional respiratory viruses including MERS-CoV.
IMPORTANCE
Proteolytic cleavage of viral envelope proteins by host cell proteases is essential for the infectivity of many viruses and relevant proteases provide promising drug targets. The transmembrane serine protease 2 (TMPRSS2) has been identified as a major activating protease of several respiratory viruses, including influenza A virus. TMPRSS2 was previously shown to interact with angiotensin-converting enzyme 2 (ACE2). Here, we report the mechanistic details of this interaction. We demonstrate that ACE2 increases or stabilizes the enzymatic activity of TMPRSS2. Furthermore, we describe ACE2 involvement in TMPRSS2-catalyzed cleavage of the influenza A virus hemagglutinin and MERS-CoV spike protein in human airway cells. These findings expand our knowledge of the activation of respiratory viruses by TMPRSS2 and the host cell factors involved. In addition, our results could help to elucidate a physiological role for TMPRSS2.
KEYWORDS: TMPRSS2, ACE2, influenza A virus, hemagglutinin cleavage, virus activation, host cell proteases
INTRODUCTION
Influenza A virus (IAV) is a global health problem that causes significant morbidity, mortality, and economic losses annually. In addition, the recurrent transmission of avian IAV to other host species provides the basis for the emergence of new viruses posing unpredictable public health threats and is likely to engender future influenza pandemics. IAVs belong to the family Orthomyxoviridae and contain a segmented, negative-sense, single-stranded RNA genome. IAVs circulate in a wide range of animal species with wild aquatic birds being the natural reservoir [reviewed in (1)].
IAV infection is initiated by the viral surface glycoprotein hemagglutinin (HA) through binding to cellular receptors and mediating the fusion of the viral lipid envelope with the endosomal membrane following endocytosis of the virus (reviewed in reference (2)). Like most viral fusion proteins, HA is synthesized as a fusion-incompetent precursor and must be cleaved by a host cell protease to acquire its fusion capacity. Proteolytic cleavage is essential for virus infectivity and spread and the proteases responsible represent potential drug targets [reviewed in reference (3)]. The HA of most IAVs including human and other mammalian IAVs as well as low pathogenic avian IAVs is cleaved at a single arginine or rarely a lysine residue designated as “monobasic cleavage site” by trypsin-like proteases (4, 5). By contrast, HA of highly pathogenic avian influenza viruses of subtypes H5 and H7 that cause fowl plague (bird flu) possesses multiple basic amino acids at the cleavage site. HA with such a “multibasic cleavage site” is cleaved by ubiquitously expressed furin and related proprotein convertases, supporting the systemic spread of infection with often fatal outcomes (6, 7).
We first identified the trypsin-like transmembrane serine protease 2 (TMPRSS2) as a protease present in the human airways that cleaves the HA of human IAV with a monobasic cleavage site (8). Subsequently, TMPRSS2 was shown to activate the fusion proteins of a number of other respiratory viruses such as human metapneumovirus, human parainfluenza viruses as well as coronaviruses (CoVs) including severe acute respiratory syndrome (SARS) CoV, Middle East respiratory syndrome (MERS) CoV, and more recently SARS-CoV-2 (9–11). Remarkably, TMPRSS2-deficient mice are protected from pathogenesis upon infection with various IAVs as well as SARS-CoV, MERS-CoV, and SARS-CoV-2 due to inhibition of the activation of progeny virus and consequently inhibition of virus spread along the respiratory tract (11–14). Moreover, we demonstrated that TMPRSS2 is the major activating protease of IAV with monobasic HA cleavage site in human airways and of influenza B virus in human lungs (15, 16). The physiological role of TMPRSS2, however, is still unclear.
In 2003, angiotensin-converting enzyme 2 (ACE2) was shown to be the functional entry receptor of SARS-CoV (17). Later, ACE2 was identified as an entry receptor for human coronavirus (HCoV) NL63 and more recently for SARS-CoV-2 (10, 18). ACE2 is a zinc metalloproteinase and carboxypeptidase that plays a central role in the renin-angiotensin system (RAS) that maintains blood pressure and fluid balance. ACE2 converts the vasoconstrictor angiotensin II into the vasodilatory heptapeptide Ang 1–7, thereby counterbalancing the action of ACE (19, 20). In addition, ACE2 hydrolyzes a number of other bioactive peptides including neurotensin, ghrelin, and apelin (19–22). Interestingly, ACE2 has also been shown to perform non-catalytic physiological functions, such as regulating the transport of neutral amino acids in the intestine by acting as a chaperone for the trafficking of the neutral amino acid transporter B0AT1 (23, 24). ACE2 is widely expressed in many different tissues with high expression in the intestinal tract, testis, kidney, heart, thyroid, and adipose tissue (25–27). It is also found in the liver and brain, albeit at typically lower levels overall (27–31). In the respiratory tract, ACE2 is highly expressed in the upper airways, while expression in the lung is low and mainly confined to type II pneumocytes (25, 32). ACE2 is a type I transmembrane protein comprised of a short cytoplasmic domain, a transmembrane domain, and a large ectodomain consisting of an N-terminal protease domain followed by a collectrin domain (Fig. 1) (22, 26, 33). It is shed from the plasma membrane as a catalytically active form after cleavage by ADAM17 (also known as TACE) within the collectrin domain (34).
Fig 1.

Schematic domain structures of ACE2 and TMPRSS2. ACE2 is a type I transmembrane metalloproteinase comprised of a short cytoplasmic domain and a large ectodomain that contains the collectrin domain (CD) and the carboxypeptidase domain (PD) carrying a zinc-binding motif (HEMGH). ACE2 can be cleaved by ADAM17 or TMPRSS2 enabling ectodomain shedding. TMPRSS2 is a type II transmembrane serine protease with a stem region comprising a low-density lipoprotein receptor class A domain (LDLRA), a scavenger receptor cysteine-rich domain (SRCR), and a serine protease domain containing the catalytic triad histidine (H), aspartic acid (D), and serine (S). TMPRSS2 undergoes autocatalytic activation by cleavage at R255 (indicated by an arrowhead). TM: transmembrane domain, SP: signal peptide. ADAM17: a disintegrin and metallopeptidase domain 17. Gray numbers indicate the amino acids.
Interestingly, previous studies demonstrated that ACE2 and TMPRSS2 interact with each other and that TMPRSS2 cleaves ACE2 within the collectrin domain (Fig. 1) (35, 36). Furthermore, ACE2 cleavage by TMPRSS2 did not enhance ACE2 shedding, rather it interfered with the cleavage and shedding of ACE2 by ADAM17 (36).
We were intrigued that ACE2 and TMPRSS2 interact and wondered whether this interaction produced an enhancement of TMPRSS2-mediated events. In the present study, we further characterized ACE2-TMPRSS2 interaction by enzyme kinetic measurements and co-immunoprecipitation analyses. Furthermore, we developed peptide-conjugated phosphorodiamidate morpholino oligomers (PPMO) to knockdown ACE2 expression in Calu-3 human airway cells and to investigate IAV replication and HA cleavage in the absence and presence of ACE2 expression. We demonstrate that ACE2 acts as a co-factor or stabilizer of TMPRSS2 activity in a non-catalytic manner and is involved in TMPRSS2-mediated IAV HA cleavage in Calu-3 human airway cells. Our data also suggest that ACE2 is involved in TMPRSS2-mediated activation of certain CoVs, exemplified here for MERS-CoV.
RESULTS
ACE2 increases the enzymatic activity of recombinant TMPRSS2
One possible mechanism of how TMPRSS2 may benefit from interaction with ACE2 might be via affecting or regulating its enzymatic activity. To examine whether ACE2 has an effect on enzymatic TMPRSS2 activity, we performed protease activity assays using recombinant TMPRSS2 and ACE2 proteins. Recombinant soluble TMPRSS2 comprising the LDLRA domain, the SRCR domain, and the catalytic domain (amino acids 109–492; see Fig. 1) was mixed with recombinant soluble ACE2 containing the peptidase domain and collectrin domain (amino acids 18–740, see Fig. 1) and incubated with a fluorogenic peptide substrate to measure TMPRSS2 activity. The substrate was efficiently cleaved by TMPRSS2, whereas ACE2 was unable to hydrolyze the substrate, as expected (Fig. 2A). Interestingly, ACE2 markedly increased the enzymatic activity of TMPRSS2. Inhibition of ACE2 carboxypeptidase activity by the peptide inhibitor DX600 (37) had no effect on ACE2-mediated enhancement of TMPRSS2 activity. The data show that ACE2 markedly increases the enzymatic activity of TMPRSS2 independent of its carboxypeptidase activity. In addition, we determined TMPRSS2 activity in the presence or absence of ACE2 at 37°C. In the absence of ACE2, TMPRSS2 initially hydrolyzed the substrate efficiently, but the activity decreased considerably over time, suggesting that TMPRSS2 was not stable under these conditions (Fig. 2B). However, in the presence of ACE2, efficient and constant hydrolysis of the substrate by TMPRSS2 was observed and was not inhibited by DX600. Thus, the data indicate that ACE2 may stabilize the proteolytically active form of TMPRSS2.
Fig 2.

ACE2 increases the enzymatic activity of recombinant TMPRSS2 in a non-catalytic manner. (A and B) Kinetic measurement of TMPRSS2 activity in the absence and presence of ACE2. Recombinant human TMPRSS2 (0.12 nM) was mixed with recombinant human ACE2 (0.8 µU) prior to the addition of fluorogenic TMPRSS2 substrate Boc-Gln-Ala-Arg-AMC. Fluorescence was measured over 1 h at room temperature (RT) (A) or at 37°C (B). When indicated, ACE2 carboxypeptidase activity was inhibited by the addition of DX600 (10 µM). (C) ACE2 activity in the presence of TMPRSS2. Recombinant ACE2 (0.8 µU) was mixed with recombinant TMPRSS2 (0.12 nM) in the absence or presence of DX600 (10 µM) prior to the addition of a quenched fluorogenic ACE2 substrate. The cleavage rate of the substrate was measured over 1h. RFU: relative fluorescence units. All data are mean values ± standard deviations (SD) of two independent experiments performed in duplicate or triplicate.
We further examined whether vice versa TMPRSS2 affects the enzymatic activity of ACE2. Recombinant TMPRSS2 and ACE2 were mixed and incubated with a fluorogenic peptide that is cleaved specifically by ACE2. As shown in Fig. 2C, ACE2 efficiently hydrolyzed the peptide, and ACE2 activity was completely inhibited by DX600. The addition of TMPRSS2 decreased the activity of ACE2 by approximately 10%.
Together, our data show that ACE2 acts as a regulator or stabilizer of TMPRSS2 protease activity independent of its carboxypeptidase activity.
Knockdown of ACE2 expression in Calu-3 cells using PPMO
Next, we sought to investigate whether ACE2 also has an effect on TMPRSS2 activity in human respiratory cells. For this purpose, we examined TMPRSS2-mediated HA cleavage in the presence or absence of ACE2 in Calu-3 human airway cells. Calu-3 cells provide a suitable human airway model to study respiratory virus-host interactions, and we have previously demonstrated that TMPRSS2 is the major HA-activating protease in Calu-3 cells (9, 15, 16, 38). We used PPMO to knockdown ACE2 expression. PPMOs are nucleic acid-like antisense agents composed of a morpholino oligomer covalently conjugated to a cell-penetrating peptide (39). PPMO interferes with gene expression by sterically blocking complementary sequences in target RNAs. We designed three different PPMOs to knockdown ACE2 expression in Calu-3 cells. The PPMO ACE2-UTR and ACE2-AUG were designed to interfere with ACE2 translation via blockade of the 5′ untranslated region (UTR) or the translation start-site region of the ACE2-mRNA, respectively. PPMO ACE2-e5i5 was designed to complementarily bind to the exon-intron splice site of exon 5 and intron 5 of the ACE2 pre-mRNA with the intent to remove exon 5 from the mature transcript. Translation of this affected ACE2-mRNA would likely result in a truncated, nonfunctional ACE2 protein or cause a frameshift in the final ACE2 transcript potentially introducing an earlier stop codon, resulting in nonsense-mediated decay of the transcript.
To examine the efficacy of the different PPMO to knockdown ACE2 expression in Calu-3 cells, the cells were treated with 25 µM PPMO for 24 h. Cell lysates were subjected to SDS-PAGE and western blot analysis with an ACE2-specific antibody. A PPMO with a random sequence designated as “scramble” was used as a negative control PPMO. Treatment of Calu-3 cells with all three ACE2-specific PPMO efficiently reduced ACE2 expression (Fig. 3A). Scramble PPMO treatment did not alter ACE2 expression compared to untreated cells, as expected. In addition, we evaluated whether PPMO treatment affects cell viability. As shown in Fig. 3B, Calu-3 cells treated with 25 µM of the respective ACE2-specific PPMO displayed no loss in viability (Fig. 3B). By contrast, 5% ethanol, which was used as a control to prove the functionality of the test, reduced the viability of the cells to 54%. In sum, PPMO ACE2-AUG, ACE2-UTR, and ACE2-e5i5 support efficient knockdown of ACE2 expression in Calu-3 cells without affecting cell viability.
Fig 3.

Knockdown of ACE2 suppresses cleavage of influenza A virus HA with monobasic cleavage site in Calu-3 airway cells. (A) Knockdown of ACE2 expression in Calu-3 cells using PPMO. Calu-3 cells were treated with 25 µM of ACE2-specific PPMO or a nonsense-sequence negative-control PPMO (scramble) or remained untreated (w/o) for 24 h. Cell lysates were subjected to SDS-PAGE and western blot analysis using an ACE2-specific antibody. Actin was used as a loading control. (B) Evaluation of the effect of PPMO treatment on cell viability. Calu-3 cells were treated with 25 µM of PPMO for 24 h. Cell viability of untreated cells (w/o) was set as 100%. Results are mean values + SD of two independent experiments performed in triplicate. Treatment with 5% ethanol (EtOH) was used as a control. (C) Calu-3 cells were treated with 25 µM PPMO or remained untreated for 24 h. Cells were then inoculated with H1N1pdm at a low MOI and incubated for 72 h without further PPMO treatment. Virus titers were determined by plaque assay at indicated time points. Data shown are mean values ± SD of three independent experiments. (D) Calu-3 cells treated with 25 µM PPMO for 24 h were inoculated with H1N1pdm at a MOI of 1. Cell lysates were subjected to SDS-PAGE and western blotting using ACE2- or HA-specific antibodies at 24 h p.i. (E/G) Calu-3 cells were treated with 25 µM PPMO for 24 h, then infected with H3N2 (E) or H7N7 (G) at a low MOI and incubated in the absence of PPMO for 72 h. At indicated time points, virus titers in supernatants were determined by plaque assay. (F) Calu-3 cells treated with PPMO for 24 h were infected with H3N2 at a MOI of 1. At 24 h p.i., cell lysates were analyzed by SDS-PAGE and western blotting using antibodies against ACE2 and H3. Uninfected cells (mock) served as control. (H) Lysates of H7N7 infected cells described above were subjected to SDS-PAGE and western blot analysis with ACE2- and H7-specific antibodies at 72 h p.i. (I) Calu-3 cells were infected with H1N1pdm at a low MOI, then treated with 5 µM ACE2 inhibitor DX600 or DMSO or remained untreated and incubated for 72 h. Virus replication was determined by plaque titration at indicated time points p.i. Data are mean ± SD of three independent experiments. (J) At 72 h p.i., cell lysates were analyzed for ACE2 expression and HA cleavage by SDS-PAGE and immunoblotting. DX600 concentration: 1 and 5 µM, respectively. One representative blot from three independent experiments is shown (D, F, H, J). Actin was used as a loading control.
Knockdown of ACE2 expression in Calu-3 cells suppresses multicycle replication of influenza A virus by interfering with cleavage of HA with a monobasic cleavage site
To investigate whether ACE2 affects TMPRSS2-mediated IAV activation in Calu-3 cells, we performed virus growth kinetics and HA cleavage analysis in cells with and without ACE2 knockdown. Calu-3 cells were treated with 25 µM of ACE2-AUG, ACE2-UTR, ACE2-e5i5, or control PPMO for 24 h. Cells were then inoculated with human H1N1 2009 pandemic IAV (H1N1pdm) at a low MOI and incubated in the absence of PPMO for 72 h. Virus replication was analyzed by plaque titration of cell supernatants from indicated time points. Multicycle replication of H1N1pdm IAV was markedly reduced in ACE2-AUG PPMO-treated cells with a 2 to 2.5 log reduction in virus titer (Fig. 3C). Treatment with ACE2-UTR and ACE2-e5i5 also reduced H1N1pdm virus titers, although to a less pronounced degree than ACE2-AUG treatment.
To analyze HA cleavage, Calu-3 cells were treated with 25 µM PPMO for 24 h, then inoculated with H1N1pdm at a MOI of 1 and incubated for a further 24 h. Cell lysates were subjected to SDS-PAGE and western blotting using HA-specific antibodies. HA0 was cleaved into HA1 and HA2 in scramble PPMO-treated control cells (Fig. 3D). By contrast, no cleavage of HA0 was observed in ACE2-AUG or ACE2-UTR-treated cells. HA0 cleavage was also strongly inhibited in cells treated with ACE2-e5i5 and only low amounts of HA1 were detected. Together, the data show that ACE2 knockdown interferes with HA cleavage in Calu-3 cells.
To further verify that the knockdown of ACE2 affects HA cleavage by TMPRSS2, we analyzed multicycle replication and HA cleavage of two additional viruses: (i) another human IAV (H3N2) that is activated by TMPRSS2 in Calu-3 cells and (ii) H7N7 IAV that possesses a multibasic cleavage site and is therefore activated by furin. The experiments were carried out only with the most effective PPMO, ACE2-AUG. Calu-3 cells were treated with ACE2-AUG or scramble PPMO or left without treatment for 24 h as described above. Cells were then inoculated with H3N2 at a low MOI to determine virus growth kinetics for 72 h (Fig. 3E) or at a MOI of 1 to analyze HA cleavage at 24 h p.i. (Fig. 3F; Fig. S1). Titers of H3N2 were strongly reduced (1.000-fold) in Calu-3 cells by knockdown of ACE2 similar to what was observed for H1N1pdm. Cleavage of HA0 into HA1 and HA2 (not detected by the antibody) was inhibited in ACE2-AUG-treated cells (Fig. 3F). By contrast, multicycle replication of H7N7 was not markedly affected by ACE2 knockdown (Fig. 3G) and efficient cleavage of HA0 was observed in both ACE2-AUG-treated and control cells (Fig. 3H).
Together, the data show that in Calu-3 cells knockdown of ACE2 strongly suppresses proteolytic activation and replication of IAV with monobasic HA cleavage site, whereas cleavage of HA with multibasic cleavage site is not reduced. Thus, the data indicate that ACE2 is involved in TMPRSS2-mediated HA cleavage in Calu-3 airway cells.
Inhibition of ACE2 carboxypeptidase activity does not affect influenza virus replication and HA cleavage in Calu-3 cells
Our data showed that ACE2 carboxypeptidase activity is not required for the enhancement of recombinant TMPRSS2 activity (Fig. 2A and B). Therefore, we examined whether the ACE2 carboxypeptidase activity is also dispensable for HA cleavage in Calu-3 cells. Cells were infected with H1N1pdm at a low MOI and then incubated in the absence or presence of the ACE2 inhibitor DX600 for 72 h. As shown in Fig. 3I, multicycle replication of H1N1pdm was not affected by DX600 in Calu-3 cells. Analysis of cell lysates by SDS-PAGE and western blotting at 72 h p.i. revealed that HA was cleaved in DX600-treated cells (Fig. 3J). Together, the data show that the ACE2 carboxypeptidase is not required for TMPRSS2-mediated HA cleavage in Calu-3 cells. Thus, ACE2 is involved in TMPRSS2-mediated HA cleavage in a non-catalytic manner.
Transient expression of ACE2 in Caco-2 cells increases H1N1pdm IAV titers
The knockdown of ACE2 markedly reduced the multicycle replication of H1N1pdm and H3N2 IAV in Calu-3 cells. In addition, we investigated whether the transient expression of ACE2 in ACE2-deficient cells that express TMPRSS2 conversely leads to increased IAV activation. TMPRSS2 has been shown to support HA cleavage in Caco-2 human colorectal adenocarcinoma cells (40). No ACE2 expression at the protein level could be detected in Caco-2 cells unless the cells were transfected with an ACE2-encoding plasmid (Fig. 4A, upper panel). To determine whether ACE2 enhances HA activation in Caco-2 cells, transient ACE2 expressing cells or cells transfected with empty vector were infected with H1N1pdm at a low MOI. Virus titers were analyzed at 24 and 48 h p.i. and cell lysates were subjected to SDS-PAGE and western blot to examine HA cleavage and ACE2 expression. A slight increase in HA0 cleavage into HA1 and HA2 was detected in ACE2-expressing cells compared to control cells (Fig. 4A, lower panel). However, a marked increase in virus titers (fourfold and sevenfold) at 24 and 48 h p.i., respectively, was observed in ACE2-expressing cells (Fig. 4B). The data show that ACE2 can enhance TMPRSS2-mediated virus activation and replication in Caco-2 cells and further support the observation that ACE2 can act as a regulator of TMPRSS2 activity.
Fig 4.
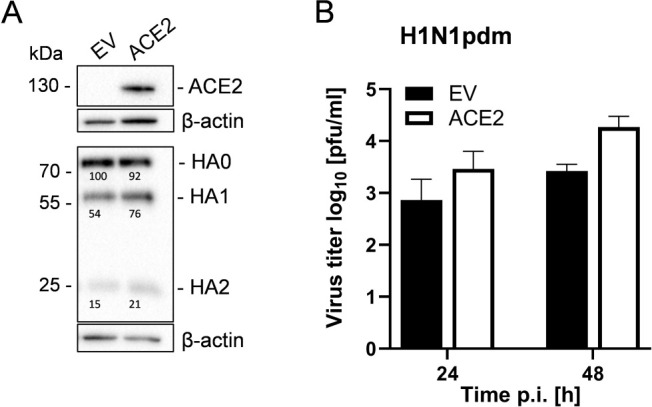
ACE2 increases H1N1pdm activation and replication in Caco-2 cells. (A) Caco-2 cells were transfected with an ACE2-encoding plasmid or empty vector (EV). At 24 h post-transfection cells were infected with H1N1pdm at an MOI of 0.001 and incubated for 72 h. Cell lysates were analyzed by SDS-PAGE and immunoblot using antibodies against HA and ACE2, respectively. Actin was used as a loading control. Numbers indicate quantification of HA with HA0 in EV-transfected cells set as 100%. (B) Virus titers in supernatants were determined by plaque assay. Data are mean values + SD of n = 3 (24 h) and n = 2 (48 h) independent experiments.
Analysis of TMPRSS2-ACE2 interaction by co-immunoprecipitation
Previous studies demonstrated that TMPRSS2 and ACE2 interact with each other and that TMPRSS2 cleaves ACE2 in the collectrin domain resulting in a 115 kDa ACE2 protein (comprising the ectodomain) and a ca. 15 kDa cytosolic domain (35, 36) (see Fig. 1). The recombinant ACE2 used here comprised the carboxypeptidase domain and the collectrin domain. To further characterize the ACE2-TMPRSS2 interaction and domains involved, we carried out co-immunoprecipitation analysis using a panel of ACE2 mutants lacking the peptidase domain (ACE2-∆PD), the collectrin domain (ACE2-∆CD) or the collectrin and transmembrane domain (ACE2-∆CD∆TM) (Fig. 5A). All ACE2 mutants were expressed with a C-terminal HA-tag to facilitate detection of un-cleaved ACE2 and the cytoplasmic domain upon ACE2 cleavage by TMPRSS2 by western blot analysis. Furthermore, we used two enzymatically inactive mutants of TMPRSS2 for the interaction studies: TMPRSS2(S441A), in which the serine residue of the catalytic triad is substituted by alanine, and a zymogen-locked mutant of TMPRSS2, TMPRSS2(R255Q), that is not able to undergo autocatalytic activation via cleavage at R255↓I256 (41). All TMPRSS2 constructs were expressed with a C-terminal 3xFLAG epitope to facilitate detection of the zymogen (70 kDa) and the catalytic domain of the autocatalytically cleaved mature form (28 kDa) by immunoblotting.
Fig 5.

Analysis of ACE2-TMPRSS2 interaction by co-immunoprecipitation. (A) Scheme of the domain structure of ACE2 mutants and TMPRSS2 mutants used in this study. Wildtype ACE2 and mutants were expressed with a C-terminal HA-tag, TMPRSS2 and mutants TMPRSS2(S441A) and TMPRSS2(R255Q) with a C-terminal 3x FLAG-tag. Autocatalytic activation of TMPRSS2 at R255 (arrowhead) and cleavage of ACE2 by TMPRSS2 (scissor) are indicated. TM: transmembrane domain, CD: collectrin domain, LDLRA: LDL receptor class A domain, SRCR: scavenger receptor cysteine-rich domain. (B) Co-immunoprecipitation analysis of TMPRSS2 and ACE2 mutants. HEK293 cells were transiently co-transfected with plasmids encoding wild-type ACE2 or the indicated mutants jointly with plasmids encoding TMPRSS2 or TMPRSS2 mutants or empty plasmid as control. At 48 h post-transfection, cells were lysed and subjected to HA-tag- and FLAG-tag-specific immunoprecipitation (IP), respectively, followed by western blot analysis of lysates (input) and precipitates using antibodies against the HA-tag or FLAG-tag. The results are representative of two or three independent experiments. Lanes are identified by gray numbers. ACE2-∆PD is indicated by a white arrowhead. (C) Summary of the capability of TMPRSS2 to interact with (Co-IP) and/or cleave (scissor) the indicated ACE2 mutant.
Co-immunoprecipitation analysis was carried out in HEK293 cells that lack endogenous expression of both TMPRSS2 and ACE2 [(42), see also www.proteinatlas.org]. Cells were transiently transfected with plasmids encoding wild-type ACE2 or mutants together with TMPRSS2 or mutants and incubated for 48 h. Cells were lysed and then lysates were divided, with one aliquot being used as input control, and the remaining lysate being used for immunoprecipitation of HA-tagged wild-type ACE2 and mutants as well as FLAG-tagged TMPRSS2 and mutants. ACE2 mutants varied in expression; however, similar expression levels were detected in cells co-expressing TMPRSS2 (for quantification see Fig. S2). Analysis of cell lysates revealed that full-length ACE2 (130 kDa) was cleaved upon co-expression of TMPRSS2 and a 15 kDa ACE2 cytosolic domain was detected (Fig. 5B, lane 7 and lane 20). By contrast, no cleavage of ACE2 was observed in cells co-expressing TMPRSS2(S441A) or TMPRSS2(R255Q) (Fig. 5B, lane 8; Fig. S3). In accordance with previous data on ACE2 cleavage by TMPRSS2 within amino acids 697 to 716, ACE2-∆CD∆TMD mutant was not cleaved upon co-expression of TMPRSS2 (Fig. 5B, lane 10), while ACE2-∆PD mutant (ca. 20 kDa, Fig. 5B, lane 3) was cleaved by TMPRSS2 (Fig. 5B, lane 9). However, ACE2-∆CD lacking the collectrin domain but containing the transmembrane domain was cleaved by TMPRSS2 upon co-expression, indicating that TMPRSS2 can also cleave ACE2 outside the collectrin domain (Fig. 5B, lane 15). Conversely, co-expression of ACE2 or any of the ACE2 mutants did not result in additional processing of TMPRSS2 or inactive mutants TMPRSS2(S441A) and TMPRSS2(R255Q) nor had an effect on autocatalytic activation of TMPRSS2 (Fig. 5B; Fig. S3). Immunoprecipitation of TMPRSS2 using anti-FLAG affinity gel resulted in co-precipitation of ACE2, whereas only little ACE2 was pulled down with inactive mutants TMPRSS2(S441A) or TMPRSS2(R255Q) (Fig. 5B, lane 8; Fig. S3). ACE2-∆CD∆TMD and ACE2-∆CD, but not ACE2-∆PD were co-precipitated with TMPRSS2 (Fig. 5B, lanes 10, 15, 9). By performing the reverse experiment immunoprecipitation of ACE2 resulted in co-precipitation of TMPRSS2, whereas only a little enzymatically inactive TMPRSS2(S441A) was pulled down with ACE2 (Fig. 5B, lane 8). Immunoprecipitation of ACE2-∆CD∆TMD and ACE2-∆CD, but not ACE2-∆PD lacking the peptidase domain resulted in co-precipitation of TMPRSS2 (lanes 10, 15, and 9). The results are summarized in Fig. 5C. Together, the data indicate that the peptidase domain of ACE2 interacts with TMPRSS2, whereas the collectrin domain and the transmembrane domain of ACE2 are dispensable for the interaction. Furthermore, the data show that the enzymatic activity of TMPRSS2 is required for interaction with ACE2. Notably, cleavage of ACE2 mutants by TMPRSS2 and interaction with TMPRSS2 was not necessarily correlated with each other since ACE2-∆PD was cleaved by TMPRSS2 but not co-precipitated, while ACE2-∆CD∆TMD was co-precipitated but not cleaved by TMPRSS2 (Fig. 5C).
We also generated an ACE2 mutant lacking the seven N-glycosylation sites of the extracellular domain (ACE2-∆Glyco7). This non-glycosylated ACE2 has been shown to accumulate in the endoplasmic reticulum (ER) instead of being localized at the plasma membrane, without impairing the enzymatic activity (43) (Fig. S4). Co-immunoprecipitation analysis showed that TMPRSS2 and ACE2-∆Glyco7 interact with each other and that ACE2-∆Glyco7 is cleaved by TMPRSS2 (Fig. 5B, lane 21). The data indicate that ACE2 and TMPRSS2 do not interact at the cell surface only but also during transport along the secretory pathway.
Knockdown of ACE2 expression suppresses multicycle replication and spike protein cleavage of MERS-CoV in Calu-3 cells
Finally, we aimed to examine whether ACE2 is involved in the TMPRSS2-catalyzed activation of another respiratory virus. We chose MERS-CoV because it is activated by TMPRSS2 but, unlike SARS-CoV and SARS-CoV-2, uses dipeptidyl peptidase 4 (DPP4) rather than ACE2 as a receptor (44, 45). The MERS-CoV spike protein S requires proteolytic cleavage at two sites, designated as S1/S2 and S2′ site, to be primed for membrane fusion. TMPRSS2 has been shown to cleave MERS-CoV S at the S2′ site in vitro and to play a crucial role in MERS-CoV replication and pathogenesis in mice in vivo (14, 45, 46). Calu-3 cells were treated with 25 µM ACE2-AUG PPMO or remained untreated for 24 h. Cells were then infected with MERS-CoV at a low MOI of 0.001 and incubated without additional PPMO treatment. At 24 and 48 h p.i., virus titers were determined by TCID50 (Fig. 6A). Interestingly, MERS-CoV titers were reduced ca. 100-fold compared to untreated control cells. To analyze S cleavage, cells were treated with ACE2-AUG or scramble PPMO or remained untreated for 24 h and were then infected with MERS-CoV as described above. At 72 h p.i., cell lysates were subjected to SDS-PAGE and immunoblotting using MERS-CoV S-specific antibodies. Reduced amounts of viral S were detected in ACE2-AUG-treated cells compared to control cells and the majority of S was non-cleaved S0 (Fig. 6B). Low amounts of S1/S2 (resulting probably from S0 cleavage by furin at the S1/S2 site) but no S2′ were detected in AUG-ACE2-treated cells, indicating that cleavage at the S2’′ site by TMPRSS2 was inhibited in the cells. The data suggest that ACE2 is also involved in TMPRSS2-catalyzed cleavage of MERS-CoV S protein. It should be mentioned that the CoV S protein is cleaved by TMPRSS2 at or close to the cell surface upon virus entry (11). Thus, our results indicate that involvement of ACE2 in TMPRSS2-mediated virus activation can occur both on the cell surface and during transport along the secretory pathway during the later stages of the virus life cycle in the cell.
Fig 6.

Knockdown of ACE2 suppresses multicycle replication of MERS-CoV and cleavage of S in Calu-3 airway cells. (A) Calu-3 cells were treated with ACE2-AUG PPMO or remained untreated (w/o) for 24 h. Cells were then inoculated with MERS-CoV at a MOI of 0.001. Virus titers in supernatants were determined by TCID50 at indicated time points post-infection. The mean + SD of three independent experiments are shown. (B) Calu-3 cells treated with ACE2-AUG or scramble PPMO or without PPMO treatment were infected with MERS-CoV as described above. At 72 h p.i., cell lysates were subjected to SDS-PAGE and western blot analysis using MERS-CoV S2-specific antibodies. Uninfected cells served as control (mock).
Taken together, our data identify ACE2 as a novel co-factor or stabilizer of TMPRSS2 enzymatic activity and as a host cell factor involved in proteolytic activation of IAV HA with monobasic cleavage site in human airway cells. Our data show that ACE2 increases the enzymatic activity of TMPRSS2 in an ACE2 activity-independent manner. Our results also suggest that ACE2 is involved in TMPRSS2-mediated activation of MERS-CoV.
DISCUSSION
During the SARS-CoV-2 pandemic, TMPRSS2 and ACE2 received considerable attention as viral entry factors. Interestingly, previous studies showed that ACE2 and TMPRSS2 interact with each other (35, 36). Here, we demonstrate that ACE2 increases the enzymatic activity of TMPRSS2 in a protease activity assay and is involved in TMPRSS2-mediated IAV HA cleavage in Calu-3 human airway cells. Transient-expressed ACE2 was also able to enhance TMPRSS2-mediated IAV activation in Caco-2 human colon carcinoma cells. Furthermore, we show that knockdown of ACE2 inhibits MERS-CoV S cleavage and reduces virus titers in Calu-3 cells, indicating that ACE2 is involved in the proteolytic activation of different respiratory viruses by TMPRSS2.
Knockdown of ACE2 expression inhibited cleavage of HA with monobasic cleavage site and strongly reduced virus titers. Cleavage of HA by TMPRSS2 takes place intracellularly prior to IAV release from the infected cell, most likely in the trans-Golgi network (TGN) or during transport to the plasma membrane (47–49). We found that the ACE2-∆Glyco7 mutant, which is not transported to the cell surface but accumulates in the ER, is cleaved by TMPRSS2 and co-precipitated with TMPRSS2, suggesting that these proteins already interact with each other within the cell. Thus, the knockdown of ACE2 in Calu-3 cells likely results in decreased TMPRSS2 activity in the TGN, reducing canonical HA cleavage (Fig. 7A and B). By contrast, proteolytic activation and multicycle replication of the H7N7 IAV were not substantially reduced by the knockdown of ACE2, suggesting that furin and related proprotein convertases do not interact with ACE2 in a manner similar to that of TMPRSS2. Furthermore, inhibition of MERS-CoV S activation and virus replication by ACE2 knockdown in Calu-3 cells suggest that ACE2-mediated regulation of TMPRSS2 enzymatic activity occurs, at least to some extent, on the cell surface (Fig. 7C).
Fig 7.

Model depicting how ACE2 expression may regulate the cleavage of IAV HA or MERS-CoV S by TMPRSS2. (A) Co-expression of ACE2 and TMPRSS2 in the trans-Golgi network (TGN) increases or stabilizes TMPRSS2 activity and supports efficient HA cleavage by TMPRSS2. (B) ACE2 knockdown results in reduced TMPRSS2 activity or stability in the TGN and thereby prevents cleavage of newly synthesized HA0 into HA1 + HA2. NA: neuraminidase. (C) ACE2 may regulate TMPRSS2 activity at the cell surface. Knockdown of ACE2 reduces the activity or stability of TMPRSS2 at the plasma membrane and inhibits proteolytic activation of MERS-CoV S upon entry.
Although the molecular mechanism underlying ACE2-mediated enhancement of TMPRSS2 activity has yet to be established, this study helps to elucidate the biochemical basis. In the enzyme kinetic measurements at 37°C, a strong decrease in TMPRSS2 activity was observed if ACE2 was absent. Presumably, TMPRSS2 was less stable under these experimental conditions. At room temperature, the marked decrease in TMPRSS2 activity was not observed. Interestingly, the activity of TMPRSS2 was increased to the same extent at both room temperature and 37°C if ACE2 was present. Thus, our data indicate that ACE2 may somehow stabilize the TMPRSS2 protein overall or its enzymatic activity in some respect. In addition, our data show that the presence of ACE2, but not its carboxypeptidase activity, is relevant for supporting enhanced TMPRSS2 activity, further indicating that ACE2 functions as a chaperone required for stabilization, trafficking, or membrane internalization of TMPRSS2. Although the best-known function of ACE2 is to counterbalance ACE by decreasing Ang II levels, it performs a number of other functions independent of the renin-angiotensin system. ACE2 is a chimeric protein: its catalytic domain is homologous with ACE and its membrane-proximal domain is homologous with collectrin (50). Similar to its renal paralogue collectrin, ACE2 acts as a molecular chaperone of the neutral amino acid transporter B0AT1 (also designated as SLC6A19) in the small intestine and is responsible for the trafficking of B0AT1 to the surface of intestinal epithelial cells (23, 24, 51). Thereby, ACE2 and B0AT1 regulate the uptake of neutral amino acids in the intestine. Hence, ACE2 may act as a trafficking chaperone for TMPRSS2 in airway cells. However, this hypothesis warrants further investigation. Interestingly, cleavage of ACE2 by TMPRSS2 has been shown to interfere with ACE2 cleavage by ADAM17 and subsequent ACE2 shedding (34, 36). Based on our results here, one could speculate that the TMPRSS2-ACE2 interaction prevents ACE2 shedding to promote TMPRSS2 activity. However, the exact nature of how this interaction affects the subcellular localization of both proteins remains to be investigated.
Furthermore, it remains to be determined whether cleavage of ACE2 by TMPRSS2 is required for ACE2-mediated enhancement of TMPRSS2 activity. TMPRSS2 was shown to cleave ACE2 at amino acids 697–716 within the collectrin domain (36). Here, we found that ACE2 lacking the collectrin domain is still cleaved by TMPRSS2, whereas ACE2 lacking both the collectrin and transmembrane domain remains un-cleaved. Thus, ACE2 cleavage by TMPRSS2 appears to have flexibility. Our co-IP analysis revealed that the (i) enzymatic activity of TMPRSS2 and (ii) the peptidase domain of ACE2 are crucial for ACE2-TMPRSS2 interaction, whereas the collectrin domain and the transmembrane domain of ACE2 were found to be dispensable. Notably, we observed that cleavage of ACE2 by TMPRSS2 and interaction of ACE2 with TMPRSS2 were not necessarily correlated with each other. ACE2-∆PD mutant was cleaved by TMPRSS2 but not co-precipitated, while ACE2∆CD∆TMD mutant was co-precipitated, but not cleaved by TMPRSS2.
In the protease activity assay, we observed a slight reduction in ACE2 activity in the presence of TMPRSS2. It is possible that the interaction of TMPRSS2 with the peptidase domain of ACE2 sterically influences ACE2 activity, and we hope to investigate this in future studies. Yan et al. solved the cryo-electron microscopy structure of the ACE2-B0AT1 complex and demonstrated that it is assembled as a dimer of heterodimers (52). Dimerization is entirely mediated by ACE2, which forms homodimers mainly via amino acids 616 to 726 of its collectrin domain (52, 53). Thus, cleavage of ACE2 by TMPRSS2 at amino acids 697–716 could interfere with ACE2 dimerization (36, 52).
In the present study, we focused primarily on the influence of ACE2 on TMPRSS2-mediated activation of IAV. However, we were able to show that ACE2 is involved in S cleavage and multicycle replication of MERS-CoV S in Calu-3 cells. Thus, it will be interesting to determine whether ACE2 is involved in TMPRSS2-mediated activation of other respiratory viruses such as human metapneumovirus, human parainfluenza viruses, and further CoVs including SARS-CoV-2 (9–11).
Protection of TMPRSS2-deficient mice from pathogenesis after infection with IAV of various HA subtypes as well as with CoVs including MERS-CoV and SARS-CoV-2 impressively demonstrated that TMPRSS2 represents a promising drug target for the treatment of respiratory virus infections (reviewed in references (11, 12). A number of broad-spectrum serine protease inhibitors, such as aprotinin and camostat as well as potent peptide or peptide-mimetic inhibitors of TMPRSS2, were shown to prevent IAV activation and multicycle replication in cell cultures during the last decade (reviewed in references (54, 55)). Finally, the critical role of TMPRSS2 in SARS-CoV-2 activation has further accelerated the search for TMPRSS2 inhibitors, and several promising candidates for further drug development to treat both influenza and COVID-19 have been described (9, 56, 57). The physiological role of TMPRSS2 in humans is not yet clear. TMPRSS2-deficient mice do not exhibit a phenotype, suggesting a non-essential function or functional redundancy (58). Hence, inhibition of TMPRSS2 during acute infection is likely to be well-tolerated. Although the molecular basis of ACE2-TMPRSS2 interaction and its effect on TMPRSS2 activity remains unclear, our results may help to identify a physiological role for TMPRSS2.
Our data immediately raise the question of whether ACE2 is also a promising target for inhibition of HA cleavage; the answer is probably no. In the lung, Ang II is known to promote the development of pulmonary hypertension and pulmonary fibrosis, and ACE2 was shown to play a critical role in the development of acute respiratory distress syndrome (ARDS) (59–61). ACE2-knockout mice show more severe ARDS symptoms when compared to wild-type mice, whereas ACE2 overexpression or administration of recombinant ACE2 was shown to be protective in rodent models (59, 60). Downregulation of ACE2 by the viral spike protein (possibly via internalization of ACE2 upon virus attachment) and accumulation of Ang II is also associated with ARDS and acute lung failure in SARS-CoV and SARS-CoV-2 infections (62, 63). Conversely, administration of recombinant ACE2, which provides both a decoy to neutralize the virus and a supplement to ACE2 carboxypeptidase activity, was found to ameliorate virus-induced lung injury in mice. Meanwhile, recombinant soluble human ACE2 is under development for the treatment of moderate to severe COVID-19 infections as well as acute lung injury, ARDS and pulmonary arterial hypertension (64–66).
To summarize, we were able to show that ACE2 increases or stabilizes the enzymatic activity of TMPRSS2 and is involved in TMPRSS2-mediated IAV HA cleavage in human airway cells. Thus, our data identify ACE2 as a new host cell factor of IAV replication. Our data also suggest that ACE2 is involved in proteolytic activation of other respiratory viruses by TMPRSS2, exemplified here for MERS-Co V. In addition, our results may also stimulate further studies addressing the physiological function of the “virus-activating protease” TMPRSS2.
MATERIAL AND METHODS
Cells and viruses
Propagation of all cells was carried out at 37°C and 5% CO2. Calu-3 human airway epithelial cells (ATCC number HTB55) were cultured in DMEM/F-12 Ham (1:1) (Gibco) supplemented with 10% fetal calf serum (FCS), penicillin, streptomycin, and glutamine, with fresh culture medium replenished every 2 to 3 days. HEK293 human embryonic kidney cells, Caco-2 human colorectal adenocarcinoma cells, and Madin-Darby canine kidney II (MDCK(II)) cells were maintained in DMEM supplemented with 10% FCS, antibiotics, and glutamine.
IAVs used here were recombinant Aichi/H3N2/WSN (HA and NA of A/Aichi/2/68 (H3N2) and six genes of A/WSN/33 (H1N1), kindly provided by Mikhail Matrosovich, designated as H3N2), recombinant H7N7/SC35M (reverse genetics system kindly provided by Jürgen Stech) and 2009 H1N1 isolate A/Hamburg/5/09 (H1N1pdm). Human IAVs were propagated in MDCK(II) cells in infection medium (DMEM supplemented with 0.1% bovine serum albumin (BSA), glutamine, and antibiotics) containing 1 µg/mL tosyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (Sigma), H7N7/SC35M was propagated in MDCK(II) cells in infection medium without the addition of TPCK-trypsin. MERS-CoV (kindly provided by Lucie Sauerhering, Institute of Virology, Marburg) was propagated in Calu-3 cells in DMEM supplemented with 3% FCS, penicillin, streptomycin, and glutamine (DMEM+++). Cell supernatants were cleared from cell debris by low-speed centrifugation and stored as virus stocks at −80°C or in liquid nitrogen (MERS-CoV). Experiments with MERS-CoV were conducted under BSL4 conditions.
Plasmids
pcDNA3.1(+) plasmids encoding TMPRSS2 or TMPRSS2(S441A) with a C-terminal 3 x FLAG epitope and ACE2, ACE2-∆CD∆TMD, ACE2-∆PD, or ACE2-∆Glyco7 (with amino acid substitutions N53Q, N90Q, N103Q, N322Q, N432Q, N546Q, N690Q) with a HA-tag were commercially obtained from GeneART. pcDNA3.1(+) encoding TMPRSS2(R255Q)−3x FLAG and ACE2-∆CD-HA, respectively, were generated by site-directed mutagenesis using the Q5 Site-Directed Mutagenesis Kit (New England biolabs) according to the supplier’s protocol. All primer sequences are available upon request.
PPMO
Phosphorodiamidate morpholino oligomers (PMO) were synthesized at Gene Tools, LLC (Philomath, OR, USA). The PMO sequences (5′ to 3′) targeted to ACE2 are as follows: ACE2-AUG (GGAAGAGCTTGACATCGTCCCCTGT), ACE2-UTR (TCACATCCACTGATGACTTTCCCT), ACE2-e5i5 (GTACAGTTCCTTGCTTACCTCTTCA), and scramble (CCTCTTACCTCAGTTACAATTTATA). The cell-penetrating peptide (RXR)4 (R = arginine and X = 6-aminohexanoic acid) was covalently conjugated to the 3′ ends of each PMO through a non-cleavable linker, to produce peptide-PMO (PPMO), by methods previously described (67).
Recombinant enzymes, protease substrates and inhibitors
Recombinant soluble human TMPRSS2 (aa 109–492; mutation SRQSR255 → DDDDK255 to avoid autocatalytic activation during overexpression and to facilitate controlled zymogen activation by enterokinase) was generated in insect cells using materials and the protocol provided by Fraser et al. (68). Briefly, the TMPRSS2 expression plasmid was transformed into DH10 Bac cells. Baculovirus DNA was transfected into ExpiSf9 cells according to the manufacturer’s protocol (Thermo Fisher Scientific). For protein production, 1 L of ExpiSf9 culture was infected with the P2 virus, and cells were harvested when viability dropped below 80%. The culture medium was incubated with nickel affinity resin. The resin was washed with PBS and eluted with 0.5M imidazole in PBS, pH 7.5. Protein was dialyzed overnight against 2 L of dialysis buffer (25 mM Tris pH 8.0, 72 mM NaCl, 2 mM CaCl2) in the presence of enterokinase (New England Biolabs). The cleaved protein was further purified on a Hiload 26/600 Superdex 75 column (Cytiva). Protein was flash-frozen and stored at −80°C until use. Recombinant soluble human ACE2 (aa 18–740) was purchased from Abcam (ab273297). Fluorogenic TMPRSS2 substrate Boc-Gln-Ala-Arg-7-amino-4-methyl coumarin (AMC) was used. ACE2 activity was measured using the Angiotensin II Converting Enzyme (ACE2) Activity Assay Kit (Abcam, ab273297) according to the supplier’s protocol. ACE2 peptide inhibitor DX600 (37) was purchased from Cayman Chemical.
Antibodies
The following antibodies were used in this study: anti-ACE2 (Abcam, ab108252), anti-beta-actin (Abcam, ab6276), anti-H1N1pdm (Sino Biological, 11085-T62), anti-H7 (Sino Biological, 40103-RP01), anti-H3 (Sino Biological, GTX 127363), anti-HA protein tag (Sigma Aldrich, H6908), anti-FLAG (Sigma Aldrich, F7425), and anti MERS-CoV-Spike-S2 (Sino Biological, 40070-T62). Species-specific horseradish-peroxidase-conjugated antibodies were purchased from DAKO.
Measurement of protease activities
To measure TMPRSS2 activity, recombinant TMPRSS2 (final assay concentration 0.12 nM) was mixed with recombinant ACE2 (0.8 µU) prior to the addition of 25 µM fluorogenic substrate Boc-Gln-Ala-Arg-AMC in 50 mM Tris HCl pH 8.0 containing 154 mM NaCl. Protease activity was measured over 1 h at room temperature (RT) or at 37°C using a BioTek Synergy H1 Plate Reader at 350/380 nm excitation and 460 nm emission. When indicated, ACE2 inhibitor DX600 (10 µM in assay) was added.
To measure ACE2 activity, the ACE2 Activity Assay Kit (Abcam, ab273297) was used according to the supplier’s protocol. Recombinant ACE2 (0.8 µU) was mixed with recombinant TMPRSS2 (0.12 nM) in the absence or presence of DX600 (10 µM) prior to the addition of a quenched fluorogenic ACE2 substrate. The cleavage rate of the substrate was measured over 1 h at RT in relative fluorescence units at 320 nm excitation and 420 nm emission. All protease activity measurements were performed in two to three independent experiments performed in duplicate or triplicate.
Cell viability assay
Confluent Calu-3 cells were treated with 25 µM of the respective PPMO in infection medium or infection medium containing 5% EtOH for 24 h at 37°C and 5% CO2. Cell viability was determined using the CellTiter-Glo Kit according to the manufacturer’s manual.
IAV infection of cells for multicycle viral replication and HA cleavage analysis
Infection experiments and PPMO treatment of Calu-3 cells were performed using an infection medium. For analysis of multicycle viral replication Calu-3 cells were seeded in 24-well plates and grown to near-confluence. Cells were incubated with 25 µM of PPMO in the infection medium for 24 h or remained untreated. The cells were then inoculated with the virus at a low MOI of 0.001 (virus growth kinetics) or an MOI of 1 (analysis of HA cleavage) in fresh infection medium without PPMO for 1 h, washed with PBS, and incubated in infection medium without PPMO for 72 h. At 16, 24, 48, and 72 h p.i., supernatants were collected, and viral titers were determined by plaque assay in MDCK(II) cells with Avicel overlay as described previously (47). Cells were subjected to SDS-PAGE and western blot as described below.
Caco-2 cells were seeded in 24-well plates and grown to 80% confluence. The cells were transfected with either plasmids encoding wild-type ACE2 or empty vector using Lipofectamin2000 (ThermoFisher) according to the manufacturer’s instructions. At 24 h post-transfection, the cells were washed once with PBS and then inoculated with the virus at a low MOI of 0.001 in the infection medium for 1 h. The cells were washed with PBS and incubated with fresh infection medium for 72 h. At 24 h and 48 h p.i., supernatants were collected, and viral titers were determined by plaque assay in MDCK(II) as described above. The cells were harvested after 72 h and subjected to SDS-PAGE and western blot as described below.
MERS-CoV infection of cells and analysis of S cleavage
PPMO treatment of Calu-3 cells 24 h prior to infection was performed as described above. The cells were then inoculated with MERS-CoV at an MOI of 0.001 under serum- and BSA-free conditions with DMEM supplemented with glutamine and penicillin/streptomycin only (DMEM++) for 1 h. Subsequently, the cells were thoroughly washed with PBS and further incubated in DMEM+++. The supernatant was sampled at indicated time points and subjected to TCID50 titration as described below to determine viral titers. At 72 h p.i., infected cells were harvested for analysis of S cleavage via SDS-PAGE and western blot as described below.
TCID50 titration
Calu-3 cells were cultivated to confluency in 96-well plates before the medium was changed to DMEM+++. Supernatants taken from infected cells at indicated time points p.i. were serially diluted from 5−1 to 5−11 in quadruplets directly on the cells. After 3–4 days, when CPE was clearly visible, viral titers were calculated with the Spearman and Kaerber algorithm (69).
SDS-PAGE and western blot analysis
Cells were washed with PBS, lysed using CellLytic M Cell Lysis Reagent (Sigma-Aldrich) for 30 min on ice, and cleared from cell debris by centrifugation. Lysates were supplemented with reducing SDS-PAGE sample buffer and heated at 95°C for 10 min. Samples produced under BSL4 conditions were heated to 100°C for 10 min twice. Proteins were subjected to SDS-PAGE (12% or 8% gel), transferred to a polyvinylidene difluoride (PVDF) membrane (GE Healthcare), and detected by incubation with primary antibodies and species-specific peroxidase-conjugated secondary antibodies. Proteins were visualized using the ChemiDoc XRS + system and Image Lab software (Bio-Rad). Using Image Lab Software (Bio-Rad), detected protein bands were relatively quantified. ACE2 bands in cell lysates expressing wt ACE2 only were set as 100% and used for the relative quantification. The relative quantifications of ACE2 +ACE2 cyto in input samples of the Co-IP were added together.
Co-immunoprecipitation assay
HEK293 cells seeded in 60 mm cell culture dishes were transfected with plasmids encoding wild-type ACE2 or the respective mutant jointly with TMPRSS2 or inactive mutant encoding plasmids or empty vector using Lipofectamin2000 (ThermoFisher) according to the manufacturer’s instructions. The plasmids were transfected at a ratio of 1:2 TMPRSS2 versus ACE2 plasmid. Cells were incubated at 37°C and 5% CO2 for 48 h. Cells were then harvested and lysed in 400 µL NP40-buffer [50 mM Tris, 150 nM NaCl, 1% Nonidet P-40, 1 mM PMSF, and 1× Protease Inhibitor Cocktail (Sigma-Aldrich)] per 60 mm cell culture dish. Lysates were precleared with 100 µL Protein-A-Sepharose (Sigma-Aldrich) for 30 min at 4°C with end-over-end rotation. The Protein-A-Sepharose was discarded, and the protein concentration of the lysates was determined using the Pierce BCA Protein Assay Kit (ThermoFisher). Lysates containing 40 µg protein were left untreated for input control. Lysates containing 1 mg protein were used for co-immunoprecipitation analysis with 30 µL of anti-HA tag agarose (Sigma-Aldrich) or ANTI FLAG M2-Affinity gel (Sigma Aldrich) incubated overnight at 4°C with end-to-end rotation. After incubation, the precipitates were washed three times with ice-cold NP-40 buffer, and bound proteins were eluted by adding SDS sample buffer + β-mercaptoethanol and boiling for 10 min. Precipitates and lysates were analyzed by SDS-PAGE and western blot using HA-tag and FLAG-tag-specific antibodies, respectively.
ACKNOWLEDGMENTS
We thank Diana Kruhl for excellent technical assistance, Lucie Sauerhering for providing the MERS-CoV, and the BSL-4 team for supporting work in the BSL4 laboratory.
This project has been funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) Project-ID 197785619—SFB 1021 (to E.B.-F.) and Project-ID 284237345—KFO309 (to E.B.-F.), State of Hesse LOEWE Center DRUID (to E.B.-F. and T.S.), and Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services, under Contract No. 75N93022C00036 (to A.DR. and B.S.). M.R.H., and E.B.-F. conceived the study and designed the experiments.
M.R.H., A.-L.R., M.S., D.B., A.H., A.DR., L.C.S., and E.B.-F. performed and analyzed the experiments. A.DR. and B.S. provided recombinant TMPRSS2. D.A.S. and H.M.M. designed and provided PPMO. B.S., T.S., and E.B.-F. provided funding for the studies. E.B.-F. wrote the manuscript. All authors contributed to the editing of the manuscript.
Contributor Information
Eva Böttcher-Friebertshäuser, Email: friebertshaeuser@staff.uni-marburg.de.
Kanta Subbarao, The Peter Doherty Institute for Infection and Immunity, Melbourne, Australia.
DATA AVAILABILITY
All data supporting the findings of this study are available within the paper and in the Supporting Information.
SUPPLEMENTAL MATERIAL
The following material is available online at https://doi.org/10.1128/jvi.00102-24.
Figures S1 to S4.
ASM does not own the copyrights to Supplemental Material that may be linked to, or accessed through, an article. The authors have granted ASM a non-exclusive, world-wide license to publish the Supplemental Material files. Please contact the corresponding author directly for reuse.
REFERENCES
- 1. Krammer F, Smith GJD, Fouchier RAM, Peiris M, Kedzierska K, Doherty PC, Palese P, Shaw ML, Treanor J, Webster RG, García-Sastre A. 2018. Influenza. Nat Rev Dis Primers 4:3. doi: 10.1038/s41572-018-0002-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Russell CJ. 2014. Acid-induced membrane fusion by the hemagglutinin protein and its role in influenza virus biology. Curr Top Microbiol Immunol 385:93–116. doi: 10.1007/82_2014_393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Böttcher-Friebertshäuser E, Garten W, Matrosovich M, Klenk HD. 2014. The hemagglutinin: a determinant of pathogenicity. Curr Top Microbiol Immunol 385:3–34. doi: 10.1007/82_2014_384 [DOI] [PubMed] [Google Scholar]
- 4. Klenk HD, Rott R, Orlich M, Blödorn J. 1975. Activation of influenza A viruses by trypsin treatment. Virology 68:426–439. doi: 10.1016/0042-6822(75)90284-6 [DOI] [PubMed] [Google Scholar]
- 5. Lazarowitz SG, Choppin PW. 1975. Enhancement of the infectivity of influenza A and B viruses by proteolytic cleavage of the hemagglutinin polypeptide. Virology 68:440–454. doi: 10.1016/0042-6822(75)90285-8 [DOI] [PubMed] [Google Scholar]
- 6. Stieneke-Gröber A, Vey M, Angliker H, Shaw E, Thomas G, Roberts C, Klenk HD, Garten W. 1992. Influenza virus hemagglutinin with multibasic cleavage site is activated by furin, a subtilisin-like endoprotease. EMBO J 11:2407–2414. doi: 10.1002/j.1460-2075.1992.tb05305.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Horimoto T, Nakayama K, Smeekens SP, Kawaoka Y. 1994. Proprotein-processing endoproteases PC6 and Furin both activate hemagglutinin of virulent avian influenza viruses. J Virol 68:6074–6078. doi: 10.1128/JVI.68.9.6074-6078.1994 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Böttcher E, Matrosovich T, Beyerle M, Klenk H-D, Garten W, Matrosovich M. 2006. Proteolytic activation of influenza viruses by serine proteases TMPRSS2 and HAT from human airway epithelium. J Virol 80:9896–9898. doi: 10.1128/JVI.01118-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bestle D, Heindl MR, Limburg H, Van Lam van T, Pilgram O, Moulton H, Stein DA, Hardes K, Eickmann M, Dolnik O, Rohde C, Klenk H-D, Garten W, Steinmetzer T, Böttcher-Friebertshäuser E. 2020. TMPRSS2 and furin are both essential for proteolytic activation of SARS-CoV-2 in human airway cells. Life Sci Alliance 3:e202000786. doi: 10.26508/lsa.202000786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. 2020. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181:271–280. doi: 10.1016/j.cell.2020.02.052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Böttcher-Friebertshäuser E. 2018. Membrane-anchored serine proteases: host cell factors in proteolytic activation of viral glycoproteins, p 153–203. In Böttcher-Friebertshäuser E, Garten W, Klenk HD (ed), Activation of viruses by host proteases. Springer International Publishing, Cham. [Google Scholar]
- 12. Iwata-Yoshikawa N, Kakizaki M, Shiwa-Sudo N, Okura T, Tahara M, Fukushi S, Maeda K, Kawase M, Asanuma H, Tomita Y, Takayama I, Matsuyama S, Shirato K, Suzuki T, Nagata N, Takeda M. 2022. Essential role of TMPRSS2 in SARS-CoV-2 infection in murine airways. Nat Commun 13:6100. doi: 10.1038/s41467-022-33911-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Metzdorf K, Jacobsen H, Greweling-Pils MC, Hoffmann M, Lüddecke T, Miller F, Melcher L, Kempf AM, Nehlmeier I, Bruder D, Widera M, Ciesek S, Pöhlmann S, Čičin-Šain L. 2023. TMPRSS2 is essential for SARS-CoV-2 beta and Omicron infection. Viruses 15:271. doi: 10.3390/v15020271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Iwata-Yoshikawa N, Okamura T, Shimizu Y, Hasegawa H, Takeda M, Nagata N. 2019. TMPRSS2 contributes to virus spread and immunopathology in the airways of murine models after coronavirus infection. J Virol 93:e01815-18. doi: 10.1128/JVI.01815-18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Limburg H, Harbig A, Bestle D, Stein DA, Moulton HM, Jaeger J, Janga H, Hardes K, Koepke J, Schulte L, Koczulla AR, Schmeck B, Klenk H-D, Böttcher-Friebertshäuser E. 2019. TMPRSS2 is the major activating protease of influenza A virus in primary human airway cells and influenza B virus in human type II pneumocytes. J Virol 93:e00649-19. doi: 10.1128/JVI.00649-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bestle D, Limburg H, Kruhl D, Harbig A, Stein DA, Moulton H, Matrosovich M, Abdelwhab EM, Stech J, Böttcher-Friebertshäuser E. 2021. Hemagglutinins of avian influenza viruses are proteolytically activated by TMPRSS2 in human and murine airway cells. J Virol 95:e0090621. doi: 10.1128/JVI.00906-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Li W, Moore MJ, Vasilieva N, Sui J, Wong SK, Berne MA, Somasundaran M, Sullivan JL, Luzuriaga K, Greenough TC, Choe H, Farzan M. 2003. Angiotensin-converting enzyme 2 is a functional receptor for the SARS coronavirus. Nature 426:450–454. doi: 10.1038/nature02145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hofmann H, Pyrc K, van der Hoek L, Geier M, Berkhout B, Pöhlmann S. 2005. Human coronavirus Nl63 employs the severe acute respiratory syndrome coronavirus receptor for cellular entry. Proc Natl Acad Sci U S A 102:7988–7993. doi: 10.1073/pnas.0409465102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Donoghue M, Hsieh F, Baronas E, Godbout K, Gosselin M, Stagliano N, Donovan M, Woolf B, Robison K, Jeyaseelan R, Breitbart RE, Acton S. 2000. A novel angiotensin-converting enzyme-related carboxypeptidase (ACE2) converts angiotensin I to angiotensin 1-9. Circ Res 87:E1–9. doi: 10.1161/01.res.87.5.e1 [DOI] [PubMed] [Google Scholar]
- 20. Vickers C, Hales P, Kaushik V, Dick L, Gavin J, Tang J, Godbout K, Parsons T, Baronas E, Hsieh F, Acton S, Patane M, Nichols A, Tummino P. 2002. Hydrolysis of biological peptides by human angiotensin-converting enzyme-related carboxypeptidase. J Biol Chem 277:14838–14843. doi: 10.1074/jbc.M200581200 [DOI] [PubMed] [Google Scholar]
- 21. Turner AJ. 2015. ACE2 cell biology, regulation, and physiological functions, p 185–189. In The protective arm of the renin angiotensin system (RAS). Elsevier. [Google Scholar]
- 22. Hamming I, Cooper ME, Haagmans BL, Hooper NM, Korstanje R, Osterhaus ADME, Timens W, Turner AJ, Navis G, van Goor H. 2007. The emerging role of ACE2 in physiology and disease. J Pathol 212:1–11. doi: 10.1002/path.2162 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kowalczuk S, Bröer A, Tietze N, Vanslambrouck JM, Rasko JEJ, Bröer S. 2008. A protein complex in the brush-border membrane explains a hartnup disorder allele. FASEB J 22:2880–2887. doi: 10.1096/fj.08-107300 [DOI] [PubMed] [Google Scholar]
- 24. Camargo SMR, Singer D, Makrides V, Huggel K, Pos KM, Wagner CA, Kuba K, Danilczyk U, Skovby F, Kleta R, Penninger JM, Verrey F. 2009. Tissue-specific amino acid transporter partners ACE2 and collectrin differentially interact with hartnup mutations. Gastroenterology 136:872–882. doi: 10.1053/j.gastro.2008.10.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hikmet F, Méar L, Edvinsson Å, Micke P, Uhlén M, Lindskog C. 2020. The protein expression profile of ACE2 in human tissues. Mol Syst Biol 16:e9610. doi: 10.15252/msb.20209610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tipnis SR, Hooper NM, Hyde R, Karran E, Christie G, Turner AJ. 2000. A human homolog of angiotensin-converting enzyme. Cloning and functional expression as a captopril-insensitive carboxypeptidase. J Biol Chem 275:33238–33243. doi: 10.1074/jbc.M002615200 [DOI] [PubMed] [Google Scholar]
- 27. Harmer D, Gilbert M, Borman R, Clark KL. 2002. Quantitative mRNA expression profiling of ACE 2, a novel homologue of angiotensin converting enzyme. FEBS Lett 532:107–110. doi: 10.1016/s0014-5793(02)03640-2 [DOI] [PubMed] [Google Scholar]
- 28. Hamming I, Timens W, Bulthuis MLC, Lely AT, Navis GJ, van Goor H. 2004. Tissue distribution of ACE2 protein, the functional receptor for SARS coronavirus. A first step in understanding SARS pathogenesis. J Pathol 203:631–637. doi: 10.1002/path.1570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Komatsu T, Suzuki Y, Imai J, Sugano S, Hida M, Tanigami A, Muroi S, Yamada Y, Hanaoka K. 2002. Molecular cloning, mRNA expression and chromosomal localization of mouse angiotensin-converting enzyme-related carboxypeptidase (mACE2). DNA Seq 13:217–220. doi: 10.1080/1042517021000021608 [DOI] [PubMed] [Google Scholar]
- 30. Zou X, Chen K, Zou J, Han P, Hao J, Han Z. 2020. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front Med 14:185–192. doi: 10.1007/s11684-020-0754-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Qi F, Qian S, Zhang S, Zhang Z. 2020. Single cell RNA sequencing of 13 human tissues identify cell types and receptors of human coronaviruses. Biochem Biophys Res Commun 526:135–140. doi: 10.1016/j.bbrc.2020.03.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lee IT, Nakayama T, Wu C-T, Goltsev Y, Jiang S, Gall PA, Liao C-K, Shih L-C, Schürch CM, McIlwain DR, et al. 2020. ACE2 localizes to the respiratory cilia and is not increased by ACE inhibitors or ARBs. Nat Commun 11:5453. doi: 10.1038/s41467-020-19145-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Zhang H, Wada J, Hida K, Tsuchiyama Y, Hiragushi K, Shikata K, Wang H, Lin S, Kanwar YS, Makino H. 2001. Collectrin, a collecting duct-specific transmembrane glycoprotein, is a novel homolog of ACE2 and is developmentally regulated in embryonic kidneys. J Biol Chem 276:17132–17139. doi: 10.1074/jbc.M006723200 [DOI] [PubMed] [Google Scholar]
- 34. Lambert DW, Yarski M, Warner FJ, Thornhill P, Parkin ET, Smith AI, Hooper NM, Turner AJ. 2005. Tumor necrosis factor-alpha convertase (ADAM17) mediates regulated ectodomain shedding of the severe-acute respiratory syndrome-coronavirus (SARS-CoV) receptor, angiotensin-converting enzyme-2 (ACE2). J Biol Chem 280:30113–30119. doi: 10.1074/jbc.M505111200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Shulla A, Heald-Sargent T, Subramanya G, Zhao J, Perlman S, Gallagher T. 2011. A transmembrane serine protease is linked to the severe acute respiratory syndrome coronavirus receptor and activates virus entry. J Virol 85:873–882. doi: 10.1128/JVI.02062-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Heurich A, Hofmann-Winkler H, Gierer S, Liepold T, Jahn O, Pöhlmann S. 2014. TMPRSS2 and ADAM17 cleave ACE2 differentially and only proteolysis by TMPRSS2 augments entry driven by the severe acute respiratory syndrome coronavirus spike protein. J Virol 88:1293–1307. doi: 10.1128/JVI.02202-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Huang L, Sexton DJ, Skogerson K, Devlin M, Smith R, Sanyal I, Parry T, Kent R, Enright J, Wu Q, Conley G, DeOliveira D, Morganelli L, Ducar M, Wescott CR, Ladner RC. 2003. Novel peptide inhibitors of angiotensin-converting enzyme 2. J Biol Chem 278:15532–15540. doi: 10.1074/jbc.M212934200 [DOI] [PubMed] [Google Scholar]
- 38. Laporte M, Stevaert A, Raeymaekers V, Boogaerts T, Nehlmeier I, Chiu W, Benkheil M, Vanaudenaerde B, Pöhlmann S, Naesens L. 2019. Hemagglutinin cleavability, acid stability, and temperature dependence optimize influenza B virus for replication in human airways. J Virol 94:e01430-19. doi: 10.1128/JVI.01430-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Moulton HM, Moulton JD. 2010. Morpholinos and their peptide conjugates: therapeutic promise and challenge for duchenne muscular dystrophy. Biochim Biophys Acta 1798:2296–2303. doi: 10.1016/j.bbamem.2010.02.012 [DOI] [PubMed] [Google Scholar]
- 40. Bertram S, Glowacka I, Blazejewska P, Soilleux E, Allen P, Danisch S, Steffen I, Choi S-Y, Park Y, Schneider H, Schughart K, Pöhlmann S. 2010. TMPRSS2 and TMPRSS4 facilitate trypsin-independent spread of influenza virus in Caco-2 cells. J Virol 84:10016–10025. doi: 10.1128/JVI.00239-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Afar DE, Vivanco I, Hubert RS, Kuo J, Chen E, Saffran DC, Raitano AB, Jakobovits A. 2001. Catalytic cleavage of the androgen-regulated TMPRSS2 protease results in its secretion by prostate and prostate cancer epithelia. Cancer Res 61:1686–1692. [PubMed] [Google Scholar]
- 42. Sherman EJ, Emmer BT. 2021. ACE2 protein expression within Isogenic cell lines is heterogeneous and associated with distinct transcriptomes. Sci Rep 11:15900. doi: 10.1038/s41598-021-95308-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Rowland R, Brandariz-Nuñez A. 2021. Analysis of the role of N-linked glycosylation in cell surface expression, function, and binding properties of SARS-CoV-2 receptor ACE2. Microbiol Spectr 9:e0119921. doi: 10.1128/Spectrum.01199-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Raj VS, Mou H, Smits SL, Dekkers DHW, Müller MA, Dijkman R, Muth D, Demmers JAA, Zaki A, Fouchier RAM, Thiel V, Drosten C, Rottier PJM, Osterhaus A, Bosch BJ, Haagmans BL. 2013. Dipeptidyl peptidase 4 is a functional receptor for the emerging human coronavirus-EMC. Nature 495:251–254. doi: 10.1038/nature12005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shirato K, Kawase M, Matsuyama S. 2013. Middle East respiratory syndrome coronavirus infection mediated by the transmembrane serine protease TMPRSS2. J Virol 87:12552–12561. doi: 10.1128/JVI.01890-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Kleine-Weber H, Elzayat MT, Hoffmann M, Pöhlmann S. 2018. Functional analysis of potential cleavage sites in the MERS-coronavirus spike protein. Sci Rep 8:16597. doi: 10.1038/s41598-018-34859-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Böttcher-Friebertshäuser E, Freuer C, Sielaff F, Schmidt S, Eickmann M, Uhlendorff J, Steinmetzer T, Klenk H-D, Garten W. 2010. Cleavage of influenza virus hemagglutinin by airway proteases TMPRSS2 and HAT differs in subcellular localization and susceptibility to protease inhibitors. J Virol 84:5605–5614. doi: 10.1128/JVI.00140-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Böttcher-Friebertshäuser E, Klenk H-D, Garten W. 2013. Activation of influenza viruses by proteases from host cells and bacteria in the human airway epithelium. Pathog Dis 69:87–100. doi: 10.1111/2049-632X.12053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zhirnov OP, Vorobjeva IV, Ovcharenko AV, Klenk HD. 2003. Intracellular cleavage of human influenza a virus hemagglutinin and its inhibition. Biochemistry (Mosc) 68:1020–1026. doi: 10.1023/A:1026020831036 [DOI] [PubMed] [Google Scholar]
- 50. Perlot T, Penninger JM. 2013. ACE2 - from the renin-angiotensin system to gut microbiota and malnutrition. Microbes Infect 15:866–873. doi: 10.1016/j.micinf.2013.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Danilczyk U, Sarao R, Remy C, Benabbas C, Stange G, Richter A, Arya S, Pospisilik JA, Singer D, Camargo SMR, Makrides V, Ramadan T, Verrey F, Wagner CA, Penninger JM. 2006. Essential role for collectrin in renal amino acid transport. Nature 444:1088–1091. doi: 10.1038/nature05475 [DOI] [PubMed] [Google Scholar]
- 52. Yan R, Zhang Y, Li Y, Xia L, Guo Y, Zhou Q. 2020. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 367:1444–1448. doi: 10.1126/science.abb2762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Barros EP, Casalino L, Gaieb Z, Dommer AC, Wang Y, Fallon L, Raguette L, Belfon K, Simmerling C, Amaro RE. 2021. The flexibility of ACE2 in the context of SARS-CoV-2 infection. Biophys J 120:1072–1084. doi: 10.1016/j.bpj.2020.10.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wettstein L, Kirchhoff F, Münch J. 2022. The transmembrane protease TMPRSS2 as a therapeutic target for COVID-19 treatment. Int J Mol Sci 23:1351. doi: 10.3390/ijms23031351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Steinmetzer T, Hardes K. 2018. The antiviral potential of host protease inhibitors, p 279–325. In Böttcher-Friebertshäuser E, Garten W, Klenk HD (ed), Activation of viruses by host proteases. Springer International Publishing, Cham. [Google Scholar]
- 56. Shapira T, Monreal IA, Dion SP, Buchholz DW, Imbiakha B, Olmstead AD, Jager M, Désilets A, Gao G, Martins M, et al. 2022. A TMPRSS2 inhibitor acts as a pan-SARS-CoV-2 prophylactic and therapeutic. Nature 605:340–348. doi: 10.1038/s41586-022-04661-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mahoney M, Damalanka VC, Tartell MA, Chung DH, Lourenço AL, Pwee D, Mayer Bridwell AE, Hoffmann M, Voss J, Karmakar P, Azouz NP, Klingler AM, Rothlauf PW, Thompson CE, Lee M, Klampfer L, Stallings CL, Rothenberg ME, Pöhlmann S, Whelan SPJ, O’Donoghue AJ, Craik CS, Janetka JW. 2021. A novel class of TMPRSS2 inhibitors potently block SARS-CoV-2 and MERS-CoV viral entry and protect human epithelial lung cells. Proc Natl Acad Sci U S A 118:e2108728118. doi: 10.1073/pnas.2108728118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Kim TS, Heinlein C, Hackman RC, Nelson PS. 2006. Phenotypic analysis of mice lacking the TMPRSS2-encoded protease. Mol Cell Biol 26:965–975. doi: 10.1128/MCB.26.3.965-975.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan B, Yang P, Sarao R, Wada T, Leong-Poi H, Crackower MA, Fukamizu A, Hui C-C, Hein L, Uhlig S, Slutsky AS, Jiang C, Penninger JM. 2005. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 436:112–116. doi: 10.1038/nature03712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ferreira AJ, Shenoy V, Yamazato Y, Sriramula S, Francis J, Yuan L, Castellano RK, Ostrov DA, Oh SP, Katovich MJ, Raizada MK. 2009. Evidence for angiotensin-converting enzyme 2 as a therapeutic target for the prevention of pulmonary hypertension. Am J Respir Crit Care Med 179:1048–1054. doi: 10.1164/rccm.200811-1678OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Kuba K, Imai Y, Ohto-Nakanishi T, Penninger JM. 2010. Trilogy of ACE2: a peptidase in the renin-angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol Ther 128:119–128. doi: 10.1016/j.pharmthera.2010.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Kuba K, Imai Y, Rao S, Gao H, Guo F, Guan B, Huan Y, Yang P, Zhang Y, Deng W, Bao L, Zhang B, Liu G, Wang Z, Chappell M, Liu Y, Zheng D, Leibbrandt A, Wada T, Slutsky AS, Liu D, Qin C, Jiang C, Penninger JM. 2005. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus-induced lung injury. Nat Med 11:875–879. doi: 10.1038/nm1267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Seltzer S. 2020. Linking ACE2 and angiotensin II to pulmonary immunovascular dysregulation in SARS-CoV-2 infection. Int J Infect Dis 101:42–45. doi: 10.1016/j.ijid.2020.09.041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Zoufaly A, Poglitsch M, Aberle JH, Hoepler W, Seitz T, Traugott M, Grieb A, Pawelka E, Laferl H, Wenisch C, Neuhold S, Haider D, Stiasny K, Bergthaler A, Puchhammer-Stoeckl E, Mirazimi A, Montserrat N, Zhang H, Slutsky AS, Penninger JM. 2020. Human recombinant soluble ACE2 in severe COVID-19. Lancet Respir Med 8:1154–1158. doi: 10.1016/S2213-2600(20)30418-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Monteil V, Kwon H, Prado P, Hagelkrüys A, Wimmer RA, Stahl M, Leopoldi A, Garreta E, Hurtado Del Pozo C, Prosper F, Romero JP, Wirnsberger G, Zhang H, Slutsky AS, Conder R, Montserrat N, Mirazimi A, Penninger JM. 2020. Inhibition of SARS-CoV-2 infections in engineered human tissues using clinical-grade soluble human ACE2. Cell 181:905–913. doi: 10.1016/j.cell.2020.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Khan A, Benthin C, Zeno B, Albertson TE, Boyd J, Christie JD, Hall R, Poirier G, Ronco JJ, Tidswell M, Hardes K, Powley WM, Wright TJ, Siederer SK, Fairman DA, Lipson DA, Bayliffe AI, Lazaar AL. 2017. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit Care 21:234. doi: 10.1186/s13054-017-1823-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Abes S, Moulton HM, Clair P, Prevot P, Youngblood DS, Wu RP, Iversen PL, Lebleu B. 2006. Vectorization of morpholino oligomers by the (R-Ahx-R)4 peptide allows efficient splicing correction in the absence of endosomolytic agents. J Control Release 116:304–313. doi: 10.1016/j.jconrel.2006.09.011 [DOI] [PubMed] [Google Scholar]
- 68. Fraser BJ, Beldar S, Seitova A, Hutchinson A, Mannar D, Li Y, Kwon D, Tan R, Wilson RP, Leopold K, Subramaniam S, Halabelian L, Arrowsmith CH, Bénard F. 2022. Structure and activity of human TMPRSS2 protease implicated in SARS-CoV-2 activation. Nat Chem Biol 18:963–971. doi: 10.1038/s41589-022-01059-7 [DOI] [PubMed] [Google Scholar]
- 69. Hierholzer JC, Killington RA. 1996. Virus isolation and quantitation, p 25–46. In Virology methods manual. Elsevier. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figures S1 to S4.
Data Availability Statement
All data supporting the findings of this study are available within the paper and in the Supporting Information.