

Abstract
Advancements in genome sequencing have led to the rapid accumulation of uncharacterized ‘hypothetical proteins’ in the public databases. Here we provide a community perspective and some best-practice approaches for the accurate functional annotation of uncharacterized genomic sequences.
Keywords: hypothetical genes, annotation, helminth, genomes, RNAi, CRISPR
The Challenges of Annotating Helminth Genomes
Nucleotide sequences are available for 370 000 described species. After initial publication, draft genomes should undergo constant improvements based on computational and functional genomics data. However, this is rarely applied to the majority of the published helminth genomes. Helminth genome projects identify between 10 000 and 20 000 protein-coding genes per helminth genome, of which half are unknown with respect to functionality. This group of genes are likely to be parasite-specific and represent genes whose biological functions are of interest for basic, as well as, applied science.
The main challenges for researchers in helminth genomics are the generation of high-quality assemblies and subsequent genome annotation. The first is largely due to the discontinuity of shotgun assemblies with short sequence reads, now being aided with current long-read sequencing platforms and additional technologies, such as optical mapping. With more precise assemblies, the challenge then becomes optimization of gene prediction and annotation tools for the exotic nature of many helminth genomes and a lack of identifiable sequence homologs or conserved protein domains in model organisms that might allow their function to be proposed. Further, there is often an inability to test functionality because the majority of helminths are presently genetically intractable and cannot be easily cultured (if at all). As many annotations are still based on primary-sequence-level search protocols, this has led to an increase in misannotation of genes as well as error propagation from previously misannotated genes [1]. Moreover, the helminth research community often uses divergent methods or tools of their own to handle hypothetical proteins, which further complicates the situation.
To improve annotation at sequence, structural, and functional levels, one solution is to consider data in a genome and proteome-wide perspective. This broadened view can improve current annotation pipelines and also highlight evolutionary processes, including adaptation mechanisms, gene family loss or gains, lateral gene transfers, structural and functional innovations, etc. The aim of this forum article is to highlight the current issues associated with annotation of helminth genomes and to promote the generation of a publicly available ‘Gold Standard’ database composed of genes/proteins based on community-driven in silico, experimental validation, and RNA sequence-based approaches.
Approaches for Annotating Genes with ‘Hypothetical’ Functions In Silico
Current eukaryotic databases and algorithms are still biased toward mammal, fly, and free-living helminth genomes. Inferring gene function for nematodes is therefore a major challenge, especially where little genomic and transcriptomic information is available. A significant bottleneck is the lack of accurate gene models for experimental design. In silico approaches are often utilized to assign functional annotation to protein coding and noncoding genes. For example, a new gene annotation algorithm has been developed that infers biological function to ‘unknown’ genes based on self-organizing map clustering of a gene set with well known function [2]. This approach, being implemented with tapeworm datasets at WormBase ParaSite (see Glossary), utilizes expression data of gene sets with well known function [Gene Ontology (GO) annotations] to annotate genes with unknown biological function.
Other approaches can improve and measure genome or proteome annotation quality (Figure 1). For example, the first step in annotation of enzymes encoded in a genome is generally leveraging homology with sequences in available databases (e.g., KEGG, UniProt, and/or BRENDA). Tools such as InterProScan integrate protein signatures from several distinct databases, providing classification based on the presence of domains and important sites, usually responsible for a particular function in the overall role of a protein. These, however, can result in false negatives due to fast sequence divergence in regions outside the active site, or convergent evolution of genes from unrelated ancestry. Such false negatives can be reduced using tools that identify enzymes via other methods, such as DETECT, PRIAM and EFICAz2.
Figure 1. Approaches for Functional Annotation of Uncharacterized Genes.
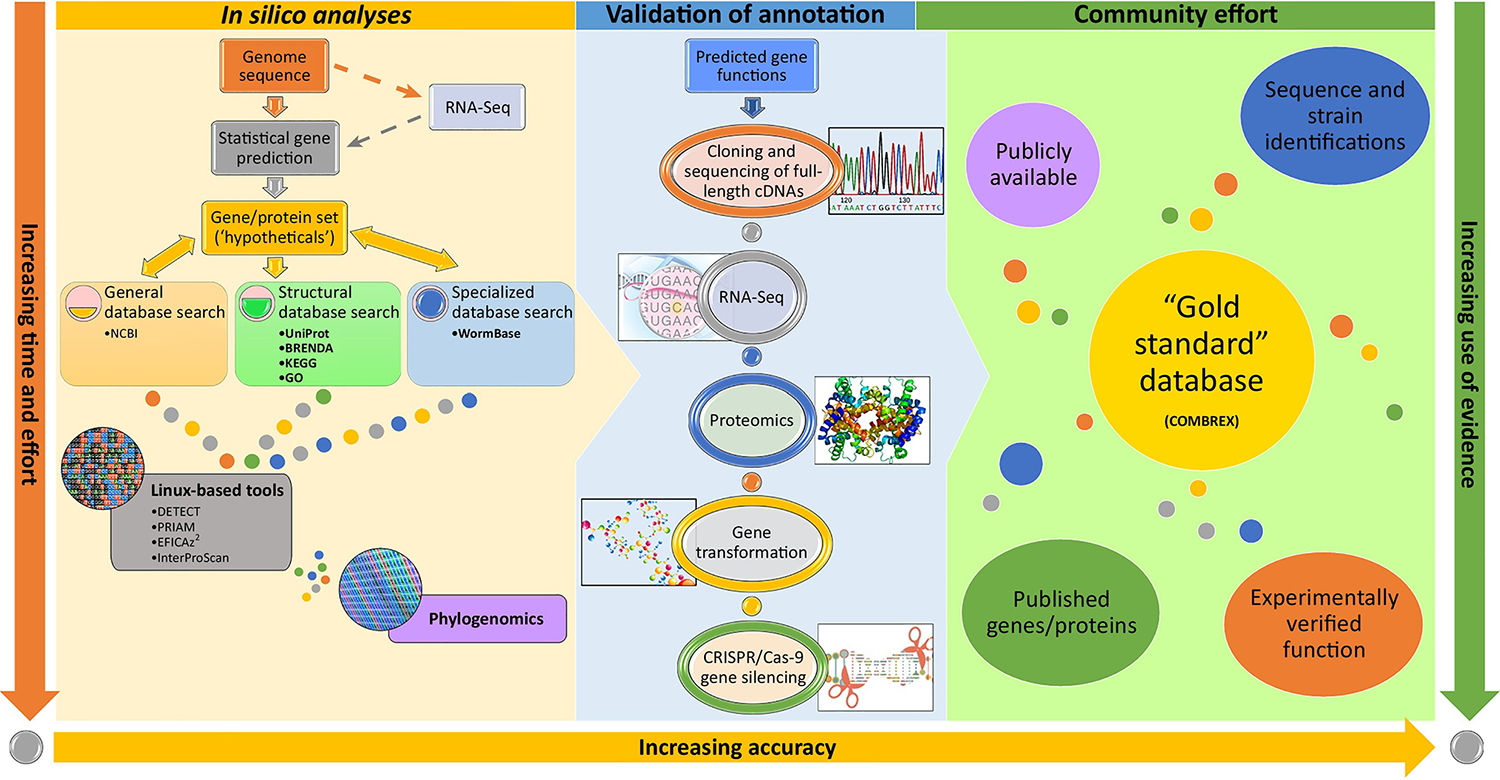
The most efficient means of investigating genes encoded in helminth genomes with the ‘hypothetical’ function annotation is to initially search the currently available sequence databases (typically, NCBI nonredundant database [https://www.ncbi.nlm.nih.gov)] for sequence similarity, using BLAST. This should be followed up by searching structural and specialized databases, for example: protein databases (such as UniProt), enzyme databases (such as BRENDA), and metabolic databases [such as KEGG and Gene Ontology (GO)], for metabolic pathway reconstruction [2]. Several linux-based tools can be used to precisely predict enzyme function, such as DETECT, PRIAM, EFICAz2, and InterProScan. Another in silico method used to improve functional annotation is phylogenomics [3], where hypothetical proteins from phylogenetically related species are compared. Once putative function is determined, cloning and sequencing of full-length cDNAs, proteomics (such as mass spectrometry), and RNA-Seq data can be used to experimentally validate annotations. Additional techniques, such as gene transformation and CRISPR/Cas-9 gene silencing, can also be applied 5, 6, 7, 8, 9, 10. The above mentioned tools and techniques should be used in concert with extensive literature mining to manually curate genomic content. The resulting genes/protein sequences should be deposited in public databases such as COMBREX and WormBase. As the research community accumulates information regarding experimentally verified and published genes/proteins along with species and strain identifications, a ‘Gold Standard’ database can emerge.
In addition to using diagnostic domains, phylogenomics can be used to improve functional annotation, which combines computational and biological sciences, taking advantage of an evolutionary perspective over comparative analyses [3]. Comparison with other hypothetical proteins from phylogenetically related species may provide an indication of positive selection. Caenorhabditis elegans, considered a model for parasitic nematodes, contains putative homologous genes from other parasitic species and demonstrates conserved gene function. It has become clear that small proteins (<30 aa) play roles in cell phenotype in prokaryotes, and significant similarity exists among the proteins in eukaryotic organisms. While focused on improved functional prediction of genes and gene products, phylogenomics can also provide information relevant to understanding processes driving the evolution of genes, genomes, and organisms. Additional informatics tools, such as Hidden Markov modeling (HMM) and 3D structural homology methods, might enable further identification of protein homologues or conserved protein domains.
High-quality annotation of both ‘unknowns’ and conserved hypotheticals can also be inferred using pathway completion and orthology considerations. Pathway reconstruction can aid recognition of gene-enzyme mapping with high confidence using pathway hole-filling, in which sequences are assigned protein functions based on a combination of nonsequence- and pathway-based information [4]. Orthology-based inference can annotate originally unannotated genes but may lead to erroneous annotation due to the multidomain structure and/or nonspecific properties of the protein.
Annotation Validation
After identification of putative proteins of interest, validation might begin by cloning and sequencing of full-length cDNAs, to confirm the sequence data available in the database. This implies a ‘gene by gene’ approach, which might not be feasible for full genomic analysis. Whole-genome- or tissue/stage-specific RNA-Seq can be also used for confirmation of annotation, which can reveal genes annotated as ‘hypothetical’. RNA-Seq library constructions with as little as 1 ng total RNA, purified mRNA, or rRNA-depleted RNA, are now feasible. Current ‘long read’ DNA sequencing technologies (e.g., Pacific Biosciences and Oxford Nanopore) are being applied to long RNA molecule sequencing. Using RNA-Seq analysis, the first in-depth gonad-specific transcriptome analysis of Schistosoma mansoni suggests that ‘hypotheticals’ possess specific and unknown functions in somatic and reproductive tissues, especially in male testes [8]. Recent developments, such as terminator exonuclease (TEX) and Cappable-Seq, methods, allow direct enrichment for the 5′ end of primary transcripts, enabling determination of transcription start sites at single-base resolution. This can lead to promoter determinations and analysis of potential functional operons.
Proteomics (in particular, mass spectrometry) also provides a significant tool which can be applied to functional analysis. Proteogenomic analysis [mass spectroscopy coupled to liquid chromatography (LC MS/MS)] can identify protein sequences that might not be in RNA-Seq or DNA-Seq databases, representing independent information or confirmation of protein presence. When possible, functional genomics tools, such as RNAi or CRISPR, can then be used to validate results. While not universally technically robust as of yet, these functional genomics tools can hopefully be applicable to other parasitic helminths to identify gene functionality in previously nontractable organisms (‘reverse genetics’). Recent work in Strongyloides stercoralis has shown that techniques applied to C. elegans, including gene transformation and CRISPR/Cas-9 gene silencing, can be adapted [6]. Currently, RNAi is the most accessible and employed tool to knockdown target genes in order to validate functions in parasite or in host–parasite interaction [7]. This approach has been efficient to validate drug targets in S. mansoni [5], Brugia malayi [9], and Onchocerca volvulus [10].
Annotation of Regulatory RNA Sequences from Genomic Data
When one considers genome annotation, it is worth considering small regulatory RNAs, particularly microRNAs (miRNAs), as key regulators of gene expression at the post-transcriptional level. Small RNA sequencing has identified various classes of regulatory RNAs from helminths. Recent work in Echinococcus [11] demonstrated a high level of expression of conserved hypothetical proteins and novel miRNAs. A computational tool was developed that identifies miRNA precursors with high confidence based on several nested self-organizing maps (SOMs). This approach was also tested with Echinococcus multilocularis and Taenia solium genome datasets and validated several of the discovered miRNAs [11]. This methodology can be adapted to any draft genome, including those from nonmodel parasitic helminths.
Small interfering RNAs (siRNAs) involved in RNAi gene silencing, and piwi-interacting RNAs (piRNAs) involved in transposon silencing, have also been identified from some nematode species. A pipeline to identify these RNA classes, as well as miRNAs from Haemonchus contortus and Brugia pahangi, has been developed where most (70%) of the miRNAs identified were unique to Haemonchus or Brugia [12]. This pipeline can be applied to other helminths, and it relied on deep sequencing, mapping reads to the available genomes and application of miRNA prediction programs.
Concluding Remarks
Many of the most interesting genes for a complete understanding of parasite life cycles, host–parasite interactions, and directed drug discovery may still encode ‘hypothetical proteins’. Ultimately, there is no substitute for biologists manually inspecting and curating their favorite genes. As more genomic data become available for parasitic and free-living nematodes, a community-driven approach can aid in curation and provide due diligence regarding deposition of novel helminth sequences into appropriate databases, such as COMBREX. A ‘Gold Standard’ database would provide a repository for annotation (and reannotation) improvements. Such a database would contain genes/proteins with published or publicly available experimentally verified function along with sequence and strain identifications. Additionally, analysis of small RNAs regulating gene expression are needed. The database would encourage involvement of scientists to test the function of high-value predictions within their area of expertise using their own laboratory assays. Other experimental possibilities can be envisioned – such as protein or RNA crystal structures, or methods such as proteome or ‘reactome’ arrays.
Acknowledgments
Finally, funding agencies should be encouraged to support methods and approaches which can help alleviate the bottleneck in our complete understanding of genomic biological function. In our opinion, funding for sequencing should be accompanied by funding for annotation to improve understanding of parasite biology.
Glossary
- BLAST (Basic Local Alignment Search Tool)
program that compares regions of similarity between biological sequences (nucleotide or protein) and calculates the statistical significance (https://blast.ncbi.nlm.nih.gov/Blast.cgi)
- BRENDA (BRaunschweig ENzyme DAtabase)
comprehensive relational database on functional and molecular information of enzymes, based on primary literature (http://www.brenda-enzymes.org)
- COMBREX (COMputational BRidges to EXperiments)
a database resource of information related to experimentally determined gene transcript and/or protein function, predicted protein function, and relationships among proteins of unknown function (http://combrex.bu.edu)
- Conserved hypotheticals
proteins that are found in organisms from several phylogenetic lineages but have not been functionally characterized.
- DETECT (Density Estimation Tool for Enzyme ClassificaTion)
a probabilistic method for enzyme prediction that accounts for varying sequence diversity in different enzyme families (http://www.compsysbio.org/projects/DETECT)
- Diagnostic domains
protein regions associated with a particular biochemical function.
- EFICAz2 (Enzyme Function Inference by a Combined Approach)
web-based resource that applies a multicomponent approach for high-precision enzyme function prediction (http://cssb.biology.gatech.edu/skolnick/webservice/EFICAz2/index.html)
- Gene Ontology (GO)
web resource that provides structured, controlled vocabularies and classifications that cover several domains of molecular and cellular biology and are freely available for community use in the annotation of genes, gene products, and sequences (http://www.geneontology.org)
- InterProScan
linux- and web-based tool that scans protein sequences against the InterPro (Integrated Resource of Protein Domains and Functional Sites) protein signature databases (http://www.ebi.ac.uk/interpro/interproscan.html)
- KEGG (Kyoto Encyclopedia of Genes and Genomes)
database resource for understanding high-level functions and utilities of the biological system, such as the cell, the organism, and the ecosystem, from molecular-level information, especially large-scale molecular datasets generated by genome sequencing and other high-throughput experimental technologies (http://www.genome.jp/kegg)
- Nested Self-Organizing Maps (SOM)
data visualization technique that reduces high dimensional data through the use of self-organizing neural networks.
- Orthology
sequences present in different species that evolved from a common ancestor by speciation. Normally, orthologs retain the same function in the course of evolution and are thus critical for reliable prediction of gene function in newly sequenced genomes.
- Phylogenomics
the application of phylogenetic analysis to annotate complete genome sequences using DNA and RNA sequences.
- PRIAM (profils pour l’i dentification a utomatisée du m étabolisme)
method for the automatic detection of likely enzymes in protein sequences using precomputed sequence profiles (http://priam.prabi.fr)
- UniProt (Universal Protein Knowledgebase)
web-based resource of comprehensive, high-quality and freely accessible protein sequences and functional information (http://www.uniprot.org)
- Unknowns
proteins for which there is no functional database assignments and no prediction of biochemical activity.
- WormBase ParaSite
web-accessible central data repository for information about Caenorhabditis elegans and related nematodes (http://www.wormbase.org)
References:
- 1.Schnoes AM, et al. Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol, 5 (2009), Article e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Leale G, et al. Inferring unknown biological functions by integration of GO annotations and gene expression data. IEEE/ACM Trans. Comput. Biol. Bioinform, 99 (2016), pp. 1–19 arXiv:1608.03672 [DOI] [PubMed] [Google Scholar]
- 3.Silva LL, et al. The Schistosoma mansoni phylome: using evolutionary genomics to gain insight into a parasite’s biology. BMC Genomics, 13 (2012), p. 617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Green ML, Karp PD. A Bayesian method for identifying missing enzymes in predicted metabolic pathway databases. BMC Bioinformatics, 5 (2004), p. 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Štefanić S, et al. RNA interference in Schistosoma mansoni Schistosomula: selectivity, sensitivity and operation for larger-scale screening. PLoS Negl. Trop. Dis., 4 (2010), Article e850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lok J, et al. Transgenesis in Strongyloides and related parasitic nematodes: historical perspectives, current functional genomic applications and progress towards gene disruption and editing. Parasitology, 144 (2017), pp. 327–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.de Moraes Mourão M, et al. Phenotypic screen of early-developing larvae of the blood fluke, Schistosoma mansoni, using RNA interference. PLoS Negl. Trop. Dis., 3 (2009), Article e502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lu Z, et al. Schistosome sex matters: a deep view into gonad-specific and pairing-dependent transcriptomes reveals a complex gender interplay. Sci. Rep, 6 (2016), Article 31150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Landmann F, et al. Efficient in vitro RNA interference and immunofluorescence-based phenotype analysis in a human parasitic nematode, Brugia malayi. Parasit. Vectors, 5 (2012), p. 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lustigman S, et al. RNA interference targeting cathepsin L and Z-like cysteine proteases of Onchocerca volvulus confirmed their essential function during L3 molting. Mol. Biochem. Parasitol, 138 (2004), pp. 165–170 [DOI] [PubMed] [Google Scholar]
- 11.Kamenetzky L, et al. MicroRNA discovery in the human parasite Echinococcus multilocularis from genome-wide data. Genomics, 107 (2016), pp. 274–280 [DOI] [PubMed] [Google Scholar]
- 12.Winter AD, et al. Diversity in parasitic nematode genomes: the microRNAs of Brugia pahangi and Haemonchus contortus are largely novel. BMC Genomics, 13 (2012), p. 4. [DOI] [PMC free article] [PubMed] [Google Scholar]