

SUMMARY
Viral rebound upon stopping combined antiretroviral therapy poses a major barrier toward an HIV cure. Cellular and anatomical sources responsible for reinitiating viral replication remain a subject of ardent debate, despite extensive research efforts. To unravel the source of rebounding viruses, we conducted a large-scale HIV-STAR (HIV-1 sequencing before analytical treatment interruption to identify the anatomically relevant HIV reservoir) clinical trial. We collected samples from 11 participants and compared the genetic composition of (pro)viruses collected under treatment from different cellular and anatomical compartments with that of plasma viruses sampled during analytical treatment interruption. We found a remarkably heterogeneous source of viral rebound. In addition, irrespective of the compartment or cell subset, genetically identical viral expansions played a significant role in viral rebound. Our study suggests that although there does not seem to be a primary source for rebound HIV, cellular proliferation is an important driver of HIV persistence and should therefore be considered in future curative strategies.
In Brief
De Scheerder et al. conduct an in-depth investigation into the origins of HIV rebound. They show that viral rebound originates from multiple compartments and cell proliferation is a driver of viral persistence. Future HIV cure strategies will need to overcome the challenges associated with heterogeneous viral rebound.
Graphical Abstract
INTRODUCTION
Although major advances have been made in identifying cells and anatomical compartments that contribute to the HIV latent reservoir, it is still unclear which of these reservoirs are responsible for viral rebound during treatment interruption in vivo (Barton et al., 2016; Chun et al., 2010). HIV is integrated into the genome of various immune cells, and considerable debate exists about which subset represents the actual latent reservoir, defined as the intact and replication-competent virus capable of viral rebound during treatment interruption (Eisele and Siliciano, 2012; Kandathil et al., 2016). The therapeutic implications of identifying such a specific reservoir are tremendous because targeting specific anatomical sites and/or cell subsets that harbor the large majority of replication-competent virus may increase the efficiency of HIV cure efforts in patients on long-term suppressive combined antiretroviral therapy (cART) (Lederman et al., 2016; Margolis et al., 2016).
Although different CD4+ T cell subsets have been identified as major contributors to the HIV-1 reservoir (Chomont et al., 2009; Hiener et al., 2017; Lee et al., 2017b) and different surface markers (CXCR3, CD30, and CD32a) (Banga et al., 2018; Descours et al., 2017; Henrich et al., 2017) and immune exhaustion markers (PD-1, TIGIT, and LAG-3) (Khoury et al., 2017; McGary et al., 2017) have emerged as potential biomarkers for latently infected cells, previous studies have not converged on a clear pattern or predominant subsets responsible for HIV persistence. Furthermore, lately, the focus has been extended from the blood compartment to cell subsets in lymph node (LN) and gut-associated lymphoid tissue (GALT), which were shown to constitute important sanctuaries for persistent HIV infection (Banga et al., 2016; Barton et al., 2016; Lorenzo-Redondo et al., 2016; Rothenberger et al., 2015). Overall, this suggests that curative strategies will most likely need to target a broad range of cells in various tissue reservoirs.
The fragmented, and to some extent, inconsistent knowledge about HIV persistence is partly due to restricted access to human tissue, limited sample sizes, and technical limitations. Moreover, viral rebound can only be truly assessed in vivo by an analytical treatment interruption trial (ATI) in fully suppressed HIV-1 infected individuals (Clarridge et al., 2018; Garner et al., 2017), and better insight in the viral reservoir can be achieved when combined with sequencing techniques to quantitatively and qualitatively assess the reservoir (Bruner et al., 2016; Hiener et al., 2017; Ho et al., 2013; Lee et al., 2017a). Due to the complexity and ethical considerations associated with ATI, replication competence has mostly been investigated in vitro using the quantitative viral outgrowth assay (qVOA) (Finzi et al., 1997). However, the in vitro replication competency might be a poor proxy of the in vivo rebound capacity (Bui et al., 2017; Ho et al., 2013; Hosmane et al., 2017).
The HIV-STAR clinical trial addresses these shortcomings by combining in-depth sampling with cell sorting in 11 chronically infected HIV-1 positive participants on long-term cART and after ATI. In order to conduct in-depth phylogenetic analyses, we generated proviral env V1–V3 sequences from different viral reservoirs (three anatomical reservoirs, seven cell subsets) on cART as well as plasma-derived env sequences before cART (T0), on cART (T1) and after ATI (T2, T3, and T4), employing single-genome sequencing (Josefsson et al., 2012). This represents the most exhaustive sampling (about >400 sequences per participant) of virus populations from different reservoirs and rebound viruses to date, allowing for a much-needed comprehensive investigation into the origins of rebound viruses.
RESULTS
In-Depth HIV Sampling before and during ATI
Twelve HIV-1 positive individuals were included in the study (STAR 1–12; Table S1, see STAR Methods for inclusion and exclusion criteria). STAR 1 was not subjected to the treatment interruption phase and therefore not included in further analyses. The study setup involved sampling of peripheral blood mononuclear cells (PBMCs), GALT, LN, plasma, and cerebrospinal fluid (CSF) on cART (time point 1, T1 in Figure 1). CD45+ cells from GALT; central (TCM), transitional (TTM), and effector (TEM) memory and naïve (TN) CD4+ T cells from PBMCs; and memory CD4+ T cell subsets (TCM and TEM) were sorted from LN (STAR Methods; Figure S5 for gating strategy). In addition, plasma was sampled prior to treatment initiation (time point 0, T0), 8 to 15 days after ATI (time point 2, T2), at the first detectable viral load (time point 3, T3), and at rebound (time point 4, T4) (Figure 1). The median time to viral rebound was 21 days (range 15–36). To determine the source of viral rebound, we characterized proviral DNA derived from a broad range of cell subsets (T1) and plasma RNA (T0-T4), and we obtained V1–V3 env sequences using single-genome sequencing (Josefsson et al., 2012). In 5 out of 11 participants, we also managed to obtain RNA sequences from CSF (Table 1), and for two participants (STAR 4 and 10), we analyzed additional sequence data generated from bone marrow (BM) samples.
Figure 1. In-Depth HIV Sampling before and during ATI.
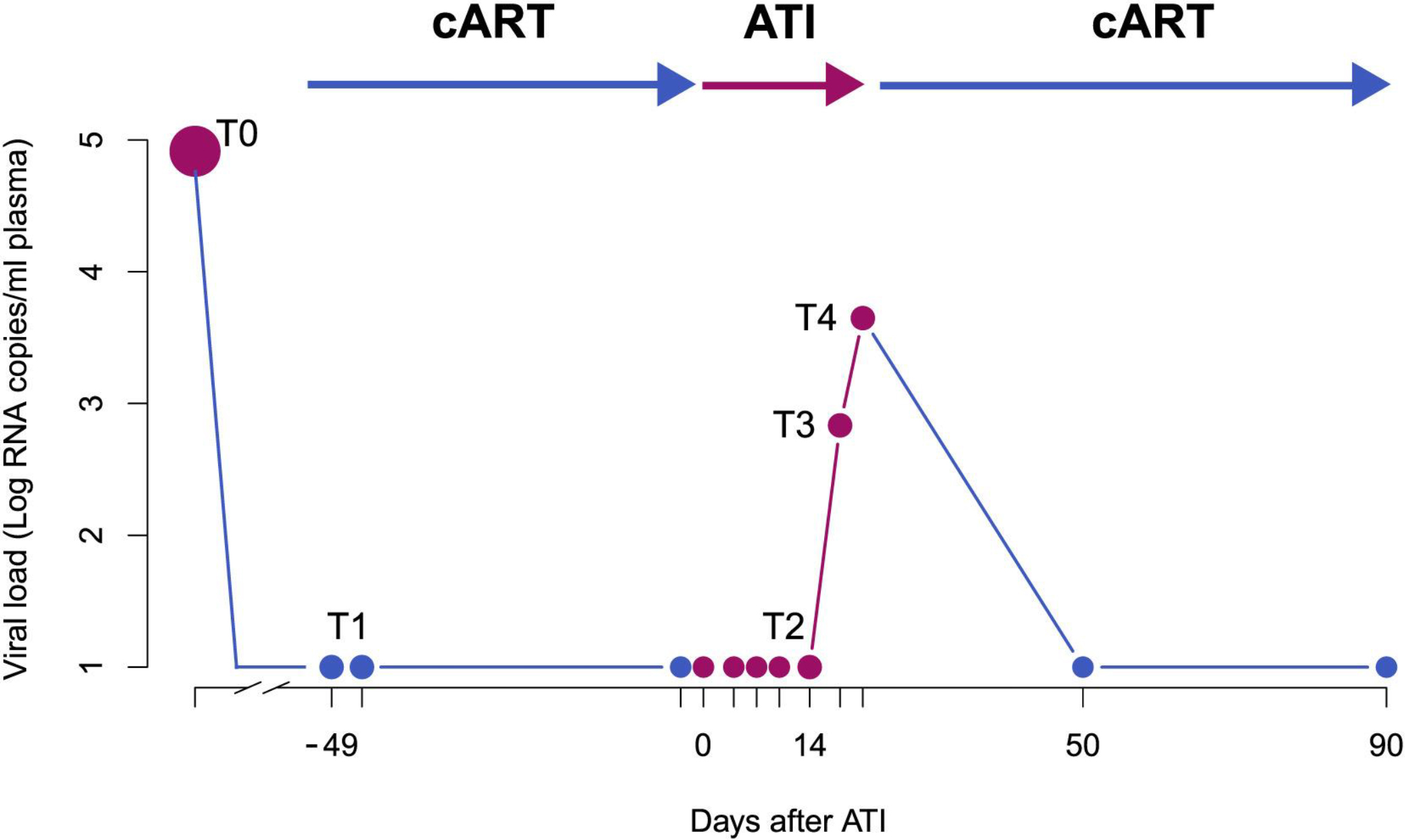
T1 represents the sampling under cART, including tissue sampling and leukapheresis within the same week; upon ATI (day 0 on the x axis), blood samples were taken every 2–3 days. T2 represents the time point of leukapheresis after ATI (8–15 days after day 0); T3 represents the first detectable viral load (>30 copies/mL) and T4 is defined as the time of viral rebound (>1,000 copies/mL or at second measurement of >200 copies/mL). Treatment was re-initiated immediately after the sampling at T4 and participants were intensively monitored until undetectable viral load in plasma was achieved (<20 copies/mL). T0 represents the time of plasma sampling prior to initial treatment initiation. The dots represent the sampling points on cART (blue), off cART (pink).
Table 1.
In-Depth HIV-1 Reservoir Characterization
| STAR 2 | STAR 3 | STAR 4 | STAR 5 | STAR 6 | STAR 7 | STAR 8 | STAR 9 | STAR 10 | STAR 11 | STAR 12 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
| ||||||||||||
| TCM blood | Total N | 30 | 34 | 48 | 37 | 31 | 41 | 40 | 35 | 39 | 41 | 34 |
| Prop intact | 0.83 | 0.91 | 0.71 | 0.62 | 0.94 | 0.61 | 0.93 | 0.74 | 1.00 | 0.88 | 0.91 | |
| Prop identical | 0.07 | 0.50 | 0.19 | 0.41 | 0.00 | 0.29 | 0.38 | 0.11 | 0.41 | 0.15 | 0.53 | |
| Infection frequency | 158 | 140 | 4658 | 970 | 397 | 219 | 190 | 2140 | 726 | 1157 | 125 | |
| TEM blood | Total N | 35 | 26 | 43 | 1 | 1 | 20 | 27 | 40 | 41 | 38 | 7 |
| Prop intact | 0.94 | 0.92 | 0.93 | 1.00 | 1.00 | 0.85 | 0.93 | 0.78 | 0.95 | 0.92 | 1.00 | |
| Prop identical | 0.49 | 0.73 | 0.63 | 0.00 | 0.00 | 0.50 | 0.59 | 0.55 | 0.83 | 0.55 | 0.57 | |
| Infection frequency | 518 | 272 | 1468 | NA | NA | 146 | 4612 | 671 | 332 | 802 | 47 | |
| TTM blood | Total N | 28 | 33 | 32 | 38 | 30 | 21 | 40 | 31 | 31 | 16 | 15 |
| Prop intact | 0.89 | 0.85 | 0.56 | 0.84 | 0.83 | 0.71 | 0.95 | 0.68 | 0.97 | 0.88 | 0.93 | |
| Prop identical | 0.29 | 0.58 | 0.41 | 0.47 | 0.20 | 0.33 | 0.28 | 0.16 | 0.35 | 0.69 | 0.67 | |
| Infection frequency | 278 | 761 | 422 | 735 | 257 | 37 | 446 | 1209 | 173 | 220 | 43 | |
| TN blood | Total N | 30 | 35 | 32 | 30 | 1 | 27 | 33 | 28 | 33 | 22 | 1 |
| Prop intact | 0.97 | 0.97 | 0.84 | 0.83 | 1.00 | 0.89 | 0.91 | 0.96 | 0.94 | 0.91 | 1.00 | |
| Prop identical | 0.23 | 0.60 | 0.28 | 0.43 | 0.00 | 0.07 | 0.55 | 0.07 | 0.73 | 0.36 | 0.00 | |
| Infection frequency | 76 | 199 | 584 | 282 | N/A | 26 | 30 | 43 | 447 | 19 | N/A | |
| TCM LN/GALT | Total N | 38 | 33 | 31 | 32 | 36 | 37 | 32 | N/A | 34 | 31 | 33 |
| Prop intact | 0.95 | 0.94 | 0.71 | 0.78 | 0.97 | 0.95 | 0.84 | N/A | 0.94 | 0.87 | 0.94 | |
| Prop identical | 0.16 | 0.64 | 0.00 | 0.25 | 0.17 | 0.05 | 0.13 | N/A | 0.38 | 0.19 | 0.67 | |
| Infection frequency | 292 | 190 | 911 | 555 | 200 | 92 | 154 | N/A | 467 | 110 | 29 | |
| TEM LN/GALT | Total N | 33 | 28 | 23 | 35 | 38 | 14 | 19 | 19 | 0 | 2 | 0 |
| Prop intact | 0.76 | 0.96 | 0.87 | 0.89 | 0.92 | 0.71 | 0.89 | 0.95 | 0.00 | 0.50 | 0.00 | |
| Prop identical | 0.45 | 0.61 | 0.52 | 0.69 | 0.24 | 0.00 | 0.68 | 0.32 | 0.00 | 0.00 | 0.00 | |
| Infection frequency | 504 | 728 | 665 | 786 | 334 | 95 | 297 | 2373 | N/A | N/A | N/A | |
| CD45 GALT | Total N | 26 | 29 | 29 | 36 | 29 | 36 | 13 | 30 | 38 | 31 | 39 |
| Prop intact | 0.92 | 0.86 | 0.59 | 0.81 | 0.93 | 0.89 | 0.92 | 0.87 | 0.95 | 0.94 | 1.00 | |
| Prop identical | 0.31 | 0.76 | 0.24 | 0.36 | 0.28 | 0.67 | 0.62 | 0.20 | 0.63 | 0.71 | 0.90 | |
| Infection frequency | 121 | 168 | 447 | 151 | 52 | 393 | 76 | 756 | 196 | 3632 | 341 | |
| Intact V1V3env RNA plasma | Total N | 163 | 102 | 72 | 108 | 125 | 125 | 104 | 101 | 133 | 106 | 72 |
| T0 | 29 | 30 | 0 | 27 | 40 | 23 | 37 | 27 | 51 | 32 | 31 | |
| T1 | 12 | 8 | 1 | 24 | 11 | 22 | 0 | 0 | 0 | 5 | 1 | |
| T2 | 8 | 5 | 12 | 0 | 0 | 19 | 0 | 4 | 21 | 2 | 0 | |
| T3 | 25 | 24 | 28 | 28 | 41 | 29 | 29 | 40 | 28 | 33 | 6 | |
| T4 | 89 | 35 | 31 | 29 | 33 | 32 | 38 | 30 | 33 | 34 | 34 | |
| Intact V1V3env RNA CSF | T1 | 0 | 1 | 2 | 0 | 0 | 2 | 1 | 0 | N/A | 1 | 0 |
| T4 | 0 | 2 | 1 | 0 | 0 | 1 | 2 | 0 | N/A | 1 | 0 | |
For each participant and sampling site, at time point 1 (compartment and cell subset) the total number of sequences (N), the number of sequences that were intact in the sequenced region (from V1 to V3) and the number of sequences that were identical within the subset is given as a proportion of the total N. The estimated infection frequency is based on the absolute cell N input and the N of sequences obtained. Infection frequency was presented as positive cells per 106. We also provide the N of intact RNA sequences from plasma and CSF at the different sampling points.
To ensure high-quality sequence data in subsequent statistical and phylogenetic analyses, we only included those sequences with a mean quality score of Q20 (99% accuracy) and excluded all defective sequences prior to and following the generation of the contigs (Barton et al., 2016; Ewing and Green, 1998; Josefsson et al., 2012; von Stockenstrom et al., 2015) (see STAR Methods, Table S3). For the phylogenetic analyses, recombinant lineages contributing to viral rebound were considered in separate downstream analyses (Martin et al., 2015). The quality control and filtering steps resulted in a total of 4,329 env sequences (an average of 393 sequences per participant, range 240–535) for further analyses.
In-Depth HIV-1 Reservoir Characterization Shows a Variability in Identical Sequence Expansions, Intact V1-V3 env Sequences, and Infection Rates between Subsets and Compartments
We first sought to identify whether substantial differences existed between cell subsets and anatomical compartments in the extent of identical sequence expansions, provirus intactness within the sequenced region, and infection frequency (Table 1; Figure 2).
Figure 2. Scatter Dot Plot Representing the Data from Table 1, Sorted by Cell Subset.
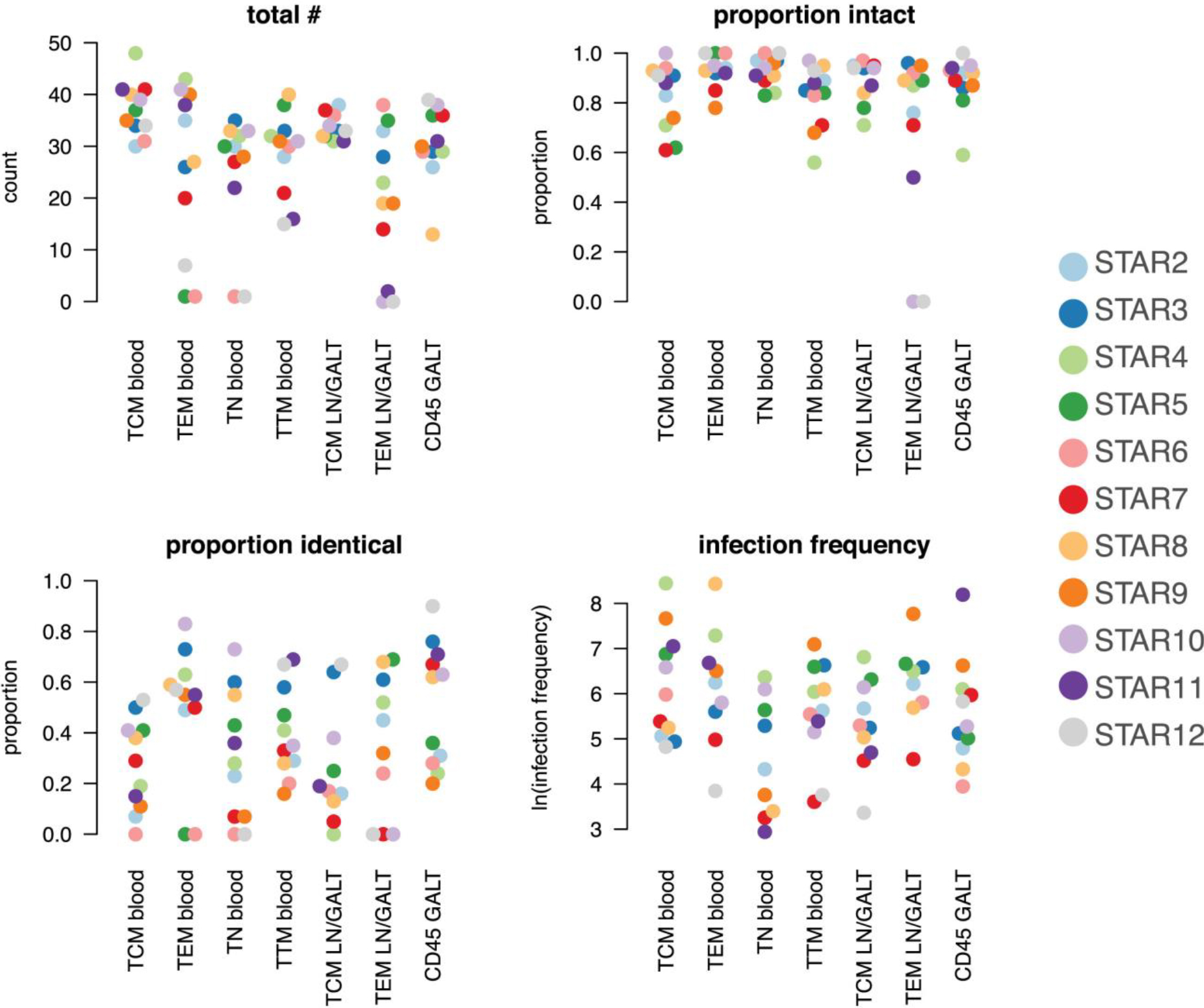
The y axis respectively shows the total N of sequences, the proportion of intact and identical sequences and the log-transformed infection frequency. The legend indicates the color used for each participant.
The contribution of the various cell subsets and anatomical compartments to the persistence of HIV infection through cellular proliferation was quantified by the proportion of identical proviral env sequences within each of the different cell subsets. This proportion and its distribution between anatomical sites and cell subsets were highly variable between participants (effect modification p < 0.001).
Across cell subsets, we found strong evidence for differences in the level of genetically identical HIV sequences (p < 0.001). Overall within the sample across participants and cell subsets TEM had the highest proportion of identical sequences (p < 0.001, OR = 2.47 95% CI 1.78–3.43); TEM from blood contained more genetically identical V1-V3 env sequences than TEM sequences from LN (p = 0.02, OR = 1.63 95% CI 1.08–2.46). At the level of anatomical compartments, we found evidence for a higher average proportion of identical V1-V3 env sequences in GALT than the LN and blood (p < 0.001, OR = 1.94 95% CI 1.51–2.50). However, this could be explained by including only CD45+ immune cells in GALT rather than differentiating between TCM and TEM.
As rebound viruses can only emerge from intact proviruses, we also scrutinized the proportion of intact V1-V3 env sequences across cell subsets and compartments. We consider the relative proportion of intact viruses in V1-V3 env to reflect relative probabilities to find intact viruses in the subsets. The proportion of genetically intact proviruses (within the V1-V3 env region) varied across the cell subsets (p < 0.001) and across participants (p = 0.04). Within the participants, TN from blood had on average the highest proportion of intact sequences (p = 0.01, OR = 1.81 95% CI 1.15–2.87) followed by TEM from blood (p = 0.02 when comparing to the other subsets excluding TN, OR = 1.70 95% CI 1.08–2.66). There was no evidence for significant variation across the anatomical compartments (p = 0.17).
Estimates of HIV infection frequency of each cell subset were also found to heavily depend on the individual participant (effect modification with cell subsets p < 0.001). On average, TEM from blood or LN and TCM from blood had the highest rate of infection (OR = 2.67 95% CI 2.45–2.92 for TEM blood or LN and TCM from blood combined against all others, p < 0.001) while TN had the lowest.
Overall, these results paint a complex reservoir picture with broad seeding and important inter-participant variability, with TEM frequently harboring high levels of genetically identical and intact proviruses.
Heterogeneous Cellular and Anatomical Reservoir Contributions to HIV Rebound
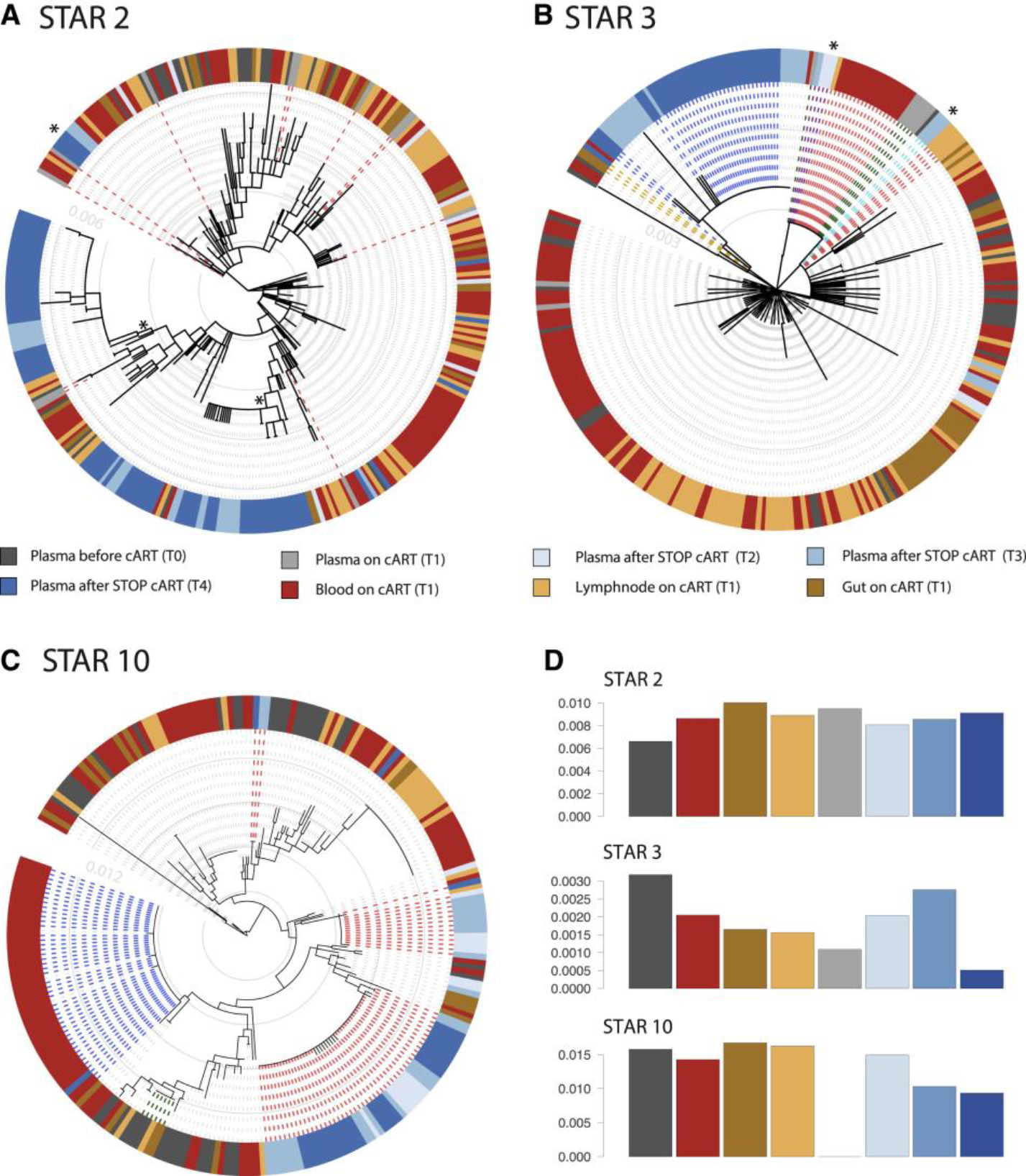
To further characterize the sources of viral rebound during ATI, we performed phylogenetic reconstruction for the viral population in each participant. Figure 3 shows the phylogenetic trees from 3 participants (trees from all participants are provided in Figure S1A). Overall, the phylogenetic diversity was relatively low, as reflected by an expected pairwise number of env V1-V3 substitutions ranging between 3 to 13 across participants, with the exception of STAR 7 with an average of 26 substitutions.
Figure 3. Heterogeneous Cellular and Anatomical Reservoir Contributions to HIV Rebound with Cellular Proliferation as a Potential Driver.
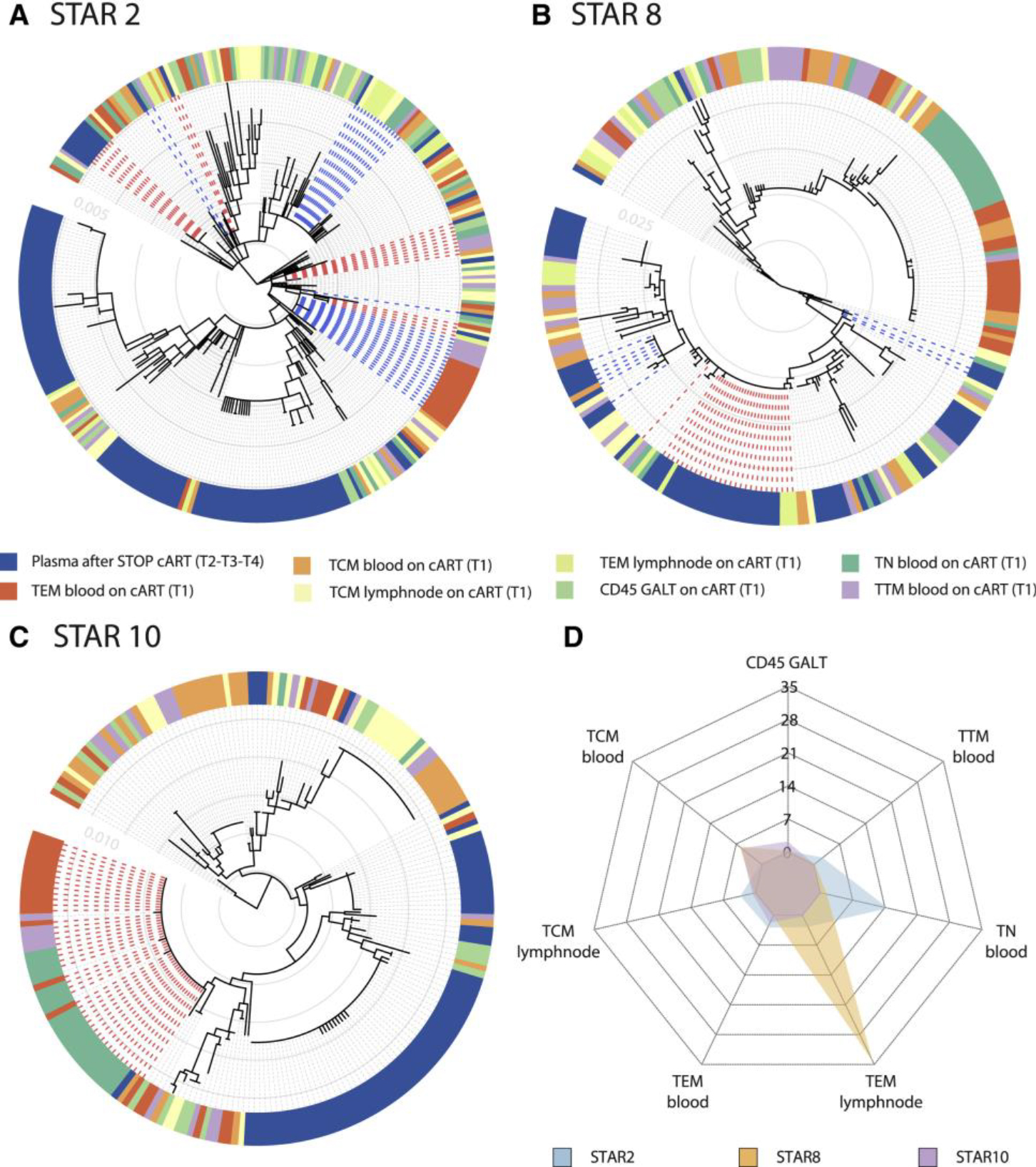
Within-host ancestor-descendant relationships between the viruses from different cell types for 3 participants and a radar plot representing the variability in viral rebound source.
(A–C) Maximum likelihood phylogenetic trees from three selected participants representing the sequences from T1 cell subsets from blood (TCM, TEM, TTM, and TN), LN (TCM and TEM), and GALT (CD45+ cells) before ATI. Plasma viruses from time points T2, T3, and T4 are grouped as plasma after STOP cART. The colored strip represents sampling origin for each sequence as indicated by the legend. The trees are drawn to scale and the gray circles represent the branch length from the root expressed as the number of substitutions per site. The scale values are given in the inset (light gray numbers). The heterogeneity in potential reservoir contribution is indicated by the color mixing in the strip. Identical cellular DNA V1–V3 sequence expansions that are identical to plasma RNA sequences after cART STOP were highlighted in the trees by the bold dashed lines, with the expansions colored in red and blue alternatingly. Trees from all participants are provided in Figure S1.
(D) The radar plot representing, for each of the 3 participants, the estimated number of times that a rebound virus lineage originates from the respective cell subsets (depicted as the numbers from 0 to 35). The legend indicates the color used for each participant. A radar plot across all participants is available in Figure S4.
The phylogenetic trees showed a variable degree of intermixing of rebound viruses with proviruses from the different cell subsets and anatomical compartments obtained during cART. Visualizing genetic similarity using haplotype networks further confirmed the diverse origins of the rebound viruses (Figure S2).
To further unravel to what extent specific compartments can act as the source of rebound viruses, we employed a phylogeographic approach that is commonly used in molecular epidemiological research (Bloomquist et al., 2010; Faria et al., 2011) and quantified the rebound virus emergence events from each cell subset. The estimated number of rebound virus emergence events for 3 participants are summarized in a radar plot in Figure 3D and across participants in Figure S4. These analyses revealed not only a large number of independent rebound events but also a high inter-participant variability in terms of which particular cell subsets were fueling rebound viruses (Figure S4). While TEM in LN dominates in one participant (STAR 8), different cell subsets appeared to contribute to rebound in other participants (STAR 2 and 10) (Figure 3D). In line with the scenario of a heterogeneous reservoir seeding and the lack of a consistent pattern in the HIV rebound dynamics, we found that there was also a substantial degree of mixing of viruses across anatomic compartments (Figures 3A–3C and S1). Using estimates of the phylogenetic association index (Wang et al., 2001), we further quantify the extent to which sequences cluster by compartment (Table S2). Phylogenetic association indices range between 0 for absolute clustering by compartment (no mixing) and 1 for randomized clustering by compartment (panmictic). In line with the degree of phylogenetic intermingling we observe in the trees, our estimates suggest a considerable degree of mixing that significantly deviates from fully structured populations by compartments but also not to the extent that mixing is consistent with randomized clustering.
Interestingly, in 6 out of 11 participants, we found CD45RA+ CD45R0− cells derived from blood to have intact V1-V3 env sequences identical to viral sequences found in plasma after viral rebound. This was observed both as part of a large expansion of genetically identical sequences shared over different cell subsets (Figures 3C and S2B; STAR 10) or as a unique sequence genetically identical to a rebound cluster (Figures 3A and 3B). Although these markers were primarily used to sort naïve T cells, we cannot exclude a small fraction of stem memory T Cells (TSCM), which were previously reported to be enriched in HIV (Buzon et al., 2014). In 2 participants (STAR 4 and 10), we further analyzed additional sequences derived from CD4+ T cells isolated from BM (Figure S1C). These sequences are dispersed across the trees and do not show any compartmental structure, nor is their clustering indicative of a particular role in viral rebound. In addition, we analyzed CSF from 5 out of 11 participants; however, the amount of sequences that we obtained was insufficient to use them for downstream analysis (Figure S1D).
Cellular Proliferation Is a Driver of Viral Rebound
To investigate the importance of cellular proliferation in the rebound dynamics, we subsequently focused on proviruses from cell subsets at T1 that were genetically identical to a plasma virus after ATI (T2-T3-T4). To illustrate this, we highlighted the identical sequence expansions that match rebound sequences from T2-T3 or T4 on the phylogenetic trees (Figures 3A–3C, red and blue dashed lines).
We provide a table in the Supplemental Information (Table S4), which represents the absolute number of sequences that are 100% identical to viral rebound for all participants. In addition, we show the number of sequences with a >99% match to take into account viral and assay error rates. Overall, these data revealed that cell subsets with large expansions of identical sequences (presumably clonally expanded) contain a higher proportion of sequences that were identical to rebound plasma virus (p = 0.003); this strongly suggests a role of cellular proliferation as a driver for viral rebound. Although we could not consistently identify a specific cellular or anatomical compartment responsible for viral rebound based on phylogenetic relationships and we observe a high inter-participant variability, these data confirm that effector memory CD4+ T cells (TEM) contribute to the latent reservoir, as this cell subset showed the highest proportion of identical sequence expansions (as previously discussed and illustrated in Table 1).
To identify differences between cellular and anatomical compartments, we looked at the magnitude of contributions to rebound as measured by the proportion of proviral sequences that are identical to plasma sequences after ATI from different anatomical compartments and different cell subsets at the individual sequence level (Table S4). When considering 100% matches, we find no evidence for a difference in rebound contributions across anatomical sites at the individual sequence level (p = 0.12). However, when viral and assay error rates > 99% were taken into account, we did find some evidence for a difference (p = 0.028). Here, we observe the blood, gut, and then LN to contribute to rebound, respectively, from low to high, but individual comparisons between these were not significant (p > 0.05). At the individual sequence level, we find evidence that rebound contributions are different across cell subsets both for 100% and for >99% matches (p < 0.001). Although, on average, we observe the order from lowest to highest as CD45 < TCM < TN < TTM < TEM for 100% matches, this was highly variable across participants (effect modification p < 0.001) and changed if >99% matches were considered instead. This result shows that all cell subsets, including TEM, play an important role in viral rebound. We conducted a second analysis where identical sequences were reduced to a single representative sequence to avoid counting the same expansion multiple times (Figure 4; Table S4). When taking into account sequence expansions so that identical sequences are only counted once, we no longer find evidence for a difference in rebound contribution across anatomical sites (p = 0.34) or cell subsets (p = 0.28), further indicating that all anatomical compartments and cell subsets are potential sources of viral rebound and that cellular proliferation is an important driver for these rebound events (Figures 3 and 4; Table S4).
Figure 4. Contributions of Anatomical and Cellular Compartments to Viral Rebound.
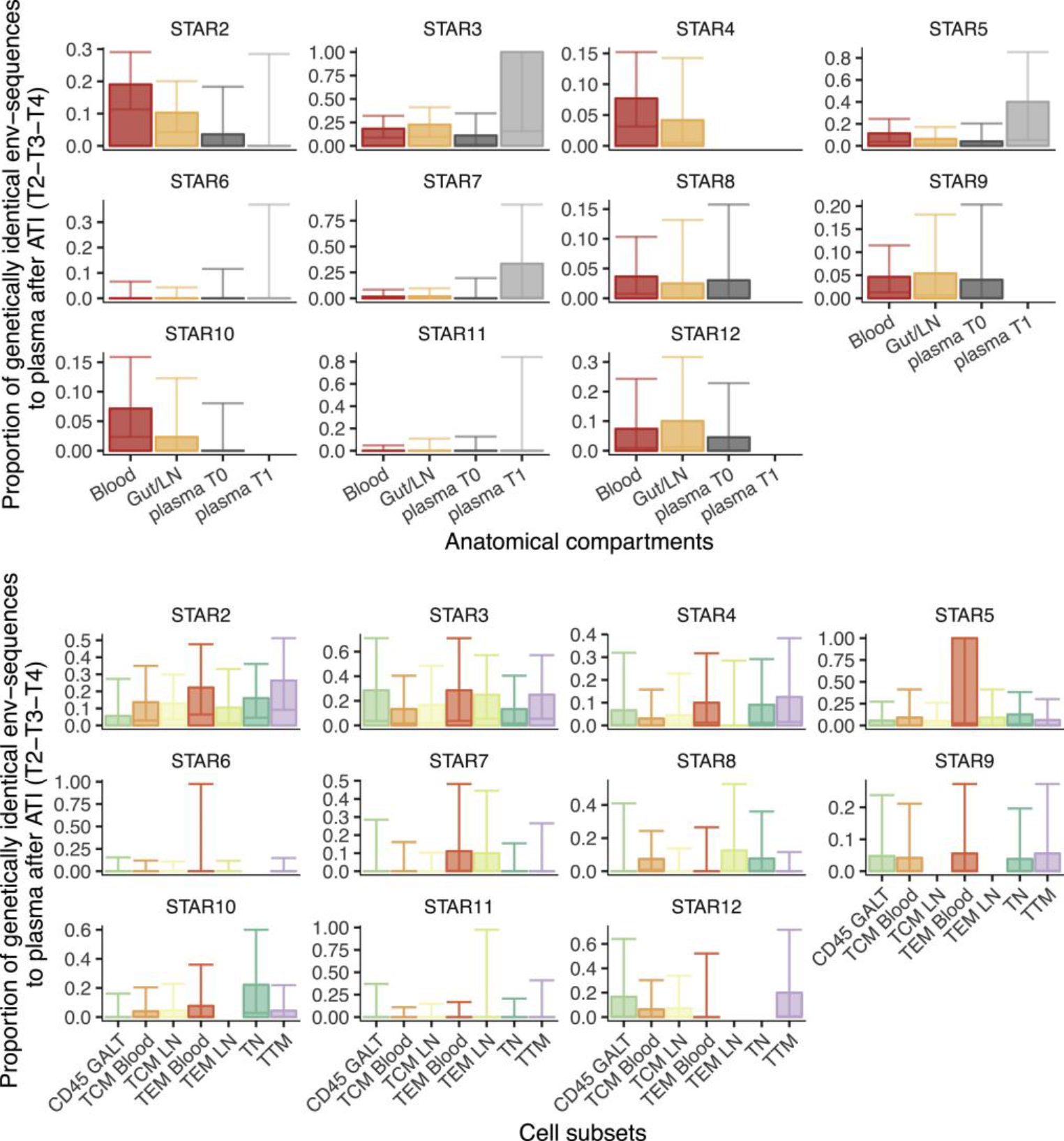
(A) Bar plots representing the proportion of env sequences from anatomical compartments T1 and plasma T0/T1 (x axis) genetically identical to plasma viruses collected after ATI (T2-T3-T4 combined) (y axis). Sequences obtained from LN and GALT were pooled together. Compartments were compared with T0 and T1 plasma-derived env sequences. Data were normalized for genetically identical expansions. Error bars represent the confidence intervals (CIs). When CIs are presented without bar, this means that we obtained sequences but none were identical. The absence of bars or CIs reflects that this particular data point was missing or we obtained too few sequences (<5) to include it in the analysis.
(B) Bar plots from STAR 2–>12 representing the proportion of proviral env sequences genetically identical to plasma virus after ATI (T2-T3-T4 combined) (y axis) between the different cell subsets (x axis). The data were normalized for genetically identical expansions within the subsets. Error bars represent the CIs. When CIs are presented without bar, this means that we obtained sequences, but none were identical. The absence of bars or CIs reflects that this particular data point was missing or we obtained too few sequences (<5) to include it in the analysis.
Evidence for Kinetic Variability and Stochastic Reactivation of Rebound Viruses after Treatment Interruption
Finally, we investigate in more detail the plasma RNA sequences collected at the different time points T0 > T4). In Figure 5, we highlight the plasma sequences at the various time points of sample collection in the phylogenetic trees for 3 participants (trees from all participants are provided in Figure S1B).
Figure 5. Evidence for Kinetic Variability and Stochastic Reactivation of Rebound Viruses after Treatment Interruption.
(A–D) Within-host ancestor-descendant relationships between the viruses from different anatomical compartments and the RNA plasma sequences collected at different time points for 3 participants and nucleotide diversity estimates. Shown are maximum likelihood phylogenetic trees highlighting the sequences obtained from plasma RNA at the different time points of sequencing T0, T1, T2, T3, and T4. (A) represents STAR 2 where the red dashed lines represent the many different plasma virus lineages detected at T1, consisting of mostly unique sequences. (B) represents STAR 3 with the green dashed lines representing identical plasma virus expansions at T1 that are identical to plasma viruses collected at later time points T2 and T3 (indicated in purple). The red dashed lines represent proviruses from the reservoir that are identical to sequences found in the plasma at T2 and T3 (indicated in purple and light blue). The blue dashed lines represent T4 rebound viruses, for which the link with the proviral reservoir is less likely in this participant than at T2 and T3. The orange dashed lines show a single rebound virus collected at T4 that is identical to a provirus from GALT. (C) represents STAR 10 where the red dashed lines represent the typical clustering of rebound viruses at T2, T3, and T4 as observed for most participants. The blue dashed lines represent a T4 rebound sequence with identical proviruses from several cell subsets but not observed earlier in the plasma at T2/T3. The green dashed line shows how expansions of virus at T0 persist over time in the reservoir. The total number of rebound events is approximated by the number of “blue blocks” (i.e., closely related rebound viruses) in the colored circle surrounding to the phylogenies, as these usually correspond to a single rebound event from the reservoir. However, when identical rebound viruses are also identical to a reservoir virus, that particular reservoir origin will be the most likely inferred state at the parental node of the terminal branches leading to these identical rebound sequences. We mark such lineages that are estimated to have arisen through multiple rebound events with an asterisk. Trees from all participants are provided in Figure S1B. (D) represents for these participants various levels of nucleotide diversity (y axis) in the different compartments and plasma time points (x axis). Nucleotide diversity for all particants is available in Figure S3.
First, we assessed whether residual viremia during cART can predict the source of viral rebound. In the 6 participants for whom we were able to sequence the plasma virus on cART at T1, we observed two distinct patterns. In most of these individuals, identical viral sequences dominate in the plasma at T1 (Figure 5B, green dashed lines), but in some individuals, we observe multiple unique variants (Figure 5A, red dashed lines). Remarkably when clones dominate the residual viremia, they were more likely to be identical to virus obtained in the plasma at later time points, suggesting that these viruses represent a part of the infectious reservoir and are able to fuel viral rebound after ATI (Figure 5B, green dashed lines; Figure S2A STAR 7; and Figure S2C STAR 7 right circle). Similarly, we found plasma-derived clonal sequences before start cART at T0 that matched intracellular HIV-1 DNA sequences at T1 and rebound viruses at later time points after ATI (Figure 5C, green dashed lines; Figure S2A STAR 12), again emphasizing the role of proliferation in the persistence of the viral reservoir and their contribution to viral rebound.
Second, we wondered whether we could observe evolution in the viral sequences collected at different time points after ATI. We found that plasma viruses collected after ATI (T2, T3, and T4) were usually intermixed (Figure 5C, red dashed lines). Confirming this mixed phylogenetic clustering, we did not find any pronounced phylogenetic association by time point of sampling (Table S2). The phylogenetic patterns indicate multiple independent rebound viruses or rebound lineages of highly variable size (Figure S4). Even when the rebound virus was largely represented by a single lineage there remained an extensive intermixing of sequences from the other compartments, which is in line with a heterogeneous reservoir origin (Figure 5B, red dashed lines; Figure S2A STAR 3 left circle). The nucleotide diversity profiles also show that the rebound virus diversity is generally comparable to or only slightly lower than the genetic diversity of the HIV-1 DNA sequences from the cell subsets and anatomical compartments, which is expected when the virus from multiple cell subsets and anatomical sites contribute to the plasma virus after ATI (Figures 5D and S3).
However, in some participants, we were able to detect distinct patterns of rebound kinetics. In STAR 3, we observe a clear outgrowth of plasma virus over time (Figure 5B, blue dashed lines; Figure S2A STAR 3 right circle). This is confirmed by the nucleotide diversity analysis, wherein some participants a decrease in nucleotide diversity between T3 and T4 is observed, suggesting enrichment of a particular viral strain over time (Figures 5D and S3). In some other participants, we observed that plasma RNA sequences at T4 are genetically identical to the HIV-1 DNA sequences sampled at T1, but no genetic match was found between the HIV-1 DNA sequences and the previous plasma-derived RNA sequences from time points T2 and T3 (Figure 5C, blue dashed lines; Figure S2B STAR 10), indicating these reservoirs reactivated at a later time point during the ATI. These individual findings suggest stochastic cellular reactivation and kinetic variability of viral rebound after treatment interruption. Plotting intact plasma-derived sequences over time (T0–T4) does not suggest any selection or enrichment of replication-competent virus across patients (Figure S4C).
DISCUSSION
Overall, the most important finding from this extensive phylogenetic and statistical analyses based on the largest and most in-depth sequencing data of the HIV reservoir is that the rebound virus can originate from several cellular and anatomical compartments after treatment interruption. The substantial inter-participant variability further supports that there is no prominent source of rebound viruses. We observed a clear link between the rebound viruses and their source in some participants, whereas in others, there were multiple potential contributors or the source was unclear. This explains why studies of only a few participants have pointed at various sources of rebound viruses, often in an inconsistent manner. The sample size in our study, in terms of the number of participants but importantly also in terms of the number of independent rebound lineages detected in each participant, offers strong support against HIV rebound seeded by a dominant reservoir.
In line with a broad seeding of the reservoir and the lack of clear structure in the HIV rebound dynamics, our results support a substantial degree of mixing of viruses across anatomical compartments. Moreover, our study revealed identical env V1-V3 sequences in different cellular and anatomical compartments, indicating dynamic interchanges between compartments and little evidence for viral evolution or clustering by site. The observed expansions of genetically identical HIV sequences during cART are the result of homeostatic and/or antigen-driven cellular proliferation, rather than resulting from ongoing viral replication (Banga et al., 2018; Kearney et al., 2017; Rosenbloom et al., 2017). In participants with groups of identical proviral env V1-V3 sequences, our data indicate that these expansions play an important role in viral rebound, confirming that cellular proliferation is a crucial driver of viral persistence, irrespective of the compartment or cell subset (Josefsson et al., 2013; von Stockenstrom et al., 2015; Wang et al., 2018). In this regard, cellular proliferation may further increase TEM contribution to rebound. Interestingly these data also showed that unique cellular sources such as CD45RO−/RA+ T cells, consisting of a majority (> 90%) naïve T cells (cf. STAR Methods), although we cannot exclude a small fraction of TSCM, which were previously reported to be enriched in HIV (Buzon et al, 2014) and unique anatomical sources such as GALT can contribute to viral rebound. We did not find clear evidence that the presence of viral production during cART can predict the dynamics of viral rebound; however, it can reveal the presence of dominant clones responsible for viral rebound.
In our unique study setup, we assess replication competence by comparing proviral sequences on cART to the rebound sequences in plasma after ATI. Therefore we focused on the highly variable V1-V3 env region (~0.8 kb) (Kearney et al., 2009; von Stockenstrom et al., 2015), and although we can therefore only account for identical and intactness of the sequenced region, modeling and previous studies have frequently used env sequences as a surrogate and showed that multiple identical env sequences provide a strong support for clonal expansion (Hosmane et al., 2017; Laskey et al., 2016). Several studies have therefore used the envelope region to identify the source of the rebound viremia (Barton et al., 2016; Lu et al., 2018).
More recently, new strategies studying near full-length proviral genome sequences are being presented and allow for even more in-depth analysis of the HIV reservoir (Bruner et al., 2016; Hiener et al., 2017; Lee et al., 2017a); however, any comparison with rebound plasma-derived HIV RNA in these studies is based on a particular HIV subgenomic region due to the fact that full-length HIV RNA assays are not currently available and therefore do not contribute to a better understanding of rebound dynamics. One of the major problems with full-length sequencing at this stage is the relative paucity of full-length HIV sequences generated, therefore requiring a much higher cellular input. This major challenge is probably the reason why no studies on tissue reservoirs employing near full-length sequencing have been published so far. Although we recognize that implementing such a technique would in theory increase resolution and provide more direct proof of intactness and clonality of the available sequences, the current state-of-the-art cannot strengthen the answer to the central research question (Wang and Palmer, 2018), and we are confident that the strategy we choose is a valuable surrogate for this extensive analysis of the viral reservoir and above all has little impact on our overall conclusion of heterogeneous viral rebound. However, we acknowledge that sequencing a subgenomic region overestimates the number of intact proviruses (Lee et al., 2017b). Furthermore, without integration site analysis, we are unable to determine if identical V1–V3 env sequences are derived from clonal expansions of the same cell (Cohn et al., 2015).
Our findings confirm the results of others (Cohen et al., 2018; Salantes et al., 2018; Winckelmann et al., 2017) that only a small fraction of plasma RNA env sequences sampled at ATI (T2-T3-T4) were genetically identical to viral sequences from the cells and anatomical sites sampled at T1 (approx. 20%, with a large range between participants). This suggests that although the origin of the rebound virus is very heterogeneous and diverse, only a small subset of this total pool of infected cells contributes to viremia (Kearney et al., 2016). Another potential explanation for mismatches between proviral and rebound sequences could be recombination, as discussed by Cohen et al. (Cohen et al., 2018). Determining the amount of viral recombination that contributes to the rebound virus within the env V1-V3 fragment is challenging to assess. Therefore, to correct for errors made during viral replication and the error rate of the assay, we determined the number of cell-derived sequences with greater than 99% identity to rebound viral sequences. In doing so, we found a significant increase in the number of genetically matching sequences (approximately 70%). However, this analysis did not change the conclusion that viral rebound is heterogeneous and cannot be attributed to one specific reservoir. By doing the >99% analysis, we furthermore took into consideration the potential assay error rate (calculated at 0.011% [Palmer et al., 2005]) and the potential bias associated with post-rebound diversification. However, based on the short post-rebound period and previous within-host evolutionary rate estimates this diversification can be considered as negligible. Indeed, if we consider evolutionary rate estimates for a similar env region obtained for 9 patients (Bielejec et al., 2016; Shankarappa et al., 1999), the highest average rate was estimated to be 0.001 subst/month (or 0.012 subst/year). So, for the longest post-rebound period of 36 days, we expect, on average, 0.0012 subst/site. For an alignment length of about 750 bp, this leads to an expectation of 0.9 substitutions. So, any sequence sampled 36 days post-rebound is expected to have accumulated only about one substitution since rebound. In other words, almost all diversity we observed in the post-rebound sequences has already been established. Depending on the participant and the cellular subsets, we see a mostly large amount of rebound lineages. Since immune pressure changes to the virus seem rather unlikely (cf. the analysis done above), we suggest that the main reason is the unique in-depth sequencing in this study providing several anatomical and cellular sources of viral rebound.
Although primer mismatch when sequencing a single and variable subgenomic region can occur, the env primers we used in our assay are HIV-1 subtype B specific and were chosen because they bind to the most conserved regions of the V1-V3 sequences in the Los Alamos HIV database and therefore are not selective for specific viral populations within the HIV-1 subtype B (Kearney et al., 2009).
While sampling bias can affect genetic analyses and one rebound event over time cannot capture the entire complexity of viral rebound, the substantial number of participants investigated and the depth at which they were sampled ensures that this will have little impact on our overall conclusion. As already suggested by others (Cohen et al., 2018; Wang et al., 2018), our data support stochastic reactivation, in line with our overall hypothesis of heterogenic contributions to rebound viruses.
In conclusion, our comprehensive study clearly demonstrates the complexity of latency mechanisms and the challenges this brings about for strategies aimed at purging the reservoir. Our data show that genetically identical viral expansions play a significant role in viral rebound. Focusing on mechanisms that drive antigenic and homeostatic proliferation of immune cells (Chomont et al., 2009; Hosmane et al., 2017) will be crucial to achieve progress toward an HIV cure. Although recent cure interventions (Mendoza et al., 2018) are promising, they also confirm that the response to treatment is heterogeneous among the trial participants and that an individual approach and a better insight into the mechanisms of viral persistence together with in-depth mapping of these latent reservoirs will be necessary to overcome the challenge of random reactivation.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact Linos Vandekerckhove (linos.vandekerckhove@ugent.be). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
The study protocol, experimental design, and recruitment strategy were approved by the Ethics Committee of the University Hospital of Ghent (Belgian registration number: B670201525474). Participants were recruited from the AIDS reference clinic at the Ghent University Hospital based on strict inclusion criteria and after intensive counseling (NCT02641756). Written informed consent was obtained from all participants. Twelve HIV-1 infected, long-term treated participants under chronic ART were included. The median age of the participants was 40y (range 32–56y). Two participants initiated cART during early infection (<6 months, STAR 3 and 5), while the remaining participants received ART during chronic infection (range 1–9 year after seroconversion). One participant was excluded from the second part of the study because of pre-existing resistance to ART, an exclusion criterion for treatment interruption. There were no drop-outs and no deviation from the protocol was reported, except for STAR 4 for which a post-hoc lower CD4 nadir was reported. The study population consisted of 11 male and 1 female subjects. Because of the lack of more female participants, we could not perform relevant analyses on the influence of gender on our study results. Participant characteristics are presented in Table S1.
METHOD DETAILS
Inclusion-and Exclusion Criteria
Inclusion and exclusion criteria are presented in NCT02641756 on clinicaltrial.gov. Twelve HIV-1 subtype B-positive individuals on long-term suppressed ART (2–11y), with stable immune reconstitution (normal CD4 count defined as > 500 CD4/μl or >30%) at time of inclusion and suppressed viral load (<20 copies/ml) for at least 2 years were recruited. All participants were treated with cART consisting of an integrase inhibitor combined with a solid backbone (in all cases 2 NRTIs). The switch to the required treatment regimen was done at least 3 months before the first sampling time point (T1, see Figure 1). Viral rebound was defined as 1 measurement with >1000 HIV RNA copies/ml or 2 consecutive measurements with >200 HIV RNA copies/ml. Randomization was not relevant for this study design.
HIV-1 subtype was defined using the Smartgene Integrated Database Network System’ (Smartgene, Lausanne, Switzerland) and viral tropism was assessed with the Web PSSM tool provided by the Mullins lab, University of Washington (Jensen et al., 2003).
Clinical Study Design And Sample Collection
A screening blood analysis (3 × 9ml EDTA tubes) was performed to assess eligibility based on general biological markers including CD4 count and viral load suppression before enrollment in the first part of the study. The total HIV-1 DNA levels in PBMCs was assessed using ddPCR (Kiselinova et al., 2016).
Under cART
(T1) participants underwent in-depth sampling consisting of lumbar puncture (LP), bone marrow (BM) aspirate, inguinal lymph node resection, bronchoscopy with bronchoalveolar lavage (BAL), gastroscopy and colonoscopy, leukapheresis, urine and vaginal fluid/sperm collection. Participants were admitted at the day-clinic of Ghent University Hospital and procedures took place consecutively. LP with collection of 100 droplets cerebrospinal fluid was done before short narcosis. Under narcosis, a BM aspirate at the posterior superior iliac crest was performed collecting 12ml in a 20ml syringe prefilled with 1 ml of heparine to prevent clotting. Afterwards a gastroscopy and colonoscopy were performed collecting punch biopsies of 3–4mm each at 3 sites (10 duodenal biopsies, 10 ileum biopsies and 20 colon biopsies). An inguinal lymph node excision (1–2 nodes) was performed after ultrasound localization and a BAL concluded the sampling procedures. The lavage was done with physiological water aiming for a 50ml recuperation after aspiration. Leukapheresis was done within one week of the in-depth sampling (+/− 3 days) and in function of participants’ availability. The leukapheresis was not combined with the other sampling procedures because of an increased bleeding risk due to potential thrombocytopenia related to the procedure. We collected an average of 5 billion PBMCs and 100ml plasma. A urine sample and semen sample were collected. Vaginal fluid collection was done at the gynecology department.
If there were medical contraindications for any of the proposed interventions, they were left out or replaced (e.g. peripheral blood draw instead of leukapheresis). If abnormalities were reported during the exams, this was communicated to the participants and adequate follow-up was arranged for.
Approximately 3 months after the initial sampling, STAR 2 to STAR 12 underwent a monitored analytical treatment interruption (ATI). A second screening blood analysis was done before the ATI to ensure that the inclusion criteria were still met. Blood draws were organized twice per week with strict follow-up of viral load and CD4 count. Participants were also interrogated on clinical symptoms related to the treatment stop. A second leukapheresis was done between day 8 and day 15 after the treatment stop (T2). At viral rebound (>1000 copies/ml or 2 consecutive measurements with >200 HIV RNA copies/ml) a less extensive sampling of blood (12× 9ml EDTA tubes), cerebrospinal fluid, urine and genital fluids was performed (T4). Treatment was immediately restarted and a follow-up blood sample was taken after 1 month to assess viral response to re-initiation of the therapy. The study was finalized with a last blood sample approx. 3 months after undetectable viral load. T3 was identified as the first detectable VL > 30 copies/ml. T0 was defined as available plasma samples before cART initiation (Figure 1).
Samples were stored on ice until processing in the appropriate collection reservoir and medium (see the Sample processing section). All samples were, on average, processed within 2 hours after collection.
Sample Processing
Experiments were not repeated due to the limited amount of sample available, however sampling processing was optimized on a trial participant (STAR 1).
Gut Associated Lymphoid Tissue (GALT):
GALT samples were collected in 15ml Falcon tubes containing 5ml of RPMI complete (10%FCS, 5% L-glutamine 100x, 5% Penicillin/Streptomycine 100x) and additional antibiotics and antifungals (Piperacillin/tazobactam 500μg/ml and amphotericin B 1.25μg/ml). Cells were isolated following protocol as previously described (Morón-López et al., 2017). Briefly, biopsies were rinsed and incubated in HBSS (Thermo Fisher) containing 1mM DTT (Sigma) and 1mM EDTA (Gibco) for 25min at room temperature and shaken to remove the epithelial layer. Then, biopsies were transferred to the complete media and cultured overnight (at 5%CO2) in 6-well low-binding plates (Costar) at 1–2 biopsies/well. Culture supernatants were collected to recover the released cells and the remaining tissue was disrupted by shaking (10min) and gently pipetting. Cell suspensions were separated from tissue debris through a 40μm mesh filter. Cells were stained for 30 min at 4°C with the following antibodies: CD45 (PercpCy5.5), CD3 (BB), CD8(PeCy7) and sorted based on CD45 expression. Cells were spun down and dry pellets were used for further analysis (see below).
Lymph Node (LN):
Lymph nodes were collected in a 50ml Falcon tube in 20ml of RPMI complete. They were transferred to a sterile plate and cleaned to remove as much fat as possible. A fraction was used for cell isolation and washed over a 70μm mesh filter. The cellular material is pushed through the filter by mechanically disrupting the tissue. The filter is washed with RPMI or PBS to ensure maximum recuperation of the cells. Cells are washed and spun down and re-suspended in the adequate volume for downstream experiments. CD4 negative selection using the BD CD4 enrichment kit was performed and CD4 cells were sorted based on the expression of the following markers: CD3 (BB), CD8 (PeCY7), CD45RO (PE) and CD27 (APC).
Peripheral Blood Mononuclear Cells (PBMCs):
PBMCs were isolated from blood or leukapheresis using leucoSep tubes and lymphoprep density gradient separation following manufacturers protocol. PBMCs were collected and counted and a fraction was used for downstream experiments. CD4 negative selection using the BD CD4 enrichment kit was performed and CD4 cells were sorted using fluorescence-activated cell-sorting, based on the expression of the following markers: CD3 (BB), CD8 (PeCY7), CD45RO (PE-PERCPCY5.5)), CD27 (APC), CD45RA (APC) and CCR7 (PE).
Cell sorting was performed using BD FACS Jazz flow cytometer, selecting the lymphocyte gate, singlets and live cells. For these experiments, we used the following sorted subsets (GALT CD45, LN TCM (CD45RO+CD27+) and TEM (CD45RO+CD27−), Blood TCM (CD45RO+CD27+), TEM (CD45RO+CD27−), TTM (CD45RO+, CD27+ CCR7−) and TN (CD45RO−, CD45RA+). The number of cells obtained after sorting ranged from 17.377–4.290.000. The average purity of the sorted subsets was 95%. Sorting strategy can be found in the Figure S5. FlowJo software was used to analyze the data.
Considering our gating strategy for T Naïve, we acknowledge that these might indeed include a small fraction of TSCM and TEMRA, however this fraction is so low, it is hardly likely to influence our results. We provide an example of the sorting strategy where we show that few cells do not express CD27 (<10%), and from the CD27+ population only a fraction were CD95 positive (<1%), therefore concluding that our target cells were indeed mostly TN cells (Figure S5).
Bone Marrow (BM):
A bone marrow aspirate was performed at the posterior iliac crest. Approximately 10–12 mL was collected in a 20ml syringe with 1 mL of heparine to prevent clothing. The aspirate was diluted with PBS and the cell fraction was isolated by ficoll gradient and washed over a 40μm mesh filter. The cells were counted and a fraction was stained for subsequent sorting of CD34+ Lin− cells and CD4+ Lin + cells.
Cerebrospinal Fluid (CSF):
RNA extraction was performed after ultracentrifugation as described below.
BAL, urine, and semen samples were not included in further downstream analysis due to the low amount of cell numbers and/or viral DNA/RNA which did not allow for sequencing analysis.
We obtained a limited amount of sequences from CSF in 5/11 participants (Table 1). These sequences were not used for downstream analysis but were represented in Figure S1 panel D.
We sequenced HIV-1 env V1-V3 from CD4+ T cells from bone marrow samples in 2 participants to illustrate that they intermingle within the phylogenetic trees and do not constitute a separate reservoir (Figure S1C) As expected, we were not able to sequence from HPC cells derived from bone marrow.
We did not obtain cells from LN in 1 participant (STAR 9). Therefore, more GALT samples were taken and sorted not only into the CD45 subset but also into TCM and TEM. We did not obtain any good sequences from TCM but did obtain sequences from TEM. These were further analysed together with the LN TEM from the other participants as illustrated in Table 1.
Single-Genome/Proviral Sequencing
We performed single-genome/proviral sequencing of HIV-1 env (V1-V3) as previously described (Josefsson et al., 2012; von Stockenstrom et al., 2015). Briefly, cell pellets were lysed in 100μl home-made lysis buffer (10mM TRisHCl, 0.5%NP-40, 0.5%Tween-20 and proteinase K 20mg/ml) and DNA was extracted by incubating the samples at 55°C for 1 h and subsequently at 85° for 15min. Virus from plasma and CSF was first concentrated by ultracentrifugation. Briefly the sample was diluted using Tris buffered saline and transferred to ultracentrifuge tubes (Optiseal). (Dahl et al., 2014; Palmer et al., 2003). After centrifugation the supernatant was removed and RNA was extracted using an in-house protocol. First TrisHCL pH8 and proteinase K were added to the pellet and incubated at 56°C for 30 min. After that Guanidium thiocyanate and Glycogen were added. After 15 min Isopropanolol was added and the sample was centrifuged for 30 min. Supernatant was then removed and two washing steps with ethanol 70% followed. cDNA was generated using SuperScript III Reverse Transcriptase after adding dNTPs (PCR nucleotide mix) and gene-specific primers (E115 for env) to the dissolved pellet. All reactions without reverse transcriptase step (no RT controls) were negative, excluding contamination by cellular debris. DNA and cDNA were serially diluted and nested PCR primers specific to the HIV env region were used to obtain PCR products derived from single HIV-1 DNA molecules (i.e. at the first dilution for which ≤30% positive reactions per cell type are observed). Additional PCRs were performed at the target dilution to obtain 30–40 single proviral sequences where possible. The following primers were used for amplification: Round 1 forward (E20) 5′-GGGCCACACATGCCTGTGTACCCACAG-3′ and reverse (E115) 5′-AGAAAAATTCCCCTCCACAATTAA-3′; round 2, forward (E30) 5′-GTGTACCCACAGACCCCAGCCCACAAG-3′ and reverse (E125) 5′-CAATTTCTGGGTCCCCTCCTGAGG-3′. The Platinum Taq PCR polymerase and reaction mix were used. For round 1 of PCR, the following thermocycler parameters were used: 94 °C for 2 min, 94 °C for 30 s, 50 °C for 30 s, 72 °C for 1 min, 44 cycles of steps 2–4 and 72 °C for 3 min. For round 2 of PCR, the following thermocycler parameters were used: 94 °C for 30 s, 55 °C for 30 s, 72 °C for 1 min, 41 cycles of steps 1–3 and 72 °C for 3 min. Read-out of positive reactions was done using E-gel 96 well 1% agarose. The PCR products representing single HIV-1 sequences were sequenced using Sanger sequencing (Australian Genome Research Facility, Sydney, Australia).
QUANTIFICATION AND STATISTICAL ANALYSIS
Phylogenetic Analysis
Low-quality sequences (rejecting sequences using a mean quality score of Q20(Ewing and Green, 1998)), potential mixtures and defective sequences were eliminated such that only high-quality contigs were used for further analysis (Table S3). Contigs were generated from the raw sequencing data using an in-house computer program written in Perl scripting language (available upon request). Sequences were aligned using MUSCLE (Edgar, 2004) in MEGA7 (http://www.megasoftware.net/) and several tools were used to eliminate potentially defective sequences (G-A Hypermutated sequences identified by the Hypermut tool; quality control tool detecting frameshifts and stop codons, available at: http://www.hiv.lanl.gov) and by visually inspecting the amino-acid sequences for premature stop codons. These procedures were consistently applied in order to only further explore intact sequences within the V1-V3 region. Neighbor joining trees were constructed including a reference HIV-1 subtype B sequence and positive controls to exclude lab contaminations and inter-participant cross-contamination. Proviral sequences obtained from cell subsets at T2 and T4 and CSF derived sequence data were excluded from downstream analyses respectively because of the risk of reinfection bias after ATI and because of their low overall abundance.
For each participant, the sequence data were aligned using TranslatorX (Abascal et al., 2010) or MAFFT (Katoh and Standley, 2013) and manually edited in AliView (Larsson, 2014). There were clear indications for superinfection of one participant (STAR6), and the rebound dynamics were independently investigated for both lineages (hereafter named clades A and B), which clustered in monophyletic groups with 100% support. We only show the phylogenies of the variant that contributed to virus rebound. Recombinant sequences and their parents were identified with RDP4 (Martin et al., 2015). We attempted to preserve an as large as possible dataset by either excluding the recombinant sequences or the major or minor parental sequences. Recombinant groups that were removed from the main alignment but that included rebound viruses were interrogated separately. Evolutionary relationships between the virus isolates was estimated with maximum likelihood (ML)methods implemented in PhyML v3.0 (Guindon and Gascuel, 2003). The substitution process was modelled as an HKY+ G process (Yang, 1994) and tree space was searched using the best of NNI and SPR rearrangements. To quantify the contribution of cell subsets and anatomical compartments to rebounding viruses, we computed estimates of the posterior expected number of times (Markov jumps) that each cell subset and compartment seeded rebound virus detected during ATI through stochastic mapping techniques (Minin and Suchard, 2008a; Minin and Suchard, 2008b) from the ML trees.
We performed phylogenetic association using BaTS (Parker et al., 2008) to assess the level of gene flow between anatomical compartments (blood, plasma, central nervous system, lymph node and gut) and between time points (T0, T1, T2, T3 and T4). This analysis is based on the phylogenetic association value (Wang et al., 2001) a measure of the degree of clustering by trait that can be rescaled in to a phylogenetic association index by dividing them by phylogenetic association values obtained for randomized traits on trees. This ratio takes on values between 0 (absolute clustering by trait) and 1 (randomized clustering by trait). The expectations for the Association index (AI) under the null hypothesis of panmixia were obtained from 1000 taxon-character randomisations, and were compared against the phylogeny-trait association measured from 500 phylogenies sampled at regular intervals from the post-burn-in posterior distribution. The latter was estimated with MrBayes v3.6.2 (Ronquist et al., 2012), specifying a HKY+Γ substitution model (Yang, 1994) and the default (unconstrained) priors on the topology and branch lengths. Mixing and convergence properties of the Markov chain were investigated with Tracer (https://github.com/beast-dev/tracer/releases/tag/v1.7.1).
Details on the data subsetting and filtering steps and the analyses performed on each data set are provided in Table S3. Phylogenies were visualized with FigTree v.1.4.3 (http://beast.community/figtree) or iTol v.3 (Letunic and Bork, 2016)
As a graphical representation alternative to maximum likelihood trees, one haplotype network was created per participant using the median-joining method (Bandelt et al., 1999) in NETWORK v.5.0.0.3 (available at http://www.fluxus-engineering.com) and plotted with the R function “networkGraph” available with the toolbox SPADS 1.0 (Dellicour and Mardulyn, 2014) (Figure S2). In these figures, each haplotype (i.e. unique sequence) is represented by a circle, the size of which is proportional to its overall sampling frequency in a given participant. The genetic relatedness between haplotypes are represented by lines. Each line segment in the networks represents a single mutational change, or multiple changes when the line is annotated by a number indicating the number of mutations. SPADS (Dellicour and Mardulyn, 2014) was also used to compute the nucleotide diversity (Nei and Li, 1979) within different sequence subsets (Figure S3).
Statistical Analysis
The sample size of this study (11 participants after exclusion, approximately >400 sequences per participant) was not determined by a sample size calculation. Rather the number of participants was limited by availability and eligibility, and the number of sequences per participant was cost and resource limited. A-prior power calculations were not possible primarily due to the unknown level of variability across participants.
For determining the sequences which were 100% genetically identical, we used Biopython to read and compare the pairwise distance between each HIV-1 env sequence within each participant’s sequence alignment (Cock et al., 2009). The sequence alignment is the input file for our in-house Biopython script and within this sequence alignment each HIV-1 env sequence has an unique identifier. The Biopython script generates an output file that lists those sequences with 100% genetic identity by their unique sequence identifier. We did not find any discrepancy when the results from the Biopython script were compared to the ElimDupes tool from Los Alamos HIV database using 100% genetic identity as the analysis parameter (https://www.hiv.lanl.gov/content/sequence/ELIMDUPES/ElimDupesExplain.html). A sequence was classified as identical if it was a 100% match against another sequences sampled from the same time point, anatomical compartment, cells subset and participant. Once identified, the proportion of identical sequences was calculated as the total number of sequences classified as identical for that group, divided by the total number of sequences for that group. ElimDupes 99% identity mentioned in this manuscript is in fact >99% which includes 99.5%, 99.9% and 100% genetic identity.
Multivariable logistic regression carried out in R version 3.4.1 using function glm, was used to compare the proportion of sequences that were intact, identical (clonal), and identical to rebound. The independent variables in these analyses were participant, cell subset, cell subset with TEM and TCM subsetted amongst anatomical locations, anatomical location, and time point of plasma collection (when appropriate). Anatomical location and cell subset frequently could not be included both due to codependency. Effect modification between an independent variable and participants was frequently strong, however models including this effect modification were unstable with poor standard errors on parameter estimates due to low sample sizes in some participant and cell subset combinations. Considering this, comparisons across subsets of the independent variable were made without considering the effect modification. Additionally, 11 participants is too limited to make population wide inferences. Therefore p-values comparing subsets of an independent variable should be interpreted as evidence for average effects within participants of this sample, rather than an indicator of evidence for population wide trends. To avoid the impact of large expansions on the proportion of identical sequences to rebound virus, clusters of identical sequences were reduced to a single sequence per subset. This reduces variability considerably, but also decreases the statistical power by reducing the number of sequences. In conducting this analysis, the TCM-derived sequences from the blood and LN were pooled and the TEM-derived sequences from the blood and LN were combined to increase sample size as there is no evidence for compartmentalization between these compartments.
The number of cells positive for HIV within a PCR/sequencing replicate was calculated as the proportion of wells positive for HIV by PCR multiplied by the proportion of sequences sequenced from the plate that were truly positive for HIV (defined as high quality forward and reverse sequences, excluding potential mixtures). For 96-well plates with no positive wells, the number of cells positive for HIV was considered as zero. Plates with positive wells of which none were sent for sequencing were excluded; these generally had too many wells positive to be informative. Estimating the true proportion of cells positive based on the Poisson distribution for these plates did not impact the results. The number of cells positive for HIV within a PCR/sequencing replicate, along with the total number of cells used within that dilution were used to estimate the proportion of cells infected with HIV. To compare the proportion of cells infected with HIV across independent variables, a logistic regression was carried out as above with the same strategy for effect modification. Odds ratios of comparisons and their confidence intervals were calculated using the General Linear Hypotheses function (glht) from R package multcomp (Hothorn Torsten and Peter, 2008);
“Presented p-values are not corrected for multiple comparisons. This is because calculated p-values (included and not-included in this manuscript) are not independent, and so multiple comparison methods would be overly conservative and bias results towards null effects. To aid interpretation this manuscript has three primary outcomes (proportion of genetically identical virus sequences; infection frequency; and proportion of sequences identical to virus found in rebound plasma) which are compared across three variables (participants, cellular subsets, and anatomical locations) making a total of 3×3=9 primary comparisons. Importantly these comparisons are not independent as all outcome and predictor variables are frequently highly correlated. We advise the reader to interpret P-value results not on a statistical significance cut-off, but rather as one aspect of a level of evidence against null effects. Further assistance in interpreting p-values can be found in references (Betensky, 2019; Wasserstein et al., 2019).
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| CD4 neg selection kit for IMag: human CD4 T cell | BD Biosciences | Cat#557939; RRID: AB_2802162 |
| Anti-human CD3 BB | BD Biosciences | Cat#564465; RRID: AB_2744386 |
| Anti-human CD8 PeCy7 | BD Biosciences | Cat#557746; RRID: AB_396852 |
| Anti-human CD45 PerCPCy5.5 | BD Biosciences | Cat#564105; RRID: AB_2744405 |
| Anti-human CD45 RO PE | BD Biosciences | Cat#555493; RRID: AB_395884 |
| Anti-human CD45 RO PerCPCy5.5 | BD Biosciences | Cat#560607; RRID: AB_1727500 |
| Anti-human CD45 RA APC | BD Biosciences | Cat#550855; RRID: AB_398468 |
| Anti-human CD27 APC | BD Biosciences | Cat#561400; RRID: AB_10645790 |
| Anti-human CD197 (CCR7) PE | BD Biosciences | Cat#560765; RRID: AB_2033949 |
| Anti-human CD34 GP 105–120 | BD Biosciences | Cat#560940; RRID: AB_10563908 |
| Lineage Cocktail 1 (lin1) | BD Biosciences | Cat#340546; RRID: AB_400053 |
| Anti-human CD206 | BD Biosciences | Cat#555954; RRID: AB_396250 |
| Viability stain 780 | BD Biosciences | Cat#565388 |
| FCR blocking reagent | Milteyni Biotec | Cat#130–059-901 |
|
| ||
| Chemicals, Peptides, and Recombinant Proteins | ||
|
| ||
| Leukosept tubes | Fisher Scientific | Cat#10349081 |
| Lymphoprep | Elitech | AXI-1114547 |
| RPMI | Thermo Fisher | Cat#21875034 |
| EDTA | Thermo Fisher | Cat#15575020 |
| PBS PH 7,2 | Thermo Fisher | Cat#20012019 |
| L-glutamine | Thermo Fisher | Cat#25030024 |
| Penicilin/Streptomycine | Thermo Fisher | Cat#15140122 |
| HBSS | Thermo Fisher | Cat#14175053 |
| DTT | Thermo Fisher | Cat#20290 |
| Costar 6 well plates | Sigma Aldrich Costar | SKU CLS3471–24EA |
| Lysis buffer (10mM TRisHCl, 0.5%NP-40, 0.5% Tween-20) | Made in the lab following protocol | N/A |
| Proteinase K 20mg/ml | Ambion /Lifetechnologies | Cat#AM2546 |
| PCR nucleotide mix 10mM 1ml | Promega | Cat#C1145 |
| 6M guanidinium thiocyanate BioUltr | Sigma Aldrich | SKU 50980–50ml; CAS 593–84-0 |
| Glycogen (from mussels) 20mg | Roche | SKU 10901393001 |
| Tris buffered saline (tablets) | Sigma Aldrich | T5030–100Tab |
| Isopropanol (2-propanolol) for molecular biology | Sigma Aldrich | I9516; CAS 67–63-0 |
| Ultracentrifuge tubes (optiseal, polypropylene 8,9, 16x60mm, (qty56) | Beckman Coulter | 361623 |
| Ethanol 100%(ethyl alcohol, Pure 200 proof, for molecular biology) | Sigma Aldrich | E7023–1L; CAS 64–17-5 |
| Ultrapure 1MTrisHCL PH 8 | Thermo Fisher Scientific | Cat#15568–025 |
| Corning Cell strainers 40μm | Sigma Aldrich | CLS 431750–50AE |
| Corning Cell strainers 70μm | Sigma Aldrich | CLS 431751–50AE |
| Fetal Calf Serum (FCS) | Hyclone RVM | 35888 |
|
| ||
| Critical Commercial Assays | ||
|
| ||
| Superscript III 1st strand | Invitrogen | Cat#18080051 |
| Platinum Taq PCR polymerase | Invitrogen | Cat#10966083 |
| E-Gel 96 well 1% agarose | Thermo Fisher Scientific | Cat#G700801 |
|
| ||
| Oligonucleotides | ||
|
| ||
| E20 F 5'GGGCCACACATGCCTGTGTACCCACAG 3' | Josefsson et al., 2012; von Stockenstrom et al., 2015 | N/A |
| E115 5' GAAAAATTCCCCTCCACAATTAA 3' | Josefsson et al., 2012; von Stockenstrom et al., 2015 | N/A |
| E30 F 5'GTGTACCCACAGACCCCAGCCCACAAG3' | Josefsson et al., 2012; von Stockenstrom et al., 2015 | N/A |
| E125 R 5'CAATTTCTGGGTCCCCTCCTGAGG 3' | Josefsson et al., 2012; von Stockenstrom et al., 2015 | N/A |
|
| ||
| Deposited Data | ||
|
| ||
| RNA and DNA sequencing | Genbank | MH642355 |
| RNA and DNA sequencing | Genbank | MH642607 |
| RNA and DNA sequencing | Genbank | MH642608 |
| RNA and DNA sequencing | Genbank | MH643062 |
| RNA and DNA sequencing | Genbank | MH643063 |
| RNA and DNA sequencing | Genbank | MH643573 |
|
| ||
| Software and Algorithms | ||
|
| ||
| Subtype analyses | Smartgene, Lausanne, Switzerland | https://www.smartgene.com/ |
| Viral Tropism | Web PSSM, Mullins lab, University of Washington | https://indra.mullins.microbiol.washington.edu/webpssm/ |
| Flow Jo software | BD Biosciences | https://www.flowjo.com/ |
| Perl script language | Perl | https://www.perl.org/ |
| MUSCLE | Edgar, 2004 | http://www.megasoftware.net/ |
| MAFFT | Katoh and Standley, 2013 | https://mafft.cbrc.jp/alignment/software/ |
| Aliview | Larsson, 2014 | www.ormbunkar.se/aliview/downloads |
| RDP4 | Martin et al., 2015 | http://web.cbio.uct.ac.za/~darren/rdp.html |
| Translator X | Abascal et al., 2010 | http://www.translatorx.co.uk/ |
| PhyML v3.0 | Guindon and Gascuel, 2003 | https://github.com/stephaneguindon/phyml/ |
| BaTS | Parker et al., 2008 | http://evolve.zoo.ox.ac.uk/Evolve/BaTS.html |
| Mr Bayes v3.6.2 | Ronquist et al., 2012 | http://nbisweden.github.io/MrBayes/download.html |
| Tracer | BEAST | https://github.com/beast-dev/tracer/releases/tag/v1.7.1 |
| Biopython | Cock et al., 2009 | https://biopython.org/wiki/Download |
| Elim Dupes | Los Alamos HIV Database | http://www.hiv.lanl.gov |
| R: A language and environment for statistical computing. | R script | https://www.R-project.org/ |
| General Liner hypothesis function (glht) | R package multcomp | https://cran.r-project.org/web/packages/multcomp/index.html |
| FigTree | BEAST | http://beast.community/figtree |
| iTol v3 | Letunic and Bork, 2016 | https://itol.embl.de/ |
| NETWORK v5.0.0.3 | Fluxus | http://www.fluxus-engineering.com |
| SPADS 1.0 | Dellicour and Mardulyn, 2014 | http://ebe.ulb.ac.be/ebe/SPADS.html |
|
| ||
| Others | ||
|
| ||
| Sanger sequencing | AGRF Australian Genome Research Facility, Sydney, Australia | N/A |
Highlights.
HIV-1 sequences sampled from different reservoirs were compared to rebound viruses in 11 individuals
Rebound viruses can originate from various cellular and anatomical compartments
Cellular proliferation is an important driver of HIV persistence
Cure strategies should take into account the lack of a prominent HIV rebound origin
ACKNOWLEDGMENTS
We would like to acknowledge the participants of the HIV STAR cohort; The Ghent University Hospital and all the collaborators for providing us the support and resources necessary to organize the in-depth sampling; the MDs involved at the Aids Reference Center in Ghent for recruiting of participants; and all the MDs involved in the sampling procedures, Prof. T. Kerre, Prof. D. De Looze, Dr. Malfait, Dr. D. Hemelsoet, Prof. Coppens, and all the supporting nurses and facilities, especially E. Van Caelenbergh, A. Masset, E. Caluwé, and S. Vanherreweghe.
M.-A.D.S. was granted a FWO travel grant and a Sofina Gustave-Boel fellowship. L.V. is supported by the Research Foundation Flanders (FWO 1.8.020.09.N.00), S.P. is supported by the Delaney AIDS Research Enterprise (DARE) to Find a Cure (1U19AI096109 and 1UM1AI126611–01) and the Australian National Health and Medical Research Council (APP1061681 and APP1149990), P.L. is supported by the Special Research Fund, KU Leuven (“Bijzonder Onderzoeksfonds”, KU Leuven, OT/14/115), the Research Foundation Flanders FWO G066215N,) and the European Research Council under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 725422-ReservoirDOCS). B.V. is supported by a postdoctoral fellowship from the Fund for Scientific Research (FWO) Flanders (Fonds voor Wetenschappelijk Onderzoek, Flanders, Belgium). S.D. is supported by the Fonds National de la Recherche Scientifique (FNRS, Belgium) and was previously supported by the Fund for Scientific Research (FWO) Flanders (Fonds voor Wetenschappelijk Onderzoek, Flanders, Belgium). S.R. received a strategic basic research fund of the Research Foundation Flanders (FWO 1S32916N). This work was supported by an MSD investigator grant (grant number: ISS 52777).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.chom.2019.08.003.
ADDITIONAL RESOURCES
Clinical trial registry number: NCT02641756.
Key resource table: further information on resources and materials are available in the Key resource table linked to this article.
DATA AND CODE AVAILABILITY
GenBank Accession Numbers
The sequences reported in this manuscript are available in GenBank with the following reference codes, MH642355; MH642607; MH642608; MH643062; MH643063; MH643573.
The datasets generated and/or analyzed from these sequences during the current study are available from the corresponding author on reasonable request.
Unreported custom computer code or algorithm used to generate results that are reported in the paper, are available upon request.
REFERENCES
- Abascal F, Zardoya R, and Telford MJ (2010). TranslatorX: multiple alignment of nucleotide sequences guided by amino acid translations. Nucleic Acids Res. 38, W7–w13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bandelt HJ, Forster P, and Röhl A (1999). Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 16, 37–48. [DOI] [PubMed] [Google Scholar]
- Banga R, Procopio FA, Noto A, Pollakis G, Cavassini M, Ohmiti K, Corpataux JM, de Leval L, Pantaleo G, and Perreau M (2016). PD-1(+) and follicular helper T cells are responsible for persistent HIV-1 transcription in treated aviremic individuals. Nat. Med. 22, 754–761. [DOI] [PubMed] [Google Scholar]
- Banga R, Procopio FA, Ruggiero A, Noto A, Ohmiti K, Cavassini M, Corpataux JM, Paxton WA, Pollakis G, and Perreau M (2018). Blood CXCR3(+) CD4 T cells are enriched in inducible replication competent HIV in aviremic antiretroviral therapy-treated individuals. Front. Immunol. 9, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barton K, Hiener B, Winckelmann A, Rasmussen TA, Shao W, Byth K, Lanfear R, Solomon A, McMahon J, Harrington S, et al. (2016). Broad activation of latent HIV-1 in vivo. Nat. Commun. 7, 12731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betensky RA (2019). The p-value requires context, not a threshold. Am. Stat. 73, 115–117. [Google Scholar]
- Bielejec F, Baele G, Rodrigo AG, Suchard MA, and Lemey P (2016). Identifying predictors of time-inhomogeneous viral evolutionary processes. Virus Evol. 2, vew023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloomquist EW, Lemey P, and Suchard MA (2010). Three roads diverged? Routes to phylogeographic inference. Trends Ecol. Evol. 25, 626–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruner KM, Murray AJ, Pollack RA, Soliman MG, Laskey SB, Capoferri AA, Lai J, Strain MC, Lada SM, Hoh R, et al. (2016). Defective proviruses rapidly accumulate during acute HIV-1 infection. Nat. Med. 22, 1043–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui JK, Halvas EK, Fyne E, Sobolewski MD, Koontz D, Shao W, Luke B, Hong FF, Kearney MF, and Mellors JW (2017). Ex vivo activation of CD4+ T-cells from donors on suppressive ART can lead to sustained production of infectious HIV-1 from a subset of infected cells. PLoS Pathog. 13, e1006230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzon MJ, Sun H, Li C, Shaw A, Seiss K, Ouyang Z, Martin-Gayo E, Leng J, Henrich TJ, Li JZ, et al. (2014). HIV-1 persistence in CD4+ T cells with stem cell-like properties. Nat. Med. 20, 139–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chomont N, El-Far M, Ancuta P, Trautmann L, Procopio FA, Yassine-Diab B, Boucher G, Boulassel MR, Ghattas G, Brenchley JM, et al. (2009). HIV reservoir size and persistence are driven by T cell survival and homeostatic proliferation. Nat. Med. 15, 893–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chun TW, Justement JS, Murray D, Hallahan CW, Maenza J, Collier AC, Sheth PM, Kaul R, Ostrowski M, Moir S, et al. (2010). Rebound of plasma viremia following cessation of antiretroviral therapy despite profoundly low levels of HIV reservoir: implications for eradication. AIDS 24, 2803–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarridge KE, Blazkova J, Einkauf K, Petrone M, Refsland EW, Justement JS, Shi V, Huiting ED, Seamon CA, Lee GQ, et al. (2018). Effect of analytical treatment interruption and reinitiation of antiretroviral therapy on HIV reservoirs and immunologic parameters in infected individuals. PLoS Pathog. 14, e1006792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, et al. (2009). Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen YZ, Lorenzi JCC, Krassnig L, Barton JP, Burke L, Pai J, Lu CL, Mendoza P, Oliveira TY, Sleckman C, et al. (2018). Relationship between latent and rebound viruses in a clinical trial of anti-HIV-1 antibody 3BNC117. J. Exp. Med. 215, 2311–2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohn LB, Silva IT, Oliveira TY, Rosales RA, Parrish EH, Learn GH, Hahn BH, Czartoski JL, McElrath MJ, Lehmann C, et al. (2015). HIV-1 integration landscape during latent and active infection. Cell 160, 420–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahl V, Peterson J, Fuchs D, Gisslen M, Palmer S, and Price RW (2014). Low levels of HIV-1 RNA detected in the cerebrospinal fluid after up to 10 years of suppressive therapy are associated with local immune activation. AIDS 28, 2251–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dellicour S, and Mardulyn P (2014). Spads 1.0: a toolbox to perform spatial analyses on DNA sequence data sets. Mol. Ecol. Resour. 14, 647–651. [DOI] [PubMed] [Google Scholar]
- Descours B, Petitjean G, López-Zaragoza JL, Bruel T, Raffel R, Psomas C, Reynes J, Lacabaratz C, Levy Y, Schwartz O, et al. (2017). CD32a is a marker of a CD4 T-cell HIV reservoir harbouring replication-competent proviruses. Nature 543, 564–567. [DOI] [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eisele E, and Siliciano RF (2012). Redefining the viral reservoirs that prevent HIV-1 eradication. Immunity 37, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewing B, and Green P (1998). Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genome Res. 8, 186–194. [PubMed] [Google Scholar]
- Faria NR, Suchard MA, Rambaut A, and Lemey P (2011). Toward a quantitative understanding of viral phylogeography. Curr. Opin. Virol. 1, 423–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, Chaisson RE, Quinn TC, Chadwick K, Margolick J, Brookmeyer R, et al. (1997). Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science 278, 1295–1300. [DOI] [PubMed] [Google Scholar]
- Garner SA, Rennie S, Ananworanich J, Dube K, Margolis DM, Sugarman J, Tressler R, Gilbertson A, and Dawson L (2017). Interrupting antiretroviral treatment in HIV cure research: scientific and ethical considerations. J. Virus Erad. 3, 82–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, and Gascuel O (2003). A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 52, 696–704. [DOI] [PubMed] [Google Scholar]
- Henrich TJ, Hatano H, Bacon O, Hogan LE, Rutishauser R, Hill A, Kearney MF, Anderson EM, Buchbinder SP, Cohen SE, et al. (2017). HIV-1 persistence following extremely early initiation of antiretroviral therapy (ART) during acute HIV-1 infection: an observational study. PLoS Med. 14, e1002417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiener B, Horsburgh BA, Eden JS, Barton K, Schlub TE, Lee E, von Stockenstrom S, Odevall L, Milush JM, Liegler T, et al. (2017). Identification of genetically intact HIV-1 proviruses in specific CD4(+) T cells from effectively treated participants. Cell Rep. 21, 813–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YC, Shan L, Hosmane NN, Wang J, Laskey SB, Rosenbloom DI, Lai J, Blankson JN, Siliciano JD, and Siliciano RF (2013). Replication-competent noninduced proviruses in the latent reservoir increase barrier to HIV-1 cure. Cell 155, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosmane NN, Kwon KJ, Bruner KM, Capoferri AA, Beg S, Rosenbloom DI, Keele BF, Ho YC, Siliciano JD, and Siliciano RF (2017). Proliferation of latently infected CD4(+) T cells carrying replication-competent HIV-1: potential role in latent reservoir dynamics. J. Exp. Med. 214, 959–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothorn Torsten BF, and Peter W (2008). Simultaneous inference in general parametric models. Biometrical J. 50, 346–363. [DOI] [PubMed] [Google Scholar]
- Jensen MA, Li FS, van ‘t Wout AB, Nickle DC, Shriner D, He HX, McLaughlin S, Shankarappa R, Margolick JB, and Mullins JI (2003). Improved coreceptor usage prediction and genotypic monitoring of R5-to-X4 transition by motif analysis of human immunodeficiency virus type 1 env V3 loop sequences. J. Virol. 77, 13376–13388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson L, Eriksson S, Sinclair E, Ho T, Killian M, Epling L, Shao W, Lewis B, Bacchetti P, Loeb L, et al. (2012). Hematopoietic precursor cells isolated from patients on long-term suppressive HIV therapy did not contain HIV-1 DNA. J. Infect. Dis. 206, 28–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Josefsson L, von Stockenstrom S, Faria NR, Sinclair E, Bacchetti P, Killian M, Epling L, Tan A, Ho T, Lemey P, et al. (2013). The HIV-1 reservoir in eight patients on long-term suppressive antiretroviral therapy is stable with few genetic changes over time. Proc. Natl. Acad. Sci. USA 110, E4987–E4996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandathil AJ, Sugawara S, and Balagopal A (2016). Are T cells the only HIV-1 reservoir? Retrovirology 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K, and Standley DM (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney M, Maldarelli F, Shao W, Margolick JB, Daar ES, Mellors JW, Rao V, Coffin JM, and Palmer S (2009). Human immunodeficiency virus type 1 population genetics and adaptation in newly infected individuals. J. Virol. 83, 2715–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney MF, Wiegand A, Shao W, Coffin JM, Mellors JW, Lederman M, Gandhi RT, Keele BF, and Li JZ (2016). Origin of rebound plasma HIV includes cells with identical proviruses that are transcriptionally active before stopping of antiretroviral therapy. J. Virol. 90, 1369–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney MF, Wiegand A, Shao W, McManus WR, Bale MJ, Luke B, Maldarelli F, Mellors JW, and Coffin JM (2017). Ongoing HIV replication During ART reconsidered. Open Forum Infect. Dis. 4, ofx173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khoury G, Fromentin R, Solomon A, Hartogensis W, Killian M, Hoh R, Somsouk M, Hunt PW, Girling V, Sinclair E, et al. (2017). Human immunodeficiency virus persistence and T-cell activation in blood, rectal, and lymph node tissue in human immunodeficiency virus-infected individuals receiving suppressive antiretroviral therapy. J. Infect. Dis. 215, 911–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiselinova M, De Spiegelaere W, Buzon MJ, Malatinkova E, Lichterfeld M, and Vandekerckhove L (2016). Integrated and total HIV-1 DNA predict ex vivo viral outgrowth. PLoS Pathog. 12, e1005472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsson A (2014). AliView: a fast and lightweight alignment viewer and editor for large datasets. Bioinformatics 30, 3276–3278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laskey SB, Pohlmeyer CW, Bruner KM, and Siliciano RF (2016). Evaluating clonal expansion of HIV-infected cells: optimization of PCR strategies to predict clonality. PLoS Pathog. 12, e1005689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederman MM, Cannon PM, Currier JS, June CH, Kiem HP, Kuritzkes DR, Lewin SR, Margolis DM, McCune JM, Mellors JW, et al. (2016). A cure for HIV infection: “not in my lifetime” or “just around the corner”? Pathog. Immun. 1, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee GQ, Orlova-Fink N, Einkauf K, Chowdhury FZ, Sun X, Harrington S, Kuo HH, Hua S, Chen HR, Ouyang Z, et al. (2017a). Clonal expansion of genome-intact HIV-1 in functionally polarized Th1 CD4+ T cells. J. Clin. Invest. 127, 2689–2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SK, Zhou S, Baldoni PL, Spielvogel E, Archin NM, Hudgens MG, Margolis DM, and Swanstrom R (2017b). Quantification of the latent HIV-1 reservoir using ultra deep sequencing and primer ID in a viral outgrowth assay. J. Acquir. Immune Defic. Syndr. 74, 221–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2016). Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 44, W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorenzo-Redondo R, Fryer HR, Bedford T, Kim EY, Archer J, Pond SLK, Chung YS, Penugonda S, Chipman JG, Fletcher CV, et al. (2016). Persistent HIV-1 replication maintains the tissue reservoir during therapy. Nature 530, 51–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu CL, Pai JA, Nogueira L, Mendoza P, Gruell H, Oliveira TY, Barton J, Lorenzi JCC, Cohen YZ, Cohn LB, et al. (2018). Relationship between intact HIV-1 proviruses in circulating CD4(+) T cells and rebound viruses emerging during treatment interruption. Proc. Natl. Acad. Sci. USA 115, E11341–E11348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolis DM, Garcia JV, Hazuda DJ, and Haynes BF (2016). Latency reversal and viral clearance to cure HIV-1. Science 353, aaf6517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin DP, Murrell B, Golden M, Khoosal A, and Muhire B (2015). RDP4: detection and analysis of recombination patterns in virus genomes. Virus Evol. 1, vev003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGary CS, Deleage C, Harper J, Micci L, Ribeiro SP, Paganini S, Kuri-Cervantes L, Benne C, Ryan ES, Balderas R, et al. (2017). CTLA-4(+)PD-1(−) memory CD4(+) T cells critically contribute to viral persistence in antiretroviral therapy-suppressed, SIV-infected rhesus macaques. Immunity 47, 776–788.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendoza P, Gruell H, Nogueira L, Pai JA, Butler AL, Millard K, Lehmann C, Suárez I, Oliveira TY, Lorenzi JCC, et al. (2018). Combination therapy with anti-HIV-1 antibodies maintains viral suppression. Nature 561, 479–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minin VN, and Suchard MA (2008a). Counting labeled transitions in continuous-time Markov models of evolution. J. Math. Biol. 56, 391–412. [DOI] [PubMed] [Google Scholar]
- Minin VN, and Suchard MA (2008b). Fast, accurate and simulation-free stochastic mapping. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 3985–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morón-López S, Puertas MC, Gálvez C, Navarro J, Carrasco A, Esteve M, Manyé J, Crespo M, Salgado M, and Martinez-Picado J (2017). Sensitive quantification of the HIV-1 reservoir in gut-associated lymphoid tissue. PloS One 12, e0175899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nei M, and Li WH (1979). Mathematical model for studying genetic variation in terms of restriction endonucleases. Proc. Natl. Acad. Sci. USA 76, 5269–5273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Kearney M, Maldarelli F, Halvas EK, Bixby CJ, Bazmi H, Rock D, Falloon J, Davey RT Jr., Dewar RL, et al. (2005). Multiple, linked human immunodeficiency virus type 1 drug resistance mutations in treatment-experienced patients are missed by standard genotype analysis. J. Clin. Microbiol. 43, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmer S, Wiegand AP, Maldarelli F, Bazmi H, Mican JM, Polis M, Dewar RL, Planta A, Liu S, Metcalf JA, et al. (2003). New real-time reverse transcriptase-initiated PCR assay with single-copy sensitivity for human immunodeficiency virus type 1 RNA in plasma. J. Clin. Microbiol. 41, 4531–4536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker J, Rambaut A, and Pybus OG (2008). Correlating viral phenotypes with phylogeny: accounting for phylogenetic uncertainty. Infect. Genet. Evol. 8, 239–246. [DOI] [PubMed] [Google Scholar]
- Ronquist F, Teslenko M, van der Mark P, Ayres DL, Darling A, Höhna S, Larget B, Liu L, Suchard MA, and Huelsenbeck JP (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbloom DIS, Hill AL, Laskey SB, and Siliciano RF (2017). Re-evaluating evolution in the HIV reservoir. Nature 551, E6–E9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothenberger MK, Keele BF, Wietgrefe SW, Fletcher CV, Beilman GJ, Chipman JG, Khoruts A, Estes JD, Anderson J, Callisto SP, et al. (2015). Large number of rebounding/founder HIV variants emerge from multifocal infection in lymphatic tissues after treatment interruption. Proc. Natl. Acad. Sci. USA 112, E1126–E1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salantes DB, Zheng Y, Mampe F, Srivastava T, Beg S, Lai J, Li JZ, Tressler RL, Koup RA, Hoxie J, et al. (2018). HIV-1 latent reservoir size and diversity are stable following brief treatment interruption. J. Clin. Invest. 128, 3102–3115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shankarappa R, Margolick JB, Gange SJ, Rodrigo AG, Upchurch D, Farzadegan H, Gupta P, Rinaldo CR, Learn GH, He X, et al. (1999). Consistent viral evolutionary changes associated with the progression of human immunodeficiency virus type 1 infection. J. Virol. 73, 10489–10502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Stockenstrom S, Odevall L, Lee E, Sinclair E, Bacchetti P, Killian M, Epling L, Shao W, Hoh R, Ho T, et al. (2015). Longitudinal genetic characterization reveals that cell proliferation maintains a persistent HIV Type 1 DNA pool During effective HIV therapy. J. Infect. Dis. 212, 596–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TH, Donaldson YK, Brettle RP, Bell JE, and Simmonds P (2001). Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J. Virol. 75, 11686–11699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang XQ, and Palmer S (2018). Single-molecule techniques to quantify and genetically characterise persistent HIV. Retrovirology 15, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Gurule EE, Brennan TP, Gerold JM, Kwon KJ, Hosmane NN, Kumar MR, Beg SA, Capoferri AA, Ray SC, et al. (2018). Expanded cellular clones carrying replication-competent HIV-1 persist, wax, and wane. Proc. Natl. Acad. Sci. USA 115, E2575–E2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wasserstein RL, Schirm AL, and Lazar NA (2019). Moving to a World Beyond “p < 0.05”. Am. Stat. 73, 1–19. [Google Scholar]
- Winckelmann A, Barton K, Hiener B, Schlub TE, Shao W, Rasmussen TA, Østergaard L, Søgaard OS, Tolstrup M, and Palmer S (2017). Romidepsin-induced HIV-1 viremia during effective antiretroviral therapy contains identical viral sequences with few deleterious mutations. AIDS Lond. Engl. 31, 771–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z (1994). Maximum likelihood phylogenetic estimation from DNA sequences with variable rates over sites: approximate methods. J. Mol. Evol. 39, 306–314. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
GenBank Accession Numbers
The sequences reported in this manuscript are available in GenBank with the following reference codes, MH642355; MH642607; MH642608; MH643062; MH643063; MH643573.
The datasets generated and/or analyzed from these sequences during the current study are available from the corresponding author on reasonable request.
Unreported custom computer code or algorithm used to generate results that are reported in the paper, are available upon request.