

Abstract
Enzymatic modifications of small molecules are a common phenomenon in natural product biosynthesis, leading to the production of diverse bioactive compounds. In polyketide biosynthesis, modifications commonly take place after the completion of the polyketide backbone assembly by the polyketide synthases and the mature products are released from the acyl-carrier protein (ACP). However, exceptions to this rule appear to be widespread, as on-line hydroxylation, methyl transfer, and cyclization during polyketide assembly process are common, particularly in trans-AT PKS systems. Many of these modifications are catalyzed by specific domains within the modular PKS systems. However, several of the on-line modifications are catalyzed by stand-alone proteins. Those include the on-line Baeyer-Villiger oxidation, α-hydroxylation, halogenation, epoxidation, and methyl esterification during polyketide assembly, dehydrogenation of ACP-bound short fatty acids by acyl-CoA dehydrogenase-like enzymes, and glycosylation of ACP-bound intermediates by discrete glycosyltransferase enzymes. This review article highlights some of these trans-acting proteins that catalyze enzymatic modifications of ACP-bound small molecules in natural product biosynthesis.
Keywords: acyl carrier protein, on-line modification, polyketide synthase, tailoring reaction, trans-acting enzyme
Graphical Abstract

In polyketide biosynthesis, modifications commonly occur after the completion of the polyketide backbone assembly and the mature products are released from the acyl-carrier protein (ACP). However, exceptions to this rule are widespread, and some are modified on-line by discrete enzymes. This review article highlights more recently reported trans-acting proteins that catalyze modifications of ACP-bound substrates in natural product biosynthesis.
1. Introduction
Enzymatic modifications of small molecules are a common phenomenon in secondary metabolism. Plants, fungi, and bacteria employ various biochemical reactions to modify their secondary metabolites leading to the formation of structurally diverse and biologically active natural products. Alkylation, acylation, hydroxylation, and glycosylation are among the common modifications that mostly occur after the construction of the core structures – decorating and turning them into biologically relevant compounds. As some natural products are synthesized by a suite of discrete enzymes which catalyze the stepwise reactions on free-floating, non-covalently bound substrates, modification reactions may take place at any time in the pathways. In contrast, polyketides and non-ribosomal peptides are constructed by polyketide synthases (PKS) or non-ribosomal peptide synthetases (NRPS), which process the growing polyketide or peptide chains tethered to acyl-carrier protein (ACP) or peptidyl-carrier protein (PCP), respectively, until full maturation of the polyketide or peptide backbones.[1] Consequently, modifications of polyketides or peptides generally take place after the completion of the backbone assembly and the mature products are released from the ACP or PCP.[2] These so-called post-PKS or post-NRPS tailoring processes are usually essential for the biological activity of the natural products. For instance, the hydroxylation and glycosylation of 6-deoxyerythronolide B to erythromycin D are necessary for the antibacterial activity of the compound.[3]
However, exceptions to this rule where modifications occur during the polyketide or non-ribosomal peptide assembly process, referred to as on-line modifications, appear to be widespread. For example, on-line hydroxylation and methyl transfer during polyketide assembly processes are common, particularly in trans-AT PKS systems. Cyclization during polyketide assembly, e.g., cycloether formation, lactonization, cyclopropanation, and intramolecular [4+2] cycloaddition, has been observed in several PKS pathways.[4]
While most of these modifications are catalyzed by specific (non-canonical) domains within the modular PKS systems, several are catalyzed by stand-alone proteins (e.g., the cyclase SalBIII in the salinomycin pathway, the cyclopropanase Jaw5 in the jawsamycin pathway, and the lipocalin-like protein CghA that catalyzes Diels-Alder reaction in the Sch 210972 pathway) (Figure 1).[4a–c] Outstanding reviews of many of these on-line modifications in the PKS and NRPS pathways were published by Pang et al. and Sundaram and Hertweck in 2016;[4d, 4e] therefore they will not be discussed in detail in this review.
Figure 1.
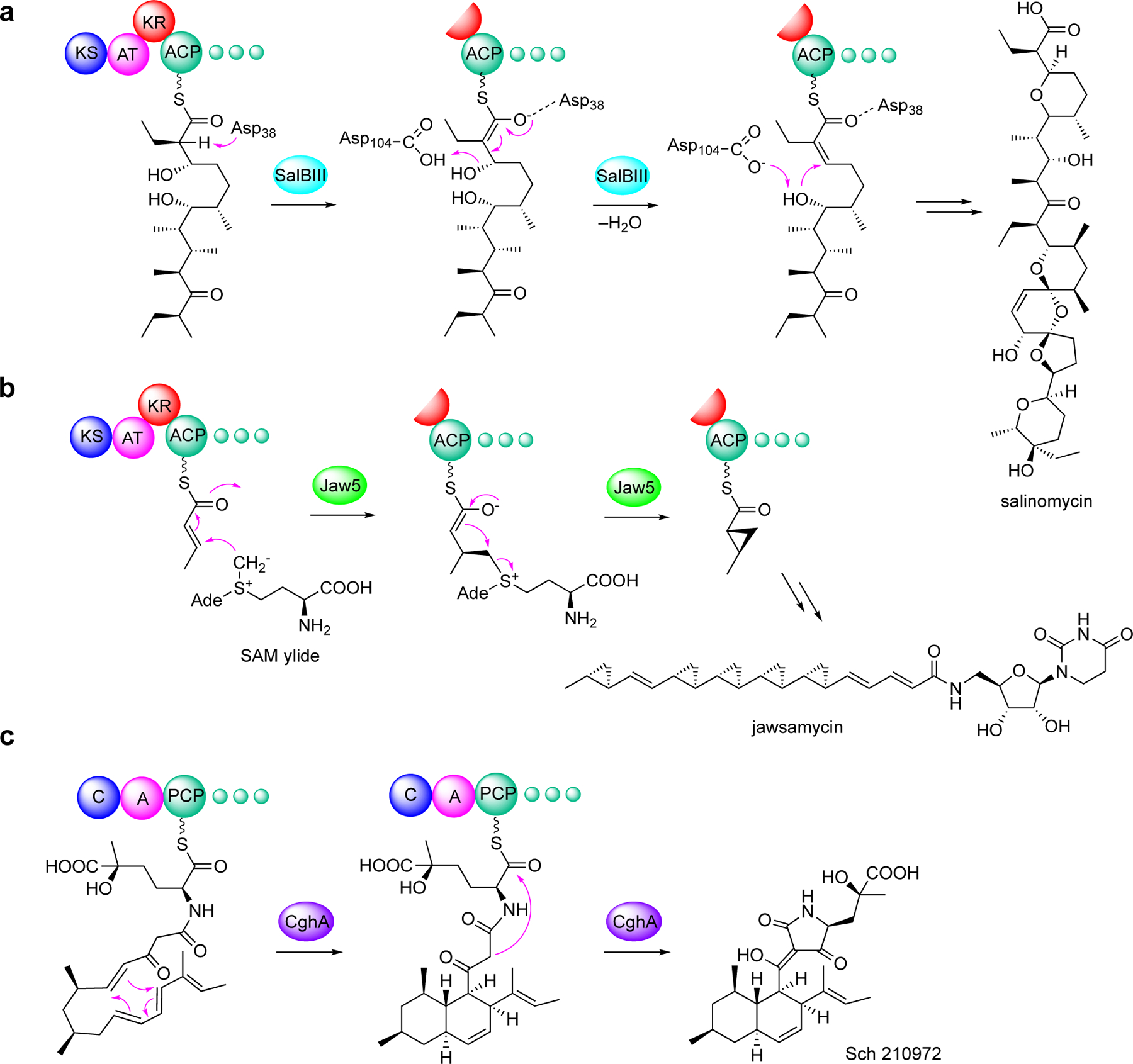
Examples of on-line modification of polyketides by discrete enzymes. (a) cycloether formation by SalBIII in salinomycin biosynthesis; (b) cyclopropanation by Jaw5 in jawsamycin biosynthesis; and (c) intramolecular [4+2] cycloaddition by CghA in Sch 210972 biosynthesis.
Since then, other examples of on-line modifications have continued to emerge, including the unprecedented use of S-adenosylmethionine (SAM) as a building block by an NRPS module and the subsequent cyclopropanation by a PKS module in the colibactin pathway,[5] and an on-line α-ketothioester decarboxylation reaction by a KS domain in the hybrid NRPS-PKS assembly line of barbamide, resulting in a one-carbon truncation of the peptide-polyketide skeleton (Figure 2).[6] The cyclopropanation by the PKS ClbI in colibactin biosynthesis is distinct from that found in other known PKS and NRPS assembly lines which produce cyclopropane-containing natural products, e.g., ambruticin, coronatine, curacin A, hormaomycin, jawsamycin, and kutzneride 2.[4b, 7] The decarboxylation of α-ketothioester by a KS domain in the barbamide pathway is a highly unusual skeleton editing system, resulting in a one-carbon truncation during the PKS-NRPS assembly. This represents yet another example of non-canonical KS functions involved in natural products biosynthesis.[8]
Figure 2.
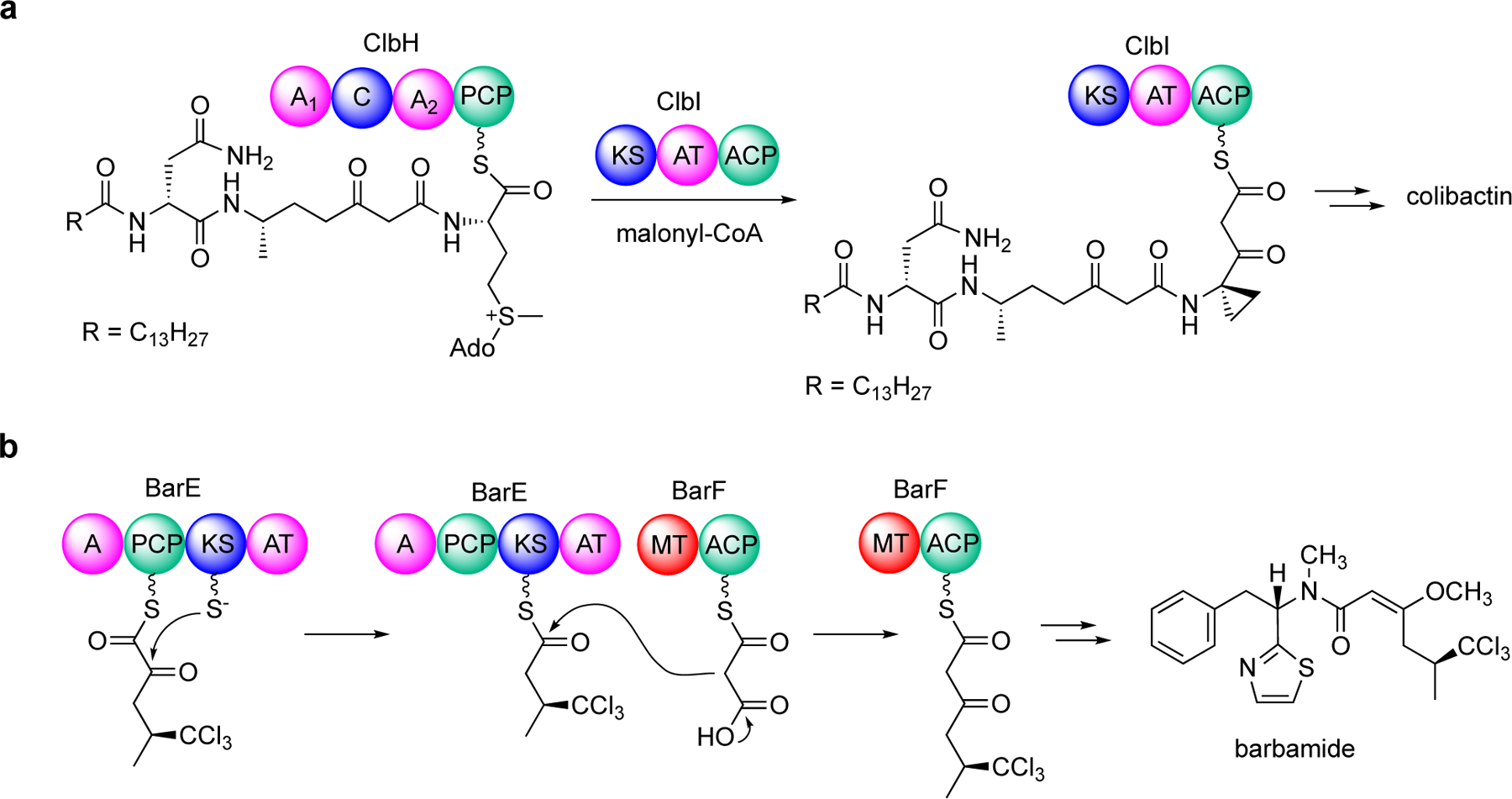
On-line modifications in colibactin and barbamide biosyntheses. (a) The use of S-adenosylmethionine as a building block and on-line cyclopropanation in the colibactin assembly line and (b) on-line α-ketothioester decarboxylation by a ketosynthase domain in barbamide biosynthesis.
Furthermore, a unique Baeyer-Villiger-type oxygen insertion into a growing polyketide chain has been shown to be catalyzed by a discrete flavin-dependent protein within the oocydin PKS system.[9] The highly versatile trans-AT oocydin PKS system can also catalyze α-hydroxylation, halogenation, and acylation all in the same polyketide assembly line.[10] While the acylation reaction is catalyzed by a non-canonical TE domain within the PKS, the Baeyer-Villiger-type oxygen insertion, the hydroxylation, and the halogenation are catalyzed by enzymes that are produced as stand-alone proteins. Other ACP-bound compound modifications involving trans-acting enzymes include the on-line methyl esterification catalyzed by a discrete SAM-dependent methyltransferase during aurantinin polyketide biosynthesis,[11] and the on-line epoxidation of a polyketide chain catalyzed by a discrete cytochrome P450 enzyme during shuangdaolide biosynthesis.[12] The previously reported variant of acyl-CoA dehydrogenase, TcsD, from the FK506 pathway catalyzes the dehydrogenation of ACP-bound short fatty acids, leading to the formation of the extender unit allylmalonyl-CoA, was re-examined.[13] This led to a revised pathway to allylmalonyl-CoA and provided a better understanding of TcsD and other bacterial acyl-CoA dehydrogenase-like enzymes.[13] A similar enzyme has also been reported in the biosynthesis of haliangicin, NFAT-133, and related compounds produced by strains of actinobacteria and myxobacteria.[13–14] The glycosylation of ACP-bound intermediates by discrete glycosyltransferase enzymes was recently reported in pactamycin and mitomycin biosynthesis, representing the first examples of glycosylation of ACP-bound small molecules.[15] This review article highlights some of the interesting discrete proteins reported more recently (2017–2023) that catalyze enzymatic modifications of protein-bound small molecules in natural product biosynthesis.
2. Modification of ACP-bound substrates by trans-acting enzymes
2.1. On-line Baeyer-Villiger-type oxidation, α-hydroxylation, halogenation, and acylation
Seminal work by Piel and co-workers interrogating oocydin biosynthesis in Serratia plymuthica 4Rx13 revealed an interesting Baeyer-Villiger-type oxygen-insertion into a growing polyketide chain catalyzed by a flavin-dependent monooxygenase, OocK.[9] Oocydins and closely related compounds are chlorinated macrolactones originally found in marine animals,[16] but were later found to be produced in Gram-negative bacteria.[17] The monooxygenase OocK is a homologue of PedG from the pederin pathway and TobD from the toblerol pathway.[9] Structurally, they contain alcohol-type termini, suggesting that they are also formed from oxygen insertion and ester cleavage. While OocK is produced as a stand-alone protein, it is proposed to work in concert with the DH0 domain of OocJ2 and the ACP and KS domains of OocL to form an oxidation module that catalyzes the oxygen insertion into the nascent polyketide chain to form an ester (Figure 3). Bioinformatic studies suggest that this type of Baeyer-Villiger oxygen insertion is more common in trans-AT PKS systems, many of which have yet to be characterized.[18] Intriguingly, the oocydin PKS system also contains a new oxygenation module (OocM), a halogenating module, and a thioesterase-like domain that functions as an acetyltransferase, all in the same polyketide assembly line, underlining the remarkable functional complexity of trans-AT PKS.[10] As described earlier, except for the TE domain, which is part of OocS, the oxidases (OocK and OocM) and the halogenase (OocP) are produced as stand-alone proteins.[10]
Figure 3.
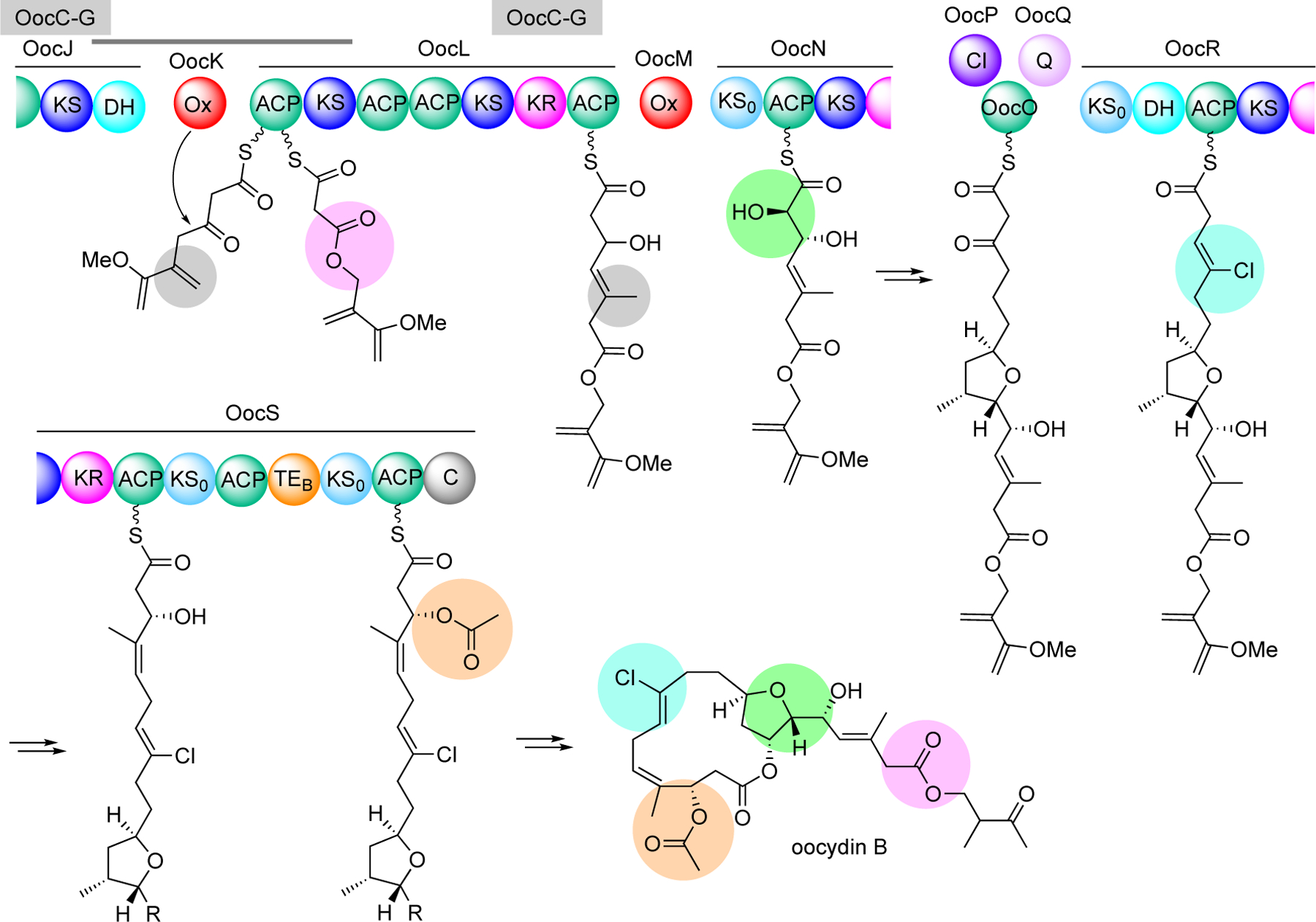
Proposed biosynthetic pathway to oocydin B. The trans-AT PKS system is highly versatile with several on-line modifications, e.g., Baeyer-Villiger oxidation (purple), α-hydroxylation (green), halogenation (aqua), and acylation (orange). OocC-G (highlighted in grey) is a β-branching cassette, which introduces methyl groups to the nascent polyketide chains. OocP is a stand-alone halogenase, whereas OocQ is an unknown protein that is proposed to stabilize an OocPQ complex.
Two homologues of OocK were found as part of the lobatamide PKS system in the Gram-negative bacterium Gynuella sunshinyii.[18] Lobatamide A is a structurally unique macrodilactone with a side chain containing an O-methyloxime terminus (Figure 4a). In contrast to OocK, the two flavin-dependent monooxygenases in the lobatamide pathway are integrated Ox domains located within the PKS proteins LbmA and LbmC. The LbmA-Ox domain was proposed to catalyze the formation of the oxime, whereas the LbmC-Ox domain catalyzes the Baeyer-Villiger-type oxygen-insertion into the nascent polyketide chain (Figure 4a). More recently, Masschelein and co-workers experimentally demonstrated the mechanism for O-methyloxime formation in the oximidine biosynthetic pathway in Pseudomonas baetica a390T involving an Ox domain similar to LbmA-Ox within the OxiB protein (Figure 4b).[19] The oximidine assembly line is highly similar to that of lobatamide except for some organizational variations. In addition, the oximidine PKS lacks a second Ox domain for the Baeyer-Villiger-type oxygen-insertion, whereas the cytochrome P450 OxiK whose gene is located right after the PKS genes in the oximidine cluster has been proposed to be involved in the post-PKS tailoring processes.[19] Consequently, while the chemical structure of oximidine is very similar to that of lobatamide, the polyketide backbone of oximidine only contains one ester bond (Figure 4b).
Figure 4.
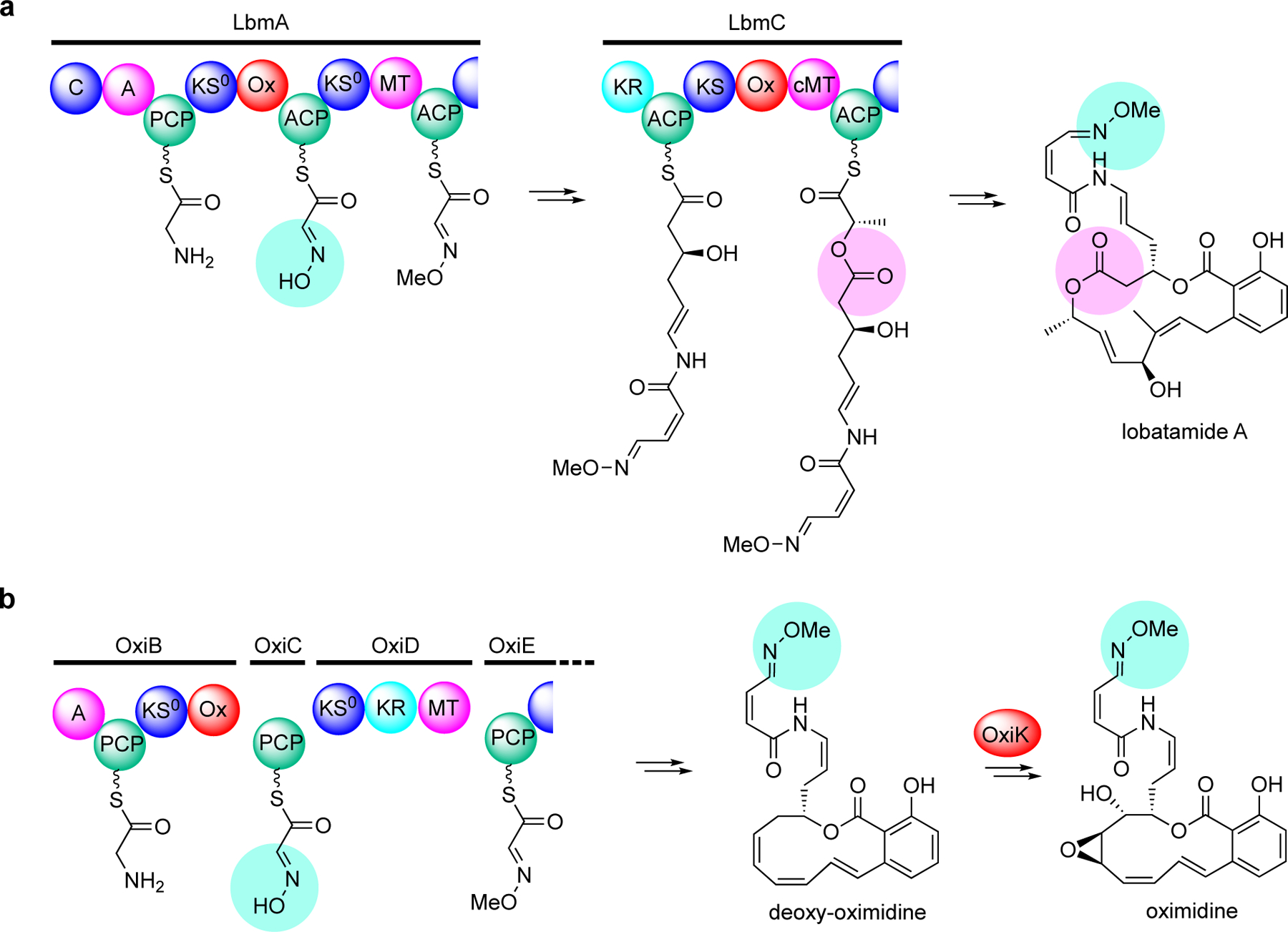
Proposed biosynthetic pathway to oximidines. (a) Two flavin-dependent monooxygenase domains, LbmA-Ox and LbmC-Ox are proposed to catalyze the formation of the oxime (aqua), whereas the LbmC-Ox domain catalyzes the Baeyer-Villiger-type oxygen-insertion into the nascent polyketide chain (pink). (b) A flavin-dependent monooxygenase domain, OxiB-Ox, is proposed to catalyze the formation of the oxime (aqua), where the P450 OxiK is proposed to install the epoxide post-assembly.
2.2. On-line methyl esterification
Although SAM-dependent methyl transfer is a common modification event in natural product biosynthesis, the enzymatic transformation of carboxylic acids to their methyl esters is relatively rare. The most common examples of such methyl esterification in nature occurs on fatty acids, sugar acids, and some linear polyketides. Recently, an unusual on-line methyl esterification has been reported in the biosynthesis of aurantinins, a group of antibacterial polyketides produced by Bacillus aurantinus strain KM-214 and B. subtilis fmb60.[11] The SAM-dependent methyltransferase Art28 was reportedly able to convert malonyl-ACP to its methyl ester, which serves as the starting point in the aurantinin PKS machinery (Figure 5). Interestingly, this methyl ester is retained until the last step of aurantinin biosynthesis, where the esterase Art9 subsequently hydrolyzes the methyl ester and converts the inactive aurantinin 9B to the active aurantinin B. Thus, the methyl ester is believed to function as a protecting group that prevents the carboxyl terminus from undesired side reactions during the skeleton assembly process, as well as a resistance strategy to protect the producing organisms from toxic aurantinin intermediates. This phenomenon appears to be widespread in polyketide biosynthesis in various bacteria.[11] In addition, the aurantinin polyketide assembly line also contains several integrated methyltransferase (MT) domains that introduce α-branching methyl groups to the growing polyketide chains, as well as a HMGS-like cassette (Art18–Art21) that is responsible for installing the β-branching methyl groups to the aurantinin polyketide skeleton, further underscoring the various strategies employed by nature to produce diverse bioactive compounds.
Figure 5.

On-line methyl esterification in aurantinin biosynthesis by the SAM-dependent protein Art28. The methyl ester is retained until the last step of the pathway, where the esterase Art9 hydrolyzes the methyl ester and converts the inactive aurantinin 9B to the active aurantinin B.
2.3. Epoxidation of ACP-bound polyketide intermediates
Another trans-AT PKS-based biosynthetic pathway was reported for shuangdaolides, a group of polyketide antibiotics obtained from cultures of Streptomyces albus J1076 harboring the sdl biosynthetic gene cluster from the marine Streptomyces sp. B59.[12] Similar compounds were also found in other strains of Streptomyces, e.g., dumulmycin from Streptomyces sp. DM28.[20] Several members of shuangdaolides, as well as dumulmycin, contain a rare internal five-membered carbocycle moiety, 2-hydroxycyclopentenone, in their skeleton. The formation of 2-hydroxycyclopentenone has been proposed to be mediated by the opening of 16,17-epoxide on the polyketide skeleton.[12] In contrast to the online epoxidation in spliceostatin biosynthesis, which is catalyzed by a FMO domain found within the PKS,[21] the epoxide moiety in the shuangdaolide pathway is installed during the polyketide chain elongation by SdlR, a multi-domain protein that is comprised of a ketoreductase (KR), an ACP, an FMO, and an FSH1 domain.[12] However, only the FMO domain, which contains a NAD(P)-binding motif that belongs to the group B subfamily of flavin proteins, appears to be responsible for the epoxidation reaction. In addition, the epoxidation catalyzed by SdlR occurs at a position seven carbon atoms away from the thioester, which is distinct from other on-line modifications by an integrated domain within the PKS, which usually modify the chain at the α, β, or γ-carbon positions (Figure 6).
Figure 6.
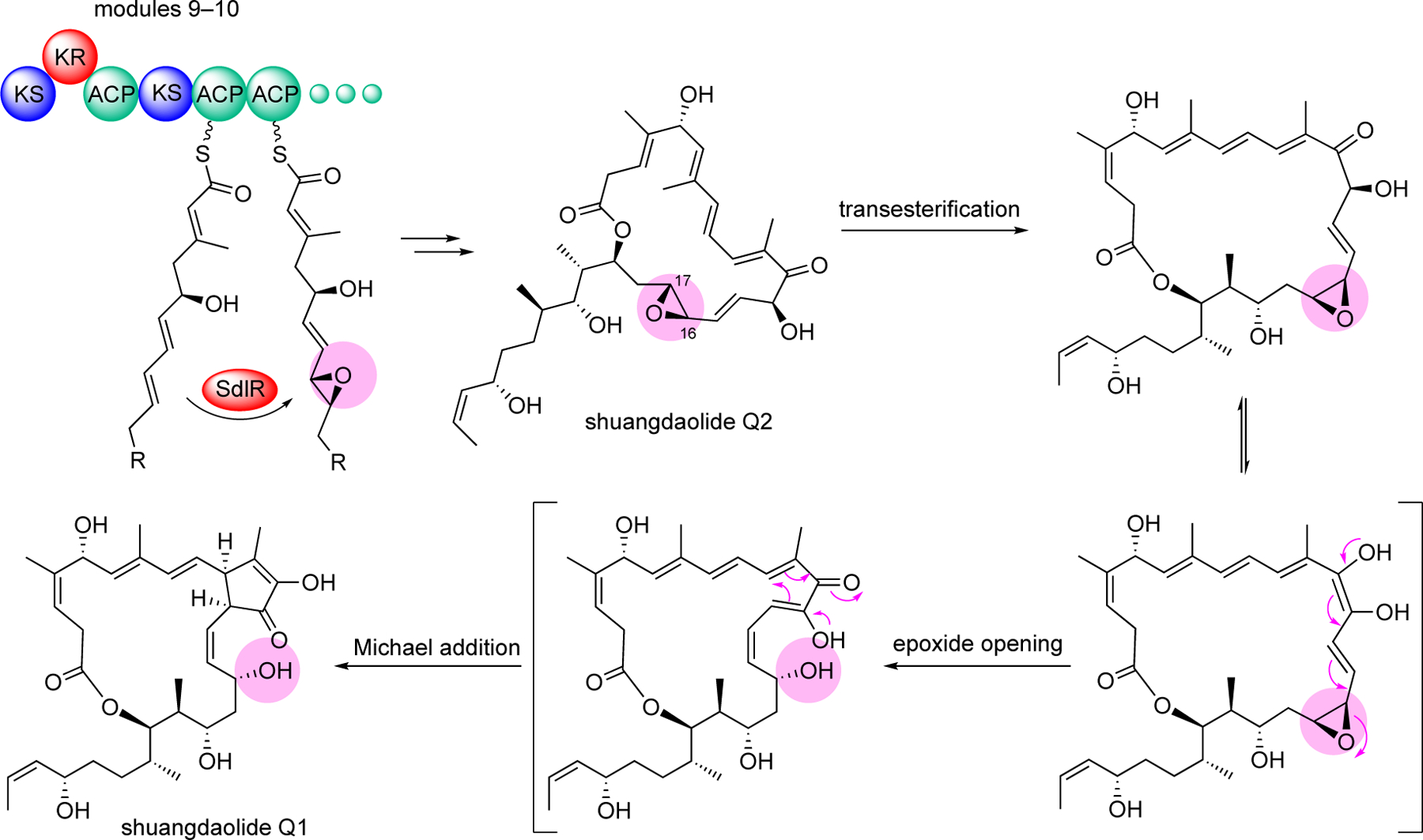
Epoxidation of an ACP-bound polyketide intermediate in shuangdaolide biosynthesis followed by the proposed chemical cascade that results in the transformation of shuangdaolide Q2 to ashuangdaolide Q1.
2.4. ACP-dependent dehydrogenation leading to terminal alkenes
Enzymatic dehydrogenation is commonly seen in the biosynthesis of unsaturated fatty acids. The enzymes, acyl-ACP desaturases, catalyze desaturation (dehydrogenation) reactions that convert acyl-ACP to their corresponding unsaturated products.[22] Acyl dehydrogenation can also occur through the action of acyl-CoA dehydrogenases, flavoenzymes that are involved in the β-oxidation of fatty acids.[23] The canonical acyl-CoA dehydrogenases catalyze dehydrogenation of acyl-CoA to form α,β-unsaturated products using FAD as co-factor. More recently, several putative acyl-CoA dehydrogenases have been reported to catalyze desaturation of ACP-bound substrates that lead to the production of polyketides with terminal alkenes. Natural products with a terminal alkene are rare, but some important polyketides, e.g., curacin A, haliangicin, tacrolimus (FK506), and tautomycetin, contain this functionality (Figure 7).[7e, 24] In polyketide biosynthesis, terminal alkenes may be formed in several ways, but they are distinct from what is normally used to generate double bonds within the polyketide chain, which involves a dehydratase (DH) domain. For example, the curacin A terminal alkene is reportedly formed through an unusual off-loading mechanism involving a sulfotransferase (ST) and thioesterase (TE) domain.[25] The tautomycetin terminal alkene is formed after the product is released from the PKS and involves the tailoring enzyme TtnD (a prenylated FMN-dependent decarboxylase belonging to the UbiD family) in the presence of TtnC (a flavin prenyltransferase).[24c]
Figure 7.

Chemical structures of several natural products with a terminal alkene.
The terminal alkene in FK506 is incorporated into the molecule through an extender unit, allylmalonyl-CoA.[26] This extender unit was proposed to be derived from trans-2-pentenyl-ACP catalyzed by TcsC (a crotonyl-CoA reductase) and TcsD (a variant of acyl-CoA dehydrogenases).[26] While both TcsC and TcsD were able to convert trans-2-pentenyl-ACP to propylmalonyl-ACP and 2,4-pentadienyl-ACP, respectively, due to a low rate conversion by TcsD, it was proposed that the major biosynthetic pathway to allylmalonyl-ACP proceeds through propylmalonyl-ACP (Figure 8a, gray bold arrows).[26] While TcsD was shown to convert propylmalonyl-ACP to allylmalonyl-ACP, the in vitro reaction was only analyzed by mass spectrometry and HPLC, with only 2 atom mass unit difference between the protein-bound substrate and the product and a slight shift in retention time, posing some concerns on their analytical accuracy. Later, Ojika and co-workers showed that HliR, a homologue of TcsD from the haliangicin pathway, was highly specific to α,β-unsaturated short chain acyl substrates (e.g., trans-2-pentenyl-SNAC) and did not consume saturated or γ,δ-unsaturated acyl substrates (Figure 8b).[14e] However, as the in vitro experiments were conducted using N-acetylcysteamine (NAC) thioesters, it is unclear whether the actual substrate of HliR is 2-methylpent-2-enoyl-CoA, as proposed, or 2-methylpent-2-enoyl-ACP.
Figure 8.
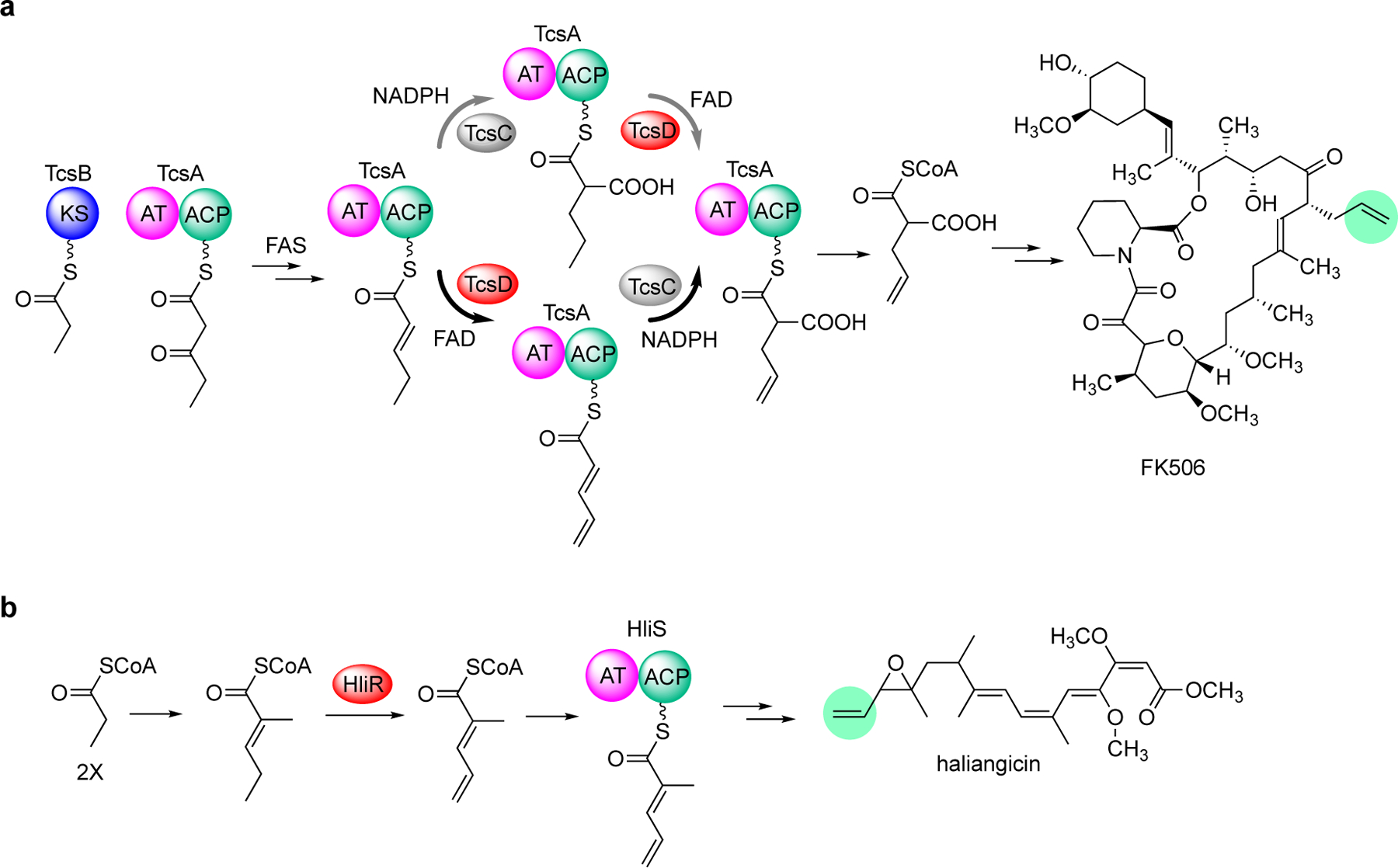
Variants of acyl-CoA dehydrogenases are involved in the formation of the terminal akenes in FK506 and haliangicin biosyntheses. (a) The γ,δ-variant of acyl-CoA dehydrogenase TcsD is involved in the formation of allylmalonyl-CoA, an extender unit in FK506 biosynthesis. An earlier study suggested that the crotonyl-CoA reductase TcsC precedes TcsD (gray bold arrows). A more recent study showed that TscD precedes TcsC (black bold arrows); (b) The γ,δ-acyl-CoA dehydrogenase variant HliR is involved in haliangicin biosynthesis.
More recently, Keasling and co-workers revisited the catalytic activity of TcsD and found that the enzyme is highly selective toward short-chain α,β-unsaturated acyl-ACP substrates.[13] The enzyme can efficiently convert 2-pentenoyl-TcsA to 2,4-pentadienoyl-TcsA in the presence of ferrocenium hexafluorophosphate to facilitate enzyme turnover, as analyzed by targeted LC–MS/MS after protease digestion generating the characteristic phosphopantetheine ejection ion.[27] Interestingly, in contrast to that previously reported, TcsD was unable to convert propylmalonyl-TcsA to allylmalonyl-TcsA under the same assay conditions, suggesting that the biosynthesis of allylmalonyl-TcsA can only proceed via 2,4-pentadienoyl-TcsA, which is then converted to allylmalonyl-TcsA by the crotonyl-CoA reductase TcsC (Figure 8a, black bold arrows). This γ,δ-variant of acyl-CoA dehydrogenase is distinct from the canonical α,β-acyl-CoA dehydrogenases involved in fatty acid β-oxidation. The X-ray crystal structure of TcsD (PDB ID code 6U1V) revealed the presence of amino acid residues Phe79, Leu83, and Ile363 that form a bulky wall in the active site of the enzyme to prevent the entrance of long fatty acid substrates.[13] On the other hand, the enzyme regioselectivity is proposed to be due to a shift in the positioning of the FAD cofactor. Based on the knowledge obtained from these biochemical and structural studies, genome mining was performed to identify numerous putative γ,δ-variants of acyl-CoA dehydrogenases that were previously unidentified or misannotated, many of which are thought to be involved in natural products biosynthesis.[13]
Another putative γ,δ-variant of acyl-CoA dehydrogenase, NftN, has been identified in the NFAT-133 biosynthetic pathways in S. pactum and S. conglobatus.[14a, 14c, 28] A homologous protein (VerN) was found in the veramycin pathway in Streptomyces sp. ST157608.[14d] NFAT-133 and related compounds (e.g., benwamycins, panowamycins, veramycins) are produced by several strains of Streptomyces and known to have immunosuppressant and anti-diabetic activities.[14a, 14d, 28–29] Although NFAT-133 and its other major analogues do not contain a terminal alkene, several of the minor analogues and intermediates obtained from knocked-out mutants, e.g., TM-125, TM-126, TM-131, and TM-132, have a terminal alkene, suggesting that NFAT-133 is synthesized from a polyketide backbone that contains a terminal alkene (Figure 9a).[14b–d] Similar to that observed in FK506 biosynthesis, the γ,δ-variant of acyl-CoA dehydrogenase NftN has been proposed to catalyze the conversion of 2-pentenoyl-ACP to 2,4-pentadienoyl-ACP (Figure 9b).[14c] Inactivation of NftN resulted in a mutant that no longer produced NFAT-133 but generated its corresponding saturated analogues, TM-136 and TM-137 (Figure 9a). However, no in vitro biochemical data for NftN are currently available to confirm its natural substrate.
Figure 9.
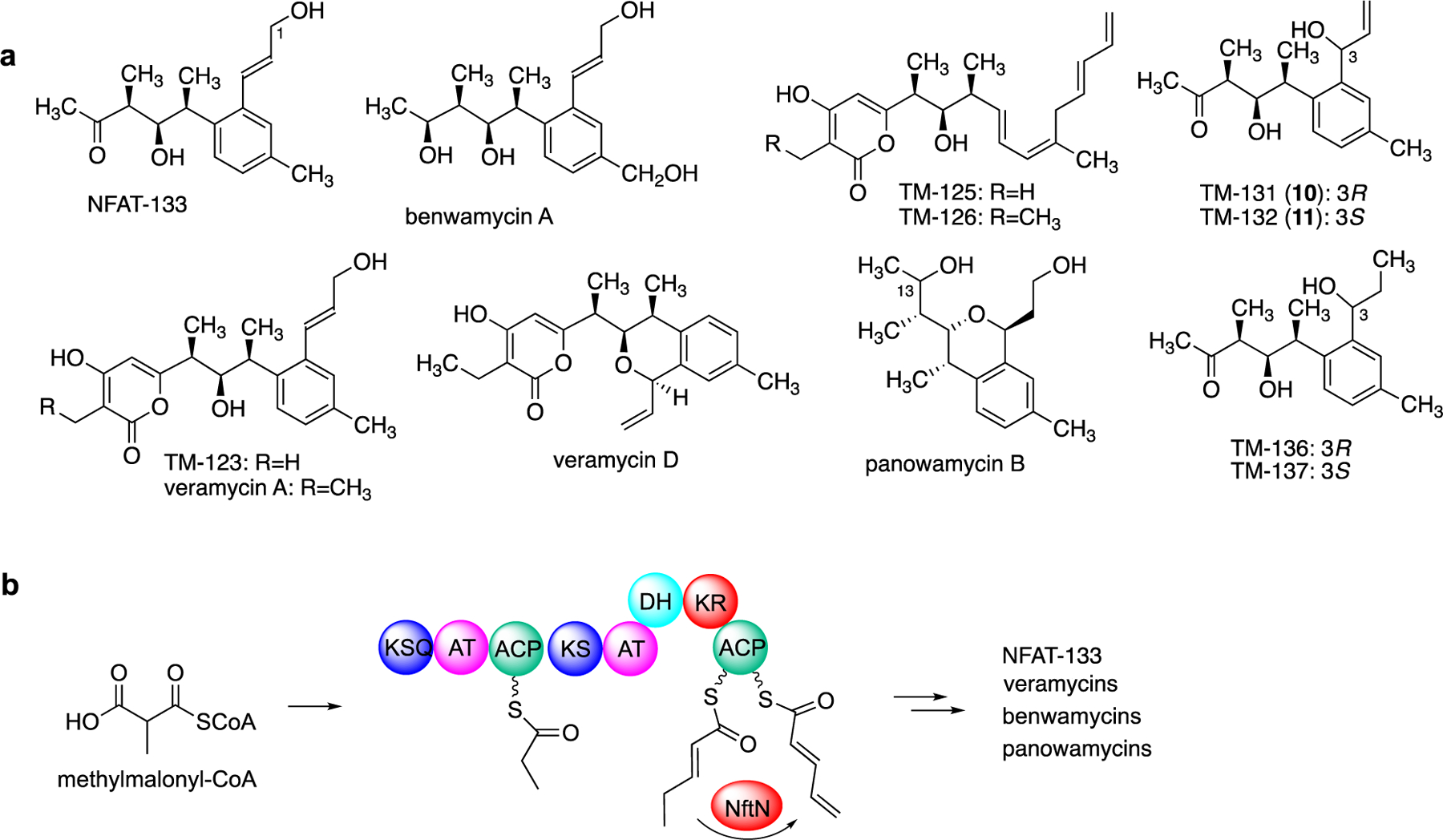
ACP-dependent dehydrogenation involved in the biosynthesis of NFAT-133 and its analogues. (a) Chemical structures of NFAT-133 and its analogues; (b) A variant of acyl-CoA dehydrogenase, NftN, may catalyze the conversion of 2-pentenoyl-ACP to 2,4-pentadienoyl-ACP during NFAT-133 polyketide assembly.
2.5. Glycosylation of an ACP-bound substrates
Another unusual modification reported recently is the glycosylation of ACP-bound substrates in the pactamycin and mitomycin pathways.[15] Pactamycin is an aminocyclitol-containing antitumor antibiotic produced by Streptomyces pactum that has strong inhibitory activity against both eukaryotic and prokaryotic ribosomes. Its structure consists of an aminocyclopentitol core decorated with a 3-aminoacetophenone, a 6-methylsalicylyl, and a urea moiety. The biosynthesis of pactamycin has been studied by several groups, and many aspects of the biosynthetic pathway have been reported.[30] However, details on the mode of formation of the aminocyclopentitiol core remains elusive.
Early work by Liu and co-workers has shown that the desosamine glycosyltransferase DesVII was able to glycosylate N-acetyl-cysteamine (NAC) thioesters of polyketide intermediates, suggesting that some glycosyltransferases may be able to act on growing polyketide chains.[31] At this time, there were no reports of glycosylation taking place on-line during the polyketide assembly process. However, our group and the Eguchi group independently reported unique glycosylation of ACP-bound substrates catalyzed by a glycosyltransferase enzyme from the pactamycin pathway. The Eguchi group reported that the glycosyltransferase PctL (PtmJ) from the pactamycin pathway in S. pactum NBRC 13433 was able to catalyze the glycosylation of ACP-bound 3-aminobenzoic acid (3ABA).[15b] 3ABA is first activated by the AMP-ligase PctU (PtmS) in the presence of ATP to give 3ABA-AMP, which is then loaded to the acyl carrier protein PctK (PtmI) to give 3ABA-PctK (Figure 10).[15a, 15b, 32] Independently, we demonstrated that holo-PtmI can also be loaded with a malonyl extender unit to give malonyl-PtmI. Subsequently, decarboxylative Claisen condensation between 3ABA-PtmI and malonyl-PtmI catalyzed by the putative β-ketoacyl-ACP synthase PtmK produces a β-ketoacyl intermediate, 3-[3-aminophenyl]3-oxopropionyl-PtmI (Figure 10).[15a] Gene inactivation experiments of the pactamycin cluster in S. pactum ATCC 27456 revealed that the three PKS proteins PtmS, PtmI, and PtmK function in a coordinated fashion.[33] The ACP-bound intermediate is then glycosylated by the N-glycosyltransferase PtmJ (PctL) to give GlcNAc-3-[3-aminophenyl]3-oxopropionyl-PtmI.[15a] PtmJ has been shown to be a promiscuous enzyme that can glycosylate both small molecules and ACP-bound substrates. In addition to 3-[3-aminophenyl]3-oxopropionyl-PtmI, it can also glycosylate 3ABA-PtmI to give GlcNAc-3ABA-PtmI.[15a, 15b] However, GlcNAc-3ABA-PtmI was not recognized as a substrate by the ketosynthase PtmK, suggesting that glycosylation most likely take place after the polyketide chain extension. The glycosylated compounds are further modified by other enzymes while tethered to an acyl-carrier protein, representing one of unique phenomena that can occur in natural product biosynthesis.
Figure 10.
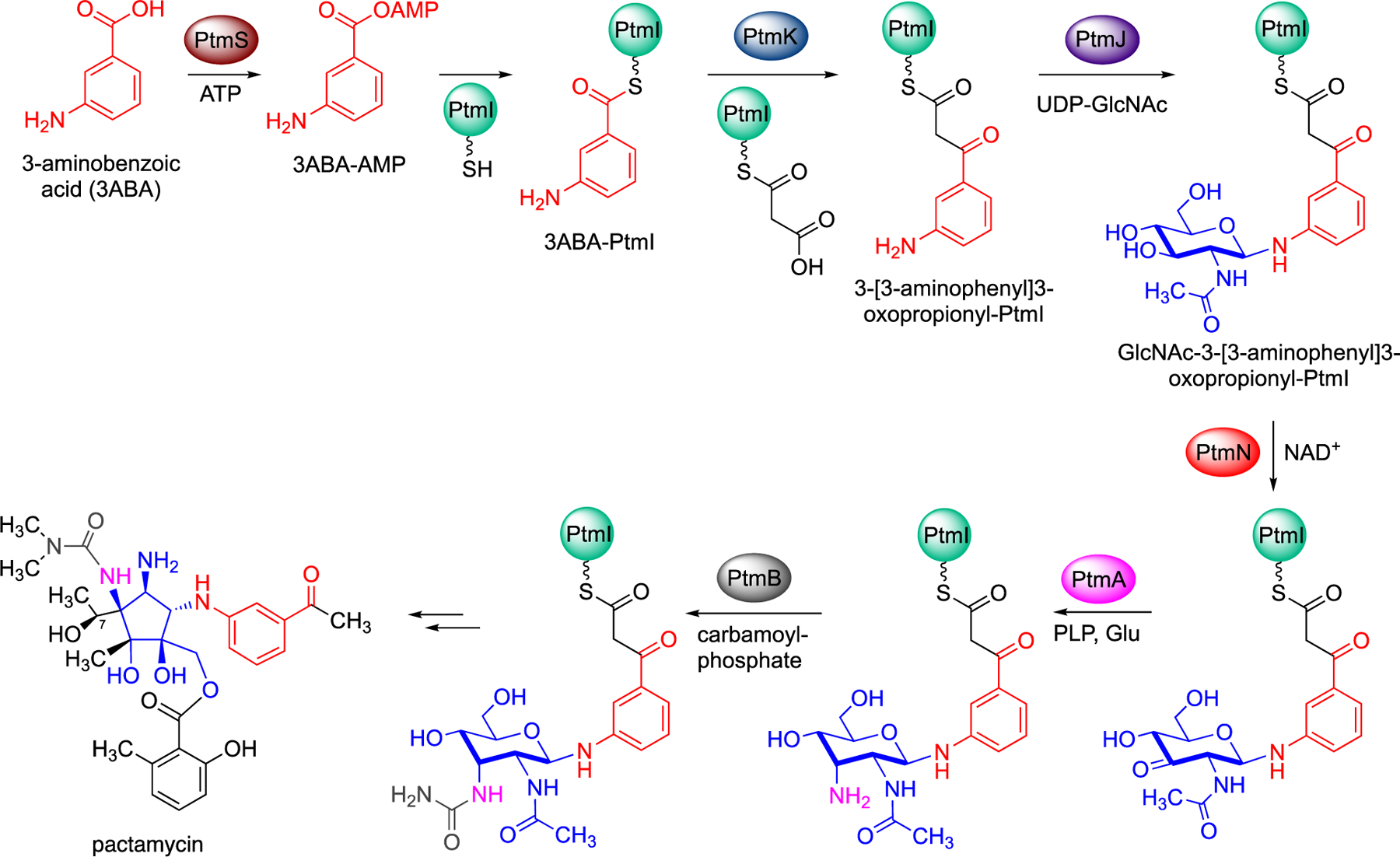
Glycosylation of ACP-bound polyketide intermediate and subsequent modifications in pactamycin biosynthesis.
2.6. Sugar modifications of ACP-bound substrates
Within the pactamycin gene cluster, there are a number of genes that encode for proteins annotated as sugar modifying enzymes. Those include PtmN (an oxidoreductase), PtmA (an aminotransferase), PtmB (a putative carbamoyltransferase), and PtmG (a putative deacetylase). This subset of proteins appears to be similar to those involved in lipid A biosynthesis in Acidithiobacillus ferroxidans. PtmN (a.k.a. PctP) and PtmA (PctC) have been previously shown to transform GlcNAc-3AAP to its corresponding 3´-amino derivative,[34] indicating their substrate promiscuity. PtmN (PctP) can also oxidize GlcNAc-3ABA and GlcNAc-3-aminophenyl-β-oxopropanoic acid ethyl ester, but does not accept UDP-GlcNAc, glucosaminyl-3AAP, and 4´-C-methyl-N-acetyl-D-galactosaminyl-3-aminoacetophenone as substrates.[34] The fact that PtmN recognizes GlcNAc-3ABA and GlcNAc-3AAP but not UDP-GlcNAc as substrates suggests that the oxidation and the subsequent transamination reactions occur after a glycosylation reaction.
Further investigations of pactamycin biosynthesis confirmed that the post-glycosylation modifications of the sugar moiety occur while it is still attached to the carrier protein.[35] These events were unprecedented and opened-up a new chapter in natural product biosynthesis. In vitro reconstitution of PtmS, PtmI, PtmJ, PtmN (an oxidoreductase), PtmA (an aminotransferase), and PtmB (a putative carbamoyltransferase) showed that the N-acetyl-D-glucosamine moiety of the glycosylated polyketide is oxidized by PtmN and transaminated by PtmA to give ACP-bound 3-amino-3-deoxy-N-acetyl-D-glucosaminyl-polyketide (Figure 10).[35] The amino group is then coupled with carbamoyl phosphate by PtmB to give a urea functionality.
The putative hydrolase PtmG was found to be a deacetylase that hydrolyses the C-2 N-acetyl group of carbamoyl-3-amino-3-deoxy-N-acetyl-D-galactosaminyl-3-[3-aminoacetophenone] or carbamoyl-3-amino-3-deoxy-N-acetyl-D-galactosaminyl-3-[3-aminobenzoyl]-SNAC to give their corresponding free amines.[35] However, due to analytical challenges, this transformation could only be demonstrated using free compounds or NAC thioesters, not the ACP-bound substrates. Therefore, whether PtmG can process an ACP-bound substrate remains undetermined.
2.7. Glycosylation of ACP-bound 3-amino-5-hydroxy-benzoic acid (AHBA)
Another example of ACP-dependent glycosylation was observed in the biosynthesis of mitomycins, mitosane-containing antitumor antibiotics produced by several strains of actinomycetes. Sherman and co-workers identified the mitomycin biosynthetic gene cluster in Streptomyces lavandulae NRRL 2564 and through gene inactivation confirmed the involvement of MitA (an AHBA synthase) and MitB (a glycosyltransferase) in mitomycin biosynthesis.[36] While this further confirmed the results of earlier studies on the biosynthetic origin of mitomycin being from 3-amino-5-hydroxybenzoic acid (AHBA) and N-acetylglucosamine, how the unique mitosane core structure is formed remained unclear. Interestingly, despite the significant structural differences between mitomycin and pactamycin, their biosynthetic pathways share a set of very similar enzymes.[30a] Particularly, the acyl carrier protein PtmI, the glycosyltransferase PtmJ, the deacetylase PtmG, and the radical-SAM enzyme PtmC from the pactamycin pathway are highly similar to MmcB, MitB, MitC, and MitD, respectively, in the mitomycin pathway.
A subsequent study by Nguyen and Yokoyama, and independently by Dairi and their co-workers, revealed the glycosylation of ACP-bound AHBA in mitomycin biosynthesis (Figure 11a).[15c, 15d] They systematically demonstrated that AHBA is first activated by the AMP-ligase MitE in the presence of ATP and then attached to the acyl carrier protein MmcB. The resulting AHBA-ACP is then coupled with UDP-GlcNAc by the glycosyltransferase MitB to give GlcNAc-AHBA-MmcB. This finding rendered MitB to be one of the only two known glycosyltransferases – the other is PtmJ (PctL) in the pactamycin pathway – that glycosylate small molecules while they are still tethered to the ACP.
Figure 11.

Glycosylation of ACP-bound AHBA and subsequent modification in mitomycin biosynthesis. (a) attachment of AHBA to the acyl carrier protein MmcB and glycosylation by the glycosyltransferase MitB; (b) deacetylation of GlcNAc-AHBA-MmcB to GlcN-AHBA-MmcB and transformation of the sugar moiety to a linear aminodiol that terminates with an epoxyethane catalyzed by the NADPH-dependent protein MitF and the radical SAM protein MitD.
2.8. Processing of GlcNAc-AHBA-ACP and proposed enzymatic steps for mitosane formation
Although the characterization of MitB provided a significant advancement in our understanding of mitomycin biosynthesis, the mode of formation of mitosane was still unclear. However, by tracing the mitomycin intermediates that are covalently tethered to the ACP MmcB, Liu and co-workers were able to identify additional enzymatic steps downstream of the pathway (Figure 11b).[37] Following the formation of GlcNAc-AHBA-MmcB, deacetylation by MitC takes place to give ACP-bound AHBA−GlcN. Subsequently, the glucosamine moiety is transformed into a linear aminodiol that terminates with an epoxyethane. This unusual transformation relies on the functional association of a NADPH-dependent protein MitF with a radical SAM protein MitD. MitC is unable to convert UDP-GlcNAc to UDP-GlcN, indicating that deacetylation indeed occurs after glycosylation. The deacetylation reaction by MitC is similar to that observed for PtmG in the pactamycin pathway; therefore, it is tempting to believe that the natural substate for PtmG is also an ACP-bound substrate. However, whether PtmG can catalyze deacetylation of an ACP-bound substrate remains to be seen.
3. Summary and Outlook
Over the eons, nature has evolved in a myriad of ways, and with contemporary tools and technologies, new discoveries beyond paradigms continue to emerge. This is particularly true with the genes and proteins that are involved in primary and secondary metabolism. Constant mutations in the genetic codes of proteins have generated countless variants of enzymes with altered catalytic functions. Some of which are embedded into large proteins or mega-synthases, e.g., as domains in modular PKS, whereas others operate discretely but have the ability to modify substrates that are attached to PKSs or stand-alone ACPs. The on-line methyl esterification in aurantinin biosynthesis, the epoxidation of a polyketide intermediate in shuangdaolide biosynthesis, and the glycosylation of ACP-bound substrates in the pactamycin and mitomycin pathways are among the known examples of modifications to PKS/ACP-bound compounds by discrete enzymes. These rather unusual traits are not easily discovered without the careful characterizations of the pathways. Consequently, while functional predictions through bioinformatics and computational approaches have significantly ameliorated our ability to elucidate biosynthetic pathways,[38] detailed investigations of the enzymes involved in natural products biosynthetic pathways remain crucial to the discovery of these unique properties. Also, how those discrete enzymes interact with the ACPs or the PKSs in a selective manner and alter the small molecules that are tethered to the proteins is generally unknown and remains to be investigated. More studies on these unusual enzymes will lead to new knowledge that may further the advancement of natural product biotechnology.
Acknowledgements
This work was supported by grant AI129957 from the National Institute of Allergy and Infectious Diseases. Leigh E. Skala was supported by Grant T32 AT010131 from the National Center for Complementary and Integrative Health (NCCIH). The content is solely the responsibility of the authors and does not represent the official views of the National Institute of Allergy and Infectious Diseases, the National Center for Complementary and Integrative Health, or the National Institutes of Health (NIH).
Biographies

Leigh Skala is currently pursuing a Ph.D. in Pharmaceutical Sciences with a focus on drug discovery at Oregon State University. She received a bachelor’s degree in chemistry and minor in toxicology in 2021 from Oregon State University. During her time as an undergraduate, she started working in Professor Taifo Mahmud’s laboratory, where she developed a passion for natural products research. Her research interests include the biosynthesis and regulation of natural products and developing new technologies to activate silent gene clusters.

Benjamin (BJ) Philmus is an Associate Professor at Oregon State University. He obtained his B.S. from Southampton College in marine biology. He took his love of the ocean to the University of Hawaií and earned his Ph.D. under the mentorship of Prof. Thomas Hemscheidt where he studied cyanobacterial natural products and biosynthesis. He joined Texas A&M University as a postdoctoral researcher with Prof. Tadhg Begley and studied enzyme mechanisms including radical SAM enzymes. He joined the faculty at OSU in 2013 where his group uses genome mining, synthetic biology, and heterologous expression to understand cyanobacterial natural products.

Taifo Mahmud is a Professor of Natural Products and Medicinal Chemistry at Oregon State University. He received a M.S. and a Ph.D. in Pharmaceutical Sciences from Osaka University, Japan, and was a Postdoctoral Research Associate with Prof. Heinz G. Floss and a Research Assistant Professor of Chemistry at the University of Washington. After six enjoyable years living in Seattle, he moved to Corvallis, the home of the Oregon State Beavers. Dr Mahmud’s research interests are broadly in medicinal chemistry and natural products biosynthesis. He uses molecular genetics, enzymology, and chemistry approaches to create and develop new biologically active compounds.
Footnotes
Institute and/or researcher Twitter usernames: @OSUPharmacy and @PhilmusLab
References
- [1].a) Grininger M, Nat Chem Biol 2023, 19, 401–415; [DOI] [PubMed] [Google Scholar]; b) Katsuyama Y, Miyanaga A, Curr Opin Chem Biol 2022, 71, 102223; [DOI] [PubMed] [Google Scholar]; c) Weissman KJ, Nat Chem Biol 2015, 11, 660–670. [DOI] [PubMed] [Google Scholar]
- [2].a) Walsh CT, Chen H, Keating TA, Hubbard BK, Losey HC, Luo L, Marshall CG, Miller DA, Patel HM, Curr Opin Chem Biol 2001, 5, 525–534; [DOI] [PubMed] [Google Scholar]; b) Xu J, Wan E, Kim CJ, Floss HG, Mahmud T, Microbiology 2005, 151, 2515–2528; [DOI] [PubMed] [Google Scholar]; c) Rix U, Fischer C, Remsing LL, Rohr J, Nat Prod Rep 2002, 19, 542–580. [DOI] [PubMed] [Google Scholar]
- [3].Staunton J, Wilkinson B, Chem Rev 1997, 97, 2611–2630. [DOI] [PubMed] [Google Scholar]
- [4].a) Luhavaya H, Dias MV, Williams SR, Hong H, de Oliveira LG, Leadlay PF, Angew Chem Int Ed Engl 2015, 54, 13622–13625; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Hiratsuka T, Suzuki H, Kariya R, Seo T, Minami A, Oikawa H, Angew Chem Int Ed Engl 2014, 53, 5423–5426; [DOI] [PubMed] [Google Scholar]; c) Sato M, Yagishita F, Mino T, Uchiyama N, Patel A, Chooi YH, Goda Y, Xu W, Noguchi H, Yamamoto T, Hotta K, Houk KN, Tang Y, Watanabe K, ChemBioChem 2015, 16, 2294–2298; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Sundaram S, Hertweck C, Curr Opin Chem Biol 2016, 31, 82–94; [DOI] [PubMed] [Google Scholar]; e) Pang B, Wang M, Liu W, Nat Prod Rep 2016, 33, 162–173. [DOI] [PubMed] [Google Scholar]
- [5].Zha L, Jiang Y, Henke MT, Wilson MR, Wang JX, Kelleher NL, Balskus EP, Nat Chem Biol 2017, 13, 1063–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Guo S, Sang Y, Zheng C, Xue XS, Tang Z, Liu W, J Am Chem Soc 2023, 145, 5017–5028. [DOI] [PubMed] [Google Scholar]
- [7].a) Julien B, Tian ZQ, Reid R, Reeves CD, Chem Biol 2006, 13, 1277–1286; [DOI] [PubMed] [Google Scholar]; b) Vaillancourt FH, Yeh E, Vosburg DA, O’Connor SE, Walsh CT, Nature 2005, 436, 1191–1194; [DOI] [PubMed] [Google Scholar]; c) Ullrich M, Bender CL, J Bacteriol 1994, 176, 7574–7586; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Couch R, O’Connor SE, Seidle H, Walsh CT, Parry R, J Bacteriol 2004, 186, 35–42; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Chang Z, Sitachitta N, Rossi JV, Roberts MA, Flatt PM, Jia J, Sherman DH, Gerwick WH, J Nat Prod 2004, 67, 1356–1367; [DOI] [PubMed] [Google Scholar]; f) Li X, Shimaya R, Dairi T, Chang WC, Ogasawara Y, Angew Chem Int Ed Engl 2022, 61, e202113189; [DOI] [PMC free article] [PubMed] [Google Scholar]; g) Shimo S, Ushimaru R, Engelbrecht A, Harada M, Miyamoto K, Kulik A, Uchiyama M, Kaysser L, Abe I, J Am Chem Soc 2021, 143, 18413–18418; [DOI] [PubMed] [Google Scholar]; h) Neumann CS, Walsh CT, J Am Chem Soc 2008, 130, 14022–14023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Bretschneider T, Heim JB, Heine D, Winkler R, Busch B, Kusebauch B, Stehle T, Zocher G, Hertweck C, Nature 2013, 502, 124–128; [DOI] [PubMed] [Google Scholar]; b) Sundaram S, Heine D, Hertweck C, Nat Chem Biol 2015, 11, 949–951; [DOI] [PubMed] [Google Scholar]; c) Yun CS, Motoyama T, Osada H, Nat Commun 2015, 6, 8758; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Bauman KD, Shende VV, Chen PY, Trivella DBB, Gulder TAM, Vellalath S, Romo D, Moore BS, Nat Chem Biol 2022, 18, 538–546; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Abugrain ME, Brumsted CJ, Osborn AR, Philmus B, Mahmud T, ACS Chem Biol 2017, 12, 362–366; [DOI] [PubMed] [Google Scholar]; f) Nofiani R, Philmus B, Nindita Y, Mahmud T, MedChemComm 2019, 10, 1517–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Meoded RA, Ueoka R, Helfrich EJN, Jensen K, Magnus N, Piechulla B, Piel J, Angew Chem Int Ed Engl 2018, 57, 11644–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hemmerling F, Meoded RA, Fraley AE, Minas HA, Dieterich CL, Rust M, Ueoka R, Jensen K, Helfrich EJN, Bergande C, Biedermann M, Magnus N, Piechulla B, Piel J, Angew Chem Int Ed Engl 2022, 61, e202116614. [DOI] [PubMed] [Google Scholar]
- [11].Li P, Chen M, Tang W, Guo Z, Zhang Y, Wang M, Horsman GP, Zhong J, Lu Z, Chen Y, Nat Commun 2021, 12, 4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Liu Y, Zhou H, Zhao S, Hao X, Dai G, Zhong L, Ren X, Sui H, Zhang Y, Yan F, Bian X, ACS Chem Biol 2023, 18, 2474–2484. [DOI] [PubMed] [Google Scholar]
- [13].Blake-Hedges JM, Pereira JH, Cruz-Morales P, Thompson MG, Barajas JF, Chen J, Krishna RN, Chan LJG, Nimlos D, Alonso-Martinez C, Baidoo EEK, Chen Y, Gin JW, Katz L, Petzold CJ, Adams PD, Keasling JD, J Am Chem Soc 2020, 142, 835–846. [DOI] [PubMed] [Google Scholar]
- [14].a) Zhou W, Posri P, Abugrain ME, Weisberg AJ, Chang JH, Mahmud T, ACS Chem Biol 2020, 15, 3217–3226; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou W, Posri P, Liu XJ, Ju Z, Lan WJ, Mahmud T, J Nat Prod 2021, 84, 2411–2419; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zhou W, Alharbi HA, Hummingbird E, Keatinge-Clay AT, Mahmud T, ACS Chem Biol 2022, 17, 2039–2045; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Dardic D, Bohringer N, Plaza A, Zubeil F, Pohl J, Sommer S, Padva L, Becker J, Patras MA, Bill MK, Kurz M, Toti L, Gorgens SW, Schuler SMM, Billion A, Schwengers O, Wohlfart P, Goesmann A, Tennagels N, Vilcinskas A, Hammann PE, Schaberle TF, Bauer A, Org Chem Front 2022, 9, 1604–1615; [Google Scholar]; e) Sun Y, Feng Z, Tomura T, Suzuki A, Miyano S, Tsuge T, Mori H, Suh JW, Iizuka T, Fudou R, Ojika M, Sci Rep 2016, 6, 22091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Eida AA, Abugrain ME, Brumsted CJ, Mahmud T, Nat Chem Biol 2019, 15, 795–802; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Kudo F, Zhang J, Sato S, Hirayama A, Eguchi T, ChemBioChem 2019, 20, 2458–2462; [DOI] [PubMed] [Google Scholar]; c) Ogasawara Y, Nakagawa Y, Maruyama C, Hamano Y, Dairi T, Bioorg Med Chem Lett 2019, 29, 2076–2078; [DOI] [PubMed] [Google Scholar]; d) Nguyen HP, Yokoyama K, Biochemistry 2019, 58, 2804–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].a) Takada N, Sato H, Suenaga K, Arimoto H, Yamada K, Ueda K, Uemura D, Tetrahedron Lett 1999, 40, 6309–6312; [Google Scholar]; b) Teruya T, Suenaga K, Maruyama S, Kurotaki M, Kigoshi H, Tetrahedron 2005, 61, 6561–6567. [Google Scholar]
- [17].a) Yamaoka M, Sato K, Kobayashi M, Nishio N, Ohkubo M, Fujii T, Nakajima H, J Antibiot 2005, 58, 654–662; [DOI] [PubMed] [Google Scholar]; b) Matilla MA, Leeper FJ, Salmond GP, Environ Microbiol 2015, 17, 2993–3008; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Levenfors JJ, Hedman R, Thaning C, Gerhardson B, Welch CJ, Soil Biol. Biochem 2004, 36, 677–685; [Google Scholar]; d) Strobel G, Li JY, Sugawara F, Koshino H, Harper J, Hess WM, Microbiology 1999, 145 (Pt 12), 3557–3564. [DOI] [PubMed] [Google Scholar]
- [18].Ueoka R, Meoded RA, Gran-Scheuch A, Bhushan A, Fraaije MW, Piel J, Angew Chem Int Ed Engl 2020, 59, 7761–7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Vriens E, De Ruysscher D, Weir ANM, Dekimpe S, Steurs G, Shemy A, Persoons L, Santos AR, Williams C, Daelemans D, Crump MP, Voet A, De Borggraeve W, Lescrinier E, Masschelein J, Angew Chem Int Ed Engl 2023, 62, e202304476. [DOI] [PubMed] [Google Scholar]
- [20].An JS, Shin B, Kim TH, Hwang S, Shin YH, Cui J, Du YE, Yi J, Nam SJ, Hong S, Shin J, Jang J, Yoon YJ, Oh DC, Org Lett 2021, 23, 3359–3363. [DOI] [PubMed] [Google Scholar]
- [21].Eustaquio AS, Janso JE, Ratnayake AS, O’Donnell CJ, Koehn FE, Proc Natl Acad Sci U S A 2014, 111, E3376–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Whittle EJ, Tremblay AE, Buist PH, Shanklin J, Proc Natl Acad Sci U S A 2008, 105, 14738–14743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Heinzelmann E, Berger S, Muller C, Hartner T, Poralla K, Wohlleben W, Schwartz D, Microbiology 2005, 151, 1963–1974. [DOI] [PubMed] [Google Scholar]
- [24].a) Fudou R, Iizuka T, Sato S, Ando T, Shimba N, Yamanaka S, J Antibiot 2001, 54, 153–156; [DOI] [PubMed] [Google Scholar]; b) Goranovic D, Kosec G, Mrak P, Fujs S, Horvat J, Kuscer E, Kopitar G, Petkovic H, J Biol Chem 2010, 285, 14292–14300; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Annaval T, Han L, Rudolf JD, Xie G, Yang D, Chang CY, Ma M, Crnovcic I, Miller MD, Soman J, Xu W, Phillips GN Jr., Shen B, ACS Chem Biol 2018, 13, 2728–2738. [DOI] [PubMed] [Google Scholar]
- [25].McCarthy JG, Eisman EB, Kulkarni S, Gerwick L, Gerwick WH, Wipf P, Sherman DH, Smith JL, ACS Chem Biol 2012, 7, 1994–2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mo S, Kim DH, Lee JH, Park JW, Basnet DB, Ban YH, Yoo YJ, Chen SW, Park SR, Choi EA, Kim E, Jin YY, Lee SK, Park JY, Liu Y, Lee MO, Lee KS, Kim SJ, Kim D, Park BC, Lee SG, Kwon HJ, Suh JW, Moore BS, Lim SK, Yoon YJ, J Am Chem Soc 2011, 133, 976–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Dorrestein PC, Bumpus SB, Calderone CT, Garneau-Tsodikova S, Aron ZD, Straight PD, Kolter R, Walsh CT, Kelleher NL, Biochemistry 2006, 45, 12756–12766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang M, Li W, Zhou L, Lin X, Zhang W, Shen Y, Deng H, Lin HW, Zhou Y, Synth Syst Biotechnol 2023, 8, 349–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].a) Burres NS, Premachandran U, Hoselton S, Cwik D, Hochlowski JE, Ye Q, Sunga GN, Karwowski JP, Jackson M, Whittern DN, et al. , J Antibiot 1995, 48, 380–386; [DOI] [PubMed] [Google Scholar]; b) Hashida J, Niitsuma M, Iwatsuki M, Mori M, Ishiyama A, Namatame M, Nishihara-Tsukashima A, Matsumoto A, Ara I, Takahashi Y, Yamada H, Otoguro K, Shiomi K, Omura S, J Antibiot 2012, 65, 197–202; [DOI] [PubMed] [Google Scholar]; c) Yang FX, Huang JP, Liu Z, Wang Z, Yang J, Tang J, Yu Z, Yan Y, Kai G, Huang SX, J Nat Prod 2020, 83, 111–117. [DOI] [PubMed] [Google Scholar]
- [30].a) Ito T, Roongsawang N, Shirasaka N, Lu W, Flatt PM, Kasanah N, Miranda C, Mahmud T, ChemBioChem 2009, 10, 2253–2265; [DOI] [PubMed] [Google Scholar]; b) Kudo F, Kasama Y, Hirayama T, Eguchi T, J Antibiot 2007, 60, 492–503; [DOI] [PubMed] [Google Scholar]; c) Lu W, Roongsawang N, Mahmud T, Chem Biol 2011, 18, 425–431; [DOI] [PubMed] [Google Scholar]; d) Hirayama A, Miyanaga A, Kudo F, Eguchi T, ChemBioChem 2015, 16, 2484–2490; [DOI] [PubMed] [Google Scholar]; e) Abugrain ME, Lu W, Li Y, Serrill JD, Brumsted CJ, Osborn AR, Alani A, Ishmael JE, Kelly JX, Mahmud T, ChemBioChem 2016, 17, 1585–1588. [DOI] [PubMed] [Google Scholar]
- [31].a) Kao CL, Borisova SA, Kim HJ, Liu HW, J Am Chem Soc 2006, 128, 5606–5607; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Borisova SA, Kim HJ, Pu X, Liu HW, ChemBioChem 2008, 9, 1554–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Eida AA, Mahmud T, Appl Microbiol Biotechnol 2019, 103, 4337–4345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Abugrain ME, Brumsted CJ, Osborn AR, Philmus B, Mahmud T, ACS Chem Biol 2017, 12, 362–366. [DOI] [PubMed] [Google Scholar]
- [34].Hirayama A, Chu J, Goto E, Kudo F, Eguchi T, ChemBioChem 2018, 19, 126–130. [DOI] [PubMed] [Google Scholar]
- [35].Eida AA, Samadi A, Tsunoda T, Mahmud T, Chemistry 2023, e202301056. [DOI] [PMC free article] [PubMed]
- [36].a) Mao Y, Varoglu M, Sherman DH, Chem Biol 1999, 6, 251–263; [DOI] [PubMed] [Google Scholar]; b) Mao Y, Varoglu M, Sherman DH, J Bacteriol 1999, 181, 2199–2208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wang S, Cheng Y, Wang X, Yang Q, Liu W, J Am Chem Soc 2022, 144, 14945–14956. [DOI] [PubMed] [Google Scholar]
- [38].a) Blin K, Shaw S, Augustijn HE, Reitz ZL, Biermann F, Alanjary M, Fetter A, Terlouw BR, Metcalf WW, Helfrich EJN, van Wezel GP, Medema MH, Weber T, Nucleic Acids Res 2023, 51, W46–W50; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Medema MH, Kottmann R, Yilmaz P, Cummings M, Biggins JB, Blin K, de Bruijn I, Chooi YH, Claesen J, Coates RC, Cruz-Morales P, Duddela S, Dusterhus S, Edwards DJ, Fewer DP, Garg N, Geiger C, Gomez-Escribano JP, Greule A, Hadjithomas M, Haines AS, Helfrich EJ, Hillwig ML, Ishida K, Jones AC, Jones CS, Jungmann K, Kegler C, Kim HU, Kotter P, Krug D, Masschelein J, Melnik AV, Mantovani SM, Monroe EA, Moore M, Moss N, Nutzmann HW, Pan G, Pati A, Petras D, Reen FJ, Rosconi F, Rui Z, Tian Z, Tobias NJ, Tsunematsu Y, Wiemann P, Wyckoff E, Yan X, Yim G, Yu F, Xie Y, Aigle B, Apel AK, Balibar CJ, Balskus EP, Barona-Gomez F, Bechthold A, Bode HB, Borriss R, Brady SF, Brakhage AA, Caffrey P, Cheng YQ, Clardy J, Cox RJ, De Mot R, Donadio S, Donia MS, van der Donk WA, Dorrestein PC, Doyle S, Driessen AJ, Ehling-Schulz M, Entian KD, Fischbach MA, Gerwick L, Gerwick WH, Gross H, Gust B, Hertweck C, Hofte M, Jensen SE, Ju J, Katz L, Kaysser L, Klassen JL, Keller NP, Kormanec J, Kuipers OP, Kuzuyama T, Kyrpides NC, Kwon HJ, Lautru S, Lavigne R, Lee CY, Linquan B, Liu X, Liu W, et al. , Nat Chem Biol 2015, 11, 625–631; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Terlouw BR, Blin K, Navarro-Munoz JC, Avalon NE, Chevrette MG, Egbert S, Lee S, Meijer D, Recchia MJJ, Reitz ZL, van Santen JA, Selem-Mojica N, Torring T, Zaroubi L, Alanjary M, Aleti G, Aguilar C, Al-Salihi SAA, Augustijn HE, Avelar-Rivas JA, Avitia-Dominguez LA, Barona-Gomez F, Bernaldo-Aguero J, Bielinski VA, Biermann F, Booth TJ, Carrion Bravo VJ, Castelo-Branco R, Chagas FO, Cruz-Morales P, Du C, Duncan KR, Gavriilidou A, Gayrard D, Gutierrez-Garcia K, Haslinger K, Helfrich EJN, van der Hooft JJJ, Jati AP, Kalkreuter E, Kalyvas N, Kang KB, Kautsar S, Kim W, Kunjapur AM, Li YX, Lin GM, Loureiro C, Louwen JJR, Louwen NLL, Lund G, Parra J, Philmus B, Pourmohsenin B, Pronk LJU, Rego A, Rex DAB, Robinson S, Rosas-Becerra LR, Roxborough ET, Schorn MA, Scobie DJ, Singh KS, Sokolova N, Tang X, Udwary D, Vigneshwari A, Vind K, Vromans S, Waschulin V, Williams SE, Winter JM, Witte TE, Xie H, Yang D, Yu J, Zdouc M, Zhong Z, Collemare J, Linington RG, Weber T, Medema MH, Nucleic Acids Res 2023, 51, D603–D610. [DOI] [PMC free article] [PubMed] [Google Scholar]