

Abstract
Mixed-stack complexes which comprise columns of alternating donors and acceptors are organic conductors with typically poor electrical conductivity because they are either in a neutral or highly ionic state. This indicates that conductive carriers are insufficient or are mainly localized. In this study, mixed-stack complexes that uniquely exist at the neutral–ionic boundary were synthesized by combining donors (bis(3,4-ethylenedichalcogenothiophene)) and acceptors (fluorinated tetracyanoquinodimethanes) with similar energy levels and orbital symmetry between the highest occupied molecular orbital of the donor and the lowest unoccupied molecular orbital of the acceptor. Surprisingly, the orbitals were highly hybridized in the single-crystal complexes, enhancing the room-temperature conductivity (10−4–0.1 S cm−1) of mixed-stack complexes. Specifically, the maximum conductivity was the highest reported for single-crystal mixed-stack complexes under ambient pressures. The unique electronic structures at the neutral–ionic boundary exhibited structural perturbations between their electron-itinerant and localized states, causing abrupt temperature-dependent changes in their electrical, optical, dielectric, and magnetic properties.
Subject terms: Electronic materials, Electron transfer
Mixed-stack complexes consisting of alternating donors and acceptors typically show low electrical conductivity. Here, the authors improved the conductivity by hybridizing the frontier orbitals of the donor and acceptor at the neutral–ionic boundary.
Introduction
Organic conductors that are lightweight, flexible, and have excellent molecular designability are widely used as essential materials in modern organic electronic devices. The most industrially successful materials are polymer conductors owing to their high conductivities and ease of synthesis1. However, analyzing their atomic-level structures and conduction mechanisms is still challenging because of the inhomogeneity of polymers with different chain lengths. To address this issue, researchers are investigating molecular conductors with precisely defined molecular weights2–6. The crystal-based structures help to understand the structure–conductivity relationships and address the conduction mechanism. Charge-transfer complexes3–6 are typical molecular conductors and are categorized as either “segregated-stack complexes,” in which donors (D) and acceptors (A) are separately stacked (DDDD…/AAAA…) or “(alternatingly) mixed-stack complexes,” in which donors and acceptors are alternately stacked (DADADADA….; Fig. 1a). The difference in the stacking forms of complexes composed of the common donors and acceptors dramatically affects the conductivities. Segregated-stack complexes, such as tetrathiafulvalene (TTF) analogs–7,7,8,8-tetracyano-p-quinodimethane (TCNQ) analogs (the molecular structures are shown in Supplementary Fig. 1)7,8, hexamethylenetetraselenafulvalene–TCNQ9, and TTF analogs–metal dithiolenes10, exhibit excellent room-temperature conductivities (σrt) of up to 103 S cm–17,9, although variations on such highly conducting examples are still limited5,6. Several hybrid materials between DD-stack and DA-mixed-stack complexes have shown relatively high σrt (10–2 S cm–1)11–14. In contrast, mixed-stack complexes are typically poor electrical conductors that show low σrt (below 10–4 S cm–1)15–21 (Fig. 1b), although they are more frequently constructed, possibly owing to the Madelung energy gain between charged donor and acceptor. Generally, mixed-stack complexes are either in a neutral state (charge-transfer degree from donor to acceptor δ < 0.54, mostly 0 < δ < 0.45,6) with insufficient conductive carriers and weak intermolecular interactions for carrier conduction, or are in a highly ionic state (mostly δ > 0.75)5,6, wherein the carriers are nearly localized and have low mobility, thereby suppressing conductivity. In the early 2000s, a few neutral complexes showed high conductivity22,23, possibly due to the structural disorders within the contaminated ionic domains. However, the structural details and mechanisms remained hidden as the CCDC data were unavailable. We hypothesized that the mixed-stack complexes at the neutral–ionic (N–I) boundary (0.59 < δ < 0.74)5 could have numerous mobile electrons and display excellent electrical conductivity24. The hypothesis was supported by the discovery of segregated-stack complexes with exceptional conductivity in the same boundary area5–10. Preceding studies24–26 predicted that charge transfer to form mixed-stack complexes with δ at the N–I boundary could be realized by engineering appropriate energy gaps between the highest occupied molecular orbital (HOMO) of donors and the lowest unoccupied molecular orbital (LUMO) of acceptors that satisfy the equation ID – EA (= ΔEREDOX) ≈ EM, where ID is the ionization potential of the donor (related to its HOMO of the donor), EA is the electron affinity of the acceptor (related to its LUMO of the acceptor), and EM is the electrostatic Madelung energy of the donor–acceptor complex (approximately 0.2 eV; Fig. 2). In addition, a consistent orbital symmetry between the donor HOMO and acceptor LUMO27 may be necessary to facilitate the hybridization of frontier orbitals and enable efficient charge transfer. Thus far, a few mixed-stack complexes have partly fulfilled these two requirements: 1) similar energy levels between the donor HOMO and the acceptor LUMO, with the appropriate energy gaps of approximately 0.2 eV, and 2) a consistent orbital symmetry between them; for example, tetrakis(methyltelluro)tetrathiafulvalene (TTeC1-TTF)–TCNQ (δ = 0.56; d’ in Fig. 1b)21 and 3,3’,5,5’-tetramethylbenzidine (TMB)–TCNQ (δ = 0.49; c’ in Fig. 1b)16,28 close to the N–I boundary exhibited relatively high σrt values of 10−2 and 10−5 S cm−1, respectively. Under high-pressure conditions (~9 kbar), δ of TTF–p-chloranil (CA) (v in Fig. 1b) was transiently increased to ≈0.6, exhibiting excellent σrt (7 S cm−1)29, although it exhibits δ ≈ 0.2 and σrt = 10–5 S cm–1 under ambient pressures.
Fig. 1. Structures and electrical conductivities of mixed-stack complexes.
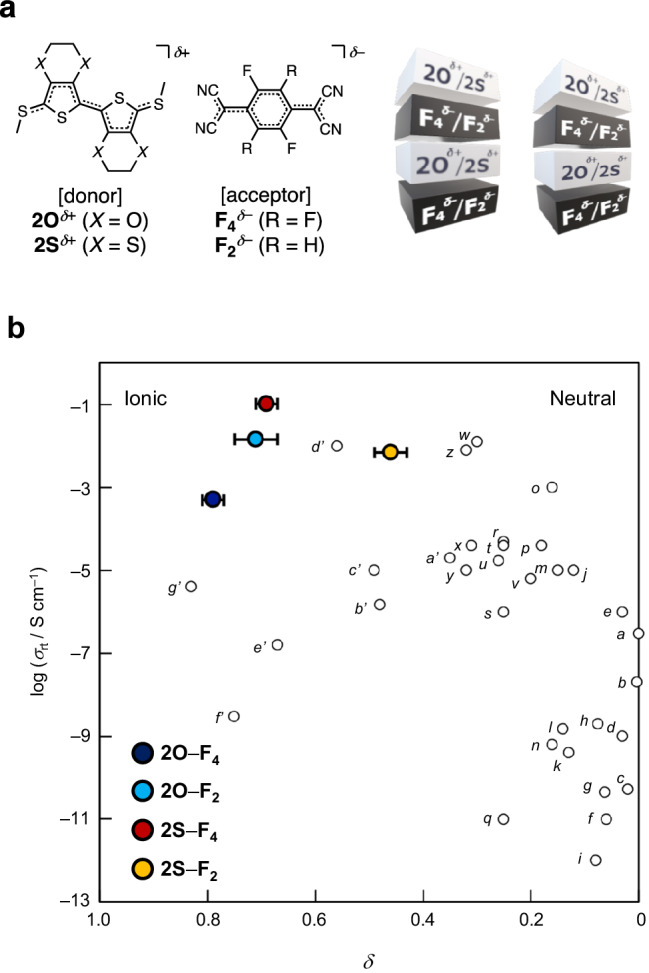
a Molecular structures of donors (2X; X = O, S) and acceptors (Fn; n = 4, 2) in the mixed-stack complexes 2X–Fn. An illustration of the 1D packing mode of 2X–Fn is shown in the right. b σrt of 1D mixed-stack complexes as a function of charge-transfer degree from donor to acceptor (δ) determined for single-crystal structures. a: Benzothieno[3,2-b]benzothiophene (BTBT)–F415. b: 1,2-Di(2-thienyl)ethylene (DTE)–7,7,8,8-tetracyano-p-quinodimethane (TCNQ)5. c: Naphthalene–TCNQ5. d: Perylene–TCNQ5. e: Dithieno[3,2-b:2’,3’-d]thiophene (DTT)–TCNQ5. f: Anthracene–TCNQ5. g: trans-Stilbene–TCNQ5. h: trans-2,2’,5,5’-Tetramethoxystilbene–TCNQ5. i: Pyrene–TCNQ5. j: Bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF)–TCNQ5. k: 2,3,7,8-Tetramethoxydibenzo[b,e][1,4]thiaselenine (Vn2SSe)–TCNQ6. l: 3,3’,5,5’-Tetramethylbenzidine (TMB)–p-chloranil (CA)16. m: Tetramethyltetraselenafulvalene (TMTSF)–TCNQ6. n: 2,3,7,8-Tetramethoxyselenanthrene (Vn2Se2)–TCNQ6. o: 1,6-Diaminopyrene (DAP)–p-bromanil (BA)22. p: 4,4’-Dimethyltetrathiafulvalene (DMTTF)–BA20. q: Coronene–TCNQ6. r: Dibenzotetrathiafulvalene (DB-TTF)–TCNQ5. s: Tetrathiafulvalene (TTF)–TCNQ17. t: DMTTF–CA20. u: 5,10-Dihydro-5-10-dimethylphenazine (DHMP)–TCNQ6. v: TTF–CA18. w: 2,5-Dichloro-1,4-phenylenediamine (Cl2PD)–2-chloro-5-methyl-7,7,8,8-tetracyano-p-quinodimethane (ClMeTCNQ)20. x: 2-Chloro-5-methyl-1,4-benzenediamine (ClMePD)–2,5-diethyl-7,7,8,8-tetracyano-p-quinodimethane (Et2TCNQ)20. y: α,α’-Dimethylquaterthiophene (DMQtT)–F419. z: ClMePD–2,5-dimethyl-N,N’-dicyanoquinone diimine (DMeDCNQI)20. a’: Tetrakis(methylthio)tetrathiafulvalene (TTC1-TTF)–2-fluoro-5-methyl-7,7,8,8-tetracyano-p-quinodimethane (FMeTCNQ)20. b’: TMB–2,5-dimethyl-7,7,8,8-tetracyano-p-quinodimethane (Me2TCNQ)16. c’: TMB–TCNQ16. d’: Tetrakis(methyltelluro)tetrathiafulvalene (TTeC1-TTF)–TCNQ21. e’: TMB–F216. f’: Phenazine–TCNQ5. g’: TMB–F416. The molecular structures are shown in Supplementary Fig. 1. The δ values for 2X–Fn are shown with error bars (standard deviation).
Fig. 2. Electronic and optical properties of mixed-stack complexes.
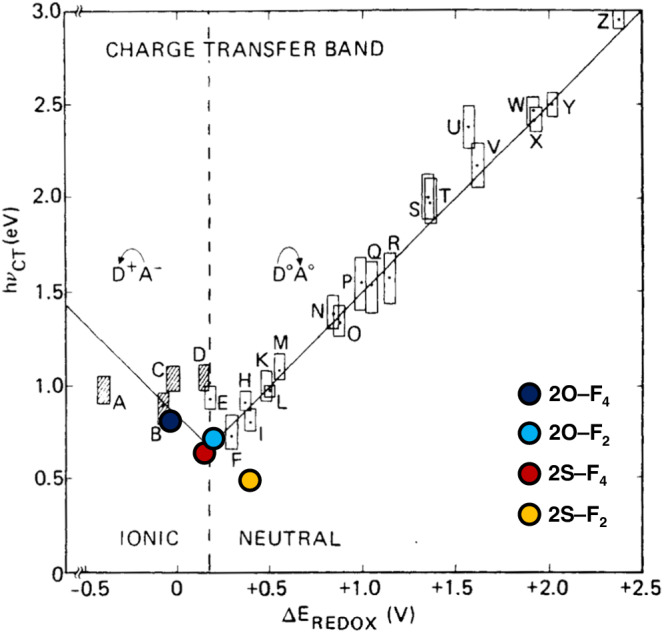
Relationship between ΔEREDOX and hνCT that has two separate linear correlations in the neutral and ionic regions; the two straight lines intersect when ΔEREDOX ≈ EM, resulting in the hνCT value reaches to the minimum (approximately 0.6 eV). The vertical dashed line indicates the neutral–ionic boundary (Reprinted and modified from Reference26, Copyright 1985, from Taylor & Francis). A: N,N,N’,N’-Tetramethyl-p-phenylenediamine (TMPD)–F4. B: DHMP–TCNQ. C: TMPD–TCNQ. D: TMPD–CA. E: N,N,N’,N’-Tetramethyl-1,6-pyrenediamine (TMDAP)–TCNQ. F: TTF–CA. G: TTF–p-fluoranil (FA). H: Dibenzene TTF–TCNQ. I: Diethyldimethyltetraselenafulvalene–Et2TCNQ. J: TMDAP–FA. K: TTF–dichlorobenzoquinone. L: Perylene–F4. M: Perylene–2,3-dichloro-5,6-dicyano-p-benzoquinone. N: Perylene–tetracyanoethylene. O: Perylene–TCNQ. P: TTF–dinitrobenzene. Q: Perylene–CA. R: Pyrene–tetracyanoethylene. S: Pyrene–CA. T: Anthracene–CA. U: Hexamethylbenzene–CA. V: Naphthalene–TCNE. X: Anthracene–pyromellitic dianhydride (PMDA). Y: Anthracene–tetracyanobenzenze. Z: Phenanthrene–PMDA. The molecular structures are shown in Supplementary Fig. 1.
In this study, we designed and synthesized mixed-stack complexes uniquely located at and near the N–I boundary, enhancing the conductivities of mixed-stack complexes. The combination of the oligo(3,4-ethylenedioxythiophene) analog 2O30,31 or its oxygen/sulfur-substituted analog 2S with tetra- or difluorinated TCNQs Fn fulfills the two requirements for electronic structures (i.e., 2X–Fn, X = O, S in Fig. 1a). Surprisingly, the donor HOMO and acceptor LUMO with comparable energy levels and well-matched orbital symmetries were highly hybridized in the complexes, enhancing the σrt values of mixed-stack complexes (10−4–0.1 S cm−1). The highest σrt value (0.1 S cm−1) observed for 2S–F4 among the combinations of donors 2X and acceptors Fn, which is located at the N–I boundary, is the highest value reported for a structurally defined 1D mixed-stack complex under ambient pressure. Furthermore, 2S–F4 exhibited a transition in electrical conductivity to a slightly less conductive phase accompanied by significant changes in its optical, dielectric, and magnetic properties. This emphasizes the unique electronic structure of the mixed-stack complex at the N–I boundary that displays a structural perturbation between electron-itinerant and localized states.
Results and discussion
Molecular electronic structures
First, 2S with non-bulky methylthio groups was synthesized in 52% yield after three-step transformations from 2,2’-bi(3,4-ethylenedithiothiophene) 132 (Fig. 3a, Supplementary Note 3, Supplementary Figs. 7, 41, 42, and Supplementary Table 1). The electrochemical properties of 2O30 and 2S were analyzed via cyclic voltammetry (CV). The voltammograms revealed the first oxidation potentials at 0.622 and 0.805 V (vs. Ag/AgCl) for 2O and 2S, respectively (Supplementary Note 4 and Supplementary Fig. 3), estimating HOMO levels of 2O/2S (−4.94/−5.13 eV) that are comparable to the LUMO levels of F4/F2 (−4.98/−4.74 eV). The gaps between the donor HOMO and acceptor LUMO (i.e., ΔEREDOX in Fig. 2) are estimated to be in the range of −0.04 to 0.39 eV (Table 1), which is consistent with those predicted by density functional theory (DFT) calculations (Fig. 4a, Supplementary Note 5, Supplementary Figs. 4, 6, Supplementary Tables 6, 7, 9, and 11). The 1e−-oxidized 2O/2S structures show highly symmetrical molecular orbital shapes spread over the entire molecules and consistent horizontally nodal patterns with an average periodicity of 2.0 Å that correspond well with those of 1e−-reduced F4/F2 with an average periodicity of 1.7–1.8 Å (Fig. 4b, Supplementary Fig. 5, Supplementary Tables 8, 10, and 12). Therefore, the combination of 2O/2S as donors and F4/F2 as acceptors ideally has similar energy levels and orbital symmetries between the HOMO of the donor and LUMO of the acceptor, possibly leading to strong hybridization between their orbitals during complexation.
Fig. 3. Synthesis of mixed-stack complexes 2X–Fn.

a Synthesis of donor 2S by bromination followed by lithiation and methylthiolation. b Synthesis of mixed-stack complexes 2X–Fn by chemical oxidation of donor (2O and 2S) by acceptors (F4 and F2). The δ values were determined using Kistenmacher’s equation37. NBS N-bromosuccinimide, nBu n-butyl, THF tetrahydrofuran, Me methyl.
Table 1.
Structural information and physical properties of mixed-stack complexes
| Donor (D)–acceptor (A) | 2O–F4 | 2O–F2 | 2S–F4 | 2S–F2 |
|---|---|---|---|---|
| Experimental data | ||||
| ΔEREDOX ( = E1/21(D) − E1/21 (A)) (V)a | −0.04 | 0.20 | 0.15 | 0.39 |
| D–A interplanar distance (Å)b | 3.361 | 3.329 | 3.406 | 3.398 |
| δ from bond length analyses in A | 0.79(2) | 0.71(4) | 0.69(2) | 0.46(3) |
| σ at 300 K (σrt; S cm−1) | 4.9 × 10−4 | 1.4 × 10−2 | 0.10 | 6.9 × 10−3 |
| hνCT (eV) | 0.83 | 0.73 | 0.64 | 0.50 |
| Ea for high temperature region (eV)c | 0.113(1) (290–315 K) | 0.178(1) (259–337 K) | 0.200(1) (288–340 K) | 0.112(1) (288–312 K) |
| Ea for low temperature region (eV)c | 0.215(2) (238–258 K) | 0.225(4) (219–231 K) | 0.277(3) (228–273 K) | 0.0902(17) (221–244 K) |
| Calculated data | ||||
| Eg (eV)d | 0.05 | <0.01e | <0.01 | 0.02e |
| W (eV)d | 0.86 | 0.89e | 0.88 | 0.89e |
| tDA (eV)f | 0.203 | 0.209e | 0.208 | 0.206e |
| Ueff (D) (eV)f | 2.31 | 2.28e | 1.89 | 1.89e |
| Ueff (A) (eV)f | 2.52 | 2.48e | 2.19 | 2.22e |
aEstimated from the cyclic voltammograms (see Supplementary Fig. 3 for details). The values were calibrated using the energy level of ferrocene/ferrocenium (Fc/Fc+ vs. Ag/AgCl/1 M KCl measured under identical conditions) as the reference.
bDetermined by measuring the distances between the centroid of the ten atoms of the bithiophene in the donor and the mean plane for the eight quinoid carbons in the acceptor in the single-crystal structures.
cDetermined from ρ–T plots using the Arrhenius equation.
dCalculated by OpenMX38,39 as a sum of dispersions for the bonding and antibonding bands between donor HOMO and acceptor LUMO (i.e., WB and WA, respectively) and energy gap between bonding and antibonding bands (i.e., Eg), if present (i.e., bandwidth W = WB + WA + Eg).
eStructural data with major occupancy were used in the calculations for 2O–F2 and 2S–F2.
Fig. 4. Electronic structure of donor 2S, acceptor F4, and the mixed-stack complex 2S–F4 according to theoretical calculations.

a Energy levels of orbitals for neutral 2S donor and F4 acceptor calculated by the Gaussian09 program. The energy levels of the donor’s highest occupied molecular orbital (HOMO) and acceptor’s lowest unoccupied molecular orbital (LUMO) were comparable, which is appropriate for their strong hybridization. b Singly occupied molecular orbitals (SOMOs) of 1e–-oxidized 2S donor and 1e–-reduced F4 acceptor calculated by Gaussian09 program. These orbitals have horizontally nodal patterns with an average periodicity of 1.8–2.0 Å that correspond well with each other (Supplementary Fig. 5). c Band structure calculated by OpenMX38,39. The bandwidth (W) value was determined from the calculated density of states (DOS). The highest occupied crystal orbital (HOCO) and lowest unoccupied crystal orbital (LUCO) at the Γ-point are depicted in red and blue dots, respectively. Γ (0,0,0), P (–0.5,0,0.5), Q (–0.5,0.5,0.5), Y (0,0.5,0). d Highly hybridized HOCO and LUCO between donor and acceptor at the Γ-point calculated by OpenMX38,39. Atoms were colored as follows; white: hydrogen; gray: carbon; blue: nitrogen, red: oxygen; light green: fluorine; yellow: sulfur. Molecular and crystal orbitals with positive and negative phases were colored with magenta and navy, respectively.
Single-crystal X-ray diffraction analyses
Motivated by the potentially ideal combination of 2O/2S donors and F4/F2 acceptors in the electronic structures, we investigated their complexation. The donors and acceptors were mixed in dichloromethane or THF, which led to a gradual color change of the solutions from yellow to dark green, indicative of charge transfer from the donor to the acceptor. After slow solvent evaporation over more than three days, dark-green needle-like single crystals of complexes 2O–F4, 2O–F2, 2S–F4, and 2S–F2 were isolated (Fig. 3b). These syntheses were highly reproducible and scalable to a yield of several milligrams in a single sequence of operations. The salts had an astonishingly high solubility in organic solvents such as acetonitrile and were stable for at least several weeks under atmospheric conditions.
X-ray diffraction (XRD) analyses determined that all single-crystal structures belonged to the P21/n space group with inversion centers at the center of gravity of the donors and acceptors and the glide plane symmetry on the 1/2 translation operation along the (a + c)/2 direction (Supplementary Note 6, Supplementary Figs. 9, 10, 12, 14, and Supplementary Table 2). The 2O and 2S donors in single-crystal complexes are nearly planar (Supplementary Fig. 11), similar to the charge-transfer salts of 2O and 2S (i.e., 2O•BF430 and 2S•BF4; Supplementary Figs. 2, 8, and Supplementary Table 1). The fluorine atoms in 2O–F2 and 2S–F2 exhibited positional disorder, with an occupancy of 94:6 and 86:14, respectively (Supplementary Fig. 9b, d). The donors and acceptors are uniformly and alternatingly π-stacked in columns along the a-axis. The donor–acceptor interplanar distances depend on the molecular size of the donor (i.e., 2O < 2S) and acceptor (i.e., F2 < F4); the distances increased in the order 2O–F2 (3.329 Å) < 2O–F4 (3.361 Å) < 2S–F2 (3.398 Å) < 2S–F4 (3.406 Å) (Table 1). Significant intracolumnar short contacts were not observed, suggesting the existence of 1D electronic structures favorable for strong intermolecular interactions. Notably, X-ray diffuse scattering was observed, reminiscent of structural fluctuation (Supplementary Note 6 and Supplementary Fig. 14).
Charge-transfer degree of mixed-stack complexes
The δ values of the complexes were determined using the conventional bond length analyses of the single-crystal structures for donors and acceptors6 in the mixed-stack complexes. An increase in the δ value of the donor from 0 (i.e., a neutral form with a benzenoid structure) to 1 (a radical cation form with a resonance structure) weakens bond alternation (i.e., C–C bonds b and d and C–S bonds g are shortened, while C=C bonds a and c are lengthened; Fig. 5a, b, c, e, f), as is apparent from the single crystals for neutral 2X (X = O30 or S, δ = 0; Supplementary Fig. 7 and Supplementary Table 1) and its 1e−-oxidized form 2X•BF430 (δ = 1; Supplementary Fig. 8 and Supplementary Table 1). All bond length alterations of the 2O and 2S donors in the mixed-stack complexes are located between those of 2X and 2X•BF4, indicative of intermediate δ values for the complexes between 0 and 1. Commonly, F4 complexes exhibited weaker bond length alternation than F2 complexes, indicating that the F4 complexes have higher δ values than the F2 complexes. Likewise, the acceptor molecules Fn exhibited weakening of the bond alternation (i.e., C–C bonds i, k, and m are shortened, while C=C bonds j and l are lengthened; Fig. 5a, d, g) upon reduction from neutral33,34 (δ = 0) to their 1e−–reduced forms35,36 (δ = 1), suggesting the intermediate δ. Notably, the 2O complexes exhibited weaker bond length alternation than the 2S complexes, suggesting that the 2O complexes have higher δ values than the 2S complexes. These tendencies are consistent with the δ values determined using Kistenmacher’s equation37, which assumes that the bond length changes linearly as δ increases from 0 to 1. The δ values of 2O–F4, 2O–F2, 2S–F4, and 2S–F2 were determined to be 0.79(2), 0.71(4), 0.69(2), and 0.46(3), respectively (Table 1). These values are close to the N–I boundary (0.59 < δ < 0.74), whereas that of 2S–F2 lies at the nearly neutral state, as predicted by the comparable energy levels of the donor HOMO and acceptor LUMO by CV analyses (Supplementary Fig. 3) and DFT calculations (Fig. 4a and Supplementary Fig. 6).
Fig. 5. Bond length analysis of mixed-stack complexes.

a Bond labels of 2O, 2S, F4, and F2. b–g Comparison of C–C and C–S bond lengths (dCC and dCS, respectively) of donors and acceptors in single crystals of mixed-stack complexes, with error bars (standard deviation). The letters along the x-axes of the plots correspond to the bonds depicted in a. As a reference, the bond lengths in single crystals of neutral donors or acceptors (i.e., δ = 0) are shown with dotted lines and 1e−-oxidized donors or 1e−-reduced acceptors (i.e., δ = 1) are shown with solid lines. Observed dCC of 2O30 (b), dCC of 2S (c), dCC of F433,35 (d), dCS of 2O30 (e), dCS of 2S (f), and dCC of F234,36 (g). i = (i1 + i2)/2.
Electronic structures of mixed-stack complexes
Based on the single-crystal structures of 2X–Fn as the average structures, we then obtained the band structures using first-principles calculations (OpenMX software; Supplementary Note 7)38,39, wherein the contributions from Coulomb repulsion between the carriers (discussed later) are ignored. All complexes showed band structures with highly dispersed bonding and antibonding orbital-derived bands formed by the donor HOMO and acceptor LUMO (Fig. 4c and Supplementary Fig. 18). The bands were exclusively dispersed along the a-axis (i.e., Γ–X corresponding to the π-stacking direction). The band structures had small or negligible energy gaps (Eg) in proximity to the Fermi level (0.02–0.05 eV for 2O–F4 and 2S–F2 and <0.01 eV for 2O–F2 and 2S–F4; Table 1 and Supplementary Fig. 18). The density of states (DOS) was calculated to determine the bandwidth (W) of the complexes in the major occupancies, the values of which were 0.86 eV (2O–F4), 0.89 eV (2O–F2), 0.88 eV (2S–F4), and 0.89 eV (2S–F2) (Table 1, Fig. 4c, and Supplementary Fig. 18), where W = WB + WA + Eg; WB and WA are the band dispersions of the bonding and antibonding orbitals, respectively. Among typical organic conductors2, high W values may be characteristic of 1D electronic structures, especially given the comparable energy levels of the donor HOMO and acceptor LUMO and their well-matched orbital symmetries, indicating strong intracolumnar interactions. We next quantified the transfer integrals between a donor and the six neighboring molecules in the single-crystal structures using combined Quantum ESPRESSO (QE)40,41 and RESPACK42 (Supplementary Fig. 27, and Supplementary Table 3). These calculations also identified prominent intracolumnar donor–acceptor interactions (tDA ≈ 0.21 eV in Table 1; t1 and t4 in Supplementary Table 3 and Supplementary Fig. 27), corresponding to W values based on the 1D tight-binding model, and negligible intercolumnar donor–acceptor and donor–donor interactions for all complexes. These findings support their 1D electronic structures, which were also suggested by the band structures. The W and intracolumnar tDA values follow nearly consistent trends: F4 complexes < F2 complexes, possibly because the donor–acceptor interplanar distances are shorter for F2 complexes (as shown in Table 1). Despite the longer interplanar distances, the trend 2O complexes < 2S complexes is primarily due to the presence of multiple S atoms with significant orbitals in the donor.
Notably, we have made a significant discovery using OpenMX calculations38,39, which identified that the HOMO of the donor and LUMO of the acceptor are highly hybridized. The calculated highest occupied crystal orbital (HOCO) and lowest unoccupied crystal orbital (LUCO) at the Γ point of 2X–Fn appeared equivalently on the donor and acceptor, respectively (Fig. 4d and Supplementary Fig. 19). This contrasts with neutral complexes3, where HOCOs and LUCOs were localized on the donors and acceptors, respectively. This finding supports that 2X–Fn complexes around the N–I boundary have different electronic structures from previously reported neutral or ionic mixed-stack complexes5,6,15–23, leading to significant W values based on their 1D electronic structures and unique electronic structures as discussed later.
Polarized reflectivity measurements
The polarized reflectivity spectra further support the observation that the δ values of 2X–Fn are close to the N–I boundary. The spectra obtained for the π-stacking direction of the single crystals have peak energies based on the charge-transfer band (hνCT) at 0.50 eV (2S–F2) < 0.64 eV (2S–F4) < 0.73 eV (2O–F2) < 0.83 eV (2O–F4) (Table 1 and Fig. 6a; complexes are located exactly at the N–I boundary, whereas 2S–F2 is nearly neutral). The spectrum of 2S–F2 has a shoulder peak at approximately 0.64 eV. The shape of this spectrum reflects the neutral-to-N–I boundary state43 (δ = 0.46(3)) or donor–acceptor π-dimerized form43,44 (discussed later), possibly causing the plot for 2S–F2 to deviate from the predicted value by δ in Fig. 2. The significant positional disordering of fluorine atoms in 2S–F2 may also affect the shape of the spectrum.
Fig. 6. Polarized reflectivity of mixed-stack complexes.

a Spectra of mixed-stack complexes obtained in the electric field of light parallel to the π-stacking direction. Bars indicate the top of the peaks. The spectrum of the relatively thin single-crystal 2O–F4 exhibits stripe-pattern signals in the low-energy region due to interference from the backside (the raw data is shown in Supplementary Fig. 34a), and thus this part of the spectrum was deleted for clarity. b Temperature-dependent reflectivity of 2S–F4 in the electric field of light parallel (//) and perpendicular (⊥) to the π-stacking directions for the low-energy region.
Given that the strong hybridization between the donor HOMO and acceptor LUMO in the band structures with nearly negligible Eg, we considered the electronic structures of 2X–Fn to be like homogeneous donor- or acceptor-stacked charge-transfer salts, with the average structure2. Under this consideration, the observed hνCT may correspond to the difference between the bonding and antibonding orbital bands separated by carrier-to-carrier Coulomb repulsions in the solid states (Ueff), rather than the donor HOMO–acceptor LUMO energy gap for neutral complexes. Accordingly, the lower hνCT for the 2S complexes than that for the 2O complexes may correspond to lower Ueff for the 2S complexes. Quantum calculations provided further insights into electronic structures. The Ueff values for the complexes were quantified for donors and acceptors (i.e., Ueff (D) and Ueff (A), respectively) by combining first-principles calculations (QE40,41/RESPACK42 packages) based on the average single-crystal structures. These calculations identified the far lower Ueff (D) and Ueff (A) for 2S complexes compared to those of 2O complexes (Table 1, Supplementary Figs. 20–26, and 28–33), emphasizing that the combined effects of smaller δ value and the larger π-conjugate area of the donor in the complexes may favorably contribute to lowering Ueff. The cooperative reduction in Ueff (D) and Ueff (A) supports the significant orbital hybridization of donors and acceptors in single crystals. According to these calculations, the 2S complexes have slightly larger W and far lower Ueff than the 2O complexes, conferring superior electrical conductivities45–48 upon the 2O complexes. However, it is not negligible that nearly neutral 2S–F2 has insufficient conductive carriers and positional disorders of fluorine atoms, which may impact the electronic structures that contribute to the conductivity (Supplementary Figs. 18–20, 26, 27, 33, and Supplementary Table 3).
Structural fluctuations involved in π-dimerization
It is also notable that the polarized infrared reflectivity spectra of the complexes along the π-stacking direction exhibited multiple sharp peaks in the low-energy region of 0.14–0.19 eV, except for nearly neutral 2S–F2 (Fig. 6b, Supplementary Figs. 34, and 35a). The optical conductivity spectrum of 2S–F4 (Supplementary Fig. 35b) derived from the infrared spectrum showed a coincidence of the shapes to the Raman spectrum at 293 K (Supplementary Fig. 35d), with varying intensities. Considering the inversion center of the single-crystal structure in a P21/n space group with uniform π-stacked donors and acceptors along the C2 glide symmetry, the Raman-active modes should not be visible in IR, while the IR-active modes should not be visible in Raman. The coincidence of the spectral shapes indicates the dynamic fluctuating of donor–acceptor-π-stacking dimerization based on the electron-molecular vibration (EMV) couplings43,44. We performed a computational study by numerical simulations using the donor–acceptor dimer model49, confirming that the EMV coupling effect is responsible for these signals (Supplementary Note 8, Supplementary Fig. 35c, Supplementary Tables 4, and 5). These results suggest that there is inhomogeneity in the one-dimensional molecular stacking, contradicting the uniform columnar structure predicted by single-crystal XRD (Supplementary Table 2 and Supplementary Fig. 10). This inhomogeneity likely indicates dynamic fluctuations from the average structure, which is supported by the X-ray diffuse scattering observed in XRD at 300 K (Supplementary Fig. 14).
Electrical conductivity measurements
Motivated by the proximity of the δ value to the N–I boundary, high W, and low Ueff for 2S complexes, we subsequently investigated the electrical conductivity of single-crystal mixed-stack complexes (Supplementary Note 9). The σrt values (300 K) determined via the direct-current method along the π-stacking direction (the a-axis) were superior by a few orders of magnitude to those of previously reported typical mixed-stack complexes with σrt (300 K) below 10–4 S cm–1: 4.9 × 10−4 S cm−1 (2O–F4), 1.4 × 10−2 S cm−1 (2O–F2), 0.10 S cm−1 (2S–F4), and 6.9 × 10−3 S cm−1 (2S–F2) (Fig. 1b and Table 1) within the ohmic region (Supplementary Fig. 36). Except for the nearly neutral 2S–F2, the trend of σrt (2S–F4 > 2O–F2 > 2O–F4) indicates that the closeness of these complexes to the N–I boundary dominantly affected the conductivity. The σrt of 2S–F4 (0.10 S cm−1) is the highest value reported to date for a single crystal of a 1D mixed-stack complex, and it is an order of magnitude higher than the previously highest value of 1.0 × 10−2 S cm−1 21. The low Ueff based on the large conjugate area may cooperatively contribute to the high σrt46–48, unlike those of the other complexes at the N–I boundary (e.g., complexes e’ and f’ in Fig. 1b).
The activation energies (Ea) near room temperature were determined from the temperature (T) dependence of the resistivity (ρ = σ−1) using the Arrhenius equation. The ρ–T plots are indicative of semiconducting behavior with relatively small Ea values: 0.113(1) eV (2O–F4), 0.178(1) eV (2O–F2), 0.200(1) eV (2S–F4), and 0.112(1) eV (2S–F2) around room temperature (Figs. 7, and 8a, and Table 1), indicating that these complexes are narrow-bandgap semiconductors. The Ea trend was inconsistent with those of σrt, the calculated Eg, and Ueff. Except for the nearly neutral 2S–F2, the Ea values increased: 2O–F4 < 2O–F2 < 2S–F4. The trend is explained by the proximity of δ to the N–I boundary which induces the π-dimerization fluctuation between the donor and acceptor; 2S–F4 at the N–I boundary showed largest increase in ρ, and highest Ea among the three complexes.
Fig. 7. Temperature-dependent electrical conductivities of mixed-stack complexes.
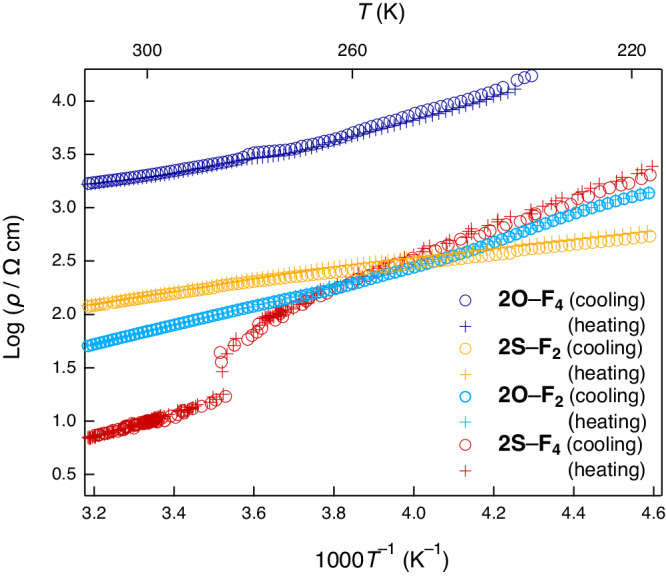
ρ–T plots obtained by a four-probe method for cooling (circles) and heating (plus marks) processes are shown.
Fig. 8. Temperature-dependent physical properties of single-crystal 2S–F4 upon cooling.
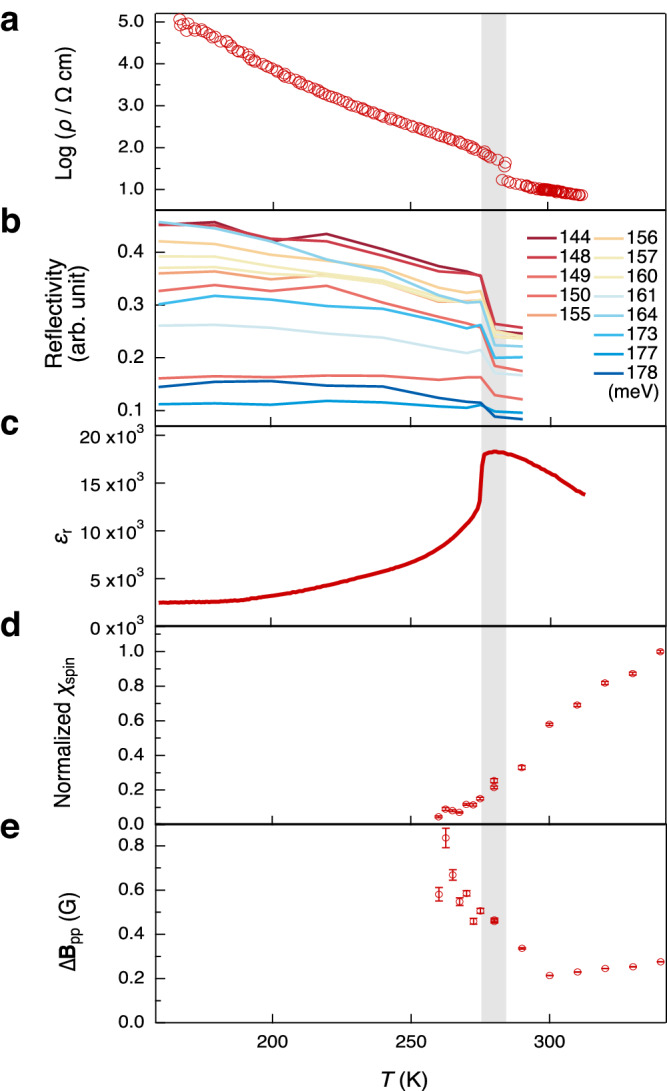
a Electrical resistivity. b Polarized reflectivity for photon energies of 144–178 meV in the electric field of light parallel to the π-stacking direction. c Real part of the dielectric constant εr at 1.0 MHz. d Normalized χspin determined from the calculation using the intensity and ΔBpp of the electron spin resonance (ESR) signals at g ≈ 2.003. The values are normalized by the value at 340 K, with error bars (standard deviation). e ΔBpp of the ESR signals at g ≈ 2.003, with error bars (standard deviation). Gray panel indicates the transition temperature around 282 K.
Abrupt changes in physical properties of 2S–F4 around room temperature
The mixed-stack complex with the highest conductivity at the N–I boundary, 2S–F4, exhibited a unique transition with a sharp increase in ρ at a transition temperature (Tc) of 282 K. The transition was followed by an increase in Ea from 0.200(1) eV at high temperatures (288–340 K) to 0.277(3) eV at low temperatures (228–273 K) without significant hysteresis upon cooling and heating (Figs. 7, 8a, and Table 1). The XRD pattern in P21/n space group at 300 K showed significant X-ray diffuse scattering (Supplementary Fig. 14), possibly due to the precursor phenomenon for the π-dimerization, which may result in EMV coupling-based signals at room temperature (Fig. 6b, Supplementary Figs. 34, and 35a). On the other hand, the scattering disappeared (Supplementary Figs. 15 and 16) at 200 K, and the XRD showed superlattice patterns with dimensions of a × 2b × 2c (Supplementary Figs. 13 and 17), possibly implying the π-dimerization along the a-axis (Supplementary Note 6). The broken symmetry upon the possible π-dimerization of the complex may induce the markedly intensified EMV coupling-based signals (e.g., from 0.20 to 0.25) near 282 K during the cooling process while maintaining the energy (Figs. 6b, 8b, and Supplementary Fig. 35a), as well as the large anomaly in the dielectric constants50 (the real part of the dielectric constant is shown in Fig. 8c). The transition events did not cause any significant hysteresis for the cooling and heating processes.
Change in the magnetic characteristics at the transition
The magnetic characteristics revealed via temperature-dependent electron spin resonance (ESR) measurements helped us to address the insights into the electronic structures of the mixed-stack complexes (Supplementary Note 10). The spectra of 2S–F4 exhibited a narrow Lorentzian signal with a peak-to-peak width (ΔBpp) of ≈0.2 G at g ≈ 2.003, which is typical for π-electrons2 (Supplementary Figs. 37–39). At 282 K, the magnetic susceptibility (χspin) quantified from the spectra decreased sharply to reach a nearly nonmagnetic state (Fig. 8d and Supplementary Fig. 39b), suggesting a spin-Peierls-like singlet formation based on the 1D electronic structure51 during the π-dimerization. This χspin decrease was accompanied by an increase in ΔBpp (Fig. 8e and Supplementary Fig. 39c, d), supporting increased antiferromagnetic interactions in the singlet ground state in the low-temperature region. A decrease in χspin and increase in ΔBpp upon cooling were likewise observed for 2O–F2 and 2O–F4 at approximately 240 and 200 K, respectively (Supplementary Fig. 39b–d), which may correspond to the increase in Ea in the electrical conductivity at low temperatures (Figs. 7, 8a, and Table 1). The signals of 2S–F2 were negligible mainly due to the relatively poor electron spins in the nearly neutral state. These magnetic behaviors were also consistent with the decrease in the magnetic susceptibilities (χ) of the polycrystals of 2S–F4 and 2O–F4 around the temperatures using a superconducting quantum interface device (SQUID) magnetometer (Supplementary Note 11 and Supplementary Fig. 40). The χ values of 2S–F4 and 2O–F4 were quantified to be 1.2 × 10−4 and 2.6 × 10−4 emu mol−1, respectively at 293 K. The χ values were relatively high compared to those of previously reported mixed-stack complexes18,50,52, most of which are in nearly nonmagnetic states. These values are comparable to those of typical organic conductors2, emphasizing the presence of abundant conductive electrons.
Surprisingly, in the case of 2S–F4, another broad signal with a ΔBpp value of ~3 G appeared at g ≈ 2.004 below 282 K (Supplementary Figs. 37–39), and overlapped with the narrow signal at g ≈ 2.003. This suggests the appearance of nearly non-correlated spins (e.g., spin solitons) originating from unpaired donors or acceptors during transient partial-π-dimerization between donor and acceptor. This observation suggests that the conductive mechanism of 2S–F4 at the N–I boundary may involve spin solitons that exist because of topological excitations29 that contribute to its high conductivity.
The abrupt change in the electrical, optical, dielectric, and magnetic properties of 2S–F4 at high Tc (282 K) under ambient pressures (Fig. 8) may reflect structural perturbation, which can be characteristic of the mixed-stack complexes at the N–I boundary. The neutral20,53 or highly ionic complexes50 transitioned based on π-dimerization to form a structure with more strongly ionic behavior at lower Tc values of 49–81 K. The complexes around the N–I boundary (δ = 0.3–0.5) had relatively high Tc values of 115–220 K20,52 under ambient pressures. Under high-pressure conditions (approximately 9 kbar), TTF–CA (δ ≈ 0.6) transiently exhibits excellent conductivity (7 S cm–1) and a high Tc at approximately 270 K29, whereas it has a low Tc of 81 K under ambient pressures (δ = 0.2, σrt = 10–5 S cm–1; v in Fig. 1b). The high Tc (282 K) and high σrt (0.1 S cm−1) even at ambient pressures of 2S–F4, for which δ is exactly at the N–I boundary (δ = 0.69(2)) with non-hysterical changes in their electronic functionalities such as electrical conductivity, reflectivity, dielectricity, and magnetism may involve the unique electronic perturbation at the N–I boundary between the electron-itinerant and localized states.
In conclusion, we developed highly conductive 1D mixed-stack complexes for which δ is close to the N–I boundary by combining a donor HOMO and acceptor LUMO that have similar energy levels and well-matched orbital symmetries. These complexes have highly hybridized orbitals between the donor and acceptor and exhibited high σrt (10−4 to 0.1 S cm−1) under ambient conditions; the top σrt (0.1 S cm−1) for 2S–F4 at δ = 0.69(2) is the highest value reported to date for a single-crystal 1D mixed-stack complex. 2S–F4 uniquely exhibited a non-hysterical change in its electrical conductivity at high Tc (282 K) under ambient pressure to a less conductive phase below this temperature. This behavior was simultaneously accompanied by abrupt changes in the low-energy EMV coupling-based reflectivity, dielectric constant, and magnetic properties, reminiscent of the π-dimerization fluctuation that spatiotemporally breaks the symmetry and may widen the band gap to form a band-insulating state and increase Ea. Furthermore, the appearance of a broad ESR signal suggests non-correlated spins of unpaired donors or acceptors, which may enhance the conductivity of 2S–F4 through mechanisms such as topological excitation29. In addition, mixed-stack complexes have unique advantages as conductor materials, such as high potential for solution processing and scalable synthesis without special equipment. The high conductivity, high solubility, and air stability of the conductors may provide scope for further investigation of the fundamental physical properties and applications using various external stimuli29,50,53,54. The rich variations in the structure-determining factors in oligomer donors, e.g., monomer unit variations, oligomer length55,56, sequences comprising multiple units56, and end-capping structures could enable precise tuning of the electronic structures toward highly functional mixed-stack complexes.
Methods
Apparatus
Proton (1H) and carbon (13C) nuclear magnetic resonance (NMR) spectra were obtained on a JEOL JNM-AL300 (1H NMR: 300 MHz; 13C NMR: 75 MHz) spectrometer. 1H NMR spectra in CDCl3 were referenced internally to tetramethylsilane as a standard. 13C NMR spectra in CDCl3 were referenced to the solvent resonance. High-resolution mass spectra (HRMS) were obtained on a JEOL JMS-AX500 with a field desorption (FD) probe in the positive mode using cholesterol as an internal standard. Single-crystal XRD measurements were performed using a Rigaku MercuryII CCD X-ray diffractometer (Mo Kα, λ = 0.71073 Å) and BL02B1 (λ = 0.30960 Å) at a synchrotron facility SPring-8 in Japan. A N2-gas-blowing device was employed for the low-temperature measurements. Cyclic voltammetry was performed using an ALS 610DB electrochemical analyzer. Microscopic mid-infrared (mid-IR) and ultraviolet-visible (UV-vis) measurements were performed on a JASCO FT/IR-6100 and FT/IR-6200 and MSV-5200TSO, both of which are equipped with a JASCO IRT-5000 FT/IR microscope. Raman spectroscopy was performed on a RENISHAW inVia Reflex. Dielectric constant measurements were performed using a custom-built alternating-current (AC) impedance probe with a Solartron Impedance Analyzer SI 1260, a Dielectric Interface 1296, and a Quantum Design SQUID magnetometer (MPMS-XL) as the temperature controller. Resistivity measurements were performed on a Quantum Design Physical Properties Measurement System (PPMS) and HUSO HECS 994C with a high-resistance-low-current electrometer KEYTHLEY 6517B equipped with an ADVANTEST R6142 power supply, an ADVANTEST R7210 scanner, and a KEYTHELY 2001 digital multimeter. X-band continuous wave ESR measurements were performed using a Bruker EMXmicro and EMX spectrometers with a N2 flow variable temperature unit ER4131VT and E500 spectrometer with a He flow cryostat ESR900. Static magnetic susceptibility measurements were performed using a MPMS-XL.
Synthesis
Synthesis of 5,5’-bis(methylthio)-2,2’-bi(3,4-ethylenedithiothiophene) 2S
General synthetic procedure and materials sources are shown in Supplementary Notes 1 and 2. To a solution of 2,2’-bi(3,4-ethylenedithiothiophene)32 1 (200 mg, 0.576 mmol) in dichloromethane (20 mL) was dropwise added at 0 °C a solution of N-bromosuccinimide (NBS; 238 mg, 1.34 mmol) in dichloromethane (6 mL), and the reaction mixture was stirred for 6 h. After the addition of a saturated aqueous solution of sodium bicarbonate (100 mL), the mixture was extracted with dichloromethane (3 × 100 mL). The combined organic layer was dried over sodium sulfate and concentrated in vacuo to afford a crude material containing 2 (250 mg) which was used for the subsequent reaction without further purification. Thus, to a solution of the mixture (250 mg) in THF (12 mL) was added nBuLi (1.6 M in hexane, 1.70 mL, 2.68 mmol) at −80 °C. The reaction mixture was stirred for 2 h and then allowed to warm to ambient temperature. To the mixture was dropwise added dimethyl disulfide (500 µL, 5.63 mmol), and the mixture was stirred at ambient temperature for 3 h. After the removal of volatile materials in vacuo and the subsequent addition of a saturated aqueous solution of sodium bicarbonate (100 mL), the mixture was extracted with dichloromethane (3 × 100 mL). The combined organic solvents were dried over sodium sulfate, concentrated in vacuo, and purified by GPC (eluent: chloroform) to afford 2S (131 mg, 0.298 mmol) as a yellowish white powder in 52% total yield for three-step transformations from 1. Physical data of 2S: 1H NMR (CDCl3, 300 MHz) δ 2.44 (s, 6H), 3.14–3.23 (m, 4H), 3.25–3.34 (m, 4H); 13C NMR (CDCl3, 75 MHz) δ 20.1, 27.6, 27.7, 126.6, 126.6, 127.8, 130.5; MS (FD) calcd for C14H14S8 [M+•] 437.8834, found 437.8856. The structural integrity and purity were identified by the NMR spectra (Supplementary Figs. 41 and 42). Block-like orange single crystals were obtained by the liquid–liquid diffusion method with dichloromethane/hexane (1:1, v/v). The single-crystal XRD analyses revealed the structural details (Supplementary Table 1 and Supplementary Fig. 7), confirming a nearly planar structure. In contrast, other solvent combinations afforded crystal-like solids, while their XRD patterns were too complicated to be resolved, suggesting that neutral 2S is polymorphic and could have multiple structures, potentially influenced by molecular twisting.
Typical procedure of mixed-stack complexes
A donor (2O30 or 2S, 10 µmol) was placed on one side of an H-shaped cell and an acceptor (F4 or F2, 10 µmol) was placed on the other side of the cell, respectively. To the cell were slowly added THF (8 mL for 2O–F4 and 2O–F2) or dichloromethane (8 mL for 2S–F4 and 2S–F2), and the mixture was kept at 25 °C in the dark. The slow evaporation of solvents from the mixture over more than three days gave dark green needle-like single crystals which were collected by paper filtration and washed with acetonitrile or 1:1 v/v dichloromethane/hexane (typical size of ~500 × ~40 × ~20 µm3). The single-crystal structures and chemical compositions were identified by single-crystal X-ray structural analyses (Supplementary Figs. 9–17 and Supplementary Table 2).
CV measurements
We performed CV in a degassed 0.6 mM solution of 2O30 or 2S in dichloromethane containing 100 mM nBu4N•PF6. We used a glassy carbon as the working electrode, a platinum wire as the counter electrode, and a silver–silver chloride electrode (Ag/AgCl in 1 M KCl) as the reference electrode (Supplementary Fig. 3).
Theoretical calculations
Energy levels and shapes of orbitals for donors and acceptors
The energy levels of orbitals for neutral donors (2O30 and 2S), one-electron-oxidized donors (2O•+ and 2S•+), neutral acceptors (F4 and F2), and one-electron-reduced acceptors (F4•– and F2•–) were calculated. The calculations were performed using the optimized structures on the Gaussian09 program (Revision D.01, Gaussian, Inc., Wallingford CT, 2016. https://gaussian.com/) at the DFT level with the (unrestricted) B3LYP functional, the gradient correction of the exchange functional by Becke57,58 and the correlation functional by Lee, Yang and Parr59 and the 6–31G(d) split valence plus polarization basis set60–62.
Band structure calculations
All the periodic DFT calculations were performed with the single-crystal structures of mixed-stack complexes as the average structures at 300 K. The calculations were performed by the OpenMX software38,39, based on optimized localized basis functions and pseudopotentials. The basis functions used were H6.0-s2p1, C6.0-s2p2d1, O6.0-s2p2d1, S7.0-s2p2d1f1, N6.0-s2p2d1, and F6.0-s2p2d1 for hydrogen, carbon, oxygen, sulfur, nitrogen, and fluorine, respectively, wherein the abbreviation of basis functions such as C6.0-s2p2d1, C stands for the atomic symbol, 6.0 the cutoff radius (Bohr) in the generation by the confinement scheme, and s2p2d1 means the employment of two, two, and one optimized radial functions for the s-, p-, and d-orbitals, respectively. The radial functions were optimized by a variational optimization method38,39. As valence electrons in the pseudopotentials (PPs), we included 1 s for hydrogen, 2 s and 2p for carbon, oxygen, nitrogen, and fluorine, and 3 s and 3p for sulfur, respectively. All the PPs and pseudo-atomic orbitals (PAOs) we used in the study were taken from the database (2019) in the OpenMX website (https://www.openmx-square.org/), which was benchmarked by the delta gauge method63. Real-space grid techniques were used for the numerical integrations and the solution of the Poisson equation using FFT with the energy cutoff of 220 Ryd64. We performed Brillouin-zone integrations on a 2 × 1 × 2 k-grid; the Fermi-Dirac distribution function at 300 K is employed as a smeared occupation function. We used a generalized gradient approximation (GGA) proposed by Perdew, Burke, and Ernzerhof to the exchange-correlation functional65. The real parts of the HOCO and LUCO at the Γ point (0,0,0) were visualized by VESTA66 (Fig. 4d and Supplementary Fig. 19).
Calculations of transfer integrals
The first-principles calculations were performed using the QE package40,41 with the GGA as the exchange-correlation function65 based on the single-crystal structure data of 2O–F4, 2O–F2, 2S–F4, and 2S–F2 at 300 K. In the calculations, SG15 Optimized Norm-Conserving Vanderbilt (ONCV) pseudopotentials67 were used as the pseudopotentials, in which 428 bands corresponding to the electron numbers were considered. The cutoff kinetic energies for wave functions, charge densities, and the mesh of the wave numbers were set as 80, 320 Ry, and 5 × 3 × 3, respectively. After the calculations, the maximally localized Wannier functions (MLWFs) were obtained using RESPACK42, and four bands near the Fermi energy were selected to reproduce the MLWFs (Supplementary Figs. 21–26). Initial coordinates of the MLWFs were located at the center of gravity of crystallographically independent two donors and two acceptors in the unit cell, respectively. Supplementary Fig. 20 shows the energy bands near the Fermi energy obtained by QE40,41 and the Wannier interpolation. Based on the MLWFs, transfer integrals (t in Supplementary Fig. 27) were calculated as summarized in Table 1 and Supplementary Table 3.
Calculations of Coulomb interactions
Effective direct Coulomb interactions for donors and acceptors (i.e., Ueff (D) and Ueff (A)) were calculated by RESPACK42, in which the Coulomb interaction was reduced due to screening effects from bands other than the target band. Using a constrained random phase approximation (cRPA) method, we obtained the screened Coulomb interactions by incorporating screening effects from non-target bands. Through previous reports of such applications to different organic material systems46–48, we know that the effective model Hamiltonians derived by the cRPA method succeed in evaluating the stability of competing phases in electron-correlated systems. Thus, we estimated the Coulomb interactions by using the cRPA method. The energy cutoff for the dielectric function was set as 5.0 Ry. Static screened direct integrals were calculated as the matrix element of the shielded Coulomb interaction between the Wannier functions on the donors and acceptors, respectively (Supplementary Figs. 21–26)42. The values at 0 Å were used as the Ueff (D) and Ueff (A) (Table 1 and Supplementary Figs. 28–33).
Optical reflection spectroscopy measurements
Steady-state polarized reflectivity spectra of mixed-stack complexes (2O–F4, 2O–F2, 2S–F4, and 2O–F2) in the mid-IR region (0.12–0.97 eV) and UV-vis-near IR region (0.46–1.35 eV) were performed using mid-IR and UV-vis spectrometers equipped with a polarizer by applying an electric field (E) along the π-stacking directions (i.e., the a-axis) and perpendicular to the directions (Figs. 6, 8b, Supplementary Figs. 34, and 35a, b). Raman spectroscopy of 2S–F4 was performed using an excitation wavelength of 532 nm (Supplementary Fig. 35d).
Dielectric constant measurements
AC impedance spectroscopy measurements to determine dielectric constant were carried out by the quasi-four-probe method in the frequency range of 1.0 MHz for 160–312 K. A direction parallel to the long axis direction of the crystal shape, corresponding to the π-stacking, was chosen for the AC impedance measurements. Electrodes were made on the two opposite sides of the samples using silver paste and gold wires (15 μm diameter).
Electrical resistivity measurements
Electrical resistivity (ρ) measurements of the single-crystal mixed-stack complexes were performed by the conventional a four-probe method along the long-axis direction of the crystal shape, corresponding to the π-stacking direction. Samples were prepared by attaching gold wires (15 µm diameter) of a single crystal with a conductive carbon paste. The ρ and conductivity (σ) values were derived from the following equation: ρ = σ–1 = R × S / L (S: cross-section, L: length). The typical size of samples was ~500 × ~40 × ~20 µm3.
ESR measurements
The X-band (∼9.4 GHz) continuous wave ESR experiments were performed on single-crystal 2O–F4, 2O–F2, 2S–F4, and 2S–F2 (Fig. 8d, e and Supplementary Figs. 37–39). The ESR signals were measured by setting the long axis of the crystal (i.e., the stacking a-axis) along the direction parallel to the magnetic field (i.e., φ ~ 0° or 180°). The typical size of samples: ~500 × ~40 × ~20 µm3.
SQUID measurements
The static magnetic susceptibility of polycrystals 2O–F4 and 2S–F4 was measured upon cooling from 300 or 320 K to 2 K by <1 K min–1 applying the static magnetic field of 10,000 Oe (Supplementary Fig. 40). The absence of ferromagnetic impurity in the synthesized samples was confirmed from the obtained M–H curve at 2 K around −55,000 to 55,000 Oe, which follows a typical Brillouin function. The obtained magnetic susceptibilities (χexp) were plotted after subtracting the contribution of the Curie impurity for S = 1/2 (χcw; 0.3% for 2O–F4 and 0.2% for 2S–F4) and the contribution from core diamagnetism of 2O–F4 and 2S–F4 (–3.14 × 10–4 and –3.74 × 10–4 emu mol–1, respectively) estimated from Pascal’s law68.
Supplementary information
Acknowledgements
Professor Taisuke Ozaki (The University of Tokyo), Professor Takehiko Mori (Tokyo Institute of Technology), Dr. Yusuke Tsutsui (Kyoto University), and Dr. Kazuma Nakamura (Kyushu Institute of Technology) for the helpful discussions related to theoretical calculations; Prof. Hidefumi Akiyama and Dr. Keisei Shibata (The University of Tokyo) for Raman spectroscopy; Ms. Yuka Ito (The University of Tokyo) for technical support in electrical conductivity measurements; Idemitsu Kosan Co., Ltd. for their support throughout the study. We also thank the Supercomputer Center, the Institute for Solid State Physics, and the University of Tokyo for providing their facilities for use. The computation for simulating EMV coupling was performed using Research Center for Computational Science, Okazaki, Japan (Project No. 23-IMS-C276). The synchrotron radiation experiments were performed at SPring-8 with the approval of the Japan Synchrotron Radiation Research Institute (JASRI) (Proposal No. 2023B0304). This work was partially supported by JST PRESTO (JPMJPR22Q8 to T.F.), JSPS Grants-in-Aid for Scientific Research (No. JP20H05206, JP21K05018, JP22H04523 to T.F., JP18H05225, JP21K18597, JP22H00106 to H.M., JP20K15240 to S.D., JP21H04988 to H.O., JP21H01041, JP22K03526 to K.Yoshimi), MEXT Grants-in-Aid for Scientific Research on Innovative Areas “Hydrogenomics” (JP18H05516 to H.M.), a research grant from Iketani Sci. Technol. Foundation, the Naito Foundation, the Kao Foundation for Arts and Science to T.F., and the Noguchi Institute to S.D. Part of this work was conducted at the Institute for Molecular Science, supported by Advanced Research Infrastructure for Materials and Nanotechnology (JPMX1222MS1002) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT), Japan.
Author contributions
T.F. conceived the project and H.M. directed it. T.F., R.K., K.O., and K.M. performed the synthesis procedures. T.F. and S.D performed electrical conductivity and dielectric constant measurements. T.F., T.M., Z.G., and H.O. performed the optical measurements. T.F., K.O., and T.N. performed magnetic measurements. T.F. and K.Yoshimi performed the theoretical calculations for the band structures and Ueff. T.F. and H.Sato. performed in-house single-crystal XRD analyses; S.K., T.A., H.Sawa, and Y.N. performed the synchrotron single-crystal XRD analyses. K.Yamamoto., A.T., and T.F. performed simulation for EMV coupling. T.F. wrote the manuscript, and all authors discussed the results and commented on the manuscript.
Peer review
Peer review information
Nature Communications thanks the anonymous reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Data availability
The crystallographic data (CIF files) for the structures reported in this Article have been deposited with the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2264341 (2S), 2264342 (2S•BF4), 2264325 (2O–F4), 2264326 (2O–F2), 2264327 (2S–F4), and 2264331 (2S–F2). These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033. All experimental data within the article and its Supplementary Information are available from the corresponding authors upon request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Tomoko Fujino, Email: fujino@issp.u-tokyo.ac.jp.
Hatsumi Mori, Email: hmori@issp.u-tokyo.ac.jp.
Supplementary information
The online version contains supplementary material available at 10.1038/s41467-024-47298-1.
References
- 1.Facchetti A. π-Conjugated polymers for organic electronics and photovoltaic cell applications. Chem. Mater. 2011;23:733–758. doi: 10.1021/cm102419z. [DOI] [Google Scholar]
- 2.Kagoshima, S., Nagasawa, H. & Sambongi, T. One-dimensional conductors, pp. 48–105 (Springer-Verlag, 1988).
- 3.Zhang J, Xu W, Sheng P, Zhao G, Zhu D. Organic donor−acceptor complexes as novel organic semiconductors. Acc. Chem. Res. 2017;50:1654–1662. doi: 10.1021/acs.accounts.7b00124. [DOI] [PubMed] [Google Scholar]
- 4.Saito G, Yoshida Y. Development of conductive organic molecular assemblies: organic metals, superconductors, and exotic functional materials. Bull. Chem. Soc. Jpn. 2007;80:1–137. doi: 10.1246/bcsj.80.1. [DOI] [Google Scholar]
- 5.Goetz KP, et al. Charge-transfer complexes: new perspectives on an old class of compounds. J. Mater. Chem. C. 2014;2:3065–3076. doi: 10.1039/C3TC32062F. [DOI] [Google Scholar]
- 6.Jiang H, et al. Tuning of the degree of charge transfer and the electronic properties in organic binary compounds by crystal engineering: a perspective. J. Mater. Chem. C. 2018;6:1884–1902. doi: 10.1039/C7TC04982J. [DOI] [Google Scholar]
- 7.Jérome D. Organic conductors: from charge density wave TTF-TCNQ to superconducting (TMTSF)2PF6. Chem. Rev. 2004;104:5565–5592. doi: 10.1021/cr030652g. [DOI] [PubMed] [Google Scholar]
- 8.Horiuchi S, Yamochi H, Saito G, Sakaguchi K, Kusunoki M. Nature and origin of stable metallic state in organic charge-transfer complexes of bis(ethylenedioxy)tetrathiafulvalene. J. Am. Chem. Soc. 1996;118:8604–8622. doi: 10.1021/ja960393v. [DOI] [Google Scholar]
- 9.Bloch AN, et al. Low-temperature metallic behavior and resistance minimum in a new quasi one-dimensional organic conductor. Phys. Rev. Lett. 1975;34:1561–1564. doi: 10.1103/PhysRevLett.34.1561. [DOI] [Google Scholar]
- 10.Kato R. Conducting metal dithiolene complexes: structural and electronic properties. Chem. Rev. 2004;104:5319–5346. doi: 10.1021/cr030655t. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi H, Kato R, Kobayashi A, Sasaki Y. The crystal structure of [BEDT-TTF][Ni(dmit)2]. A new route to design of organic conductors. Chem. Lett. 1985;14:191–194. doi: 10.1246/cl.1985.191. [DOI] [Google Scholar]
- 12.Kato R, et al. New molecular conductors, α- and β-(EDT-TTF)[Ni(dmit)2] metal with anomalous resistivity maximum vs. semiconductor with mixed stacks. Chem. Lett. 1989;18:1839–1842. doi: 10.1246/cl.1989.1839. [DOI] [Google Scholar]
- 13.Jacobsen CS, Yartsev VM, Tanner DB, Bechgaard K. Infrared near-isotropy in a mixed stack organic conductor: OMTSF-Ni(DMIT)2. Synth. Met. 1993;56:1925–1930. doi: 10.1016/0379-6779(93)90349-2. [DOI] [Google Scholar]
- 14.Tarrés J, et al. Synthesis, structure and physical properties of charge-transfer complexes based on BET-TTF and M(mnt)2 (M = Au, Pt) J. Mater. Chem. 1995;5:1653–1658. doi: 10.1039/JM9950501653. [DOI] [Google Scholar]
- 15.Sato R, et al. Charge-transfer complexes of benzothienobenzothiophene with tetracyanoquinodimethane and the n‐channel organic field-effect transistors. J. Phys. Chem. C. 2017;121:6561–6568. doi: 10.1021/acs.jpcc.7b00902. [DOI] [Google Scholar]
- 16.Uekusa T, Sato R, Yoo D, Kawamoto T, Mori T. Transistor characteristics of charge-transfer complexes observed across a neutral–ionic transition. ACS Appl. Mater. Interfaces. 2020;12:24174–24183. doi: 10.1021/acsami.0c03898. [DOI] [PubMed] [Google Scholar]
- 17.Mori T, Inokuchi H. Crystal structure of the mixed-stacked salt of bis(ethylenedithio)tetrathiafulvalene (BEDT-TTF) and tetracyanoquinodimethane (TCNQ) Bull. Chem. Soc. Jpn. 1987;60:402–404. doi: 10.1246/bcsj.60.402. [DOI] [Google Scholar]
- 18.Mitani T, Kaneko Y, Tanuma S, Tokura Y, Koda T, Saito G. Electric conductivity and phase diagram of a mixed-stack charge-transfer crystal: tetrathiafulvalene-p-chloranil. Phys. Rev. B. 1987;35:427–429. doi: 10.1103/PhysRevB.35.427. [DOI] [PubMed] [Google Scholar]
- 19.Hotta S, Kobayashi H. Crystal and molecular structure of a charge-transfer complex between α,α’-dimethylquaterthiophene and 2,3,5,6-tetrafluoro-7,7,8,8-tetracyanoquinodimethane. Synth. Met. 1994;66:117–122. doi: 10.1016/0379-6779(94)90087-6. [DOI] [Google Scholar]
- 20.Horiuchi S, Kumai R, Okimoto Y, Tokura Y. Chemical approach to neutral–ionic valence instability, quantum phase transition, and relaxor ferroelectricity in organic charge-transfer complexes. Chem. Phys. 2006;325:78–91. doi: 10.1016/j.chemphys.2005.09.025. [DOI] [Google Scholar]
- 21.Iwasawa N, et al. Quasi three-dimensional electrical conductor having alternating mixed stacks: tetrakis(methyltelluro)tetrathiafulvalene (TTeC1-TTF)•TCNQ complex. Chem. Lett. 1988;17:215–218. doi: 10.1246/cl.1988.215. [DOI] [Google Scholar]
- 22.Saito G, Pac S-S, Drozdova OO. First metallic DA alternating charge transfer complex: (HMTTeF)(Et2TCNQ)(THF)x. Synth. Met. 2001;120:667–670. doi: 10.1016/S0379-6779(00)01040-7. [DOI] [Google Scholar]
- 23.Fujinawa T, et al. Unusual electrical properties of neutral mixed-stack charge-transfer complexes of DAP–BRL (DAP = 1,6-Diaminopyrene, BRL = p-Bromanil) Bull. Chem. Soc. Jpn. 1999;72:21–26. doi: 10.1246/bcsj.72.21. [DOI] [Google Scholar]
- 24.Saito G, Murata T. Mixed valency in organic charge transfer complexes. Philos. Trans. R. Soc. A. 2008;366:139–150. doi: 10.1098/rsta.2007.2146. [DOI] [PubMed] [Google Scholar]
- 25.Torrance JB, Vazquez JE, Mayerle JJ, Lee VY. Discovery of neutral-to-ionic phase transition in organic materials. Phys. Rev. Lett. 1981;46:253–257. doi: 10.1103/PhysRevLett.46.253. [DOI] [Google Scholar]
- 26.Torrance JB. An overview of organic charge-transfer solids: insulators, metals, and the neutral-ionic transition. Mol. Cryst. Liq. Cryst. 1985;126:55–67. doi: 10.1080/15421408508084154. [DOI] [Google Scholar]
- 27.Sato R, Kawamoto T, Mori T. Asymmetrical hole/electron transport in donor–acceptor mixed-stack cocrystals. J. Mater. Chem. C. 2019;7:567–577. doi: 10.1039/C8TC05190A. [DOI] [Google Scholar]
- 28.Imaeda K, Enoki T, Inokuchi H, Saito G. Electronic properties of 3,3′,5,5′-tetramethyl-benzidine complexes with TCNQ derivatives. Mol. Cryst. Liq. Cryst. 1986;141:131–140. doi: 10.1080/00268948608080204. [DOI] [Google Scholar]
- 29.Takehara R, et al. Topological charge transport by mobile dielectric-ferroelectric domain walls. Sci. Adv. 2019;5:eaax8720. doi: 10.1126/sciadv.aax8720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kameyama R, et al. The simplest model for doped poly(3,4-ethylenedioxythiophene) (PEDOT): single-crystalline EDOT dimer radical cation salts. Chem. Eur. J. 2021;27:6696–6700. doi: 10.1002/chem.202005333. [DOI] [PubMed] [Google Scholar]
- 31.Kameyama R, et al. Band-filling effects in single-crystalline oligomer models for doped PEDOT: 3,4-ethylenedioxythiophene (EDOT) dimer salt with hydrogen-bonded infinite sulfate anion chains. J. Mater. Chem. C. 2022;10:7543–7551. doi: 10.1039/D2TC01216B. [DOI] [Google Scholar]
- 32.Cortizo-Lacalle D, et al. BODIPY-based conjugated polymers for broadband light sensing and harvesting applications. J. Mater. Chem. A. 2012;22:14119–14126. doi: 10.1039/c2jm32374e. [DOI] [Google Scholar]
- 33.Emge TJ, Maxfield M, Cowan DO, Kistenmacher TJ. Solution and solid state studies of tetrafluoro-7,7,8,8-tetracyano-p-quinodimethane, TCNQF4. Evidence for long-range amphoteric intermolecular interactions and low-dimensionality in the solid state structure. Mol. Cryst. Liq. Cryst. 1981;65:161–178. doi: 10.1080/00268948108082132. [DOI] [Google Scholar]
- 34.Krupskaya Y, Gibertini M, Marzari N, Morpurgo AF. Band-like electron transport with record-high mobility in the TCNQ family. Adv. Mater. 2015;27:2453–2458. doi: 10.1002/adma.201405699. [DOI] [PubMed] [Google Scholar]
- 35.O’Kane SA, et al. New crystalline polymers of Ag(TCNQ) and Ag(TCNQF4): structures and magnetic properties. J. Solid State Chem. 2000;152:159–173. doi: 10.1006/jssc.2000.8679. [DOI] [Google Scholar]
- 36.Funasako Y, et al. Decamethyl- and octamethyl-ferrocenium salts of F1- and F2-TCNQ: effects of fluorine substitution on the crystal structures and magnetic interactions. Inorg. Chim. Acta. 2014;419:105–110. doi: 10.1016/j.ica.2014.05.011. [DOI] [Google Scholar]
- 37.Miyasaka H, et al. Control of charge transfer in a series of Ru2II,II/TCNQ two-dimensional networks by tuning the electron affinity of TCNQ units: a route to synergistic magnetic/conducting materials. J. Am. Chem. Soc. 2010;132:1532–1544. doi: 10.1021/ja909489s. [DOI] [PubMed] [Google Scholar]
- 38.Ozaki T. Variationally optimized atomic orbitals for large-scale electronic structures. Phys. Rev. B. 2003;67:155108. doi: 10.1103/PhysRevB.67.155108. [DOI] [Google Scholar]
- 39.Ozaki T, Kino H. Numerical atomic basis orbitals from H to Kr. Phys. Rev. B. 2004;69:195113. doi: 10.1103/PhysRevB.69.195113. [DOI] [Google Scholar]
- 40.Giannozzi P, et al. Quantum ESPRESSO: a Modular and open-source software project for quantum simulations of materials. J. Phys. Condens. Matter. 2009;21:395502. doi: 10.1088/0953-8984/21/39/395502. [DOI] [PubMed] [Google Scholar]
- 41.Giannozzi P, et al. Advanced capabilities for materials modelling with Quantum ESPRESSO. J. Phys. Condens. Matter. 2017;29:465901. doi: 10.1088/1361-648X/aa8f79. [DOI] [PubMed] [Google Scholar]
- 42.Nakamura K, et al. RESPACK: an ab initio tool for derivation of effective low-energy model of material. Comput. Phys. Commun. 2021;261:107781. doi: 10.1016/j.cpc.2020.107781. [DOI] [Google Scholar]
- 43.Castagnetti N, Masino M, Rizzoli C, Girlando A. Mixed stack charge transfer crystals: crossing the neutral-ionic borderline by chemical substitution. Phys. Rev. Mater. 2018;2:024602. doi: 10.1103/PhysRevMaterials.2.024602. [DOI] [Google Scholar]
- 44.Okamoto H, et al. Photoinduced phase transition in tetrathiafulvalene-p-chloranil observed in femtosecond reflection spectroscopy. Phys. Rev. B. 2004;70:165202. doi: 10.1103/PhysRevB.70.165202. [DOI] [Google Scholar]
- 45.Imada M, Fujimori A, Tokura Y. Metal-insulator transitions. Rev. Mod. Phys. 1998;70:1039–1263. doi: 10.1103/RevModPhys.70.1039. [DOI] [Google Scholar]
- 46.Ido K, Yoshimi K, Misawa T, Imada M. Unconventional dual 1D–2D quantum spin liquid revealed by ab initio studies on organic solids family. npj Quantum Mater. 2022;7:1–42. doi: 10.1038/s41535-022-00452-8. [DOI] [Google Scholar]
- 47.Ohki D, Yoshimi K, Kobayashi A, Misawa T. Gap opening mechanism for correlated Dirac electrons in organic compounds α-(BEDT-TTF)2I3 and α-(BEDT-TSeF)2I3. Phys. Rev. B. 2023;107:L041108. doi: 10.1103/PhysRevB.107.L041108. [DOI] [Google Scholar]
- 48.Yoshimi K, Misawa T, Tsumuraya T, Seo H. Comprehensive ab initio investigation of the phase diagram of quasi-one-dimensional molecular solids. Phys. Rev. Lett. 2023;131:036401. doi: 10.1103/PhysRevLett.131.036401. [DOI] [PubMed] [Google Scholar]
- 49.Rice MJ. Organic linear conductors as systems for the study of electron-phonon interactions in the organic solid state. Phys. Rev. Lett. 1976;37:36–39. doi: 10.1103/PhysRevLett.37.36. [DOI] [Google Scholar]
- 50.Kagawa F, Horiuchi S, Tokunaga M, Fujioka J, Tokura Y. Ferroelectricity in a one-dimensional organic quantum magnet. Nat. Phys. 2010;6:169–172. doi: 10.1038/nphys1503. [DOI] [Google Scholar]
- 51.Bray, J. W., Interrante, L. V., Jacobs, I. S. & Bonner, J. C. The Spin-Peierls transition. In Extended linear chain compounds, vol. 3, (ed. Miller, J. S.) pp. 353–415 (Springer-Verlag, 1983).
- 52.Iwasa Y, et al. Temperature-induced neutral-ionic transition in tetramethylbenzidine-tetracyanoquinodimethane (TMB-TCNQ) Phys. Rev. B. 1990;42:2374–2377. doi: 10.1103/PhysRevB.42.2374. [DOI] [PubMed] [Google Scholar]
- 53.Horiuchi S, Okimoto Y, Kumai R, Tokura Y. Ferroelectric valence transition and phase diagram of a series of charge-transfer complexes of 4,4’-dimethyltetrathiafulvalene and tetrahalo-p-benzoquinones. J. Am. Chem. Soc. 2001;123:665–670. doi: 10.1021/ja0016654. [DOI] [PubMed] [Google Scholar]
- 54.Morimoto T, Miyamoto T, Okamoto H. Ultrafast electron and molecular dynamics in photoinduced and electric-field-induced neutral–ionic transitions. Crystals. 2017;7:132. doi: 10.3390/cryst7050132. [DOI] [Google Scholar]
- 55.Kameyama R, Fujino T, Dekura S, Mori H. Conjugation length effect on the conducting behavior of single-crystalline oligo(3,4-ethylenedioxythiophene) (nEDOT) radical cation salts. Phys. Chem. Chem. Phys. 2022;24:9130–9134. doi: 10.1039/D2CP00250G. [DOI] [PubMed] [Google Scholar]
- 56.Onozuka K, et al. Metallic state of a mixed-sequence oligomer salt that models doped PEDOT family. J. Am. Chem. Soc. 2023;145:15152–15161. doi: 10.1021/jacs.3c01522. [DOI] [PubMed] [Google Scholar]
- 57.Becke AD. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. A. 1988;38:3098–3100. doi: 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]
- 58.Becke AD. Density‐functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993;98:5648–5652. doi: 10.1063/1.464913. [DOI] [Google Scholar]
- 59.Lee C, Yang W, Parr RG. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density. Phys. Rev. B. 1988;37:785–789. doi: 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- 60.Ditchfield R, Hehre WJ, Pople JA. Self‐consistent molecular‐orbital methods. IX. An extended Gaussian‐type basis for molecular‐orbital studies of organic molecules. J. Chem. Phys. 1971;54:724–728. doi: 10.1063/1.1674902. [DOI] [Google Scholar]
- 61.Hehre WJ, Ditchfield R, Pople JA. Self—consistent molecular orbital methods. XII. further extensions of Gaussian—type basis sets for use in molecular orbital studies of organic molecules. J. Chem. Phys. 1972;56:2257–2261. doi: 10.1063/1.1677527. [DOI] [Google Scholar]
- 62.Hariharan PC, Pople JA. Accuracy of AHn equilibrium geometries by single determinant molecular orbital theory. Mol. Phys. 1974;27:209–214. doi: 10.1080/00268977400100171. [DOI] [Google Scholar]
- 63.Lejaeghere K, et al. Reproducibility in density functional theory calculations of solids. Science. 2016;351:aad3000. doi: 10.1126/science.aad3000. [DOI] [PubMed] [Google Scholar]
- 64.Ozaki T, Kino H. Efficient projector expansion for the ab initio LCAO method. Phys. Rev. B. 2005;72:045121. doi: 10.1103/PhysRevB.72.045121. [DOI] [Google Scholar]
- 65.Perdew JP, Burke K, Ernzerhof M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996;77:3865–3868. doi: 10.1103/PhysRevLett.77.3865. [DOI] [PubMed] [Google Scholar]
- 66.Momma K, Izumi F. VESTA 3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Cryst. 2011;44:1272–1276. doi: 10.1107/S0021889811038970. [DOI] [Google Scholar]
- 67.Schlipf M, Gygi F. Optimization algorithm for the generation of ONCV pseudopotentials. Comput. Phys. Commun. 2015;196:36–44. doi: 10.1016/j.cpc.2015.05.011. [DOI] [Google Scholar]
- 68.Bain GA, Berry JF. Diamagnetic corrections and Pascal’s constants. J. Chem. Educ. 2008;85:532–536. doi: 10.1021/ed085p532. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The crystallographic data (CIF files) for the structures reported in this Article have been deposited with the Cambridge Crystallographic Data Centre (CCDC), under deposition numbers 2264341 (2S), 2264342 (2S•BF4), 2264325 (2O–F4), 2264326 (2O–F2), 2264327 (2S–F4), and 2264331 (2S–F2). These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033. All experimental data within the article and its Supplementary Information are available from the corresponding authors upon request.