

Abstract
Biallelic mutations in the coenzyme Q7 (COQ7) encoding gene were recently identified as a genetic cause of distal hereditary motor neuropathy. Here, we explored the clinical, electrophysiological, pathological, and genetic characteristics of a Chinese patient with spastic paraplegia associated with recessive variants in COQ7. This patient carried a novel c.322C>A (p.Pro108Thr) homozygous variant. Sural biopsy revealed mild mixed axonal and demyelinating degeneration. Immunoblotting showed a significant decrease in the COQ7 protein level in the patient's fibroblasts. This study confirmed that COQ7 variant as a genetic cause of HSP, and further extended spastic paraplegia to the phenotypic spectrum of COQ7‐related disorders.
Introduction
Hereditary spastic paraplegia (HSP) is a group of genetically heterogeneous diseases primarily characterized by a progressive spastic gait. The length‐dependent degeneration of the corticospinal tracts is a pathological hallmark of HSP, resulting in bilateral limb spasticity, hyperreflexia, and extensor plantar responses. 1 HSPs can be classified into pure and complicated forms, depending on whether they coexist with other symptoms such as epilepsy, ataxia, and peripheral neuropathy. Several genes associated with HSP are identified annually, and a total of 91 subtypes of HSP have been identified to date (https://neuromuscular.wustl.edu/spinal/fsp.html). Nevertheless, a significant number of patients with spastic paraplegia (SPG) remain genetically unresolved. 2
Coenzyme Q10 (CoQ10), also known as ubiquinone, functions as an electron carrier in the mitochondrial respiratory chain. 3 It shuttles electrons from Complex I or Complex II to Complex III in the inner mitochondrial membrane. 4 Primary CoQ10 deficiency is caused by mutations in the genes that are responsible for its intracellular biosynthesis. Recent studies have found that patients with biallelic mutations in COQ7, a component of CoQ10 biosynthesis, can present with symptoms resembling HSP or HSP‐like. 5 , 6 , 7 , 8 However, there are no entries in the OMIM or neuromuscular website indicating that COQ7 variants can cause HSP. In this study, we report a case of juvenile‐onset HSP, accompanied by epilepsy, distal amyotrophy, and weakness, which is associated with a homozygous variant (c.322C>A, p.Pro108Thr) in the COQ7 gene.
Materials and Methods
This study was approved by the Ethics Committee of the First Affiliated Hospital of Nanchang University [SJNKHDJ20221213160659]. Written informed consent was obtained from the participants.
DNA was extracted from the patient's peripheral blood leukocytes. The causative gene in the index patient (IV:2) was explored using a whole exome sequence (WES). Sanger sequencing with specific primers was performed to confirm the COQ7 variant in the parents and available sibling. Homozygous regions were analyzed using Automap software (https://automap.iob.ch/).
Sural nerve biopsy was conducted in the index patient. The nerve was fixed in 4% formaldehyde, paraffin‐embedded, cut into 4 μm sections, and stained by hematoxylin–eosin (HE) staining. Immunostaining with neurofilament (NF) and myelin basic protein (MBP) antibodies was conducted. The rest of the nerve specimen was fixed in 3% glutaraldehyde and embedded in Epon 812. Semithin sections were stained with toluidine blue.
Total cell lysates were subjected to sodium dodecyl sulfate–polyacrylamide gel electrophoresis and immunoblotting. Antibodies were targeted against COQ7 (Proteintech) and tubulin (Proteintech).
Results
The proband (Fig. 1A) was a 34‐year‐old female patient from a consanguineous family. At the age of 11, she began to feel tightness in her lower limbs, which led to frequent falls and difficulty in walking long distances. At that same year, she experienced several generalized tonic–clonic seizures. Five years later, she noticed muscle weakness and wasting in her lower limbs, which resulted in the spraining and stubbing of her toes while walking on flat roads. Recently, she had occasionally experienced urinary incontinence when she was in urgent need of urination. Her stiffness in her lower limbs had worsened. The muscle weakness and wasting in the lower limbs were more pronounced with mild muscle wasting observed in the hands (Fig. 1B). The physical examination revealed decreased muscle strength in the distal limbs and pyramidal tract signs (Table S1). There were no sensory deficits, signs of ataxia or cognitive impairments. There were no abnormal clinical symptoms or signs observed in the parents and siblings of the patient.
Figure 1.
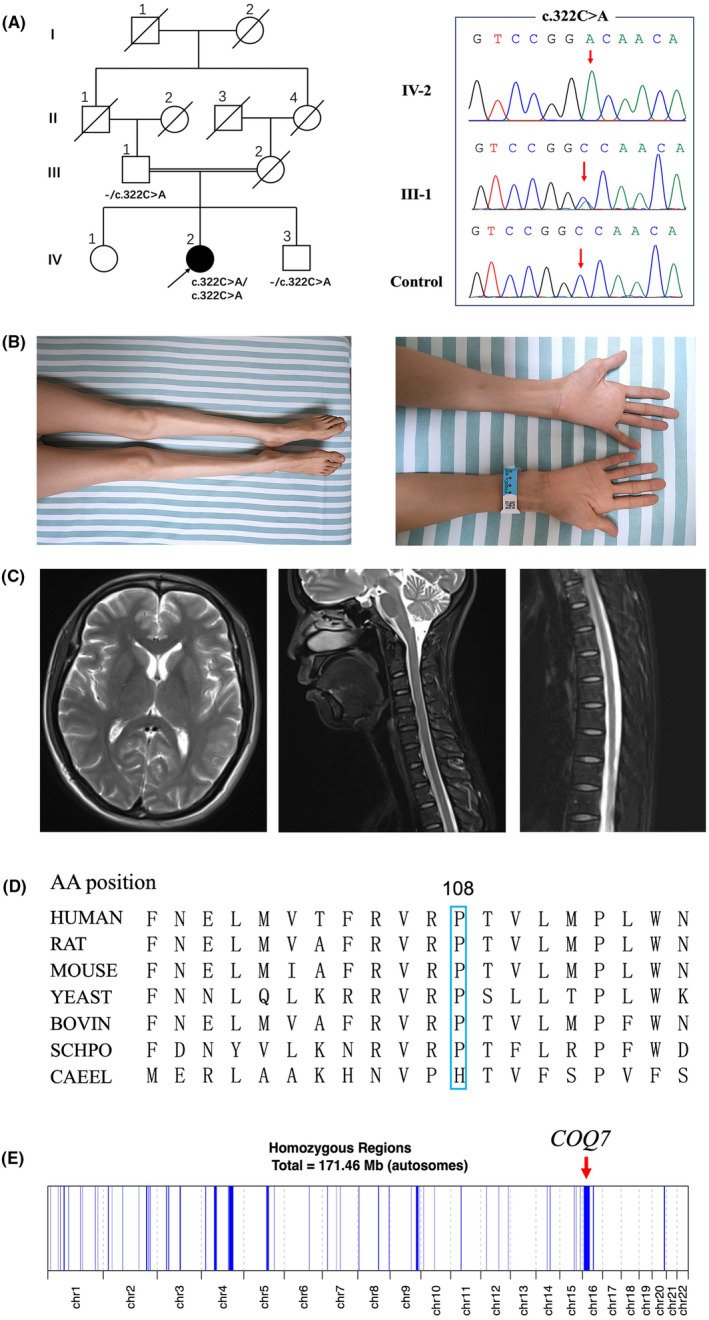
Clinical features of the index patient with COQ7 homozygous variant. (A) Left: Pedigree diagram of this family. Right: Sanger sequencing chromatograms showed homozygous and heterozygous c.322C>A variants in this family. (B) Left: Muscle wasting of the distal lower limbs. Right: Muscle wasting of distal upper limbs. (C) Magnetic resonance T2‐weighted images of the index patient's brain, cervical spinal cord, and thoracic spinal cord. (D) The conservation of proline at 108 residue in different species. (E) Chromosomal homozygous region of the patient. The red arrow indicates the region where the COQ7 is located.
Nerve conduction studies revealed a reduction in compound motor action potential in multiple motor nerves, with a more pronounced severity in the lower limbs compared to the upper limbs. However, the nerve conduction velocity and latency remained within the normal range. The sensory nerves were intact (Table S2). The needle electromyography showed a chronic active neurogenic pattern in the quadriceps femoris and anterior tibialis muscles. At this time, the electroencephalogram was found to be normal. During the hearing, fundus, and cardiac examinations, no abnormalities were detected. The cerebral MRI demonstrated a normal structure, whereas the spinal MRI revealed a mild atrophy of the thoracic spinal cord (Fig. 1C).
By analyzing the patient's WES data, we identified a novel COQ7 homozygous variant (c.322C>A, p.Pro108Thr, NM_016138.5). This variant is not found in multiple human genome databases, including 1000G, ExAC, and gnomAD. The mutant is highly conserved among species (Fig. 1D) and predicted to be harmful or probably pathogenic using MutationTaster, SIFT, Polyphen2, and CADD software. The Sanger sequence indicated a co‐segregation in the family. Using Automap software (https://automap.iob.ch/), we found that a total homozygous regions were ~ 171.46 Mb in the patient, with the variant situated within the largest homozygous region, spanning ~23.09 Mb (chr16:5533549‐28620242) (Fig. 1E).
Pathologically, no abnormalities of the sural nerve were observed on HE staining (Fig. 2A). Immunohistochemical staining of MBP antibody showed the normal structure of myelinated fibers (Fig. 2B). However, the toluidine blue staining revealed a slight decrease in the density of myelinated nerve fibers, accompanied by a few thin myelinated fibers (Fig. 2C), indicating a mild mixed axonal and demyelinating degeneration of sensory nerves. Electron microscopy demonstrated the presence of thin myelinated fibers, while the density of unmyelinated axons was slightly decreased with the formation of atypical collagen pockets (Fig. 2D).
Figure 2.
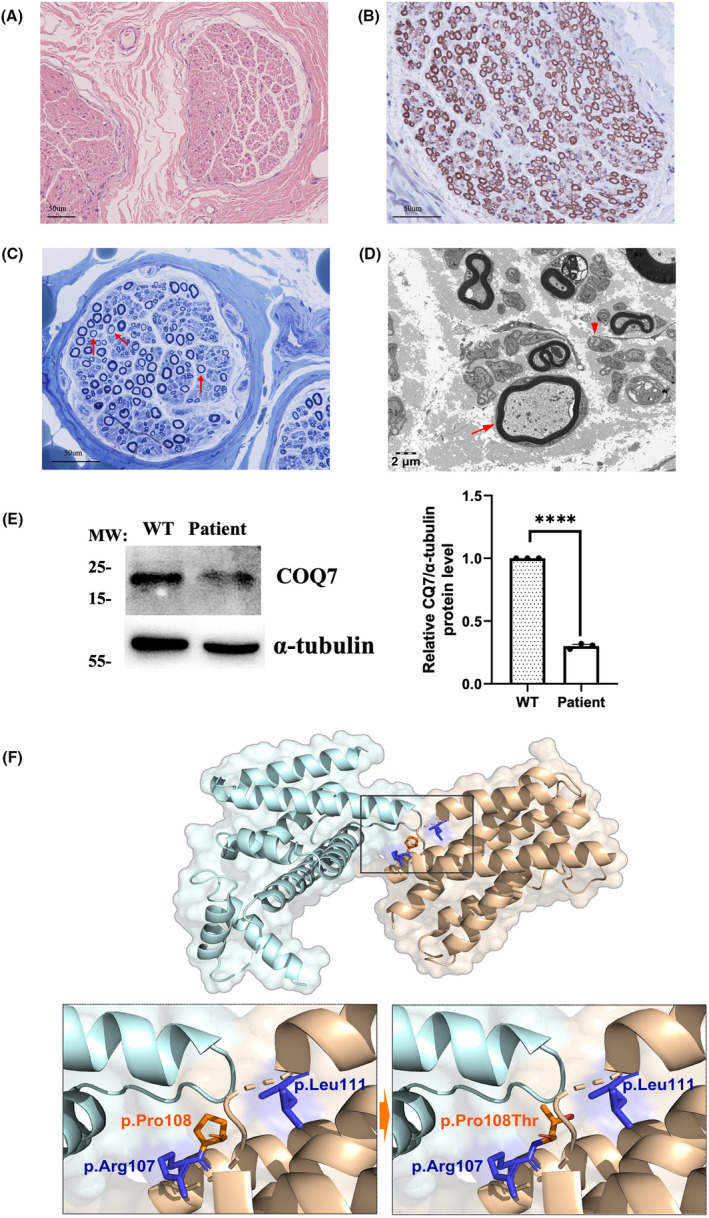
Neuropathological and molecular changes in patient and predicted structure changes of COQ7 protein. (A) HE staining revealed the normal structure of sural nerve. (B) Immunohistochemical staining of MBP antibody revealed the normal structure of myelinated fibers in sural nerve. (C) Toluidine blue staining revealed a mild reduction in the density of myelinated nerve fibers and a few thin myelinated fibers (Red arrow). (D) Electron microscopy revealed thin myelinated fibers (Red arrow) and a mild reduction of the density of unmyelinated axons with atypical collagen pockets (Red arrowhead). (E) Western blot analysis of COQ7 protein level was significantly decreased compared with the normal control in skin fibroblasts. (F) Structure of the COQ7:COQ9 dipolymer (PDB ID 7SSP) and mapped by PyMOL. Details for p.Pro108Thr mutant interefering the COQ7:COQ9 interaction.
In order to evaluate the pathogenic role of the variant, we detected the expression level of COQ7 protein from the patient's skin fibroblasts. Western blot analysis showed that compared with the normal control, the COQ7 protein level in the patient's skin fibroblasts was significantly decreased, which suggested that this homozygous variant resulted in protein instability, leading to loss‐of‐function mechanism (Fig. 2E).
Discussion
COQ7 variants are the genetic cause of a group of neurodevelopmental disorders. To date, a total of 34 patients with COQ7 mutations have been reported. 7 , 8 , 9 Despite incomplete clinical data for some patients, the most common clinical manifestations included muscle weakness (27/29), muscle atrophy (26/29), increased knee reflexes (22/32), and increased muscle tone of lower limbs (11/21). Our patient presented with juvenile‐onset HSP, characterized by spastic gait, limb rigidity, hyperreflexia, patellar clonus, extensor plantar responses, and bladder dysfunction. Additionally, she was accompanied by epilepsy and distal motor neuropathy (dHMN), which can be categorized as a complex HSP.
Recently, several articles have reported that biallelic mutations in the COQ7 gene caused dHMN with or without upper motor neuron signs. The axonal degeneration of motor nerves and corticospinal tracts was a continuous spectrum of axonopathy, so it was reasonable for some patients with COQ7‐related disorder to exhibit predominant dHMN accompanied by upper motor neuron signs. Vice versa, it was also possible for some patients could exhibit a dominance of HSP along with dHMN. Additionally, at least 16 subtypes of complex HSP could also exhibit spasticity combined with motor neuropathy, such as SPG17 and SPG38, 10 , 11 among others. Their clinical manifestations were very similar to those of patients with COQ7 variants. Therefore, the clinical phenotypes of COQ7‐related disorders can be divided into three distinct subgroups (Table 1). The first was a developmental encephalomyopathy (5/34). The second group exhibited milder forms of peripheral neuropathy with or without upper motor neuron signs (24/34). The final group comprised patients with either pure or complex HSP (5/34).
Table 1.
The summary of reported patients with COQ7 mutations.
| Phenotype | Variants | Origin | Gender | Family | Age at onset (years) | Manifestations | Reference |
|---|---|---|---|---|---|---|---|
| Encephalomyopathy | |||||||
| Encephalomyopathy | c.599_600delins/c.319C>T | China | Male | Consanguineous | After birth | Fatal developmental encephalomyopathy | Kwong, et al. (2019) |
| Encephalomyopathy | Homozygous c.422T>A | Syrian | Male | Consanguineous | After birth | Progressive encephalo‐neuro‐nephro‐cardiopathy | Freyer et al. (2015) |
| Encephalomyopathy | c.308C>T/c.332T>C | Canada | Female | Consanguineous | 2 | Global developmental delay with spastic paraplegia | Wang et al. (2017) |
| Encephalomyopathy | Homozygous c.161G>A | Turkey | Female | Nonconsanguineous | 5 | Language and motor developmental delay | Wang et al. (2022) |
| Encephalomyopathy | c.446A>G/c.161G>A | European | Male | Nonconsanguineous | After birth | Developmental encephalomyopathy | Parith et al. (2023) |
| Peripheral neuropathy | |||||||
| dHMN with UMN signs | Homozygous c.3G>T | Portugal | Male | Nonconsanguineous | 12 | Progressive atrophy and weakness of the distal muscles | Jacquier et al. (2023) |
| dHMN with UMN signs | Homozygous c.3G>T | Portugal | Female | Nonconsanguineous | 9 | Progressive atrophy and weakness of the distal muscles | Jacquier et al. (2023) |
| dHMN | Homozygous c.3G>T | Portugal | Male | Nonconsanguineous | 10 | Progressive atrophy and weakness of the distal muscles | Jacquier et al. (2023) |
| dHMN | c.253‐2A>T/c.467T>A | China | Male | Nonconsanguineous | 14 | Progressive atrophy and weakness of the distal muscles | Liu et al. (2023) |
| dHMN | c.160C>T/c.467T>G | China | Male | Nonconsanguineous | 15 | Progressive atrophy and weakness of the distal muscles | Liu et al. (2023) |
| dHMN | Homozygous c.1A>G | Syria | Male | Consanguineous | 10 | Progressive atrophy and weakness of the distal muscles | Smith et al. (2023) |
| dHMN | Homozygous c.1A>G | Syria | Female | Consanguineous | 10 | Progressive atrophy and weakness of the distal muscles | Smith et al. (2023) |
| dHMN | Homozygous c.1A>G | Syria | Male | Consanguineous | 10 | Progressive atrophy and weakness of the distal muscles | Smith et al. (2023) |
| dHMN | c.197T>A/c.446A>G | USA | Male | Nonconsanguineous | Mid‐teens | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN | c.197T>A/c.446A>G | USA | Male | Nonconsanguineous | Childhood | Never able to run, walking difficulties | Rebelo et al. (2023) |
| dHMN | c.197T>A/c.319C>T | Brazil | Male | Nonconsanguineous | <10 | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN with UMN signs | Homozygous c.161G>A | Brazil | Female | Nonconsanguineous | 3 | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN with UMN signs | Homozygous c.3G>T | Brazil | Female | Nonconsanguineous | 5 | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN with UMN signs | Homozygous c.3G>T | Brazil | Female | Nonconsanguineous | 10 | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN | Homozygous c.3G>T | Brazil | Female | Nonconsanguineous | 1 | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN | Homozygous c.3G>T | Brazil | Female | Consanguineous | School age | Walking difficulties | Rebelo et al. (2023) |
| dHMN | Homozygous c.3G>T | Brazil | Male | Consanguineous | 4 | Walking and running difficulties | Rebelo et al. (2023) |
| dHMN | Homozygous c.3G>T | Brazil | Female | Consanguineous | 10 | Walking and running difficulties | Rebelo et al. (2023) |
| CMT2 | Homozygous c.319C>T | Wales | Male | Consanguineous | 8–10 | Walking difficulties | Rebelo et al. (2023) |
| CMT2 | c.467T>G/c.599_600delins | China | Male | Nonconsanguineous | 6 | Progressive atrophy and weakness of the distal muscles | Zhang et al. (2023) |
| CMT | c.446A>G/c.3G>T | NA | Female | NA | 12 | Progressive weakness of lower limbs | Parith et al. (2023) |
| CMT | Homozygous c.161G>A | Brazil | Female | NA | 3 | Equine posturing and pes cavus deformity | Parith et al. (2023) |
| CMT | c.197T>A/c.446A>G | Netherlands | NA | Nonconsanguineous | NA | Axonal neuropathy, mild neurodegenerative disorder | Tom et al. (2017) |
| CMT | c.197T>A/c.446A>G | Netherlands | NA | Nonconsanguineous | NA | Axonal neuropathy, mild neurodegenerative disorder | Tom et al. (2017) |
| HSP | |||||||
| HSP‐like | Homozygous c.332T>C | Iran | Male | Consanguineous | 1 | Moderate progressive spastic paraparesis | Sadr et al. (2023) |
| HSP‐like | Homozygous c.161G>A | Brazil | Female | Consanguineous | 3 | Progressive spastic paraplegia | Parith et al. (2023) |
| HSP‐like | Homozygous c.3G>T | Brazil | Female | Consanguineous | 5 | Progressive spastic paraplegia | Parith et al. (2023) |
| HSP‐like | c.308C>T/c.332T>C | Iran | Female | Consanguineous | 2 | Moderate to severe progressive spastic paraplegia | Hashemi et al. (2021) |
| HSP‐like | Homozygous c.322C>A | China | Female | Consanguineous | 11 | HSP with epilepsy, distal amyotrophy, and weakness | This study |
Abbreviations: CMT, Charcot‐Marie‐Tooth; dHMN, distal hereditary motor neuropathy; HSP, hereditary spastic paraplegia; NA, not available; UMN, upper motor neuron.
Patients with COQ7 mutations primarily exhibited axonopathy in the nervous system; however, there were no autopsy reports for these individuals. This study provided the first case (albeit of a sensory nerve) of neuropathological findings, characterized by decreased axonal density, thin myelin sheath of large myelinated fibers, and degeneration of unmyelinated fibers. Pathologically, we speculated that our patient might eventually develop axonal Charcot‐Marie‐Tooth (CMT2), despite currently presenting with dHMN at electrophysiological level.
COQ7 plays a key role in the penultimate step of COQ10 biosynthesis. The structure of COQ7 contains six helices (α1‐α6), helices α1, α3, α4, and α6 form a hydrophobic surface and a large hydrophobic channel. 12 COQ7 binds to COQ9 at this hydrophobic surface to form a complex, the reported mutations associated with HSP phenotype are located on this hydrophobic surface (p.Arg107Trp, p.Pro108Thr, and p.Leu111Pro) 5 , 13 (Fig. 2F). We speculated that these mutants near this structure might affect the binding between COQ7 and COQ9, which might potentially modify the clinical manifestations. Mutations in COQ4, another gene involved in the biosynthesis of CoQ10, have been reported to cause HSP and epilepsy. COQ4 protein levels in fibroblasts derived from patients with COQ4‐related HSP were reduced, but only slightly reducing CoQ10 levels. A similar metabolic pattern had also been observed in a case with COQ7 p.Leu111Pro variant. In this study, we also observed that COQ7 protein levels in fibroblasts derived from patient with the COQ7‐related HSP were significantly decreased. Although the p.Pro108Thr variant (1995683) had been reported as uncertain significance on ClinVar, considering the preliminary functional studies, this missense variant could be classified as likely pathogenic. It can be speculated that the milder clinical phenotype may be associated with a mild reduction rather than a severe reduction in CoQ10. Given that neurons were highly sensitive to energy restrictions, the mitochondrial dysfunction caused by the slight reduction in CoQ10 might be more susceptible to affecting neurons.
In summary, while only one family was included in this study, the patient exhibited a typical phenotype of complex HSP and was associated with a novel homozygous variant in COQ7, which expanded the spectrum of pathogenic variants and clinical phenotypes of COQ7‐related disorders. The COQ7 gene should be included in the genetic workflow for the diagnosis of HSP.
Author Contributions
D.H. and Q.Y. contributed to the conception and design of the study; Y.X. and M.Z. contributed to the acquisition and analysis of data; L.W. and D.T. contributed to drafting the text and preparing the figures.
Funding Information
This work was supported by the National Natural Science Foundation of China (Grants 82160252 and 82271439), Natural Science Foundation of Jiangxi province (20224ACB206015), and Double thousand talents program of Jiangxi province (jxsq2019101021).
Conflict of Interest Statement
The authors have no conflicts of interest to declare.
Supporting information
Table S1.
Acknowledgment
We thank the patients and their families for their participation in this study. This work was supported by the National Natural Science Foundation of China (Grants 82160252 and 82271439), Natural Science Foundation of Jiangxi province (20224ACB206015), and Double thousand talents program of Jiangxi province (jxsq2019101021).
Funding Statement
This work was funded by National Natural Science Foundation of China grants 82160252 and 82271439; Natural Science Foundation of Jiangxi province grant 20224ACB206015; Double thousand talents program of Jiangxi province grant jxsq2019101021.
References
- 1. Shribman S, Reid E, Crosby AH, Houlden H, Warner TT. Hereditary spastic paraplegia: from diagnosis to emerging therapeutic approaches. Lancet Neurol. 2019;18(12):1136‐1146. [DOI] [PubMed] [Google Scholar]
- 2. Schüle R, Wiethoff S, Martus P, et al. Hereditary spastic paraplegia: Clinicogenetic lessons from 608 patients. Ann Neurol. 2016;79(4):646‐658. [DOI] [PubMed] [Google Scholar]
- 3. Staiano C, García‐Corzo L, Mantle D, et al. Biosynthesis, deficiency, and supplementation of coenzyme Q. Antioxidants (Basel). 2023;12(7):1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Guerra RM, Pagliarini DJ. Coenzyme Q biochemistry and biosynthesis. Trends Biochem Sci. 2023;48(5):463‐476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hashemi SS, Zare‐Abdollahi D, Bakhshandeh MK, et al. Clinical spectrum in multiple families with primary COQ(10) deficiency. Am J Med Genet A. 2021;185(2):440‐452. [DOI] [PubMed] [Google Scholar]
- 6. Liu XX, Wang N, Chen YK, et al. Biallelic variants in the COQ7 gene cause distal hereditary motor neuropathy in two Chinese families. Brain. 2023;146(5):e27‐e30. [DOI] [PubMed] [Google Scholar]
- 7. Zhang XY, Dong HL, Wu ZY. Axonal Charcot‐Marie‐tooth disease due to COQ7 mutation: expanding the genetic and clinical spectrum. Brain. 2023;146:e117‐e119. [DOI] [PubMed] [Google Scholar]
- 8. Rebelo AP, Tomaselli PJ, Medina J, et al. Biallelic variants in COQ7 cause distal hereditary motor neuropathy with upper motor neuron signs. Brain. 2023;146(10):4191‐4199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wongkittichote P, Duque Lasio ML, Magistrati M, et al. Phenotypic, molecular, and functional characterization of COQ7‐related primary CoQ(10) deficiency: hypomorphic variants and two distinct disease entities. Mol Genet Metab. 2023;139(4):107630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Windpassinger C, Auer‐Grumbach M, Irobi J, et al. Heterozygous missense mutations in BSCL2 are associated with distal hereditary motor neuropathy and silver syndrome. Nat Genet. 2004;36(3):271‐276. [DOI] [PubMed] [Google Scholar]
- 11. Kropatsch R, Schmidt HM, Buttkereit P, Epplen JT, Hoffjan S. BICD2 mutational analysis in hereditary spastic paraplegia and hereditary motor and sensory neuropathy. Muscle Nerve. 2019;59(4):484‐486. [DOI] [PubMed] [Google Scholar]
- 12. Manicki M, Aydin H, Abriata LA, et al. Structure and functionality of a multimeric human COQ7:COQ9 complex. Mol Cell. 2022;82(22):4307‐4323.e4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Sadr Z, Zare‐Abdollahi D, Rohani M, Alavi A. A founder mutation in COQ7, p.(Leu111Pro), causes pure hereditary spastic paraplegia (HSP) in the Iranian population. Neurol Sci. 2023;44(7):2599‐2602. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1.