

Abstract
Key Clinical Message
High‐dose intravenous immunoglobulin exhibits great potential in the treatment of Netherton syndrome.
Abstract
Netherton syndrome (NS) is a rare autosomal recessive genodermatosis (OMIM #256500) characterized by superficial scaling, atopic manifestations, and multisystemic complications. It is caused by loss‐of‐function mutations in the SPINK5 gene, which encode a key kallikrein protease inhibitor. There are two subtypes of the syndrome that differ in clinical presentation and immune profile: ichthyosiform erythroderma and ichthyosis linearis circumflexa. NS is a multisystemic disease with numerous extracutaneous manifestations. Current therapy for patients with NS is mainly supportive, as there is no curative or specific treatment, especially for children with NS, but targeted therapies are being developed. We describe an 8‐year‐old boy with genetically proven NS treated with intravenous immunoglobulin for recurrent skin and systemic infections from infancy, growth retardation, and associated erythroderma. Under this therapy, his skin status, infectious exacerbations, and quality of life all improved. Knowledge of the cytokine‐mediated pathogenesis of NS and the development of new biologic drugs open new possibilities for NS patients. However, the different therapeutic options have been applied in a limited number of cases, and variable responses have been shown. Randomized controlled trials with a sufficient number of patients stratified and treated according to their specific immune profile and clinical phenotype are needed to evaluate the safety and efficacy of treatment options for patients with NS.
Keywords: children, ichthyosiform erythroderma, ichthyosis linearis circumflexa, infections, Netherton syndrome, treatment
INTRODUCTION
Netherton syndrome (NS) is a rare autosomal recessive genodermatosis (OMIM #256500) characterized by superficial scaling, atopic manifestations, and multisystemic complications, a triad of congenital ichthyosiform erythroderma (IE) or ichthyosis linearis circumflexa (ILC), hair shaft abnormalities, and immune dysregulation. Its incidence is one in 200,000 newborns. It may represent up to 18% of infants with congenital erythroderma. 1 NS is caused by loss‐of‐function mutations in serine protease inhibitor Kazal‐type 5 (SPINK5) gene, which is located on chromosome 5q31‐32 and encodes lymphoepithelial Kazal‐type‐related protease inhibitor (LEKTI). 2 To date, a total of 108 variants have been found, encompassing all types of mutations. 3 LEKTI, expressed in the thymus and epithelia, is a direct inhibitor of kallikrein‐related peptidases and cathepsin G and affects epidermal desquamation and altered skin barrier function. Skin manifestations in NS include IE or ILC, a specific hair shaft defect called trichorrhexis invaginata (bamboo hair), and an atopic diathesis. Indeed, NS is a multisystemic disease with characteristic cutaneous and numerous extracutaneous manifestations. 4 Diagnosis of NS can be enhanced with histologic examination 5 ; immunostaining shows the absence or reduction of LEKTI expression in the granular layer of the epidermis and in the hair follicles. 6 Abnormalities of the hair shaft could be detected in only 20%–50% of affected hairs. 7 Laboratory findings show peripheral eosinophilia and elevated IgE levels. It is very difficult to distinguish atypical NS from other types of ichthyosis; detection of gene mutations in SPINK5 is crucial for diagnosis. Current therapy for patients with NS is mainly supportive, as there is no curative or specific treatment, especially for children with NS, but targeted therapies are being developed.
We describe a child with genetically proven NS, for whom intravenous immunoglobulin for 5 months has proven beneficial.
CASE HISTORY/EXAMINATION
Аn 8‐year‐old boy, born from a second normal pregnancy, with no history of collodion membrane at birth, was admitted to the neonatology ward at 5 days of age for generalized skin erythema with fine scaling. His cutaneous eruption was complicated by a secondary infection. A diagnosis of erythrodermia ichthyosiformis congenita was rendered (Figure 1). Subsequently, he was repeatedly hospitalized for cutaneous flares, which manifested with severe erythroderma with areas with fissures and erosions with serous exudate (Figure 2). Because of the severe pruritus, intravenous treatments with antibiotics and human albumin, topical corticosteroids, emollients, and oral antihistamines were administered. Despite this treatment, the pruritus and the severe erythrodermic state involving the trunk, limbs, the head, and ears were unaltered. The child's hair was sparse, lusterless, brittle, and fragile. Trichoscopy revealed trichorrhexis invaginata, plus golf tee and matchstick hairs (Figure 3). The nails were without eponychium. Over the years, he suffered from acute tubulointerstitial nephritis, chickenpox, bronchiolitis, and pneumonia. Genetic testing subsequently confirmed two mutations in the SPINK5 gene: the pathogenic variant NM _006846.3:c. 1530C > A p.(Cys510Ter) and the probable pathogenic variant c.420del p(Ser141ProfaTer5). Systemic treatment with acitretin, oral cholecaciferol, antihistamines and emollients was initiated. After 3 months of therapy with acitretin, only worsening of the dermatologic condition was observed, and this therapy was terminated.
FIGURE 1.
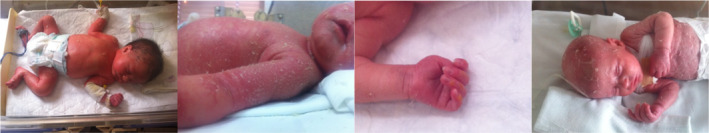
Photos taken during the stay in the neonatology ward showing diffuse redness and scaling all over the body.
FIGURE 2.
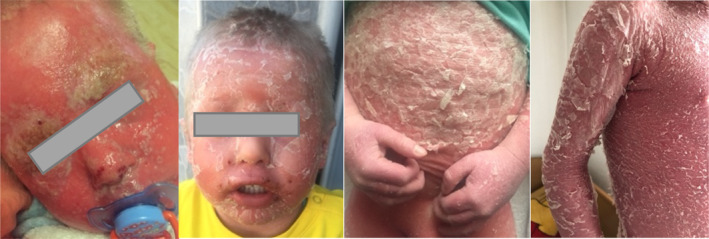
Photos taken over the years during exacerbations.
FIGURE 3.
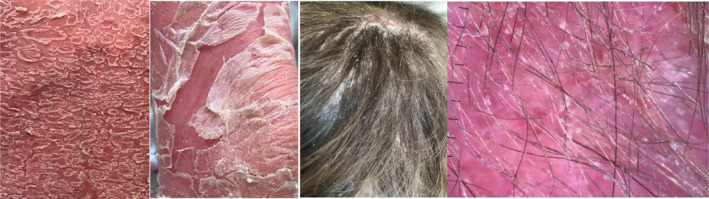
Trichoscopy and clinical photos taken at admission to the pediatric clinic.
At 8 years of age, the child was readmitted to the pediatric clinic with fever up to 39°C for 2 weeks, with a flare of his cutaneous eruption, evidence of leukocytosis and increased inflammatory activity, with inconclusive evidence of pneumonia from the radiographs. The boy weighed 21 kg (5 P, −1.68 z) with a height of 122 cm (11 P, −1.23 z), and a BMI of 11 P, −1. Axillary and inguinal lymph nodes were enlarged in the form of protruding bundles, with a soft‐elastic consistency, slightly painful on palpation. Respiration was attenuated vesicular, with fine crackles in the left base. The heart rate and rhythm were normal, with a holosystolic murmur 3/6 degrees at the left sternal border. There was a hepatomegaly with a sharp liver border at 3 cm below the costal margin, firm consistency, not painful on palpation. The spleen was not enlarged.
METHODS (DIFFERENTIAL DIAGNOSIS, INVESTIGATION AND TREATMENT)
Laboratory tests revealed a fivefold increase in liver transaminases, and a decrease in serum albumin, creatinine, and uric acid levels. Furthermore, discrete increase in D‐dimers and fibrinogen, and very low levels of 25‐OH vitamin D (15 nmol/L) were observed. Elevation of procalcitonin (three times above normal), C‐reactive protein and serum IgE level (>2500 IU/mL) were noted. The ALEX allergy test (295 allergens) showed elevated IgE levels for birch (Bet v 1, PR‐10), alder (Aln g 1, PR‐10), hazelnut (Cor a 1.0103, PR‐10), beech (Fag s 1, PR‐10), strawberry (Fra a 1 + 3, PR‐10 + LTP), dog (Can f 1, Lipocalin), and cat (Fel d 7, Lipocalin). The presence of PR‐10 allergens explains the IgE cross‐reactivity between hazel, alder, beech, and oak pollen. No allergic symptoms were observed in the child during the pollen season or allergic reactions to food or medication. The child has no atopic predisposition, that is, there is no family history of asthma, eczema, or hay fever. Citrobacter spp. was isolated from nasal and wound swabs examined as an outpatient. Staphylococcus aureus was isolated from throat swabs and hemoculture during hospitalization. Consultation with a pediatric gastroenterologist revealed that the increase in serum transaminases was most likely during the course of infection. Echocardiography showed mild subaortic stenosis, not advanced in its dynamics, with mild hypertrophy of the left ventricle and upper diastolic size of the same. Mild aortic regurgitation, mitral valve prolapse with mild mitral regurgitation; preserved ventricular function. The child was examined by an ophthalmologist as hypermetropia and a POS‐stromal lesion of the cornea of the right eye were diagnosed. The child has no cognitive impairment or developmental delay.
Treatment with an intravenous antibiotic for 7 days, an oral antihistamine, and emollients was given. It was continued at home with an oral antibiotic and antifungal, a probiotic, and oral vitamin D administration. Because of the subjective discomfort the child experienced with topical application of methylprednisolone aceponate milk, therapy with a cooling cream containing clobetasol propionate cream once daily and an emollient once daily was recommended. Administration of IVIG was suggested as part of the systemic therapy.
In a period without underlying infection, the immunologic status was examined, which showed a total T lymphocyte count in the reference range with an increase in the cytotoxic suppressor subpopulation. While B lymphocytes and NK cells were within reference ranges, there was increased percentage of CD57+ CD8+ cells. Serum immunoglobulins were normal with the exception of greatly increased IgE levels. The accumulation of CD8+ CD57+ T cells was attributed to repeated antigen stimulation during chronic infection, these cells being the most effective ones to fight chronic viral infections due to their high expression of interferon‐γ, granzyme B, and perforin, as well as their marked cytolytic activity. 8
RESULTS (OUTCOME AND FOLLOW‐UP)
Due to recurrent skin and systemic infections in infancy and associated erythroderma, the child was started on intravenous immunoglobulin, as subcutaneous administration was judged not possible in this child, to reduce infectious exacerbations, improve local skin status, and improve quality of life. Our patient, like most NS patients, has normal serum IgG levels, suggesting that the improvement with replacement therapy in these patients is more likely due to the immunomodulatory effects of IVIG. Recent data show that the number of pediatric patients with NS treated with immunoglobulins was 14, of whom 12 showed a positive clinical effect, with the dose administered ranging from 400 to 500 mg/kg/month and the duration of treatment from 2 months to 5 years. 9 A recent meta‐analysis comparing different IVIG infusion regimens in patients with Kawasaki disease found that high‐dose IVIG regimens are likely to be more effective and may even reduce the need for additional treatment compared to medium‐ or low‐dose IVIG regimens, with little or no difference in the number of adverse events between groups. 10 We decided to test whether the higher IVIG dose would not be more effective in patients with NS. We started with 250 mg/kg/day for 3 days or 750 mg/kg/month administration and have not observed any adverse effects so far.
During the follow‐up period, normalization of liver transaminases was noted, and another interesting phenomenon was the sustained decrease in total IgE (>2500–>484. 5–>256–>183. 3–>160. 2–>129.2 IU/mL). It may represent a marker of favorable therapeutic response. No such positive trend was observed with vitamin D supplementation, and the child maintained reduced levels (51 nmol/L) despite oral supplementation. In the last 5 months since receiving replacement therapy, the child gained two kilograms and grew three centimeters (BMI 18 P, −0.91 z). Furthermore, decrease in the incidence of infectious exacerbations was noted in addition to a significant reduction of the Netherton Area and Severity Assessment (NASA) score (66–36 points) (Figure 4).
FIGURE 4.
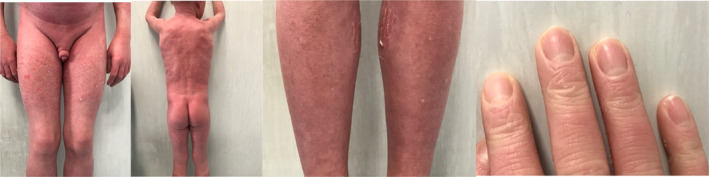
Photos from the hospital stay for the fourth infusion.
DISCUSSION
Netherton syndrome is a rare and severe autosomal recessive genodermatosis in the group of congenital ichthyosis. Clinically, it is first evident as the triad of IE, trichorrhexis invaginata (bamboo hair), and atopic diathesis. 4 IE is present at birth or shortly thereafter as generalized erythroderma with desquamation. In some individuals, the erythrodermic endotype may persist throughout life, but it more often is periodically or completely replaced by another endotype called ILC, manifested by polycyclic and serpiginous migratory erythematous plaques with double‐edged scales at the periphery.
Due to the disrupted epithelial barrier, the neonatal period is crucial and life‐threatening complications such as electrolyte imbalances, including dehydration, hypothermia, gastrointestinal symptoms, skin/respiratory/systemic infections, and sepsis may occur due to increased transepidermal water loss and superficial scaling of the skin. 4 Patients are highly susceptible to cutaneous and systemic infections. Immunophenotyping of patients from NS revealed no evidence of immunodeficiency. Variable T‐ and B‐cell imbalances are found in a limited number of patients, but these changes are likely secondary to chronic skin infections and inflammation. 11 However, the most recent International Union of Immunological Societies (IUIS) classification of inborn errors of immunity places NS in the group of hyper‐IgE syndromes, which are combined immunodeficiencies with associated or syndromic features. 12 Due to increased catabolic rate, malabsorption, chronic inflammation, and recurrent infections, these children suffer from failure to thrive, growth retardation, and short stature. Extracutaneous manifestations such as acute pancreatitis, hypothyroidism, thymic atrophy, and acute bilateral renal vein thrombosis have been rarely reported. 4 Cognitive impairment and developmental delays have been identified in some patients. Adult patients may develop cutaneous squamous cell carcinoma. 13
The diagnosis of NS can be enhanced with histologic examination of involved skin showing detachment of the stratum corneum, hyperkeratosis, acanthosis, focal hypergranulosis, and superficial perivascular lymphocytic infiltration. 5 Laboratory findings show peripheral eosinophilia and elevated IgE levels. It is very difficult to distinguish atypical NS from other types of ichthyosis; detection of gene mutations in SPINK5 is essential for diagnosis.
There is currently no ideal treatment, particularly in childhood. 14 The first successful therapy of a genetic disorder, specifically ILC (NS), was achieved by the legendary Edmund Klein and associates, including a coauthor of this current paper (RAS), 15 in the late 1970s utilizing oral low dose cyclophosphamide, possibly related to selective effects of this agent on lymphocyte subpopulations and portending many other options. However, this syndrome tends to improve as patients age, but the clinical course may be disturbed by intermittent exacerbations. The predominant therapy for patients with NS is mostly supportive, as there is neither curative nor specific therapy specifically for children with NS, but targeted therapies are currently in development. The challenge is to find therapies that both repair the disrupted skin barrier and stop chronic inflammation. Current treatment options include skin cleansing, bleach baths, emollients/moisturizers, calcineurin inhibitors, and oral antihistamines. Antibiotics, topical corticosteroids, and intravenous immunoglobulins (IVIG) can be used during exacerbations. The likely mechanism of action of intravenous immunoglobulin is the ability of IgG to opsonize bacteria, stimulate phagocytosis, and reduce the incidence of infection and inflammation. Immunoglobulin replacement therapy is preferred in severe cases of NS with frequent infections and failure to thrive and can be used at any age. Many reports describe a remarkable clinical response with no significant adverse effects. 14 Primarily, a reduction in recurrent infections and improvement in skin condition have been reported, as the therapy is well tolerated by patients (mainly neonates and children with NS). The disadvantage of the therapy is the cost and the fact that it has to be performed in a hospital. 4 Systemic therapy in NS patients with chemically synthesized vitamin A derivatives has shown a variable degree of efficacy and tolerability. Partial improvement or exacerbation has been reported for acitretin or isotretinoin. The side effects with bone toxicity and teratogenicity and questionable therapeutic response outweigh the benefits of taking these drugs. 4 In our patient, treatment with acitretin was also discontinued due to worsening of the condition. Another modern approach to consider is the use of topical tacrolimus. 16 There is a wide range of potential target therapies such as kallikrein inhibitors, monoclonal antibodies blocking IL‐36, IL‐17, IL‐12/IL‐23, IL‐4/IL‐13 (targeting allergic manifestations), IL‐9, JAK/STAT inhibitors, and complement inactivation. None of these treatment options represents a complete cure from NS. Of the therapies used to date, anti‐IL‐17A therapy may be the most effective. Currently, some gene therapy approaches are being developed.
The best therapeutic strategy for the moment may be a combination of anti‐inflammatory therapy and therapy that restores the disrupted skin barrier in NS patients. Unfortunately, all therapeutic options have been applied only in a limited number of cases, and variable responses have been shown. 15 Limited data published to date suggest that biologics therapy is safe and often effective in adults and children with NS, but long‐term efficacy and tolerability remain unknown. 14 Randomized controlled trials with sufficient numbers of patients stratified and treated according to their specific immune profile and clinical phenotype are needed to assess the safety and efficacy of treatment options for patients with NS. 16 Studies based on stratification of patients using omics‐based techniques such as genomics, transcriptomics, proteomics, metabolomics, and bioinformatics would thus be a step toward precision medicine in NS.
CONCLUSION
Thus, NS is multisystemic disease with considerable clinical variability. Based on the reported cases and the authors' recommendations, we recommend consideration of IVIG therapy, based on this child's favorable response noting reduced recurrent severe infections and enhanced growth. If his condition worsens, we would consider therapy blocking IL‐17A, which may be the best therapeutic option for improving persistent inflammatory skin diseases and/or preventing acute relapses. Insights into the cytokine‐mediated pathogenesis of NS and the increasing use of biologic therapies are opening new possibilities for NS patients. However, the various therapeutic options have been used in a limited number of cases and have shown variable responses. There is a great need for randomized controlled trials with enough patients stratified and treated according to their specific immune profile and clinical phenotype to evaluate the safety and efficacy of treatment alternatives for patients with NS. 17
AUTHOR CONTRIBUTIONS
Polina Kostova: Conceptualization; data curation; formal analysis; methodology; supervision; visualization; writing – original draft; writing – review and editing. Guergana Petrova: Writing – original draft; writing – review and editing. Martin Shahid: Writing – original draft; writing – review and editing. Vera Papochieva: Writing – original draft; writing – review and editing. Dimitrinka Miteva: Writing – original draft; writing – review and editing. Ivelina Yordanova: Writing – original draft; writing – review and editing. Kossara Drenovska: Data curation; investigation; project administration; supervision; validation; writing – original draft; writing – review and editing. Irena Bradinova: Writing – original draft; writing – review and editing. Camila K. Janniger: Writing – original draft; writing – review and editing. Robert A. Schwartz: Supervision; writing – original draft; writing – review and editing. Snejina Vassileva: Conceptualization; data curation; formal analysis; investigation; methodology; project administration; resources; supervision; writing – original draft; writing – review and editing.
FUNDING INFORMATION
The authors did not receive any financial support for this case report.
CONFLICT OF INTEREST STATEMENT
The authors declare that the article was written without financial or commercial motives that could represent a potential conflict of interest.
CONSENT
Written informed consent was obtained from the patient to publish this report in accordance with the journal's patient consent policy.
ACKNOWLEDGMENTS
The authors thank the patient and his relatives for permission to publish this case report. Adherence to treatment would not have been possible without the support of the child's family. It is to the family's credit that the child can enjoy lasting well‐being. Respect for their endless love and care every day.
Kostova P, Petrova G, Shahid M, et al. Netherton syndrome—A therapeutic challenge in childhood. Clin Case Rep. 2024;12:e8770. doi: 10.1002/ccr3.8770
DATA AVAILABILITY STATEMENT
The medical records for this article are not publicly available for reasons of patient anonymity.
REFERENCES
- 1. Pruszkowski A, Bodemer C, Fraitag S, Teillac‐Hamel D, Amoric JC, de Prost Y. Neonatal and infantile erythrodermas: a retrospective study of 51 patients. Arch Dermatol. 2000;136(7):875‐880. [DOI] [PubMed] [Google Scholar]
- 2. Chavanas S, Bodemer C, Rochat A, et al. Mutations in SPINK5, encoding a serine protease inhibitor, cause Netherton syndrome. Nat Genet. 2000;25(2):141‐142. [DOI] [PubMed] [Google Scholar]
- 3. The Human Gene Mutation Database (HGMD). Accessed March 20, 2023. https://www.hgmd.cf.ac.uk/ac/gene.php?gene=SPINK5
- 4. Petrova E, Hovnanian A. Advances in understanding of Netherton syndrome and therapeutic implications. Expert Opin Orphan Drugs. 2020;8(11):455‐487. [Google Scholar]
- 5. Hovnanian A. Netherton syndrome: skin inflammation and allergy by loss of protease inhibition. Cell Tissue Res. 2013;351(2):289‐300. [DOI] [PubMed] [Google Scholar]
- 6. Bitoun E, Micheloni A, Lamant L, et al. LEKTI proteolytic processing in human primary keratinocytes, tissue distribution and defective expression in Netherton syndrome. Hum Mol Genet. 2003;12(19):2417‐2430. [DOI] [PubMed] [Google Scholar]
- 7. Srinivas SM, Hiremagalore R, Suryanarayan S, Budamakuntala L. Netherton syndrome with pili torti. Int J Trichology. 2013;5(4):225‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Huang B, Liu R, Wang P, et al. CD8+CD57+ T cells exhibit distinct features in human non‐small cell lung cancer. J Immunother Cancer. 2020;8(1):e000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nouwen AEM, Schappin R, Nguyen NT, et al. Outcomes of systemic treatment in children and adults with Netherton syndrome: a systematic review. Front Immunol. 2022;13:864449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Broderick C, Kobayashi S, Suto M, Ito S, Kobayashi T. Intravenous immunoglobulin for the treatment of Kawasaki disease. Cochrane Database Syst Rev. 2023;1(1):CD014884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Eränkö E, Ilander M, Tuomiranta M, et al. Immune cell phenotype and functional defects in Netherton syndrome. Orphanet J Rare Dis. 2018;13(1):213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Tangye SG, Al‐Herz W, Bousfiha A, et al. Human inborn errors of immunity: 2022 update on the classification from the International Union of Immunological Societies Expert Committee. J Clin Immunol. 2022;42(7):1473‐1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. van der Voort EAM, Prens EP. Netherton syndrome with multiple non‐melanoma skin cancers. Acta Derm Venereol. 2013;93(6):727‐728. [DOI] [PubMed] [Google Scholar]
- 14. Pontone M, Giovannini M, Filippeschi C, et al. Biological treatments for pediatric Netherton syndrome. Front Pediatr. 2022;10:1074243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Klein E, Hahn GM, Solomon JA, et al. Explorations of antimitotic agents in the treatment of a congenital disease, ichthyosis linearis circumflexa. J Surg Oncol. 1979;11(12):85‐88. [DOI] [PubMed] [Google Scholar]
- 16. Suga Y, Tsuboi R, Hashimoto Y, Yoshiike T, Ogawa H. A case of ichthyosis linearis circumflexa successfully treated with topical tacrolimus. J Am Acad Dermatol. 2000;42(3):520‐522. [DOI] [PubMed] [Google Scholar]
- 17. Clinical trials register—search for Netherton syndrome. Accessed March 20, 2023. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=Netherton+Syndrome
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The medical records for this article are not publicly available for reasons of patient anonymity.