

Abstract
Canavan disease is a leukodystrophy caused by ASPA mutations that diminish oligodendroglial aspartoacylase activity, and is characterized by markedly elevated brain concentrations of the aspartoacylase substrate N‐acetyl‐l‐aspartate (NAA) and by astroglial and intramyelinic vacuolation. Astroglia express NaDC3 (encoded by SLC13A3), a sodium‐coupled transporter for NAA and other dicarboxylates. Astroglial conditional Slc13a3 deletion in aspartoacylase‐deficient Canavan disease model mice (“CD mice”) reversed brain NAA elevation and improved motor function. These results demonstrate that astroglial NaDC3 contributes to brain NAA elevation in CD mice, and suggest that suppressing astroglial NaDC3 activity would ameliorate human Canavan disease.
Introduction
Canavan disease is a recessively inherited early onset leukodystrophy caused by inactivating ASPA mutations that diminish oligodendroglial aspartoacylase activity. 1 The concentration of the aspartoacylase substrate N‐acetyl‐l‐aspartate in brain ([NAAB]) is elevated in these patients, who fail to attain or maintain motor and intellectual milestones, are often neonatally megalencephalic, and frequently develop seizures. 1 , 2 Neuroimaging reveals brain white matter signal abnormalities consistent with cytotoxic edema, and autopsy demonstrates astroglial and intramyelinic vacuolation. 3 , 4 Mice homozygous for the Aspa nonsense mutation nur7 (“CD mice”) lack detectable aspartoacylase, demonstrate a typically twofold or greater elevation in [NAAB], and develop motor deficits by postnatal day 21. 5
NaDC3, encoded by SLC13A3 (Slc13a3 in mice), is a plasma membrane Na+‐coupled dicarboxylate transporter that can mediate intracellular accumulation of NAA and Na+ from the medium by cultured rat astroglia. 6 , 7 Slc13a3 is also expressed by meningeal cells and renal proximal tubular epithelium. 6 , 8 To explore the specific pathophysiological significance of astroglial NaDC3 in Canavan disease, we examined the effects of astroglial Slc13a3 conditional knockout on [NAAB] and motor function in young adult CD mice.
Methods
Slc13a3 flox mice were derived from Slc13a3 knockout first mice (MMRRC.049665‐UCD) by crossing with Flp1 deleter mice (Jackson Laboratory #009086). The Slc13a3 flox mice were then crossed with Aspa nur7 mice (Jackson Laboratory #008607) and with GFAPcreER T2 mice (Jackson Laboratory #012849) to yield the “CDcKO” mice used in this study, which were homozygous for Aspa nur7 and Slc13a3 flox and carried one copy of the GFAPcreER T2 transgene (Fig. 1A). Control (“CDctrl”) mice were bred to also be homozygous for Aspa nur7 , but to lack either GFAPcreER T2 or Slc13a3 flox alleles. Mouse genotypes were validated by qPCR. All mice were maintained on a C57BL6 background in an AAALAC‐certified vivarium. Both male and female mice were employed. All animal procedures were conducted in accord with a UC Davis IACUC‐approved protocol.
Figure 1.
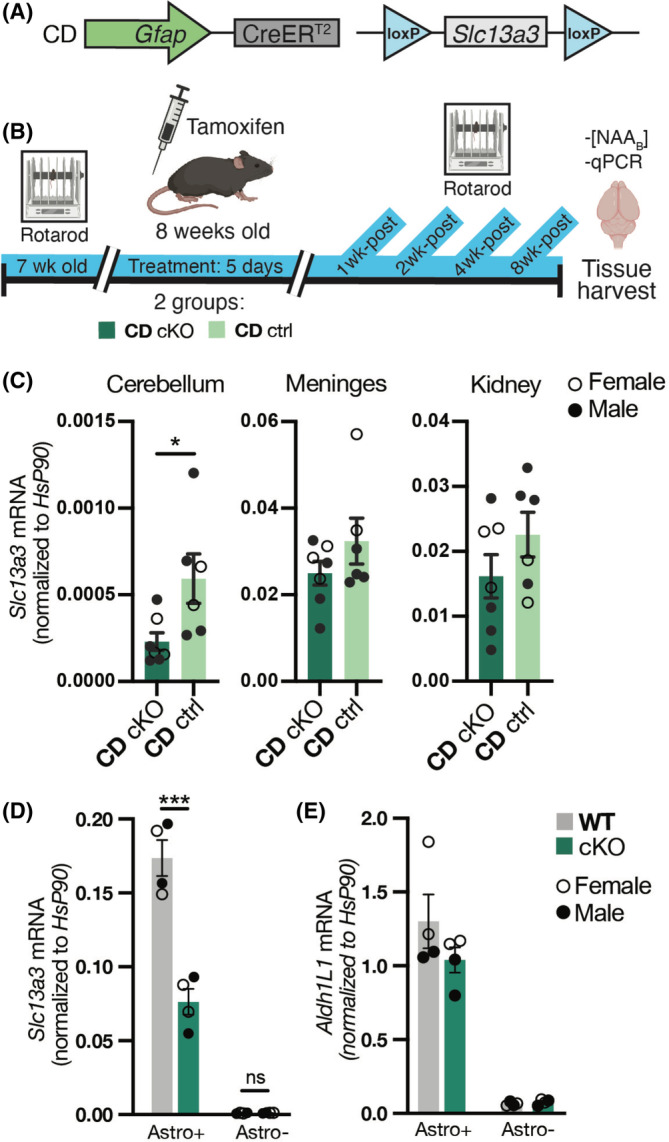
Panel A: Slc13a3 conditional deletion construct. Panel B: Experimental design. Panel C: qRT/PCR Slc13a3 mRNA results in cerebellum, meninges, and kidney of CDcKO (dark green) and CDctrl (light green) mice, normalized to Hsp90 mRNA. Panel D: Astrocytes were isolated using MACS with Anti‐ACSA‐2 MicroBeads (ACSA‐2: astrocyte cell surface antigen‐2) according to the manufacturer's instruction (Miltenyi Biotec) The flow‐through, devoid of ACSA‐2‐positive astrocytes, was collected as the astrocyte‐depleted fraction. Panel E: Quantitative PCR was used to quantify the astrocyte marker Aldh1L1 mRNA 9 (forward primer 5′‐CAGGAGGTTTACTGCCAGCTA‐3′ and reverse primer 5′‐CACGTTGAGTTCTGCACCCA‐3′) and Slc13a3 mRNA (see Methods) in the astrocyte‐enriched and astrocyte‐depleted fractions. Each circle represents results in an individual mouse (solid circles male and open circles female). Column heights represent means, and vertical brackets denote SEMs. *p = 0.0265, ***p = 0.0003, Student's t‐test.
An accelerating rotarod apparatus was used to assess motor function in non‐tamoxifen‐treated CD mice, tamoxifen‐treated CD/GFAPcreER T2 /Slc13a3 flox/flox mice (“CDcKO” mice), and tamoxifen‐treated CDctrl mice just before and at intervals after a 5 day course of tamoxifen (1.5 mg/50 μL sunflower oil/day) (Fig. 1B). 10 Eight weeks post‐tamoxifen, the mice were euthanized; meninges were carefully dissected away from underlying brain; and cerebella, forebrain, meninges, and kidneys were separately flash‐frozen and stored at −80°C. Slc13a3 recombination in cerebellar parenchyma, meninges, and kidney was assessed by qPCR, using the Slc13a3 cDNA primers (forward primer 5′‐TCTCAGTGTAAGAAGCGC‐3′ and reverse primer 5′‐CACTGCCTGGGTTCAAAGTC‐3′). Results were normalized to Hsp90 mRNA (forward primer 5′‐AAACAAGGAGATTTTCCTCCGC‐3′ and reverse primer 5′‐CCGTCAGGCTCTCATATCGAAT‐3′). Forebrain samples from euthanized 16 week old CDcKO and CDctrl mice were assayed for NAA by HPLC 8 or enzymatically dissociated prior to magnetic activated cell sorting (MACS) 11 to prepare astrocyte‐enriched and astrocyte‐depleted cell fractions for qPCR quantifications of Slc13a3 mRNA and of Aldh1L1 mRNA. Cryostat forebrain and cerebellar sections from 16‐week‐old wild‐type, CDcKO and CDctrl mice were employed to quantify cerebellar vacuolation.
Results
Tamoxifen activation of GFAPcreER T2 elicited a 61% reduction in Slc13a3 mRNA abundance in cerebellar parenchyma of CDcKO mice, but did not significantly alter cerebellar parenchymal Slc13a3 mRNA abundance in CDctrl mice, nor in the meninges and kidneys of either CDcKO or CDctrl mice (Fig. 1C). Magnetic activated cell sorting was used to prepare astroglia‐enriched (“astro+”) and astrocyte‐depleted (“astro−”) cell fractions from forebrains of 16‐week‐old wild‐type (WT) mice. Abundances of both Slc13a3 mRNA and mRNA encoding the astrocyte marker Aldh1L1 9 were markedly greater in the astro+ fraction than in the astro− fraction, and Slc13a3 mRNA abundance was 56% lower in the astroglial fraction from tamoxifen‐treated than from non‐tamoxifen‐treated GFAPcre ERT2/Slc13a3 flox/flox mice (Fig. 1D).
At age 16 weeks, [NAAB] in the tamoxifen‐treated CDcKO mice was 55% below that in the tamoxifen‐treated CDctrl mice (Fig. 2A), but the extent of cerebellar vacuolation in the CDcKO mice did not differ significantly from that in the CDctrl mice (Fig. 2B). Accelerating rotarod retention times in CDcKO mice had significantly increased by 1 week after completion of tamoxifen administration, and remained substantially elevated through 7 additional weeks of observation, whereas accelerating rotarod retention times in CD mice not treated with tamoxifen and in CDctrl mice treated with tamoxifen remained consistent throughout the entire period of testing (Fig. 2C).
Figure 2.
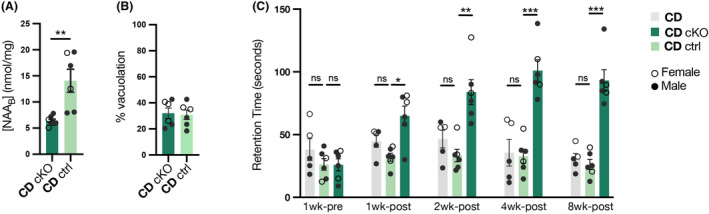
Panel A: [NAAB] of CDcKO (dark green) and CDctrl (light green) mice. **p = 0.0031, Student's t‐test. Panel B: Cerebellar vacuolation in CDcKO and CDctrl mice, showing no significant difference between the two groups. Panel C: Accelerating rotarod retention times of CDcKO and CDctrl mice. Column heights represent means, and vertical brackets denote SEMs. *p < 0.05; **p = 0.0043; ***p < 0.0001, comparisons by two‐way repeated measures ANOVA.
Discussion
Prior studies have demonstrated that lowering [NAAB] by inhibiting neuronal NAA synthesis can prevent or reverse brain vacuolation and neuronal dendritic shortening in CD mice. 12 , 13 , 14 More recently, whole body NaDC3 ablation achieved by constitutive Slc13a3 deletion was also found to prevent [NAAB] elevation and leukodystrophy in CD mice. 8 But that study did not illuminate the specific role of astroglial NaDC3, versus NaDC3 encoded by meningeal and renal Slc13a3, 8 in mediating [NAAB] elevation and eliciting clinical neurological deficits in this murine Canavan disease model. We addressed this issue by testing the effects of astroglial conditional Slc13a3 deletion in young adult CD mice on [NAAB], brain vacuolation, and motor function.
Gene recombination elicited by GFAPcreER T2 in the adult murine CNS is restricted largely to astroglia. 15 In the present study, the abundance of Slc13a3 mRNA was substantially lower in whole cerebellar parenchyma of 16‐week‐old CDcKO than CDctrl mice, but was not lowered in CDcKO meninges or kidneys. MACS results indicated that Slc13a3 mRNA expression in brain parenchyma was confined largely to astroglia, and was diminished in astroglia by more than 50% by CDcKO. We concluded, therefore, that mouse brain Slc13a3 knockdown elicited in tamoxifen‐treated CD/GFAPcreER T2 /Slc13a3 flox/flox mice was largely or wholly confined to astroglia.
The substantially lower [NAAB] in the 16 week old CDcKO than CDctrl mice supports the concept that astroglial NaDC3 activity contributes to brain NAA elevation in Canavan disease. Though astroglial conditional Slc13a3 knockout initiated at age 8 weeks did not yield a reduction in the extent of cerebellar vacuolation at age 16 weeks, accelerating rotarod analysis demonstrated a rapid and long‐lasting improvement in motor function in the CDcKO mice. A similarly rapid initial improvement in motor function was previously observed in young adult CD mice when [NAAB] was lowered by inhibiting brain NAA synthesis via intrathecal administration of an Nat8l antisense oligonucleotide. 14 Together, these observations strengthen support for the pathophysiological significance of elevated [NAAB] in Canavan disease.
Are SLC13A3 and NaDC3 suitable targets for treating Canavan disease? Constitutive Slc13a3 deletion in wild‐type mice has no obvious phenotypic effects other than increasing urinary NAA output, 8 but, in humans, mutations that inactivate both alleles of SLC13A3 increase susceptibility to viral infection‐triggered acute reversible leukoencephalopathy. 16 Thus, measures designed to suppress global, or astrocyte‐specific, SLC13A3 expression and NaDC3 activity in infants and children with Canavan disease may prove useful in limiting [NAAB] elevation and preventing or ameliorating their neurological deficits, but might also increase their risk for developing reversible leukoencephalopathy.
Author Contributions
VLH and DP wrote the first draft. All authors provided critical review and feedback and approved the final draft. VLH, YW, JM, and FG provided statistical analysis, VLH, YW, and DP designed the work. VLH, YW, JM, MZ, TB, NR, AD and DP performed and interpreted the studies.
Conflict of Interest
The authors declare no conflict of interest.
Acknowledgments
This study was supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through grant number UL1 TR001860 and linked award TL1 TR001861 (VLH); National Institutes of Health grants R21NS117386 (DP) and R21NS133881 (DP); and by funds from Shriners Hospitals for Children.
Funding Statement
This work was funded by National Institutes of Health grants R21NS117386 and R21NS133881; the National Center for Advancing Translational Sciences, National Institutes of Health grants TL1 TR001861 and UL1 TR001860; Shriners Hospitals for Children .
References
- 1. Wei H, Moffett J, Amanat M, et al. The pathogenesis of, and pharmacological treatment for, Canavan disease. Drug Discov Today. 2022;27:2467‐2483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bley A, Denecke J, Kohlschutter A, et al. The natural history of Canavan disease: 23 new cases and comparison with patients in the literature. Orphanet J Rare Dis. 2021;16:227. doi: 10.1186/s13023-020-01659-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Merrill S, Nelson G, Longo N, Nonkowsky J. Cytotoxic edema and diffusion restriction as an early pathoradiologic marker in Canavan disease; case report and review of the literature. Orphanet J Rare Dis. 2016;11:169. doi: 10.1186/s3023-016-0549-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Adachi M, Schneck L, Cara J, Volk B. Spongy degeneration of the central nervous system (Van Bogaert and Bertrand type; Canavan's disease). A review. Hum Pathol. 1973;4:331‐347. [DOI] [PubMed] [Google Scholar]
- 5. Traka M, Wollmann R, Cerda S, Dugas J, Barres BA, Popko B. Nur7 is a nonsense mutation in the mouse aspartoacylase gene that causes spongy degeneration of the CNS. J Neurosci. 2008;28:11537‐11549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Huang W, Wang H, Kekuda R, et al. Transport of N‐acetylaspartate by the Na+‐dependent high‐affinity dicarboxylate transporter NaDC3 and its relevance to the expression of the transporter in the brain. J Pharmacol Exp Ther. 2000;295:392‐403. [PubMed] [Google Scholar]
- 7. Fujita T, Tatsukawa H, Yodoya E, et al. Transport characteristics of N‐acetyl‐l‐aspartate in rat astrocytes: involvement of sodium‐coupled high‐affinity carboxylate transporter NaC3/NaDC3‐mediated transport system. J Neurochem. 2005;93:706‐714. [DOI] [PubMed] [Google Scholar]
- 8. Wang Y, Hull V, Sternbach B, et al. Ablating the transporter sodium‐dependent dicarboxylate transporter 3 prevents leukodystrophy in Canavan disease mice. Ann Neurol. 2021;90:845‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cahoy J, Emery B, Kaushal A, et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J Neurosci. 2008;28:264‐278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brooks S, Dunnett S. Tests to assess motor phenotype in mice: a user's guide. Nature Rev Neurosci. 2009;10:519‐529. [DOI] [PubMed] [Google Scholar]
- 11. Jan J, Won J. Methodological comparison of FACS and MACS isolation of enriched microglia and astrocytes from mouse brain. J Immunol Methods. 2020;486:112834. [DOI] [PubMed] [Google Scholar]
- 12. Guo F, Bannerman P, Mills Ho E, et al. Ablating N‐acetylaspartate prevents leukodystrophy in a Canavan disease model. Ann Neurol. 2015;77:884‐888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Maier H, Wang‐Eckhardt L, Hartmann D, Gieselmann V, Eckhardt M. N‐acetylaspartate synthase deficiency corrects the myelin phenotype in a Canavan disease mouse model but does not affect survival time. J Neurosci. 2015;35:14501‐14516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hull V, Wang Y, Burns T, et al. Antisense oligonuceotide reverses leukodystrophy in Canavan disease mice. Ann Neurol. 2020;87:480‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guo F, Maeda Y, Ma J, et al. Macroglial plasticity and the origins of reactive astroglia in experimental autoimmune encephalomyelitis. J Neurosci. 2011;31:11914‐11928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dewulf J, Wiame E, Dorboz I, et al. SLC13A3 variants cause reversible leukoencephalopathy and a‐ketoglutarate accumulation. Ann Neurol. 2019;85:385‐395. [DOI] [PubMed] [Google Scholar]