

Abstract
ATP1A1 encodes a sodium‐potassium ATPase that has been linked to several neurological diseases. Using exome and genome sequencing, we identified the heterozygous ATP1A1 variant NM_000701.8: c.2707G>A;p.(Gly903Arg) in two unrelated children presenting with delayed motor and speech development and autism. While absent in controls, the variant occurred de novo in one proband and co‐segregated in two affected half‐siblings, with mosaicism in the healthy mother. Using a specific ouabain resistance assay in mutant transfected HEK cells, we found significantly reduced cell viability. Demonstrating loss of ATPase function, we conclude that this novel variant is pathogenic, expanding the phenotype spectrum of ATP1A1.
Introduction
Sodium‐potassium pumps play a vital role in maintaining the electrochemical gradients across cell membranes through ATP‐dependent processes. ATP1A1 encodes the α1 subunit of Na+/K+‐ATPase, which is highly expressed in the nervous system. Heterozygous missense mutations can lead to reduced ATPase function, which has previously been associated with an axonal to intermediate neuropathy phenotype 1 , with hereditary spastic paraplegia, 2 and a complex severe intellectual disability syndrome, partially accompanied by hypomagnesemia, 3 , 4 all being inherited in an autosomal dominant manner.
Patients and Methods
Patients were examined by experienced neurologists. We obtained written informed consent from both patients' legal representatives prior to study enrollment. Exome and genome sequencing were conducted following site‐specific protocols. Sanger sequencing was used for variant confirmation and co‐segregation studies. To assess reduced ATPase function, we transfected HEK cells with a mutant ATP1A1 plasmid that otherwise confers resistance to ouabain in vitro. We measured cell viability after 72 h of treatment with the unselective ATPase inhibitor ouabain. 4
Results
We identified the variant c.2707G>A; p.(Gly903Arg) in ATP1A1 (ENST00000295598.10/NM_000701.8) (Fig. 1A) in a heterozygous state in two unrelated probands. The two individuals were male, one 10, and the other 7 years old at the time of examination. Patient 1 was born in the United States of America with mixed German/Irish and Colombian descent; patient 2 originated from France. Both patients presented with intellectual disability as the main symptom, accompanied by delayed motor and speech development. Patient 1 was diagnosed with autism; patient 2 had mild dysmorphic facial features. Variants were validated by Sanger sequencing, and co‐segregation analyses revealed de novo status in patient 2. Patient 1's unaffected mother was found to be mosaic for the variant (Fig. 1B), and his 23‐year‐old maternal half‐sister, who also has a history of developmental delay and intellectual disability, was likewise found to be heterozygous for the same ATP1A1 variant.
Figure 1.
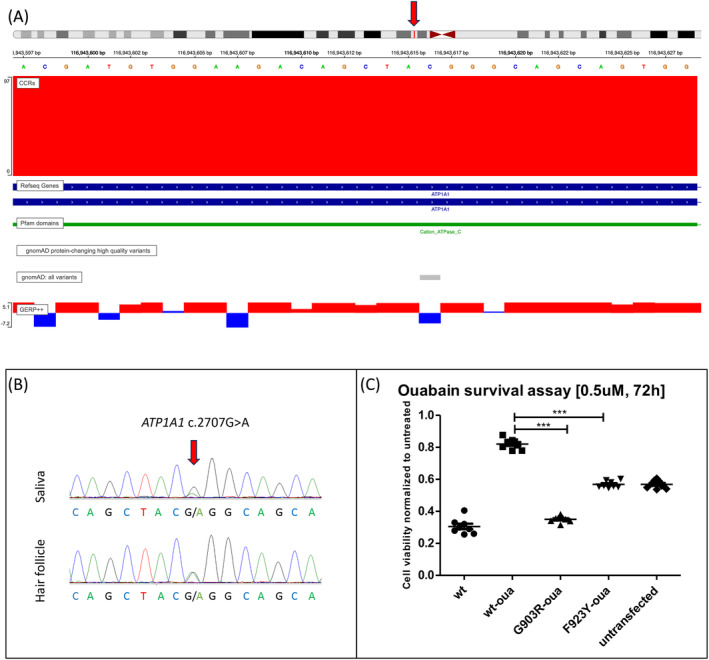
Genetic and experimental evidence for p.Gly903Arg pathogenicity. (A) Chromosomal position of the ATP1A1 variant c.2707G>A;(p.Gly903Arg) with its high regional constraint (CRR), high conservation (GERP), and absence from control databases (gnomAD). The red arrow indicates the variant's nucleotide position on chromosome 1. (B) Sanger sequencing shows c.2707G>A mosaicism in saliva and hair follicle in patient 1's unaffected mother. (C) HEK cells were transfected with ouabain‐insensitive ATP1A1 plasmids for mutant and wildtype (oua‐wt), as well as wt‐ATP1A1 (not ouabain‐insensitive) and treated with ouabain (0.5 μmol/L) for 72 h. Cell viability was measured in replicates of eight, using the luminescence‐based CellTiterGlo assay. Ratios are shown in comparison with untreated cells (mean) transfected with the same plasmid. Statistics: one‐way ANOVA, α‐error correction: Tukey's post‐test. ***P < 0.0001
The variant has not been published in association with any disease phenotype, nor has it been listed in control databases (gnomAD v2). Located in exon 19 (of 23), it affects the longest extracellular loop (52 residues) between transmembrane regions 7 and 8, which both contain other pathogenic missense variants (c.2590G>C; p.(Gly864Arg) and c.2768T>A; p.(Phe923Tyr)) that have previously been linked to intellectual disability. 4 , 5 In the same extracellular domain, another reported pathogenic variant c.2629G>A; p.(Gly877Ser) has been associated with intermediate sensorimotor neuropathy. 6 The novel p.(Gly903Arg) variant affects a highly conserved amino acid position and a highly constrained coding region (CCR > 95th percentile). 7 In silico predictions are pathogenic (CADD score: 32), and the transformer‐based neuronal network MAVERICK 8 assigned a pathogenicity probability of 91.9% for autosomal dominant inheritance.
To functionally assess the variant's pathogenicity, we transfected HEK cells with ouabain‐insensitive ATP1A1 plasmids encoding either the wildtype (wt) or the mutant, in replicates of eight. As a negative control, we used ouabain‐insensitive wt‐ATP1A1 plasmids, and as a positive control, we chose ouabain‐insensitive plasmids containing the previously reported p.Phe923Tyr variant that is located in close vicinity, as well as the ouabain‐sensitive wt, and untransfected cells. 24 h after transfection, we treated the cells with the ATPase inhibitor ouabain at a concentration of 0.5 μmol/L. We measured cell survival after 72 h, using the previously established luciferase (CellTiter‐Glo) assay. To demonstrate that cell death was not an effect of transfection, we normalized each treated mutant to untreated cells transfected with the same plasmid. Compared to the ouabain‐insensitive wt, cell viability was significantly reduced for the p.(Gly903Arg) variant (Fig. 1C), and cell survival ratios were even lower than compared to the previously reported variant p.(Phe923Tyr). This result indicates reduced ATPase function, rendering otherwise ouabain‐insensitive cells more vulnerable for cell death.
Discussion
We herein describe a novel, pathogenic ATP1A1 variant p.(Gly903Arg) causing developmental delay and intellectual disability in two unrelated families. Based on its absence from control populations and its affecting of a highly conserved position and constrained protein region, it is very likely to be pathogenic. Our established bioassay revealed a significant loss of ATPase function in cells transfected with the mutant, consistent with other pathogenic mutations reported in the literature, supporting haploinsufficiency as the pathomechanism for this variant. Considering that other missense mutations in the same gene cause late‐onset hereditary neuropathy as an allelic disorder, 1 , 6 the herein described clinical phenotype is relatively severe. Interestingly, no signs of neuropathy or spastic paraplegia have been reported in these two children, which may be attributed to the patients' young age. The specific phenotypic outcome of one point mutation over another, as well as potential modifications by other inherited or environmental factors contributing to the disease spectrum requires further investigation.
Autism is known for its broad underlying genetic heterogeneity. As commonly observed in neurodevelopmental syndromes, effects on fecundity may complicate the recognition of heritability, and large Medelian ancestries are rare to find. Like in the herein described families, de novo mutations and mosaicism are relevant mechanisms to take under consideration when searching for the underlying genetic cause. 9 To validate potentially pathogenic mutations, we therefore recommend to perform genetic sequencing on parent samples, including DNA derived from tissue other than blood.
We conclude that variant p.(Gly903Arg) is pathogenic, associated with a childhood‐onset neurological phenotype mainly manifesting with developmental delay and intellectual disability. The gene ATP1A1 should therefore be considered for genetic testing in patients with a comparable neurodevelopmental phenotype, including spectrum disorders such as autism.
Members of the Undiagnosed Diseases Network
Maria T. Acosta; David R. Adams; Ben Afzali; Aimee Allworth; Raquel L. Alvarez; Justin Alvey; Ashley Andrews; Euan A. Ashley; Carlos A. Bacino; Guney Bademci; Ashok Balasubramanyam; Dustin Baldridge; Jim Bale; Michael Bamshad; Deborah Barbouth; Pinar Bayrak‐Toydemir; Anita Beck; Alan H. Beggs; Edward Behrens; Gill Bejerano; Hugo J. Bellen; Jimmy Bennett; Jonathan A. Bernstein; Gerard T. Berry; Anna Bican; Stephanie Bivona; Elizabeth Blue; John Bohnsack; Devon Bonner; Nicholas Borja; Lorenzo Botto; Lauren C. Briere; Elizabeth A. Burke; Lindsay C. Burrage; Manish J. Butte; Peter Byers; William E. Byrd; John Carey; Thomas Cassini; Sirisak Chanprasert; Hsiao‐Tuan Chao; Ivan Chinn; Gary D. Clark; Terra R. Coakley; Laurel A. Cobban; Joy D. Cogan; Matthew Coggins; F. Sessions Cole; Heather A. Colley; Heidi Cope; Brian Corner; Rosario Corona; William J. Craigen; Andrew B. Crouse; Michael Cunningham; Hongzheng Dai; Surendra Dasari; Joie Davis; Jyoti G. Dayal; Margaret Delgado; Esteban C. Dell'Angelica; Katrina Dipple; Daniel Doherty; Naghmeh Dorrani; Argenia L. Doss; Emilie D. Douine; Precilla D'Souza; Dawn Earl; David J. Eckstein; Lisa T. Emrick; Christine M. Eng; Kimberly Ezell; Marni Falk; Elizabeth L. Fieg; Paul G. Fisher; Brent L. Fogel; Jiayu Fu; William A. Gahl; Ian Glass; Page C. Goddard; Rena A. Godfrey; Joanna M. Gonzalez; Andrea Gropman; Meghan C. Halley; Rizwan Hamid; Neil Hanchard; Kelly Hassey; Nichole Hayes; Frances High; Anne Hing; Fuki M. Hisama; Ingrid A. Holm; Jason Hom; Martha Horike‐Pyne; Yan Huang; Alden Huang; Sarah Hutchison; Wendy Introne; Kosuke Izumi; Gail P. Jarvik; Jeffrey Jarvik; Suman Jayadev; Orpa Jean‐Marie; Vaidehi Jobanputra; Emerald Kaitryn; Shamika Ketkar; Dana Kiley; Gonench Kilich; Shilpa N. Kobren; Isaac S. Kohane; Jennefer N. Kohler; Susan Korrick; Deborah Krakow; Donna M. Krasnewich; Elijah Kravets; Seema R. Lalani; Christina Lam; Brendan C. Lanpher; Ian R. Lanza; Kumarie Latchman; Kimberly LeBlanc; Brendan H. Lee; Richard A. Lewis; Pengfei Liu; Nicola Longo; Sandra K. Loo; Joseph Loscalzo; Richard L. Maas; Ellen F. Macnamara; Calum A. MacRae; Valerie V. Maduro; AudreyStephannie C. Maghiro; Rachel Mahoney; May Christine V. Malicdan; Laura A. Mamounas; Teri A. Manolio; Rong Mao; Ronit Marom; Gabor Marth; Beth A. Martin; Martin G. Martin; Julian A. Martínez‐Agosto; Shruti Marwaha; Allyn McConkie‐Rosell; Alexa T. McCray; Elisabeth McGee; Matthew Might; Danny Miller; Ghayda Mirzaa; Eva Morava; Paolo Moretti; Marie Morimoto; John J. Mulvihill; Mariko Nakano‐Okuno; Stanley F. Nelson; Serena Neumann; Shirley Nieves‐Rodriguez; Donna Novacic; Devin Oglesbee; James P. Orengo; Laura Pace; Stephen Pak; J. Carl Pallais; Jeanette C. Papp; Neil H. Parker; LéShon Peart; Leoyklang Petcharet; John A. Phillips; Jennifer E. Posey; Lorraine Potocki; Barbara N. Pusey Swerdzewski; Aaron Quinlan; Deepak A. Rao; Anna Raper; Wendy Raskind; Adriana Rebelo; Genecee Renteria; Chloe M. Reuter; Lynette Rives; Amy K. Robertson; Lance H. Rodan; Jill A. Rosenfeld; Elizabeth Rosenthal; Francis Rossignol; Maura Ruzhnikov; Marla Sabaii; Jacinda B. Sampson; Timothy Schedl; Kelly Schoch; Daryl A. Scott; Elaine Seto; Prashant Sharma; Vandana Shashi; Emily Shelkowitz; Sam Sheppeard; Jimann Shin; Edwin K. Silverman; Janet S. Sinsheimer; Kathy Sisco; Edward C. Smith; Carson A. Smith; Kevin S. Smith; Lilianna Solnica‐Krezel; Ben Solomon; Rebecca C. Spillmann; Andrew Stergachis; Joan M. Stoler; Kathleen Sullivan; Jennifer A. Sullivan; Shirley Sutton; David A. Sweetser; Virginia Sybert; Holly K. Tabor; Queenie K.‐G. Tan; Amelia L. M. Tan; Arjun Tarakad; Herman Taylor; Mustafa Tekin; Willa Thorson; Cynthia J. Tifft; Camilo Toro; Alyssa A. Tran; Rachel A. Ungar; Tiina K. Urv; Adeline Vanderver; Matt Velinder; Dave Viskochil; Tiphanie P. Vogel; Colleen E. Wahl; Melissa Walker; Nicole M. Walley; Jennifer Wambach; Jijun Wan; Michael F. Wangler; Patricia A. Ward; Daniel Wegner; Monika Weisz Hubshman; Mark Wener; Tara Wenger; Monte Westerfield; Matthew T. Wheeler; Jordan Whitlock; Lynne A. Wolfe; Kim Worley; Shinya Yamamoto; Zhe Zhang; Stephan Züchner
Conflict of interest
The authors declare that there are no conflicts of interest.
Written informed consent for publication of case
The patients' parents provided written informed consent for the publication of these cases.
Acknowledgements
We thank the patients and their parents for participating in this study. This study has been supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG010230 (GB, APR, NAB, SAB, MT, SZ) and NIH R01NS105755 (SZ). MFD has received funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, DO 2386/1‐1).
Funding Information
This study has been supported by the NIH Common Fund, through the Office of Strategic Coordination/Office of the NIH Director under Award Number U01HG010230 (GB, APR, NAB, SAB, MT, SZ) and National Institutes of Health R01NS105755 (SZ). MFD has received funding by the German Research Foundation (Deutsche Forschungsgemeinschaft, DFG, DO 2386/1‐1).
Funding Statement
This work was funded by Deutsche Forschungsgemeinschaft, DFG grant DO 2386/1‐1; National Institutes of Health grant R01NS105755; Office of Strategic Coordination/Office of the NIH Director grant U01HG010230.
Contributor Information
Undiagnosed Diseases Network:
Maria T. Acosta, David R. Adams, Ben Afzali, Aimee Allworth, Raquel L. Alvarez, Justin Alvey, Ashley Andrews, Euan A. Ashley, Carlos A. Bacino, Guney Bademci, Ashok Balasubramanyam, Dustin Baldridge, Jim Bale, Michael Bamshad, Deborah Barbouth, Pinar Bayrak‐Toydemir, Anita Beck, Alan H. Beggs, Edward Behrens, Gill Bejerano, Hugo J. Bellen, Jimmy Bennett, Jonathan A. Bernstein, Gerard T. Berry, Anna Bican, Stephanie Bivona, Elizabeth Blue, John Bohnsack, Devon Bonner, Nicholas Borja, Lorenzo Botto, Lauren C. Briere, Elizabeth A. Burke, Lindsay C. Burrage, Manish J. Butte, Peter Byers, William E. Byrd, John Carey, Thomas Cassini, Sirisak Chanprasert, Hsiao‐Tuan Chao, Ivan Chinn, Gary D. Clark, Terra R. Coakley, Laurel A. Cobban, Joy D. Cogan, Matthew Coggins, F. Sessions Cole, Heather A. Colley, Heidi Cope, Brian Corner, Rosario Corona, William J. Craigen, Andrew B. Crouse, Michael Cunningham, Hongzheng Dai, Surendra Dasari, Joie Davis, Jyoti G. Dayal, Margaret Delgado, Esteban C. Dell'Angelica, Katrina Dipple, Daniel Doherty, Naghmeh Dorrani, Argenia L. Doss, Emilie D. Douine, Precilla D'Souza, Dawn Earl, David J. Eckstein, Lisa T. Emrick, Christine M. Eng, Kimberly Ezell, Marni Falk, Elizabeth L. Fieg, Paul G. Fisher, Brent L. Fogel, Jiayu Fu, William A. Gahl, Ian Glass, Page C. Goddard, Rena A. Godfrey, Joanna M. Gonzalez, Andrea Gropman, Meghan C. Halley, Rizwan Hamid, Neil Hanchard, Kelly Hassey, Nichole Hayes, Frances High, Anne Hing, Fuki M. Hisama, Ingrid A. Holm, Jason Hom, Martha Horike‐Pyne, Yan Huang, Alden Huang, Sarah Hutchison, Wendy Introne, Kosuke Izumi, Gail P. Jarvik, Jeffrey Jarvik, Suman Jayadev, Orpa Jean‐Marie, Vaidehi Jobanputra, Emerald Kaitryn, Shamika Ketkar, Dana Kiley, Gonench Kilich, Shilpa N. Kobren, Isaac S. Kohane, Jennefer N. Kohler, Susan Korrick, Deborah Krakow, Donna M. Krasnewich, Elijah Kravets, Seema R. Lalani, Christina Lam, Brendan C. Lanpher, Ian R. Lanza, Kumarie Latchman, Kimberly LeBlanc, Brendan H. Lee, Richard A. Lewis, Pengfei Liu, Nicola Longo, Sandra K. Loo, Joseph Loscalzo, Richard L. Maas, Ellen F. Macnamara, Calum A. MacRae, Valerie V. Maduro, AudreyStephannie C. Maghiro, Rachel Mahoney, May Christine V. Malicdan, Laura A. Mamounas, Teri A. Manolio, Rong Mao, Ronit Marom, Gabor Marth, Beth A. Martin, Martin G. Martin, Julian A. Martínez‐Agosto, Shruti Marwaha, Allyn McConkie‐Rosell, Alexa T. McCray, Elisabeth McGee, Matthew Might, Danny Miller, Ghayda Mirzaa, Eva Morava, Paolo Moretti, Marie Morimoto, John J. Mulvihill, Mariko Nakano‐Okuno, Stanley F. Nelson, Serena Neumann, Shirley Nieves‐Rodriguez, Donna Novacic, Devin Oglesbee, James P. Orengo, Laura Pace, Stephen Pak, J. Carl Pallais, Jeanette C. Papp, Neil H. Parker, LéShon Peart, Leoyklang Petcharet, John A. Phillips, Jennifer E. Posey, Lorraine Potocki, Barbara N. Pusey Swerdzewski, Aaron Quinlan, Deepak A. Rao, Anna Raper, Wendy Raskind, Adriana Rebelo, Genecee Renteria, Chloe M. Reuter, Lynette Rives, Amy K. Robertson, Lance H. Rodan, Jill A. Rosenfeld, Elizabeth Rosenthal, Francis Rossignol, Maura Ruzhnikov, Marla Sabaii, Jacinda B. Sampson, Timothy Schedl, Kelly Schoch, Daryl A. Scott, Elaine Seto, Prashant Sharma, Vandana Shashi, Emily Shelkowitz, Sam Sheppeard, Jimann Shin, Edwin K. Silverman, Janet S. Sinsheimer, Kathy Sisco, Edward C. Smith, Carson A. Smith, Kevin S. Smith, Lilianna Solnica‐Krezel, Ben Solomon, Rebecca C. Spillmann, Andrew Stergachis, Joan M. Stoler, Kathleen Sullivan, Jennifer A. Sullivan, Shirley Sutton, David A. Sweetser, Virginia Sybert, Holly K. Tabor, Queenie K.‐G. Tan, Amelia L. M. Tan, Arjun Tarakad, Herman Taylor, Mustafa Tekin, Willa Thorson, Cynthia J. Tifft, Camilo Toro, Alyssa A. Tran, Rachel A. Ungar, Tiina K. Urv, Adeline Vanderver, Matt Velinder, Dave Viskochil, Tiphanie P. Vogel, Colleen E. Wahl, Melissa Walker, Nicole M. Walley, Jennifer Wambach, Jijun Wan, Michael F. Wangler, Patricia A. Ward, Daniel Wegner, Monika Weisz Hubshman, Mark Wener, Tara Wenger, Monte Westerfield, Matthew T. Wheeler, Jordan Whitlock, Lynne A. Wolfe, Kim Worley, Shinya Yamamoto, Zhe Zhang, and Stephan Züchner
Data availability statement
Original data are available upon reasonable request to the corresponding author.
References
- 1. Lassuthova P, Rebelo AP, Ravenscroft G, et al. Mutations in ATP1A1 cause dominant Charcot‐Marie‐Tooth type 2. Am J Hum Genet. 2018;102(3):505‐514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stregapede F, Travaglini L, Rebelo AP, et al. Hereditary spastic paraplegia is a novel phenotype for germline de novo ATP1A1 mutation. Clin Genet. 2020;97(3):521‐526. [DOI] [PubMed] [Google Scholar]
- 3. Schlingmann KP, Bandulik S, Mammen C, et al. Germline de novo mutations in ATP1A1 cause renal hypomagnesemia, refractory seizures, and intellectual disability. Am J Hum Genet. 2018;103(5):808‐816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dohrn MF, Rebelo AP, Srivastava S, et al. De novo ATP1A1 variants in an early‐onset complex neurodevelopmental syndrome. Neurology. 2022;98(11):440‐445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Lin Z, Li J, Ji T, Wu Y, Gao K, Jiang Y. ATP1A1 de novo mutation‐related disorders: clinical and genetic features. Front Pediatr. 2021;9:657256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. He J, Guo L, Lin S, et al. ATP1A1 mutations cause intermediate Charcot‐Marie‐Tooth disease. Hum Mutat. 2019;40(12):2334‐2343. [DOI] [PubMed] [Google Scholar]
- 7. Havrilla JM, Pedersen BS, Layer RM, Quinlan AR. (2019) a map of constrained coding regions in the human genome. Nat Genet. 2019;51(1):88‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Danzi MC, Dohrn MF, Fazal S, et al. Deep structured learning for variant prioritization in Mendelian diseases. Nat Commun. 2023;14(1):4167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. D'Gama AM, Walsh CA. Somatic mosaicism and neurodevelopmental disease. Nat Neurosci. 2018;21(11):1504‐1514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Original data are available upon reasonable request to the corresponding author.