

Abstract
The efficacy of pembrolizumab monotherapy versus chemotherapy increased with increasing programmed death ligand 1 (PD‐L1) expression, as quantified by combined positive score (CPS; PD‐L1 expression on both tumour cells and immune cells) in patients with previously treated metastatic triple‐negative breast cancer (mTNBC) in the phase 3 KEYNOTE‐119 study. This exploratory analysis was conducted to determine whether the expression of PD‐L1 on tumour cells contributes to the predictive value of PD‐L1 CPS in mTNBC. PD‐L1 expression in tumour samples was assessed using PD‐L1 IHC 22C3 pharmDx and quantified using both CPS and tumour proportion score (TPS; PD‐L1 expression on tumour cells alone). Calculated immune cell density (CID) was defined as CPS minus TPS. The ability of each scoring method (CPS, TPS, and CID) to predict clinical outcomes with pembrolizumab was evaluated. With pembrolizumab, the area under the receiver operating characteristic curve was 0.69 (95% CI = 0.58–0.80) for CPS, 0.55 (95% CI = 0.46–0.64) for TPS, and 0.67 (95% CI = 0.56–0.77) for CID. After correction for cutoff prevalence, CPS performed as well as, if not better than, CID with respect to predicting objective response rate, progression‐free survival, and overall survival. Data from this exploratory analysis suggest that, although PD‐L1 expression on immune cells alone is predictive of response to programmed death 1 blockade in mTNBC, adding tumour PD‐L1 expression assessment (i.e. CPS, which combines immune cell and tumour cell PD‐L1 expression) may improve prediction. PD‐L1 CPS thus remains an effective and broadly applicable uniform scoring system for enriching response to programmed death 1 blockade with pembrolizumab in mTNBC as well as other tumour types.
Keywords: pembrolizumab, PD‐L1, triple‐negative breast cancer
Introduction
Triple‐negative breast cancer (TNBC), which lacks oestrogen and progesterone receptor expression and does not overexpress human epidermal growth factor receptor 2, accounts for ~15–20% of all breast cancers [1]. As most TNBC tumours do not harbour targetable genetic alterations, patients with TNBC have limited treatment options [1, 2].
Programmed death 1 (PD‐1) is an immune checkpoint protein expressed on tumour‐infiltrating lymphocytes and acts on two ligands: programmed death ligand 1 (PD‐L1) and programmed death ligand 2 (PD‐L2). PD‐L1 is widely expressed, including on antigen‐presenting cells, dendritic cells, activated monocytes, B cells, and non‐lymphoid tissues of different organs [3]. Based on work by Dong et al [4], PD‐L1 overexpression on tumour cells became the focus of early clinical studies for T‐cell‐based cancer immunotherapy, an approach that was successful for non‐small cell lung cancer (NSCLC). However, as understanding of the PD‐1/L1 pathway increased, the importance of PD‐L1 expression on immune cells became clear in mechanistic terms [5], as did its significance as a biomarker [6].
PD‐L1 is expressed in approximately 20–42% of advanced TNBC [6, 7, 8]. Anti‐PD‐1/L1 monoclonal antibodies have demonstrated antitumour activity in patients with advanced or metastatic TNBC (mTNBC) and appear to exhibit an enhanced treatment benefit in PD‐L1‐positive disease [9, 10, 11, 12, 13]. In the phase 1b KEYNOTE‐012 study, patients with PD‐L1‐positive (expression in stroma or ≥1% of tumour cells by a prototype immunohistochemistry assay using the 22C3 antibody) recurrent or mTNBC who were treated with PD‐1 inhibitor pembrolizumab as monotherapy demonstrated an objective response rate (ORR) of 18.5% [9]. In the phase 3 IMpassion130 study, patients with untreated mTNBC, regardless of PD‐L1 expression, who were treated with PD‐L1 inhibitor atezolizumab plus nab‐paclitaxel demonstrated improved progression‐free survival (PFS) and ORR compared with those treated with nab‐paclitaxel [12]. Treatment benefit was enriched in the PD‐L1‐positive subgroup (expression on tumour‐infiltrating immune cells as a percentage of tumour area ≥1%), with an ORR of 58.9%, compared with 42.6% in the PD‐L1‐negative subgroup; the stratified hazard ratio (HR) for progressive disease (PD) or death was 0.62 (95% CI = 0.49–0.78; p < 0.001) in which a clinically significant improvement in overall survival (OS) was observed [12]. These findings from IMpassion130 highlight the potential clinical importance of PD‐L1 immune cell expression status alone as a biomarker predictive of response to PD‐1/L1 blockade in mTNBC.
The importance of determining PD‐L1 expression on both tumour cells and immune cells as a predictive biomarker has evolved based on the development and implementation of PD‐L1 combined positive score (CPS) [14]. In the multicohort phase 2 KEYNOTE‐086 study, single‐agent pembrolizumab as second‐line or later treatment for mTNBC regardless of PD‐L1 expression (cohort A) or first‐line treatment for PD‐L1 CPS ≥1 disease (cohort B) demonstrated ORRs of 5.7% and 21.4%, respectively, when PD‐L1 was assessed using an investigational version of PD‐L1 IHC 22C3 pharmDx [10, 11].
In the first‐line setting, the efficacy of pembrolizumab in combination with chemotherapy was also enriched with higher PD‐L1 CPS in patients with previously untreated locally advanced inoperable or mTNBC in the phase 3 KEYNOTE‐355 study [13]. In patients with CPS ≥ 10, the HR for death was 0.73 (95% CI = 0.55–0.95; p = 0.0093; boundary of p = 0.0113 met), and the HR for progressive disease (PD) or death was 0.66 (95% CI = 0.50–0.88) with pembrolizumab‐chemotherapy compared with chemotherapy at the final analysis [13]; these findings led to US Food and Drug Administration approval of pembrolizumab plus chemotherapy for patients with PD‐L1‐positive (CPS ≥ 10) TNBC. These data suggest that, in addition to PD‐L1 immune cell expression, PD‐L1 expression on tumour cells may also play a role in mTNBC with regard to the mechanism of action of pembrolizumab and as a predictive biomarker.
In the randomised, open‐label, phase 3 KEYNOTE‐119 study, pembrolizumab monotherapy did not significantly improve OS compared with chemotherapy as second‐ or third‐line treatment for patients with mTNBC [15]. However, the benefit of pembrolizumab compared with chemotherapy appeared to be greater with increasing PD‐L1 CPS, whereas outcomes with chemotherapy were unaffected by PD‐L1 expression. Herein, we evaluate whether PD‐L1 expression on both tumour cells and immune cells independently contributes to the value of PD‐L1 assessment as a predictive biomarker in patients with mTNBC in the KEYNOTE‐119 trial.
Materials and methods
Study design and patients
KEYNOTE‐119 (NCT02555657) was an international, randomised, open‐label, phase 3 trial [15]. Eligible patients had centrally confirmed mTNBC, one or two previous systemic treatments for metastatic disease, documented PD on most recent therapy, and previous treatment with an anthracycline or a taxane in the (neo)adjuvant or metastatic setting. Patients were randomly assigned 1:1 to pembrolizumab 200 mg every 3 weeks for up to 35 cycles or investigator's choice of single‐agent chemotherapy (capecitabine, eribulin, gemcitabine, or vinorelbine; 60% enrolment cap for each). The study was conducted in accordance with the protocol and its amendments, the Declaration of Helsinki, the International Conference on Harmonisation Guidelines for Good Clinical Practice, and local and national regulations. The study protocol and all amendments were approved by the institutional review board or ethics committee at each participating institution. All patients provided written informed consent.
Assessments and statistical analysis
PD‐L1 immunohistochemical evaluation of tumour samples was performed prospectively during screening at a central laboratory using an investigational version of PD‐L1 IHC 22C3 pharmDx (Agilent Technologies, Inc., Santa Clara, CA, USA). This assay was validated for use in TNBC at CPS ≥ 1 (the investigational cutoff, different from the approved cutoff of CPS ≥ 10), and pathologists were trained and certified by Agilent to score PD‐L1 in this tumour type and at each cutoff. PD‐L1 was scored using CPS, tumour proportion score (TPS), and mononuclear immune cell density score (MIDS) as previously described [14]. CPS was evaluated before randomisation and defined as the number of PD‐L1‐staining cells (tumour cells, lymphocytes, and macrophages) divided by the total number of viable tumour cells, multiplied by 100; a minimum of 100 viable tumour cells must have been present to be considered evaluable. TPS [defined as the percentage of PD‐L1‐expressing tumour cells (partial or complete membrane staining) relative to the total number of tumour cells] and MIDS (an estimate of the ratio of PD‐L1‐stained immune cells to the total number of viable tumour cells evaluated on a five‐point scale) were captured as exploratory measures, whereas calculated immune cell density (CID; defined as CPS minus TPS) was computed post hoc for the purpose of the analyses in this report.
Before this post hoc exploratory analysis, we expected that CID would provide the best estimate of immune cell PD‐L1 expression. However, there were concerns that because CPS is truncated at 100, CID might be underestimated when TPS is very high. To account for this effect, we corrected CID results according to the following rules:
If CPS = 100 and MIDS = 2, then CID is the higher of CPS − TPS or 1.
If CPS = 100 and MIDS = 3, then CID is the higher of CPS − TPS or 10.
If CPS = 100 and MIDS = 4, then CID = 100.
This correction was made after viewing results.
This post hoc exploratory analysis evaluated the ability of CPS, TPS, and CID to predict ORR with pembrolizumab using receiver operating characteristic (ROC) analysis and the ORR enrichment profile across a range of cutoffs. In addition, we evaluated the HRs for PFS and OS for pembrolizumab versus chemotherapy using a stratified Cox proportional hazard model with the Efron method of tie handling in subgroups by PD‐L1 score. For this descriptive exploratory analysis, no statistical plan was written. As a result, no hypothesis testing was performed, and thus the reporting of p values is not deemed appropriate.
Results
Between 25 November 2015 and 11 April 2017, 622 patients were randomly assigned to receive pembrolizumab or single‐agent chemotherapy. The median follow‐up [defined as time from randomisation to the date of death or database cutoff (11 April 2019)] was 9.9 months in the pembrolizumab arm and 10.9 months in the chemotherapy arm.
Among the 622 patients enrolled, tumour samples were available for the evaluation of PD‐L1 expression in 601 patients (96.6%) (pembrolizumab, n = 309; chemotherapy, n = 292). Of the 601 tumour samples included in this analysis, only four CID results were adjusted based on MIDS: one in the pembrolizumab arm and three in the chemotherapy arm. The patient in the pembrolizumab arm was a responder, and the CID changed from 0 to 1 based on CPS = 100, TPS = 100, and MIDS = 2. As expected, adjustments to the three patient scores in the chemotherapy arm lacked any meaningful impact on this analysis, as demonstrated by the following points: (1) the ROC analysis did not include patients in the chemotherapy arm and (2) sensitivity analysis confirmed that the few affected data points had negligible effects on the correlation between TPS and CID.
At the database cutoff date, the ORR was 9.7% (30 of 309 patients) in the pembrolizumab arm and 11.3% (33 of 292 patients) in the chemotherapy arm. Based on the three separate scoring systems, the area under the receiver operating characteristic (AUROC) for discriminating objective response in pembrolizumab‐treated patients was 0.69 (95% CI = 0.58–0.80) for CPS, 0.55 (95% CI = 0.46–0.64) for TPS, and 0.67 (95% CI = 0.56–0.77) for CID (Figure 1A), suggesting that CPS may have a slight advantage over CID and TPS with respect to predicting pembrolizumab response across a range of specified cutoffs (Figure 1B).
Figure 1.
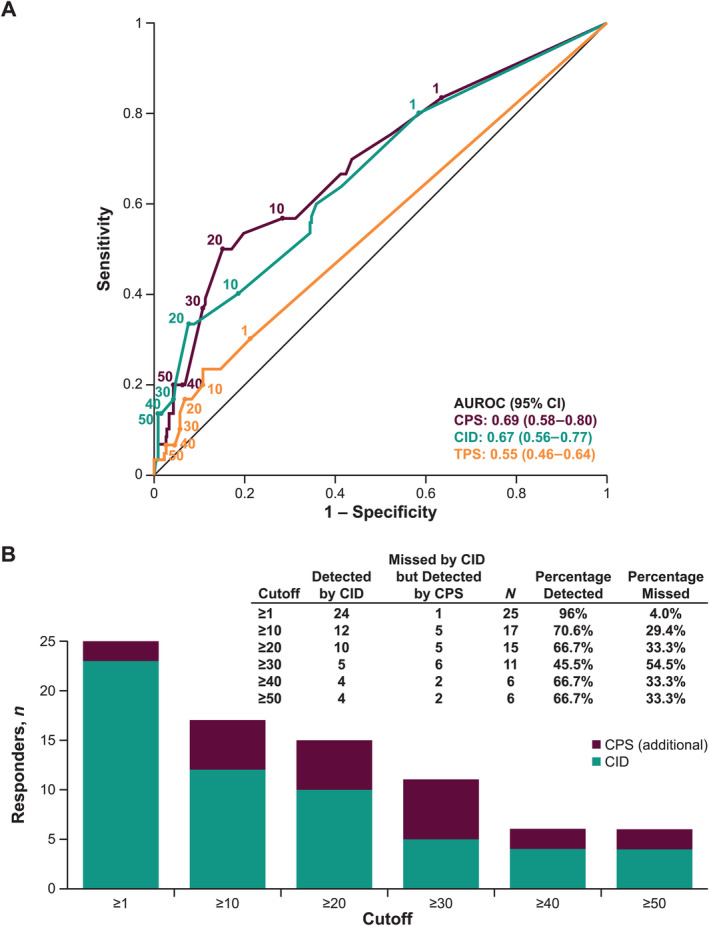
(A) ROC curves by PD‐L1 scoring method in pembrolizumab‐treated patients (n = 309). Sensitivity reports the fraction of responders detected on the y axis and 1 − specificity reports the fraction of non‐responders falsely detected on the x axis. The higher the ROC curve is on the vertical axis and the further left it is on the horizontal axis, the more valid the test. A cutoff of 0 (upper right) corresponds to the detection of all responders (sensitivity = 1) but the false detection of all non‐responders (1 − specificity = 1). As the cutoff increases, a perfect test would move horizontally to the upper left corner (sensitivity = 1; 1 − specificity = 0) and then straight down. (B) Responders by cutoffs using PD‐L1 CPS and CID in pembrolizumab‐treated patients (n = 30 responders).
At each cutoff, CID had a lower estimated sensitivity (i.e. more missed responders) than CPS. In addition, except for a cutoff of 1, CID had a lower Youden index than CPS (Table 1). Of the 30 responders in the pembrolizumab arm, the numbers of missed responders for CID compared with CPS at each cutoff were 1 (CPS ≥ 1), 5 (CPS ≥ 10), 5 (CPS ≥ 20), 6 (CPS ≥ 30), 2 (CPS ≥ 40), and 2 (CPS ≥ 50) after adjusting for one potentially truncated value.
Table 1.
ROC analysis by PD‐L1 scoring method
| PD‐L1 scoring method | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| CPS* | CID | TPS | ||||||||||
| Cutoff | Sens | Spec | YI † | Prev | Sens | Spec | YI † | Prev | Sens | Spec | YI † | Prev |
| 0 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 | 1 | 0 | 0 | 1 |
| 1 | 0.833 | 0.366 | 0.199 | 0.654 | 0.800 | 0.416 | 0.216 | 0.605 | 0.300 | 0.789 | 0.089 | 0.220 |
| 10 | 0.567 | 0.717 | 0.284 | 0.311 | 0.400 | 0.814 | 0.214 | 0.207 | 0.200 | 0.892 | 0.092 | 0.117 |
| 20 | 0.500 | 0.849 | 0.349 | 0.184 | 0.333 | 0.925 | 0.258 | 0.100 | 0.167 | 0.932 | 0.099 | 0.078 |
| 30 | 0.367 | 0.892 | 0.259 | 0.133 | 0.167 | 0.957 | 0.124 | 0.055 | 0.100 | 0.943 | 0.043 | 0.061 |
| 40 | 0.200 | 0.935 | 0.135 | 0.078 | 0.133 | 0.986 | 0.119 | 0.026 | 0.067 | 0.953 | 0.020 | 0.049 |
| 50 | 0.200 | 0.957 | 0.157 | 0.058 | 0.133 | 0.993 | 0.126 | 0.019 | 0.067 | 0.968 | 0.034 | 0.036 |
Prev, prevalence; sens, sensitivity; spec, specificity; YI, Youden index.
There are three practicable cutoffs for CPS that show equal or better YI than the cutoff for CID with the highest YI (bolded values).
YI (defined as: sensitivity + specificity − 1) is a measure of enrichment for responders.
However, as can be seen in the data presented in Table 1, for each of these three different biomarkers, the prevalence for CID is smaller for any given fixed numeric cutoff value. This is as would be expected because it is defined as CPS − TPS (leading to the prevalence for any given score of CID always being smaller than that of CPS). Evaluating CID at the same numeric value as CPS may require a higher level of immune cell staining for CID to achieve the same numeric value as CPS, i.e. yielding a more stringent CPS cutoff, with an anticipated loss in sensitivity to capture responders.
To correct for this, sensitivity and ORR enrichment profiles were plotted against the percentile of the population implied at a given cutoff for both CPS and CID. As shown in Figure 2A, in relation to immune cell scoring alone (i.e. CID), the inclusion of tumour cell assessment in the CPS scoring schema did not diminish the ability of CPS to predict ORR. Furthermore, at higher cutoffs that capture ≤35% of the study population, CPS may have a slight advantage over CID with respect to predicting response to pembrolizumab treatment. Similarly, across the range of specified cutoffs, the OS HR (Figure 2B) and the PFS HR (Figure 2C) tended to be slightly lower for CPS than for CID at the same percentage of the patient population selected. When the relationship between TPS and CID was assessed, CID appeared to be orthogonal to TPS, with a Pearson correlation of −0.015 for all patients (Figure 3; chemotherapy arm, ρ = 0.03, pembrolizumab arm, ρ = −0.06).
Figure 2.

(A) Sensitivity and ORR enrichment profile; and HR (pembrolizumab/chemotherapy) profile for (B) OS and (C) PFS according to the percentage of the population identified by cutoff. The x axis shows the percentile of positive tumours.
Figure 3.
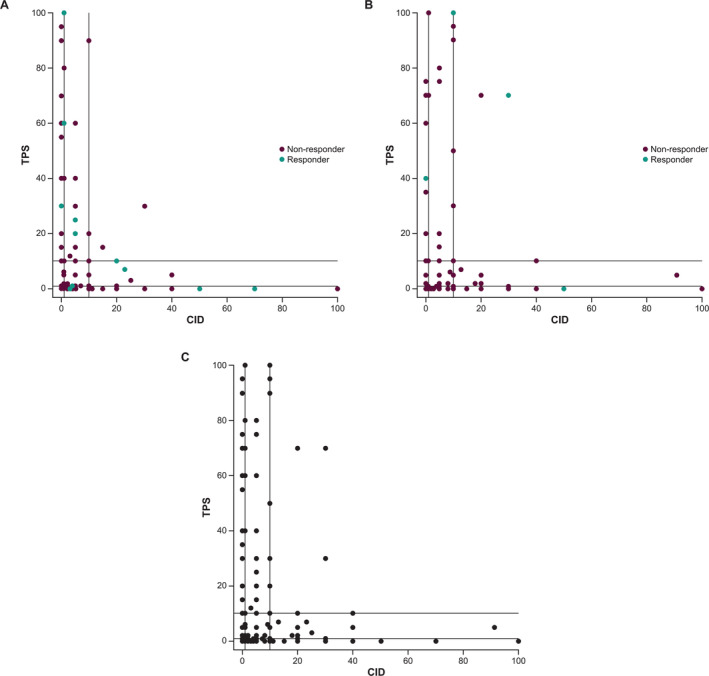
Scatterplot of PD‐L1 TPS versus CID in (A) pembrolizumab‐treated patients (n = 309), (B) chemotherapy‐treated patients (n = 292), and (C) all patients (N = 601).
Discussion
This post hoc exploratory analysis evaluated different PD‐L1 expression scoring methods and cutoffs in predicting response to second‐line or later pembrolizumab monotherapy in patients with mTNBC in the phase 3 KEYNOTE‐119 trial. The ROC analysis of response versus scoring methods showed similar AUROC when PD‐L1 expression was measured using CPS (tumour cells and immune cells), TPS (tumour cells only), or CID (immune cells only), with CPS being slightly numerically higher. At any given numeric scoring cutoff, responders were missed when PD‐L1 expression was measured with CID.
When accounting for prevalence (i.e. the percentile of the population selected), evaluation of the enrichment of ORR and sensitivity indicated that CPS performed similarly to CID, with a numeric trend towards an advantage for CPS, especially at higher cutoffs. CPS also consistently showed a small numeric advantage for the HR for OS and PFS compared with CID. As such, although the level of PD‐L1 expression on immune cells alone (i.e. CID) is clearly predictive of response to PD‐1 blockade using pembrolizumab, the association between clinical outcome and CPS appears to show an association similar to or better than that for immune cell staining alone, and no trends suggested that inclusion of tumour cell PD‐L1 expression in the assessment negatively impacted the clinical enrichment profile of the PD‐L1 CPS assay. Although different individual tumour type‐specific PD‐L1 scoring systems may possibly be similarly predictive of response to pembrolizumab (e.g. TPS for NSCLC), the use of a uniform scoring system such as CPS, with its incorporation of both tumour and immune cell assessment, has consistently proven to have clinical utility response across multiple different tumour indications. Using CPS for multiple cancers potentially reduces complexity for pathologists assessing PD‐L1 expression across multiple tumour types in their practices.
These findings were consistent with the results of an exploratory biomarker analysis of the IMpassion130 study evaluating atezolizumab plus nab‐paclitaxel versus placebo plus nab‐paclitaxel as first‐line treatment for patients with mTNBC [16]. Patients with PD‐L1 expression on ≥1% of both tumour cell and tumour‐infiltrating immune cells (HR = 0.51; 95% CI = 0.28–0.91) had a lower HR for PD or death compared with patients with PD‐L1 on ≥1% of tumour cells alone (HR = 0.56; 95% CI = 0.34–0.92) and patients with PD‐L1 expression on ≥1% of tumour‐infiltrating immune cells alone (HR = 0.65; 95% CI = 0.52–0.82) [16]. However, cross‐trial comparisons should be interpreted with caution because the present analysis was specific to pembrolizumab.
PD‐L1 expression on tumour cells and tumour‐infiltrating immune cells could have biological and clinical relevance in TNBC. PD‐L1 expression in these two cell compartments may represent distinct mechanisms that independently attenuate anticancer immunity; expression on immune cells likely reflects interferon‐γ‐induced adaptive regulation accompanied by increased tumour‐infiltrating lymphocytes and effector T cells, whereas expression on tumour cells reflects a dysregulation of the PD‐L1 gene expression accompanied by poor immune infiltration, sclerotic/desmoplastic stroma, and mesenchymal features [17]. In the current analysis of mTNBC tumour samples, CID appeared to be orthogonal to TPS, suggesting that tumours with the highest PD‐L1 expression in tumour cells and tumours with the highest PD‐L1 expression in immune cells had little overlap. This is consistent with the observations by Kowanetz et al [17] regarding PD‐L1 expression pattern in NSCLC.
In the present analysis, PD‐L1 expression by CPS was more sensitive for identifying patients with metastatic disease who were likely to respond to pembrolizumab monotherapy but less specific compared with CID. Thus, fewer responders were missed when PD‐L1 expression was measured using CPS, ensuring better identification of patients who might be appropriate for pembrolizumab monotherapy. Notably, the Youden index, an objective way to address the trade‐off between sensitivity and specificity, was also higher for CPS than for CID. Furthermore, there is a tendency to weight sensitivity more heavily than specificity in the late‐line setting when trying to identify patients who have limited treatment options.
This analysis has several limitations. KEYNOTE‐119 was a study of single‐agent pembrolizumab as second‐line or later therapy, and therefore, these results may not apply to combination therapy in the first‐line setting. Furthermore, this was a post hoc exploratory analysis, and the use of the 22C3 antibody limits the comparisons of similar studies using other anti‐PD‐L1 antibodies. The use of calculated CID in this study was also a limitation because the actual immune cell scoring was not directly evaluated. Lastly, these findings are currently of limited clinical utility in breast cancer (non‐TNBC), given that pembrolizumab monotherapy is not currently approved in this setting.
In this exploratory analysis of KEYNOTE‐119, our findings suggest that although immune cell PD‐L1 expression alone associates with pembrolizumab monotherapy efficacy in the treatment of second‐line or later mTNBC, the addition of tumour cell PD‐L1 expression assessment to that of immune cells (i.e. CPS) may enhance the ability to identify those patients with mTNBC who are most likely to benefit from pembrolizumab treatment. CPS as a single composite scoring system thus remains a highly effective biomarker predictive of response to PD‐1 blockade in mTNBC as well as across multiple other tumour types. Further exploratory analyses are needed to validate these findings with combination therapy.
Author contributions statement
PS, SO, VK, PJ, LH and KE conceived, designed or planned the study. OL, S‐AI, AG, EM‐C, KSL, KT, IW, SO, SH, KK and JC acquired the data. EM‐C, PS, LT, LH and KE analysed the data. EPW, S‐AI, EM‐C, PS, LT, VK, JM, JAM, PJ, LH, KE and JC interpreted the results. PS, PJ, LH and KE drafted the manuscript. EPW, OL, S‐AI, AG, EM‐C, KSL, PS, KT, LT, IW, SO, SH, KK, VK, JM, JAM, LH, KE and JC critically reviewed or revised the manuscript for important intellectual content. All authors had final approval of the manuscript.
Acknowledgements
The authors thank the patients and their families and caregivers as well as the primary investigators and site personnel for participating in the study. The authors also thank Zbigniew Nowecki of the Department of Soft Tissue/Bone Sarcoma and Melanoma, Maria Skłodowska‐Curie National Research Institute of Oncology, Warsaw, Poland. Medical writing and/or editorial assistance was provided by Bwalya A. Witika, PhD, Lei Bai, PhD, and Holly C. Cappelli, PhD, CMPP, of ApotheCom (Yardley, PA, USA). Funding for this study was provided by Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. The funder participated in study design, data analysis and interpretation, and manuscript writing. All authors had full access to the data and had final responsibility for the decision to submit the manuscript for publication.
Conflict of interest statement: JC reports a consulting/advisor role for AstraZeneca, Athenex, Bioasis, BioInvent, Boehringer Ingelheim, Celgene, Cellestia, Clovis Oncology, Daiichi Sankyo, Ellipses, Erytech, GEMoaB, Gilead, GSK, Hibercell, Leuko, Lilly, Menarini, Merck Sharp & Dohme, Polyphor, Reveal Genomics, Roche, Seattle Genetics, and Zymeworks; honoraria from Celgene, Daiichi Sankyo, Eisai, Lilly, Merck Sharp & Dohme, Novartis, Roche, Pfizer Inc., and Samsung Bioepis; research funding to the institution from Ariad Pharmaceuticals, AstraZeneca, Baxalta GMBH/Servier Affaires, Bayer Healthcare, Eisai, F. Hoffmann‐La Roche, Guardant Health, Merck Sharp & Dohme, Pfizer Inc., Piqur Therapeutics, Puma C, Queen Mary University of London, and Roche; stock, patents, and intellectual property ownership for Leuko (relative), MedSIR, and Nektar Pharmaceuticals; and travel, accommodation, and expenses from AstraZeneca, Daiichi Sankyo, Eisai, Novartis, Pfizer, and Roche. EPW reports consulting fees from Athenex, Carrick Therapeutics, Genentech (self and research to institute; RTI ended 1/2022), Gilead, GSK, Jounce, Leap Therapeutics, and Syros. OL has nothing to disclose. S‐AI reports an advisory role for AstraZeneca, Daiichi Sankyo, GSK, Hanmi, Idience, Lilly, MSD, Novartis, Pfizer Inc., and Roche; grants from AstraZeneca, Daewoong Pharmaceutical, Eisai, Pfizer Inc., and Roche; and other for AstraZeneca, Daiichi Sankyo, Novartis, Pfizer Inc., and Roche. AG reports consulting fees to their institution for AstraZeneca, Innate Pharma, MSD, Novartis, and Parexel, and support for attending meetings and/or travel for AstraZeneca, Menarini, MSD, Mylan, Novartis, and Roche. EM‐C reports an advisory board role for Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Pierre Fabre, Roche, and Sanofi; honoraria from Amgen, Bristol Myers Squibb, Merck Sharp & Dohme, Novartis, Pierre Fabre, and Roche; and clinical trial participation (principal investigator) for Amgen, Bristol Myers Squibb, GlaxoSmithKline, Merck Sharp & Dohme, Novartis, Pierre Fabre, Roche, and Sanofi. KSL reports personal fees from Bixink, Eisai, Everest Medicine, Daiichi Sankyo, Lilly, MSD, Novartis, Pfizer, and Roche and non‐financial support (drug supply) from Dong‐A ST Co. Ltd. PS reports consultancy fees from AstraZeneca, Bayer, Boehringer Ingelheim, Celgene, Eisai, Merck, Novartis, Pfizer Inc., Puma, and Roche; grant/funding to institution from Astellas, AstraZeneca, Genentech, Medivation, Novartis, OncoGenex, and Roche; and spousal employment for Roche. KT has nothing to disclose. LT reports grants from Novartis; personal fees from Amgen, Daiichi Sankyo, Lilly, MSD, Novartis, Pfizer Inc., and Roche; and non‐financial support from United Medical. IW reports consultancy fees from AstraZeneca, Merck, Novartis, Pfizer, and Roche and grant/funding to institution from Merck and Daiichi Sankyo. SO has nothing to disclose. SH reports employment with Agilent Technologies; stock ownership in Agilent Technologies; and a patent issued on the CPS method (‘Combined positive score for the interpretation of PD‐L1 expression on different tumour indications’) for which Agilent Technologies is the licensee. KK reports employment with Agilent Technologies; stock ownership in Agilent Technologies; and a patent issued on the CPS method (‘Combined positive score for the interpretation of PD‐L1 expression on different tumour indications’) for which Agilent Technologies is the licensee. VK, JM, LH, and SKP report employment with Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, and stock ownership in Merck & Co., Inc., Rahway, NJ, USA. JAM and PJ report employment for Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA. KE reports employment with Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA, at the time of this study and current employment with AbbVie Inc.; stock ownership in Merck & Co., Inc., Rahway, NJ, USA; spousal employment and stock ownership for BMS; stock ownership for Bayer AG and Johnson & Johnson; and an issued patent related to scoring of PD‐L1 with no personal income derived.
Data availability statement
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymised data and clinical study reports from the company's clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data‐sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country or region‐specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis‐driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data‐sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.
References
- 1. Yao H, He G, Yan S, et al. Triple‐negative breast cancer: is there a treatment on the horizon? Oncotarget 2017; 8: 1913–1924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Boyle P. Triple‐negative breast cancer: epidemiological considerations and recommendations. Ann Oncol 2012; 23: vi7–vi12. [DOI] [PubMed] [Google Scholar]
- 3. Muenst S, Schaerli AR, Gao F, et al. Expression of programmed death ligand 1 (PD‐L1) is associated with poor prognosis in human breast cancer. Breast Cancer Res Treat 2014; 146: 15–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dong H, Strome SE, Salomao DR, et al. Tumor‐associated B7‐H1 promotes T‐cell apoptosis: a potential mechanism of immune evasion. Nat Med 2002; 8: 793–800. [DOI] [PubMed] [Google Scholar]
- 5. Berraondo P. Mechanisms of action for different checkpoint inhibitors. HemaSphere 2019; 3: 28–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rozenblit M, Huang R, Danziger N, et al. Comparison of PD‐L1 protein expression between primary tumors and metastatic lesions in triple negative breast cancers. J Immunother Cancer 2020; 8: e001558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mittendorf EA, Philips AV, Meric‐Bernstam F, et al. PD‐L1 expression in triple‐negative breast cancer. Cancer Immunol Res 2014; 2: 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Botti G, Collina F, Scognamiglio G, et al. Programmed death ligand 1 (PD‐L1) tumor expression is associated with a better prognosis and diabetic disease in triple negative breast cancer patients. Int J Mol Sci 2017; 18: 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nanda R, Chow LQ, Dees EC, et al. Pembrolizumab in patients with advanced triple‐negative breast cancer: phase Ib KEYNOTE‐012 study. J Clin Oncol 2016; 34: 2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Adams S, Schmid P, Rugo HS, et al. Pembrolizumab monotherapy for previously treated metastatic triple‐negative breast cancer: cohort A of the phase II KEYNOTE‐086 study. Ann Oncol 2019; 30: 397–404. [DOI] [PubMed] [Google Scholar]
- 11. Adams S, Loi S, Toppmeyer D, et al. Pembrolizumab monotherapy for previously untreated, PD‐L1‐positive, metastatic triple‐negative breast cancer: cohort B of the phase II KEYNOTE‐086 study. Ann Oncol 2019; 30: 405–411. [DOI] [PubMed] [Google Scholar]
- 12. Schmid P, Adams S, Rugo HS, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer. N Engl J Med 2018; 379: 2108–2121. [DOI] [PubMed] [Google Scholar]
- 13. Cortes J, Cescon DW, Rugo HS, et al. Pembrolizumab plus chemotherapy versus placebo plus chemotherapy for previously untreated locally recurrent inoperable or metastatic triple‐negative breast cancer (KEYNOTE‐355): a randomised, placebo‐controlled, double‐blind, phase 3 clinical trial. Lancet 2020; 396: 1817–1828. [DOI] [PubMed] [Google Scholar]
- 14. Kulangara K, Zhang N, Corigliano E, et al. Clinical utility of the combined positive score for programmed death ligand‐1 expression and the approval of pembrolizumab for treatment of gastric cancer. Arch Pathol Lab Med 2019; 143: 330–337. [DOI] [PubMed] [Google Scholar]
- 15. Winer EP, Lipatov O, Im SA, et al. Pembrolizumab versus investigator‐choice chemotherapy for metastatic triple‐negative breast cancer (KEYNOTE‐119): a randomised, open‐label, phase 3 trial. Lancet Oncol 2021; 22: 499–511. [DOI] [PubMed] [Google Scholar]
- 16. Emens LA, Molinero L, Loi S, et al. Atezolizumab and nab‐paclitaxel in advanced triple‐negative breast cancer: biomarker evaluation of the Impassion130 study. J Natl Cancer Inst 2021; 113: 1005–1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kowanetz M, Zou W, Gettinger SN, et al. Differential regulation of PD‐L1 expression by immune and tumor cells in NSCLC and the response to treatment with atezolizumab (anti‐PD‐L1). Proc Natl Acad Sci U S A 2018; 115: E10119–E10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Merck Sharp & Dohme LLC, a subsidiary of Merck & Co., Inc., Rahway, NJ, USA (MSD) is committed to providing qualified scientific researchers access to anonymised data and clinical study reports from the company's clinical trials for the purpose of conducting legitimate scientific research. MSD is also obligated to protect the rights and privacy of trial participants and, as such, has a procedure in place for evaluating and fulfilling requests for sharing company clinical trial data with qualified external scientific researchers. The MSD data sharing website (available at: http://engagezone.msd.com/ds_documentation.php) outlines the process and requirements for submitting a data request. Applications will be promptly assessed for completeness and policy compliance. Feasible requests will be reviewed by a committee of MSD subject matter experts to assess the scientific validity of the request and the qualifications of the requestors. In line with data privacy legislation, submitters of approved requests must enter into a standard data‐sharing agreement with MSD before data access is granted. Data will be made available for request after product approval in the US and EU or after product development is discontinued. There are circumstances that may prevent MSD from sharing requested data, including country or region‐specific regulations. If the request is declined, it will be communicated to the investigator. Access to genetic or exploratory biomarker data requires a detailed, hypothesis‐driven statistical analysis plan that is collaboratively developed by the requestor and MSD subject matter experts; after approval of the statistical analysis plan and execution of a data‐sharing agreement, MSD will either perform the proposed analyses and share the results with the requestor or will construct biomarker covariates and add them to a file with clinical data that is uploaded to an analysis portal so that the requestor can perform the proposed analyses.