

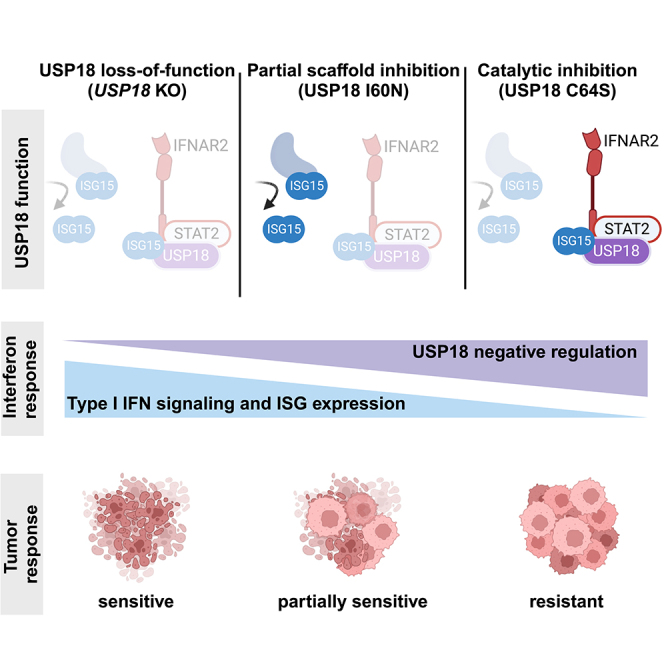
Summary
Precise regulation of Type I interferon signaling is crucial for combating infection and cancer while avoiding autoimmunity. Type I interferon signaling is negatively regulated by USP18. USP18 cleaves ISG15, an interferon-induced ubiquitin-like modification, via its canonical catalytic function, and inhibits Type I interferon receptor activity through its scaffold role. USP18 loss-of-function dramatically impacts immune regulation, pathogen susceptibility, and tumor growth. However, prior studies have reached conflicting conclusions regarding the relative importance of catalytic versus scaffold function. Here, we develop biochemical and cellular methods to systematically define the physiological role of USP18. By comparing a patient-derived mutation impairing scaffold function (I60N) to a mutation disrupting catalytic activity (C64S), we demonstrate that scaffold function is critical for cancer cell vulnerability to Type I interferon. Surprisingly, we discovered that human USP18 exhibits minimal catalytic activity, in stark contrast to mouse USP18. These findings resolve human USP18's mechanism-of-action and enable USP18-targeted therapeutics.
Subject area: Immunity, Immune response, Cell biology, Cancer
Graphical abstract
Highlights
-
•
USP18 is the primary negative regulator of Type I interferon signaling
-
•
In contrast to mouse USP18, human USP18 possesses weak enzymatic activity
-
•
USP18 acts chiefly through its scaffold function
-
•
Targeting USP18 scaffold function leads to cancer cell killing
Immunity; Immune response; Cell biology; Cancer
Introduction
Type I interferon (IFN) signaling is essential for antiviral defense and the regulation of cellular immunity.1,2 Engagement of the Type I IFN receptor, composed of IFNAR1 and IFNAR2, activates the JAK/STAT signaling cascade, resulting in the transcription of interferon-stimulated genes (ISGs). ISGs contribute to multiple pro-inflammatory pathways, including antigen processing and presentation, secretion of immunomodulatory chemokines and cytokines, and, ultimately, the induction of cell autonomous growth arrest and death.3,4,5 This coordinated inflammatory response eliminates infected and neoplastic cells, and can be harnessed therapeutically to promote anti-tumor immunity and tumor intrinsic growth arrest or death.6,7,8 If left unchecked, however, excessive pro-inflammatory signaling can damage healthy tissue, resulting in autoimmunity.9,10 To prevent excessive inflammation, Type I IFN signaling concomitantly induces negative feedback mediators such as USP18 (ubiquitin-specific protease 18) and PD-L1, which are expressed at undetectable or low levels under basal conditions.11,12,13
The central role of USP18 in IFN negative regulation is supported by human genetics.14,15,16,17 For instance, USP18 loss-of-function results in a severe interferonopathy called pseudo-TORCH syndrome 2 that limits lifespan to infancy.15,16 Affected individuals may exhibit microcephaly, intracranial calcifications, and features of severe systemic infection in the absence of a detectable infectious agent.15,16 Furthermore, the genetic ablation of USP18 in murine models results in hypersensitivity to Type I IFN.18,19,20 USP18 is thought to negatively regulate Type I IFN signaling by binding directly to STAT2, followed by recruitment to the IFN receptor subunit, IFNAR2, where it prevents downstream JAK1 signaling.21,22 Point mutations in USP18 (I60N) and STAT2 (R148Q and R148W) that disrupt this scaffold function cause a rare form of Type I interferonopathy in patients.17,23,24
In addition to the scaffold function, USP18 possesses proteolytic catalytic activity that cleaves the interferon-inducible ubiquitin-like protein, ISG15, from ISG15-conjugated (ISGylated) proteins.19,25 Like USP18, ISG15 and the enzymatic cascade required for ISGylation (E1 activating enzyme, UBA7; E2 conjugating enzyme, UBCH8/UBE2L6; E3 ligase HERC5) are ISGs and, therefore, tightly regulated upon IFNAR signaling.26 Since newly synthesized proteins are ISGylated co-translationally, ISGs are themselves ISGylated.27 Newly synthesized bacterial and viral proteins are also ISGylated if Type I IFN signaling is induced by infection.28,29,30,31
The functional consequences of ISGylation upon IFNAR engagement are not fully understood but have been best characterized in the context of viral infection. ISGylation of viral proteins is believed to block viral replication, resulting in decreased infectivity.29,30,31 While mice with impaired USP18 deISGylase activity have increased ISGylation and increased viral protection, mice lacking the E1 enzyme required for ISGylation, UBA7, exhibit decreased ISGylation and decreased viral protection.32,33,34 Furthermore, viruses such as SARS-CoV-2 have evolved deISGylases that efficiently cleave ISG15 from viral and endogenous proteins, which may serve as an immune evasion mechanism.31,35,36
In the absence of viral infection, it is less clear whether USP18 deISGylase activity can negatively regulate IFN signaling. The impact of ISGylation on endogenous proteins, including ISGs, has remained more elusive, especially in humans. Previous reports have suggested that ISGylation can alter protein stability by competing for ubiquitin-mediated degradation, or protein function by altering protein-protein interactions.31,37,38 Therefore, it has been proposed that USP18 catalytic activity can negatively regulate IFN signaling by removing ISGylation from pro-inflammatory ISGs, which in turn, reduces their levels.18,31,39,40 Our understanding of deISGylase activity has been hindered because USP18 loss-of-function studies cannot disambiguate the contribution of deISGylase and scaffold activity. USP18 scaffold function acts upstream to directly regulate the entire IFN signaling cascade, which includes the expression of USP18, ISG15, and the enzymatic cascade required for ISGylation. Since loss of scaffold function is predicted to increase ISG expression and ISGylation, changes correlated with increased ISGylation upon USP18 loss cannot be directly attributed to loss of catalytic function (Figure S1).
Given the crucial role of USP18 in immune regulation, it is of great interest to determine the mechanism by which it functions. Genetic ablation of USP18 in human cancer cells confers sensitivity to Type I IFN treatment, associated with an accumulation of ISGylated proteins, increased ISG expression, and cell autonomous growth inhibition and death.39,41 However, the relative contribution of USP18 catalytic versus scaffold function to the survival of diverse cancer lineages remains unknown. Published reports have implicated catalytic function because the modulation of USP18 increased the ISGylation of a wide range of proteins, including ISGs and tumor suppressors that can regulate tumor growth.39,42,43,44,45 For example, increased ISGylation of ISGs and the tumor suppressor p53 is hypothesized to increase tumor intrinsic IFN signaling or p53-dependent transcription, respectively, resulting in decreased tumor growth.42 Nevertheless, a causal relationship between deISGylase activity and cancer survival has not been established. To determine the relative contribution of scaffold and catalytic activity, it is necessary to develop tools that selectively impair either function, and to evaluate multiple cancer lineages.
We investigated the impact of USP18 deISGylase activity across a broad range of human tumors by introducing a deISGylation mutation into the endogenous USP18 locus. Complete loss of USP18 confers IFN sensitivity to multiple cancer cell lineages, yet strikingly, none are sensitive to loss of deISGylase activity. Furthermore, a patient-derived mutation that disrupts IFNAR2 binding, I60N, is sufficient to confer IFN sensitivity. Collectively these findings demonstrate that deISGylase activity does not repress IFNAR signaling or mediate tumor intrinsic IFN sensitivity. Insight into this key negative regulator of IFN signaling facilitates the development of therapeutics for cancer immunotherapy, acute and chronic viral infection, and autoinflammatory disorders.
Results
USP18 loss in murine CT26 tumors inhibits tumor growth in vivo
Type I IFN signaling induces the expression of ISGs that may inhibit tumor growth and promote anti-tumor immunity.6,7,8 Since USP18 is the key negative regulator of Type I IFN signaling, USP18 inhibition in tumors is predicted to increase sensitivity to endogenous Type I IFN and upregulate pro-inflammatory ISG pathways. For this proposed mechanism, tumor-intrinsic USP18 expression and local Type I IFN signaling in the tumor microenvironment (TME) are required for efficacy. To determine if USP18 loss can inhibit in vivo tumor growth in the presence of physiological Type I IFN concentrations, we genetically ablated murine Usp18 (mUsp18) in murine CT26 colorectal cancer cells (mUsp18−/− CT26). As a control, Rosa26 was genetically ablated in CT26 cells (Rosa26−/− CT26). To assess mUSP18 protein reduction in knock-out pools, we conducted proteomic analysis of expanded mUsp18−/−, Rosa26−/−, and parental WT CT26 cells prior to implantation. mUSP18 protein abundance substantially decreased by ≥ 50% in mUsp18−/− relative to Rosa26−/− and parental CT26 (Figure S2A). To functionally validate impaired mUSP18 expression in mUsp18−/− pools, we confirmed enhanced IFN-induced transcription of the ISGs Ifit1 and Isg15 compared to controls (Figure 1A). Furthermore, mUSP18 loss conferred IFN sensitivity to CT26 cancer cells in vitro. Although mUSP18 loss did not impact baseline cell viability in the absence of interferon, IFN treatment resulted in cell growth inhibition (Figures 1B and S2B) and increased apoptosis (Figure S2C). To determine the impact on tumor growth in vivo, mUsp18−/− or Rosa26−/− CT26 cells were subcutaneously injected in wild-type, immunocompetent BALB/c mice. Tumor volume was significantly reduced in BALB/c mice injected with mUsp18−/− CT26 tumors as compared to Rosa26−/− CT26 controls (Figures 1C–1E). Therefore, mUSP18 loss in CT26 tumors inhibits tumor growth in vivo.
Figure 1.

mUSP18 loss in CT26 tumors inhibits tumor growth in vivo
(A and B) In vitro characterization of mUsp18−/− and Rosa26−/− cancer cells prior to implantation. (A) Relative mRNA expression of Ifit1 (left) and Isg15 (right) to Gapdh measured by RT-qPCR after 72 h treatment with the indicated concentration of murine IFN-β. Each data point denotes the mean of n = 3 replicates ± SEM. (B) Confluence was normalized to 0 U/mL IFN control from each cell line.
(C–E) Tumor growth curves of (C,E) control Rosa26−/− and (D and E) mUsp18−/− CT26 tumors injected into wild-type BALB/c mice. Data are represented as (C and D) individual animals and (E) mean, n = 12 animals per genotype. (B) EC50 determinations for each cell line are presented in Table S1. (E) Results from nonlinear regression analysis are presented in Table S1 (slopes significantly different for each dataset, p < 0.0001). See also Figures S1 and S2.
Human USP18 loss confers interferon sensitivity across diverse cancer lineages
Previous work demonstrated that the genetic ablation of human USP18 (hUSP18) in leukemia-derived HAP1 and colorectal HCT116 cancer cells confers sensitivity to IFN treatment.39 IFN sensitivity was associated with the accumulation of ISGylated proteins, increased ISG expression, and ultimately cell growth inhibition and death.39 To determine if hUSP18 loss confers IFN sensitivity to human cancer cell lines from diverse lineages, we applied the CRISPR LAPSE method developed at Pfizer (Clustered Regularly Interspaced Short Palindromic Repeats Longitudinal Assay Profiling of Specific Edits, Tuladhar and Oyer, in preparation) (Figure 2A). Using this approach, hUSP18 knock-out (KO) pools were generated and divided into three sub-pools. One sub-pool was sequenced for initial KO efficiency, another was passaged in the presence of IFN, and a third was passaged under control conditions. After passaging, hUSP18 was sequenced, and the proportion of KO to wild-type (WT) alleles was compared to the initial time point and control conditions, thereby indicating whether hUSP18 KO resulted in IFN sensitivity (Figure 2A). Consistent with data collected from hUSP18 KO HAP139 and HCT116 clones (Figures 2B and 2C), KO allelic frequency was depleted compared to WT after IFN treatment in HAP1 and HCT116 LAPSE pools (Figure 2D). Furthermore, we observed a decrease in hUSP18 KO allelic frequency after 2 weeks of IFN treatment in HT-29 (colorectal), A549 and HCC366 (lung), and HCC1143 (breast) LAPSE pools (Figure 2D). hUSP18 loss did not impact baseline cell viability in the absence of interferon. Thus, the genetic ablation of hUSP18 confers IFN sensitivity to multiple cancer cell lines, including those derived from hematopoietic, colorectal, lung, and breast lineages.
Figure 2.

hUSP18 loss confers IFN sensitivity across diverse cancer lineages
Parental human cancer cell lines were electroporated with sgRNA targeted to hUSP18 and allelic frequency was determined by sequencing samples 72 h post-electroporation (time initial) and after 2 additional weeks of passaging cells (time final) in the presence (+) or absence (−) of human 1000 U/mL IFN-α. After 2 weeks of passaging cells, the relative ratio of KO to WT alleles at time final +IFN vs. time final -IFN indicates whether hUSP18 KO results in IFN sensitivity.
(A) Schematic of experimental design (created with BioRender.com).
(B and C) Growth rate of HCT116 wild-type (WT) and hUSP18 KO HCT116 cancer cells ±1000 U/mL human IFN-α added at time = 0 h. Each data point denotes n = 3 replicates; mean ± SEM.
(D) KO indicates frameshift mutation or in-frame mutation ≥21 bp; WT indicates no mutation or in-frame mutation <21 bp. Two independent biological replicates (top and bottom rows) were electroporated, passaged, and sequenced in parallel.
C64S impairs human USP18 catalytic function
To determine whether loss of USP18 catalytic or scaffold functions, or both, contributed to IFN sensitivity, we introduced single amino acid substitutions predicted to specifically impair each function. Mutation of the catalytic cysteine, C64, to serine (C64S) is predicted to disrupt the ability of hUSP18 to cleave human ISG15 (hISG15) from conjugated proteins while minimizing impact on tertiary protein structure. There is only a single atom change in which the hydroxyl group (-OH) of serine replaces the thiol group (-SH) of cysteine.17,46,47,48,49 A substitution of isoleucine at residue 60 to asparagine (I60N) causes Type I IFN-mediated autoimmunity in patients.17,50 The I60N mutation partially inhibits the hUSP18 scaffold function that represses IFNAR signaling while sparing deISGylase activity.17,50,51 While C64S and I60N mutations have been characterized in the literature, the functional impact of these mutations has not been directly compared using recombinant proteins or endogenously edited cell lines.
To measure hUSP18 catalytic activity independently of its scaffold function, we purified recombinant WT, C64S, and I60N hUSP18 protein for in vitro assays. Given that biochemical properties of recombinant mUSP18, but not hUSP18, have been previously characterized,25,52 we also purified mUSP18 as a positive control to benchmark in vitro assays. To determine the quality and purity of recombinant hUSP18 and mUSP18, we performed size exclusion chromatography and intact mass spectrometry and confirmed that the catalytic cysteine of hUSP18 or mUSP18 was available for conjugation to its species appropriate ISG15-PA probe (Figures S3A–S3F).52 Finally, we validated protein-protein interactions between hUSP18 and hISG15. Complex formation of purified hUSP18 and hISG15 was observed by size exclusion chromatography (Figure S3G); bio-layer interferometry experiments indicated a tight binding affinity between the two proteins (KD = 40 nM) (Figures 3A and 3B).
Figure 3.

C64S and I60N, respectively, disrupt hUSP18 catalytic and scaffold functions
(A and B) Biotinylated hUSP18 was immobilized to a streptavidin biosensor and incubated with the indicated concentration of recombinant hISG15. Protein association and dissociation were sequentially monitored for 10 min each. (B) KD = 40 nM was determined by fitting the instrumental response after association (measured in nm wavelength shift) to a standard single-site binding equation as a function of hISG15 concentration.
(C) KM(apparent) and VMAX(apparent)/[Enzyme concentration] determinations for hUSP18 (WT, I60N, or C64S) vs. hISG15-Rho110. The reported values are averaged from 2 determinations at different enzyme concentrations and rounded to 2 significant digits. N.D., not determined; signal below the level of detection.
(D) Progression curve of 1000 nM hISG15-Rho110 cleavage by 10 nM hUSP18 (WT or C64S) or no enzyme control (buffer) at RT.
(E) Cleavage of 5 μM pro-hISG15 (AA1-165) to mature hISG15 (AA1-157) was assessed by SDS-PAGE after 10 min incubation at 37°C with 1 μM of recombinant hUSP18 (WT, C64S, C64S/C64S, or I60N) or no enzyme control (−).
(F) HAP1 hUSP18 KO cells were treated for 24 h with IFN-α prior to cell lysis and lysates were incubated with 1, 10, 100, or 1000 nM of indicated recombinant protein for 1 h at RT. Lysates were analyzed by western blot for levels of ISGylated proteins and hISG15 (top) or vinculin (bottom).
(G and H) Ratio of phosphorylated STAT1 (pSTAT1) to total STAT1 levels measured by HTRF in individual HAP1 hUSP18 KO and (A) hUSP18 C64S KI or (B) hUSP18 I60N KI clones compared to parental HAP1 cells (WT). Cells were treated with 1000 U/mL human IFN-α for 4 h (IFN prime), followed by 24 h rest, and subsequent 15 min IFN-α re-stimulation (IFN pulse) at the indicated concentration prior to cell lysis (mean ± SEM, n = 3 replicates). (G) Unedited control clone underwent the same electroporation conditions as C64S KI clones, but no editing was observed at the endogenous hUSP18 locus. Results from the two-way ANOVA test with Tukey’s multiple comparisons are presented in Table S1. See also Figure S3.
USP18 exhibits peptidase19 and isopeptidase19,25 activity for ISG15 substrates, and the kinetics of mUSP18 against fluorogenic mISG15 substrate analogues have been described in the literature.25 To assess USP18 catalytic function, we incubated recombinant USP18 with recombinant ISG15 protein conjugated via a peptide bond to a Rhodamine 110 dye (ISG15-Rho110) on its C-terminus. Rho110 is quenched when conjugated to ISG15, and fluorescence increases upon cleavage. We first benchmarked the ISG15-Rho110 substrates by confirming the ability of WT mUSP18 to cleave mouse ISG15-Rho110 (mISG15-Rho110) and human ISG15-Rho110 (hISG15-Rho110) (Figures S3H and S3I). We next assessed the ability of hUSP18 variants to cleave hISG15-Rho110. WT and I60N hUSP18 exhibited comparable activity (Figure 3C), confirming that I60N does not impair catalytic activity.17 Enzymatic activity of C64S hUSP18 could not be quantified because no significant cleavage of hISG15-Rho110 was observed during the assay time course (Figure 3D). Therefore, the C64S substitution substantially reduced hUSP18 catalytic activity.
Surprisingly, WT and I60N hUSP18 cleaved hISG15-Rho110 less efficiently than mUSP18 cleaved mISG15-Rho110 (Figures 3C and S3H). During the assay time course of 80 min, enzymatic turnover of hISG15-Rho110 substrate by WT hUSP18 did not reach steady state. We hypothesized that differential catalytic activity across species may be related to substrate affinity.53 Although the catalytic site of USP18 is well conserved between mice and humans, ISG15 sequences vary.52,54 To compare interactions between USP18 and ISG15, we performed a competitive inhibition experiment with cross-species enzyme/substrate pairs. We titrated in non-fluorescent full-length hISG15 or mISG15 into a solution containing fixed enzyme concentrations of: (1) either hUSP18 or mUSP18 and (2) fluorescent substrate (mISG15-Rho110). The ability of non-fluorescent ISG15 substrate to compete with fluorescent mISG15-Rho110 substrate is a proxy for affinity between non-fluorescent ISG15 and USP18. hISG15 potently inhibited cleavage of mISG15-Rho110 by hUSP18, but not by mUSP18 (Figures S3J and S3K). mISG15 was not able to compete as efficiently with mISG15-Rho110 for both hUSP18 and mUSP18 in the reciprocal experiment (Figures S3J and S3K). These results demonstrate a surprisingly high affinity between hUSP18 and hISG15 compared to mUSP18 and mISG15. Therefore, species-specific properties for these enzyme and substrate pairs should be accounted for when comparing results obtained in human or mouse systems. Finally, WT and I60N hUSP18 were able to turnover mISG15-Rho110 (Figure S3H), demonstrating the enzyme preparations were kinetically competent with this lower affinity substrate.53 C64S hUSP18 did not cleave detectable amounts of mISG15-Rho110 (Figure S3I). Therefore, the C64S substitution effectively impairs the ability of hUSP18 to cleave both human and mouse ISG15-Rho110.
Furthermore, USP18 has peptidase catalytic activity enabling it to cleave pro-human ISG15 (pro-hISG15, AA 1–165) into mature human ISG15 (mature hISG15, AA 1–157).19 hUSP18 is sufficient to cleave pro-hISG15 in vitro, but hUSP18 is not essential for cleavage in a cellular context because mature hISG15 is observed in hUSP18 KO cell lysates. We evaluated how hUSP18 variants impact pro-hISG15 cleavage by incubating 1 μM of WT, C64S, C64S/C65S, or I60N hUSP18 with 5 μM of recombinant pro-hISG15 for 3 or 10 min. Pro-hISG15 cleavage was assessed by SDS-PAGE and mature hISG15 was detected after incubation with WT and I60N, but not C64S or C64S/C65S, hUSP18 (Figure 3E). Pro-hISG15 cleavage was not observed in the absence of recombinant hUSP18 protein (Figure 3E). The expected mass for pro- and mature hISG15 protein products was confirmed by mass spectrometry (Figures S3L and S3M).
Building on these results, we next assessed the deISGylation of endogenous ISGylated proteins by hUSP18. Since the deISGylase activity of recombinant hUSP18 has not yet been described, we first established positive controls for this in vitro deISGylase assay by incubating recombinant mUSP18 or PLpro, the SARS-CoV-2 deISGylase and deubiquitinase,35 with endogenously ISGylated proteins obtained from human and mouse cells. To induce the endogenous ISGylation of human or mouse proteins, hUSP18 KO human HAP1 cells or mUsp18 KO mouse CT26 cells, respectively, were treated with 1000 U/mL IFN for 24 h. A decrease in human and mouse ISGylated proteins was observed upon incubation with ≥10 nM of WT mUSP18 and SARS-CoV-2 PLpro, thus confirming deISGylase activity can be observed for the positive controls (Figures 3F and S3N). In agreement with the hISG15-Rho110 assay, WT hUSP18 was less efficient at deISGylating human substrates compared to WT mUSP18 and SARS-CoV-2 PLpro (Figure 3F). Yet the deISGylase activity of WT hUSP18 remained inefficient against endogenously ISGylated mouse proteins (Figure S3N), suggesting that WT hUSP18 may have distinct catalytic properties when ISG15 is conjugated via peptide (mISG15-Rho110) or isopeptide (endogenously ISGylated mouse proteins) bonds.
When deISGylase activity was compared across WT, C64S, and I60N hUSP18 variants, decreased ISGylated substrate levels were observed upon incubation with 1000 nM WT or I60N hUSP18 (Figure 3F). Although I60N hUSP18 was active in all three in vitro assays, we observed a further decrease in endogenous ISGylated protein levels upon incubation with 1000 nM WT hUSP18 as compared to incubation with 1000 nM I60N hUSP18 (Figure 3F). In agreement with the hISG15-Rho110 and pro-hISG15 assay, incubation with C64S hUSP18 did not alter deISGylation levels (Figure 3F). Together these data demonstrate that the C64S substitution impairs the established peptidase and isopeptidase catalytic functions of hUSP18.
I60N impairs human USP18 scaffold function
Having determined catalytic function with purified proteins, we next introduced C64S and I60N single amino acid mutations into the endogenous hUSP18 locus to create C64S or I60N hUSP18 knock-in (KI) cells. In its scaffold capacity, hUSP18 binds to IFNAR2 and attenuates IFNAR signaling, resulting in the decreased phosphorylation of STAT1 and STAT2 downstream of Type I IFN stimulation. To quantify the scaffold activity of hUSP18 variants, we applied Homogeneous Time-Resolved Fluorescence (HTRF) assays and measured the ratio of phosphorylated STAT1 (pSTAT1) to total STAT1 levels as a readout of IFNAR signaling. Cells were pre-treated with 1000 U/mL of IFN for 4 h, washed, and rested overnight to induce ISGs, including USP18, STAT2, and ISG15. Cells were then re-stimulated with a pulse of IFN for 15 min, and pSTAT1 and total STAT1 were measured. hUSP18 KO cells demonstrated a complete loss of scaffold function, as shown by the increase in pSTAT1/total STAT1 levels compared to WT (Figures 3G and 3H). While partial scaffold deficits were observed in I60N KI cells, C64S KI cells showed no deficits in scaffold function compared to WT hUSP18 (Figures 3G and 3H). Surprisingly, C64S KI cells exhibited a modest enhancement of scaffold function (Figure 3G). Since USP18 expression is induced post-IFN stimulation, no differences in pSTAT1/total STAT1 levels were observed between WT, hUSP18 KO, and KI cells without IFN pre-treatment (Figures 3G and 3H).
Together, these experiments demonstrate that the C64S substitution impairs catalytic function and the I60N patient-derived substitution partially impairs scaffold function. Therefore, C64S and I60N substitutions are effective tools to interrogate the impact of inhibiting catalytic or scaffold activity, respectively, in cancer cells.
Disrupting deISGylation is not sufficient to confer interferon sensitivity
It is unknown which functional activities of hUSP18 contribute to the increased ISG induction, ISGylation, and cell growth inhibition upon the IFN treatment of hUSP18 KO cells. Thus, we assayed C64S and I60N KI hUSP18 cells to determine if preventing deISGylation is sufficient to phenocopy hUSP18 KO cells. Upon 48 h of 1000 U/mL IFN treatment, ISGylated protein levels and cell growth inhibition were significantly reduced in C64S KI compared to hUSP18 KO cells (Figures 4A and 4C). Although WT and C64S KI hUSP18 cells exhibited similar phenotypes, C64S KI cells had a modest decrease in ISGylation and IFN sensitivity compared to WT cells (Figures 4A and 4C). Decreased IFN sensitivity in C64S KI cells may be due to the small enhancement of scaffold function observed in the STAT1 HTRF assay (see Figure 3G).50 Therefore, preventing deISGylation is not sufficient to phenocopy hUSP18 KO cells. I60N KI cells, however, exhibited intermediate levels of ISGylation and cell growth inhibition compared to hUSP18 KO and WT cells (Figures 4B and 4C). These results indicate a correlation between impaired scaffold function and increased IFN sensitivity. However, we cannot formally exclude the possibility that I60N disrupts additional undefined scaffold functions of USP18 beyond IFNAR2 repression.17,50,51 Nonetheless, impairing the deISGylase activity of USP18 is not sufficient to confer IFN sensitivity.
Figure 4.

Disrupting deISGylation is not sufficient to confer IFN sensitivity
(A–C) Individual HAP1 hUSP18 KO (KO) and (A and C) hUSP18 C64S KI or (B and C) hUSP18 I60N KI clones were compared to parental HAP1 cells (WT). (A and B) Whole-cell lysates were analyzed by western blot for levels of ISGylated proteins and hISG15 (top) or vinculin and hUSP18 (bottom). Cells were treated with 1000 U/mL human IFN-β for 24 h prior to cell lysis. (C) Viability of hUSP18 C64S KI (top) or I60N KI (bottom) cells was measured after 72 h treatment with the indicated concentration of human IFN-α. Viability was normalized to 0 U/mL IFN control for each cell line. Each data point denotes mean of n = 2 replicates ± SEM. EC50 determinations for each cell line are presented in Table S1. See also Figure S1.
Disrupting the ISGylation pathway does not rescue interferon sensitivity
If deISGylase activity is not implicated in IFN sensitivity, hUSP18 KO cells with defects in the ISGylation pathway should remain sensitive to IFN. To this end, we genetically ablated the human E1 ligase required for ISGylation, UBA7 (Ubiquitin-Like Modifier Activating Enzyme 7), in WT and hUSP18 KO cells to generate UBA7 KO and UBA7/USP18 double KO (UBA7/USP18 dKO) HAP1 human cells (Figures 5A and 5B). As expected, a substantial decrease in ISGylation was observed in UBA7 KO and UBA7/USP18 dKO cells upon 24 h 1000 U/mL IFN treatment (Figure 5A). Furthermore, UBA7/USP18 dKO cells remained sensitive to IFN and induced ISG expression at levels comparable to hUSP18 KO cells (Figures 5C and 5D).55 No significant differences in ISG induction or IFN sensitivity were observed between UBA7 KO cells and WT (Figures 5C and 5D). By genetically ablating the E1 ligase required for ISGylation, UBA7, we demonstrate that loss of ISGylation does not rescue IFN sensitivity upon hUSP18 KO. These results provide orthogonal evidence that the deISGylase activity of USP18 is dispensable for IFN sensitivity.
Figure 5.

Disrupting the ISGylation pathway does not rescue IFN sensitivity
UBA7 KO pools and UBA7/USP18 double KO (dKO) pools were compared to USP18 KO clone and parental HAP1 cells (WT).
(A) Whole-cell lysates were analyzed by western blot for levels of ISGylated proteins and ISG15 (top) or vinculin (bottom). Cells were treated with 1000 U/mL human IFN-β for 24 h prior to cell lysis.
(B) Schematic of the enzymatic cascade required for ISGylation (UBA7, UBCH8/UBE2L6, and HERC5) and deISGylation (USP18), created with BioRender.com.
(C) Cell confluence and (D) relative mRNA expression of IFIT1 (left) or ISG15 (right) to GAPDH measured by RT-qPCR after 48 h treatment with the indicated concentration of human IFN-β. Each data point denotes mean of n = 3 replicates ± SEM. EC50 determinations for each cell line are presented in Table S1. (C) Confluence was normalized to 0 U/mL IFN control from each cell line.
Inhibiting human USP18 deISGylase activity does not confer interferon sensitivity across diverse cancer lineages
To determine if deISGylation is dispensable for IFN sensitivity across multiple human cancer cell lines, we performed CRISPR LAPSE experiments with C64S knocked-in to the endogenous hUSP18 locus of blood (HAP1), breast (HCC1143, HCC1954, SKBR3, and MDA-MB-468), colorectal (HCT116 and HT-29), and lung (A549, NCI-H1650, and HCC366) human cancer cells. Initial C64S KI and KO efficiency in pools were measured 72 h after electroporation using genomic DNA PCR prior to 2 weeks ± IFN treatment. Consistent with the observed IFN sensitivity in HAP1 and HCT116 hUSP18 KO clones, the frequency of HAP1 and HCT116 hUSP18 KO cells decreased upon IFN stimulation in LAPSE experiments (Figures 6A and S4). Although the frequency of WT was relatively stable between HAP1 pools with or without IFN treatment, the frequency of C64S increased upon IFN treatment, in line with the observation that C64S KI cells have a modest decrease in IFN sensitivity compared to WT cells (Figures 6A and S4). The modest enhancement of C64S scaffold function observed upon brief (15 min) IFN pulses in HTRF experiments may cumulatively confer significant protection during prolonged (2 weeks) IFN selective pressure. KO cells were depleted as compared to WT upon IFN stimulation across all pools tested except for MDA-MB-468 (Figures 6A and S4). MDA-MB-468 cells contain a single nucleotide polymorphism splice variant in STAT2, which may alter Type I IFN signal transduction and/or the scaffold interaction between USP18 and STAT2 required for IFNAR repression. Importantly, the frequency of C64S cells did not decrease across all pools tested (Figures 6B and 6C). In fact, as with HAP1 cells, the frequency of C64S increased in most cancer cell lines treated with IFN as compared to no IFN controls (Figures 6 and S4). Therefore, inhibiting deISGylation is not sufficient to confer IFN sensitivity across multiple cancer cell lines, including those derived from blood, colorectal, breast, and lung lineages.
Figure 6.

deISGylation does not mediate IFN sensitivity across diverse cancer lineages
(A) Parental human cancer cell lines were electroporated with C64S donor oligo and sgRNA targeted to hUSP18. Allelic frequency was determined by sequencing samples 72 h post-electroporation (time initial) and after 2 additional weeks of passaging cells (time final) in the presence (+) or absence (−) of 1000 U/mL human IFN-α. KO indicates frameshift mutation or in-frame mutation ≥21 bp; WT indicates no mutation or in-frame mutation <21 bp; KI indicates donor oligo integration at the appropriate position. An independent biological replicate was electroporated, passaged, and sequenced in parallel (Figure S4).
(B and C) Summary of data presented in (A) and Figure S4. (B) Cumulative percent allelic frequency for each genotype (C64S KI, USP18 KO, WT) at the indicated time point, summed across all cell lines and biological replicates. (C) Percent allelic frequency for each genotype at time final -IFN was subtracted from time final +IFN. Positive or negative values indicate an increase or decrease, respectively, in allelic frequency after IFN treatment. Each square denotes the mean of 2 biological replicates. See also Figure S4.
Inhibiting murine USP18 deISGylase activity does not reduce tumor growth in vivo
Our results thus far demonstrate that hUSP18 exhibits limited catalytic activity toward hISG15 and catalytic inhibition does not confer IFN sensitivity across human cancer lineages. To determine whether catalytic inhibition is sufficient to inhibit tumor growth in vivo, we generated catalytically inactive mUSP18 CT26 clones. Given that mouse C61 is the catalytic cysteine that corresponds to human C64,19 serine was introduced into C61 at the endogenous mUsp18 locus to complement the C64S KI hUSP18 cell lines.
To bridge human cancer cell line IFN sensitivity with in vivo mouse syngeneic tumor studies, it was necessary to determine deISGylase activity and IFN sensitivity in C61S KI mUSP18 cells using the same assays as C64S KI hUSP18 cells. Since the in vitro deISGylase activity of hUSP18 was minimal, a significant accumulation of ISGylated proteins was not necessarily expected upon catalytic inhibition in C64S KI hUSP18 cells (Figure 4A). However, the catalytic inhibition of robust mUSP18 deISGyalse activity is predicted to increase ISGylated protein accumulation in C61S KI mUSP18 cells.32 We observed the expected increase in ISGylated protein and concomitant decrease in free ISG15 in C61S KI mUSP18 cells treated with IFN (Figures 7A and S5A), supporting the hypothesis that hUSP18 may be less catalytically active than mUSP18. We next determined the contribution of mUSP18 catalytic activity to IFN sensitivity. Like C64S KI hUSP18 cells, C61S KI mUSP18 cells were not sensitive to IFN in vitro (Figures 7B and S5B). Therefore, the increased ISGylation observed in C61S KI mUSP18 cells is not sufficient to confer IFN sensitivity to CT26 cancer cells.
Figure 7.

Inhibiting deISGylase activity is not sufficient to confer IFN sensitivity
Individual unedited control (unedited), mUSP18 C61S KI (C61S), and mUsp18 KO (KO) clones were compared to parental CT26 cells (WT). Unedited control clones underwent same electroporation conditions as C61S KI and KO clones, but no editing was observed at the endogenous mUsp18 locus.
(A) Cells were treated with 1000 U/mL murine IFN-α for 24 h prior to cell lysis and whole-cell lysates were analyzed by western blot for levels of mouse ISGylated proteins and free mISG15 (top) or vinculin (bottom).
(B) Cell viability (left) and confluence (right) were measured after 72 h or 84 h treatment with the indicated concentration of murine IFN-α, respectively. Viability and confluence were normalized to 0 U/mL IFN control for each cell line. Each data point denotes a mean of n = 2 replicates ± SEM. EC50 determinations for each cell line are presented in Table S1.
(C and D) Tumor growth curves of WT, C61S, or KO CT26 tumors injected into wild-type BALB/c mice. For each genotype, 3 individual clones were injected with n = 10 animals per clone (n = 30 animals per genotype). Data are represented as (C) individual animals and (D) mean tumor growth was first determined for each clone, and data for each genotype are represented as the mean of 3 clones ± SEM. Results from nonlinear regression analysis are presented in Table S1.
(E) Wild-type USP18 can negatively regulate Type I IFN responses through its catalytic function to deISGylate ISG15-conjuagated proteins and its scaffold function to repress IFNAR signaling by displacing JAK1. Negative IFN regulation by wild-type USP18 protects cancer cells from Type I IFN sensitivity. Complete loss-of-function mutations in USP18 confer sensitivity to Type I IFN, resulting in enhanced ISG induction, ISGylation, and cancer cell growth inhibition. Mutations in USP18 C64S (human) or C61S (mouse) disrupt deISGylase activity, but are not sufficient to confer IFN sensitivity across cancer lineages. However, partial impairments in scaffold function in human USP18 I60N mutants results in partial IFN sensitivity and an intermediate phenotype between WT and hUSP18 KO. Model created with BioRender.com. See also Figures S5–S7.
To determine the impact on tumor growth in vivo, WT, KO, and C61S KI mUSP18 CT26 tumors were subcutaneously injected in wild-type, immunocompetent BALB/c mice. 3 individual clones of each genotype were injected to control for clone-specific phenotypes, and the average tumor growth across the 3 clones was compared to determine genotype-specific effects. In agreement with our first in vivo study with mUsp18−/− pools (Figures 2C and 2D), tumor volume was significantly reduced in BALB/c mice injected with mUsp18 KO tumors as compared to WT controls (Figures 7C and 7D. Reduced tumor growth was not observed in C61S KI mUSP18 tumors, indicating the catalytic activity is not essential for tumor growth inhibition observed in mUsp18 KO tumors (Figures 7C and 7D). Although mUSP18 and hUSP18 have distinct catalytic properties, the inhibition of deISGylase activity is not sufficient to confer IFN sensitivity to cancer cells of either species or inhibit in vivo tumor growth in CT26 syngeneic mouse models (Figure 7E).
Discussion
USP18 is a key negative regulator of Type I IFN signaling, yet the relative contribution of catalytic versus scaffold function has not been firmly established. Published reports have implicated either activity, complicating the development of therapeutics targeting USP18.39,42,43,44,45,56 Here, we demonstrate that deISGylase activity is not required to suppress Type I IFN signaling in human cancer cells and mouse syngeneic tumor models (Figure 7).
Prior work demonstrated that the ectopic expression of hUSP18 C64R/C65R mutants could not rescue IFN sensitivity in HAP1 hUSP18 KO cells.39 However, the contribution of hUSP18 catalytic versus scaffold activity to IFN sensitivity could not be definitively determined in this system because the bulky double arginine substitutions may cause structural changes that impair hUSP18 scaffold function in addition to catalytic function. For example, cells expressing C64R/C65R exhibited increased ISG expression, suggesting impaired scaffold function.39 Furthermore, we reasoned that overexpression using an exogenous promoter would lack the contemporary regulation of other IFN-stimulated genes, which may be important to fully recapitulate hUSP18 function. In this study, we introduced a deISGylation-specific mutation into the endogenous hUSP18 locus. These results clearly demonstrate that while a broad range of cancer cell types are sensitive to IFN upon the genetic ablation of hUSP18, none are sensitive to loss of deISGylase activity.
Our biochemical results indicate that hUSP18 inefficiently deISGylates its endogenous substrates. In this context, it may not be surprising that deISGylase activity is dispensable for IFN sensitivity in human cancer cells. The contribution of deISGylase activity may differ in mice and other species because mUSP18 efficiently cleaves mISG15 from substrate proteins.25,32 Nevertheless, the deletion of mISG15 did not rescue Type I IFN hypersensitivity phenotypes in Usp18−/− mice.20 Species-specific differences in deISGylase activity may reflect evolutionary adaptations to host-pathogen interactions and contribute to divergent viral susceptibility phenotypes observed with ISG15 loss-of-function mutations.53 Although ISG15 is weakly conserved across species, the catalytic site of USP18 is well conserved.52,54 Species-specific sequence differences at the USP18-ISG15 interface may impact binding affinity and the ability of USP18 to cleave ISG15 (Figures S6, S7A, and S7B). Meanwhile, the conservation of the USP18 catalytic triad may indicate that the residues required for catalytic function are also required for proper tertiary structure and/or scaffold function(s). Alternatively, hUSP18 may have other protein substrates, in addition to conjugated ISG15, for which it is a more efficient enzyme. Indeed, other species, such as birds, do not appear to encode ISG15 in their genomes, although USP18 is conserved.57,58
Since ISGylation is tightly coupled to the scaffold role of USP18, caution must be taken when dissecting the catalytic and scaffold roles of USP18. IFN signaling induces the expression of ISGs, including all the machinery required for ISGylation (UBA7, UBCH8/UBE2L6, HERC5, and ISG15), and ISG15 conjugation occurs concurrently with protein translation.26,27 Therefore, when USP18 scaffold function is lost, more ISGs are synthesized, and these become ISGylated upon translation. Hence, loss of scaffold function results in a large increase of ISGylated proteins, many of which are ISGs themselves (Figure S1). At the same time, loss of deISGylase function can also lead to increased ISGylated proteins. In this study, we designed in vitro assays with purified recombinant protein to directly assess deISGylase activity without the confound of increased ISGylation downstream of impaired scaffold function. Our results demonstrate that I60N retains deISGylase activity, and the observed accumulation of ISGylated proteins can be decoupled from the loss of catalytic function.
Partial impairments in scaffold function by the I60N mutation were correlated with intermediate levels of ISGylation and IFN sensitivity, implicating scaffold function as the primary mechanism by which USP18 loss promotes tumor intrinsic growth arrest and cell death. The I60N mutation has been shown to disrupt USP18-ISG15-STAT2 protein-protein interactions required for scaffold function in humans.17,50,51 To help clarify the structural basis of these changes, we modeled the USP18-ISG15-STAT2 complex with AlphaFold. In this model, I60N is located near the USP18-STAT2 interface (Figures S7C and S7D), which is critical for scaffold function.23,24 Due to species-specific differences in USP18-ISG15-STAT2 protein-protein interactions, the analogous mutation in mUSP18, I57, may not result in a similar scaffold impairment.53 Future genome-wide CRISPR screening strategies paired with endogenous IPs of WT, catalytic, and scaffold mutants may help identify effector pathways and protein-protein interactions mediating USP18-dependent IFN sensitivity across cancer types. It will be of great interest to determine whether a universal effector pathway downstream of USP18 is conserved across lineages, or if USP18 can regulate multiple effector pathways that converge on tumor intrinsic growth arrest and cell death. A deeper mechanistic understanding will help predict which patients will respond to USP18 inhibition and could explain variable responses across cancer types in mouse syngeneic tumor models.41,59
Future work will be needed to define the patient stratification strategy for USP18-targeted therapies. Prior analysis of patient databases identified cancer types in which lower USP18 expression correlated with improved patient outcomes, suggesting that inhibiting USP18 may be beneficial in certain patient populations.41,60 It is reasonable to hypothesize that patients with high tumor USP18 expression would be most likely to respond to USP18 inhibition. USP18-high tumors may be more sensitive both because they express the target and because they demonstrate local IFN signaling, as implied by the expression of the prominent ISG, USP18. For many cancer types examined, USP18 expression was higher in primary cancer samples when compared to corresponding noncancerous tissue controls.41,60 Accurate patient stratification will likely require understanding the dynamic relationship between pro-inflammatory ISGs, such as CXCL10, and negative regulatory ISGs, such as USP18 and PD-L1.
While USP18 scaffold inhibition holds great therapeutic potential, it will be critical to determine what degree of inhibition is both safe and effective. Complete USP18 loss-of-function causes severe interferonopathy in humans.15,16 Therefore, complete inhibition of USP18 would not be desirable. However, individuals with partial loss of USP18 function can present with less severe symptoms, despite enhanced IFN sensitivity and ISG expression. This includes patients with the I60N mutation,17 as well as patients with ISG15 loss-of-function, which results in reduced USP18 protein expression.14,17,61 Consequently, it may be feasible to identify a safe therapeutic window for USP18 inhibition that would achieve efficacy without unmanageable immune-related adverse events.10
Understanding the regulation of Type I IFN signaling has profound implications for identifying treatments for a broad range of pathophysiological conditions. Type I IFN signaling is well-established as a critical defense against viral infection and malignant cells,1,2 and a mediator of immune homeostasis and physiological function.8,62 On the other hand, the dysregulation of Type I IFN signaling can result in severe autoimmune disease and has been linked to immunotherapy resistance.6,7,8,9 This work galvanizes our mechanistic understanding of USP18, the key regulator of Type I IFN signaling in human cells, which will facilitate USP18-targeted therapeutics to improve patients’ lives.
Limitations of the study
In this study, we demonstrated that USP18 I60N mutants exhibit partial impairments in scaffold function and partial IFN sensitivity. However, I60N could disrupt additional functions of hUSP18 that have yet to be described. For example, recent studies have described additional protein-protein interactions with NEMO in the absence of exogenous IFN and with STAT1, OAS3, MX1, and ROBO1 upon IFN stimulation.63,64 In leukemia-derived THP-1 monocytic cells, nuclear hUSP18 interacts with IRF9 and STAT2 to regulate non-canonical ISG expression and pyroptosis.41 It is unknown if these interactions are conserved across cancer cell lines. Future studies with STAT2 R148W and R148Q patient-derived mutations23,24 will provide an orthogonal approach to interrogate USP18 scaffold contribution without directly manipulating USP18 sequences.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-hISG15 (7H29L24) | ThermoFisher | Cat# 703131; RRID:AB_2784562 |
| Rabbit anti-hUSP18 (D4E7) | Cell Signaling | Cat# 4813S; RRID:AB_10614342 |
| Rabbit anti-Vinculin | Abcam | Cat# Ab129002; RRID:AB_11144129 |
| Rabbit anti-mISG15 (E5B3U) | Cell Signaling | Cat# 89771 |
| Rabbit FAB anti-mUSP18 (#5080) | BioRad | Cat# AbD55080sco-1 |
| Bacterial and virus strains | ||
| Escherichia coli BL21(DE3) | Thermo Fisher | Cat# EC0114 |
| Bac-to-Bac Baculovirus | Invitrogen | Cat# 10359016 |
| Chemicals, peptides, and recombinant proteins | ||
| Human IFN-alpha 2b | PBL Assay Science | Cat# 11105-1 |
| Mouse IFN-alpha A | PBL Assay Science | Cat# 12100-1 |
| Mouse IFN-β | PBL Assay Science | Cat# 12410-1 |
| Human IFN-β1a | PBL Assay Science | Cat# 11410-2 |
| Human ISG15-Rho110 | South Bay Bio | Cat# SBB-PS0002 |
| Mouse ISG15-Rho110 | UbiQ Bio BV | Cat# UBIQ-127 |
| Recombinant pro-human ISG15 (AA1-165) | This paper | N/A |
| Recombinant human USP18 (WT, C64S, C64S/C65S, I60N) | This paper | N/A |
| Recombinant mouse USP18 WT | This paper | N/A |
| Annexin V green dye | Sartorius | Cat# 4642 |
| Cas9 | Thermo Fisher | Cat# A36498 |
| Protease inhibitor cocktail | Sigma | Cat# 56619600 |
| Pierce universal nuclease | Thermo Fisher | Cat# 88702 |
| HDR Enhancer V2 | IDT | Cat# 10007910 |
| Human ISG15(79-C)-propargylamide(PA) (hISG15-PA) | This paper | N/A |
| Mouse ISG15(77-C)-PA (mISG15-PA) | This paper | N/A |
| Chitin-Binding beads | NEB | Cat# S6651 |
| MESNa | Sigma | Cat# 1392807 |
| PD10 desalting column | Cytiva | Cat# 17085101 |
| stain-free SDS-PAGE | BioRad | Cat# 4568086 |
| RIPA buffer | Thermo Fisher | Cat# 89900 |
| Benzonase | Sigma | Cat# E1014 |
| Tris-HCl pH 7.5 | Thermo Fisher | Cat# 15567027 |
| CHAPS | Sigma | Cat# C5070 |
| BSA | Thermo Fisher | Cat# 15260037 |
| TCEP | UBP Bio | Cat# P1021-100 |
| Critical commercial assays | ||
| Total STAT1 HTRF assay | Perkin Elmer | Cat# 63ADK096PEG |
| pSTAT1 HTRF assay | Perkin Elmer | Cat# 63ADK026PEG |
| TaqMan gene expression assay-IFIT1 Mm00515153_m1 | Thermo Fisher | Cat# 4331182 |
| TaqMan gene expression assay-ISG15 Mm01705338_s1 | Thermo Fisher | Cat# 4331182 |
| TaqMan gene expression assay-GAPDH Mm03302249_g1 | Thermo Fisher | Cat# 4331182 |
| Cells-to-CT gene expression kit | Invitrogen | Cat# AM1729 |
| DNeasy Blood & Tissue Kit | Qiagen | Cat# 69506 |
| 12-230 kDa Separation Module | ProteinSimple | Cat# PS-PP03 |
| anti-rabbit Detection Module | ProteinSimple | Cat# DM-001 |
| Pierce BCA Protein Assay Kit | Thermo Fisher | Cat# 23228 |
| CellTiter-Glo 2.0 | Promega | Cat# G9243 |
| Experimental models: Cell lines | ||
| Hap1 Parental | Horizon Discovery | Cat# C631 |
| Hap1 USP18 KO | Horizon Discovery | Cat# HZGHC000492c006 |
| Hap1 USP18 C64S | This paper | N/A |
| Hap1 USP18 I60N | This paper | N/A |
| HT-29 | ATCC | RRID: CVCL_0320 |
| HCC1143 | ATCC | RRID: CVCL_1245 |
| A549 | ATCC | RRID: CVCL_0023 |
| HCC366 | DSMZ | RRID: CVCL_2059 |
| HCC1954 | ATCC | RRID: CVCL_1259 |
| SKBR3 | ATCC | RRID: CVCL_0033 |
| MDA-MB-468 | ATCC | RRID: CVCL_0419 |
| NCI-H1650 | ATCC | RRID: CVCL_1483 |
| HCT116 | ATCC | RRID: CVCL_0291 |
| CT26 | ATCC | Cat# CRL-2638TM RRID: CVCL_7254 |
| CT26 USP18-C61S | This paper | N/A |
| CT26 Usp18 KO | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Spodoptera frugiperda Sf9 | ATCC | Cat# CRL-1711 |
| BALB/c Mice | Charles River | Strain code: 28 |
| Oligonucleotides | ||
| sgRNA for hUSP18 KO cell line generation: GCAGCCCAGAGAGCGTCCCA | This paper | N/A |
| sgRNA for hUSP18 KO LAPSE: CAGGCCCTGGGACGCTCTCT | This paper | N/A |
| PCR primers for hUSP18 KO LAPSE: Forward: 5′-TCTAAGACCTGCTCTTT GGCATCAGAA-3′ Reverse, 5′-AAAG CTCTGCGGCTTCAGAGCGGAGG-3′ |
This paper | N/A |
| sgRNA for hUSP18 C64S KI cell line generation: ACACCTGAATCAAGGAGTTA | This paper | N/A |
| ssODN template for hUSP18 C64S KI cell line generation: CTTATAGGCCTGGTTG GTTTACACAACATTGGACAGACCAGC TGTCTTAACTCCTTGATTCAGGTGTTC GTAATGAATGTGGACTTCACC |
This paper | N/A |
| PCR primers for hUSP18-C64S KI cell line generation: Forward: 5′-CGGCTGATT TTTGGATTTTTTTTGTAGAGACAT-3′ Reverse, 5′-ATAATACAAATGCTCAT CAGCCTGAA-3′ |
This paper | N/A |
| sgRNA for hUSP18 I60N KI cell line generation: CTGGTTGGTTTACACAACAT | This paper | N/A |
| ssODN template for hUSP18 I60N KI cell line generation: CTTGCCCTCAGCATTTTTTTCTC TTCCCCTTATAGGCCTGGTTGGTTTACAC AATAACGGACAGACCTGCTGCCTTAACT CCTTGATTCAGGTGTTCGT |
This paper | N/A |
| PCR primers for hUSP18-I60N KI cell line generation: Forward: 5′-CGGCTGATTT TTGGATTTTTTTTGTAGAGACAT-3′ Reverse, 5′- AAAGTACCAATACTTGG GTCGCACCC -3′ |
This paper | N/A |
| sgRNA for UBA7 KO cell line generation: CCCATCCACAGGTATGTGCT | This paper | N/A |
| PCR primers for UBA7 KO cell line generation: Forward: 5′- GACCTGGACAGCTCTGTTGA-3′ Reverse, 5′- GCCACCTACACAAAGACCC -3′ | This paper | N/A |
| sgRNA for mUsp18 KO in CT26 cells: sgRNA-mUsp18#1: TGTACAGCCC ACGCAAATC; sgRNA-mUsp18#2: AAAGTCAGCCATCCCAACGT. |
This paper | N/A |
| sgRNA for Rosa26 KO in CT26 cells: GAAGATGGGCGGGAGTCTTC | This paper | N/A |
| Recombinant DNA | ||
| pGEX6p1 | Cytiva | Cat# 28-9546-48 |
| Software and algorithms | ||
| ICE analysis tool | Synthego | https://ice.synthego.com/#/ |
| Prism Version 9 | Graphpad | https://www.graphpad.com/features |
| The PyMOL Molecular Graphics System, Version 2.5.2 | Schrödinger | https://pymol.org/2/#page-top |
| Agilent MassHunter software | Agilent | https://www.agilent.com/en/promotions/masshunter-mass-spec |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Paul Wes (paulwes@gmail.com).
Materials availability
Licenses with vendor products, including vectors and cell lines, may restrict sharing of materials derived from their products with third parties.
Data and code availability
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.
Experimental model and study participant details
In vivo animal studies
All procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by an Institutional Animal Care and Use Committee or through an ethical review process. Female specific pathogen free (SPF) BALB/c mice (strain code – 028), 6–8 weeks old (Figures 1C–1E) or 8–10 weeks old (Figure 7C), were procured from Charles River Laboratories and provided Enrich-o Cobs bedding. Mice were housed under standard 12:12 light:dark cycle in ventilated racks at a room temperature of 72°F and Relative Humidity between 30 and 70%. 5LOD PicoLab laboratory rodent diet from LabDiet and filter sterilized reverse osmosis water were provided ad libitum. All mice were housed and cared for using guidelines from the Guide for the care and use of laboratory animals and with oversight from an attending Veterinarian. All cell lines used were pathogen tested by Charles River Laboratories Mouse/Rat comprehensive Clear panel and cleared for use in vivo prior to implantation. All mice on study were randomly assigned into test groups.
Cell lines
All cell lines were cultured in media supplemented with 10% Heat inactivated FBS (ThermoFisher, 16140071) and 1% Pen/Strep (ThermoFisher, 15070063) at 37°C/90% RH/5% CO2, except for MDA-MB-468, which was cultured at 0% CO2. All cell lines are immortalized human cell lines, except for CT26, which is an immortalized mouse cell line. Sex, media, and RRID for each cell line: HAP1 [sex: male, media: IMDM (ThermoFisher, 31980030), RRID: CVCL_Y019]; HCT116 [sex: male, media: McCoy’s 5A (ThermoFisher, 16600108), RRID: CVCL_0291]; HT-29 [sex: female, media: McCoy’s 5A, RRID: CVCL_0320]; HCC1143 [sex: female, media: RPMI 1640 (ThermoFisher, A1049101), RRID: CVCL_1245]; A549 [sex: male, media: DMEM (ThermoFisher, 10569010), RRID: CVCL_0023]; HCC366 [sex: female, media: RPMI 1640, RRID: CVCL_2059]; HCC1954 [sex: female, media: RPMI 1640, RRID: CVCL_1259]; SKBR3 [sex: female, media: McCoy’s 5A, RRID: CVCL_0033]; MDA-MB-468 [sex: female, media: Leibovitz L-15 (ThermoFisher, 11415064), RRID: CVCL_0419]; NCI-H1650 [sex: male, media: RPMI 1640, RRID: CVCL_1483]; CT26 [sex: female, media: RPMI 1640, RRID: CVCL_7254]. Cell lines were tested for mycoplasma upon arrival at Pfizer and authenticated by STR analysis internally at Pfizer and/or by ATCC.
Method details
CRISPR LAPSE
For USP18 KO LAPSE 1 × 106 cells were electroporated (Lonza, 4D-Nucleofactor) using the following electroporation buffers and protocols: A549: Buffer SF, CM-130; HAP1: Buffer SE, DZ-113; HCC1143: Buffer SE, E0-117; HCC366: Buffer SE, E0-117; HCT116: Buffer SE, EN-113; HT-29: Buffer SE, CM-150. For all cell lines, ribonucleoprotein complexes consisting of 200 pmol sgRNA (Thermo Fisher, modified TrueGuide sgRNA) and 10 μg Cas9 (Thermo Fisher, A36498) were prepared in electroporation buffer to a final volume of 100 μL per sample. At 72 h post-electroporation (time initial), cell pools were mixed thoroughly and split into 2 equivalent cell pools maintained in culture for 14 days (time final), with or without 1,000 U/mL human IFN-alpha2b (pbl Assay Science, 11105-1). Media was replaced 2x per week and cells were split as needed. At time initial and time final, an aliquot of at least 100,000 cells was pelleted and genomic DNA was extracted (Fisher Scientific, NC9904870) for PCR and Sanger sequencing. Editing efficiency for time initial, time final -IFN, and time final +IFN groups were evaluated using the ICE analysis tool (Synthego). USP18 KO sgRNA were targeted to the 3′ end of USP18 exon 2, downstream of predicted start codons for both isoforms and upstream of the catalytic cysteine, C64.
USP18 KO:
[1] sgRNA: GCAGCCCAGAGAGCGTCCCA
[1.2] For HAP1 USP18 KO LAPSE experiments, the following sgRNA sequence was used instead: CAGGCCCTGGGACGCTCTCT
[2] PCR primers: (Forward, 5′-TCTAAGACCTGCTCTTTGGCATCAGAA-3’; Reverse, 5′-AAAGCTCTGCGGCTTCAGAGCGGAGG-3′)
For hUSP18 C64S KI CRISPR LAPSE, 1 × 106 cells were electroporated (Maxcyte STx) in 50 × 3 processing assemblies using electroporation buffer (Maxcyte, EPB-1) and cell line-specific protocols recommended by the manufacturer. Ribonucleoprotein complexes consisting of 200 pmol sgRNA (Synthego, CRISPRevolution sgRNA EZ kit), 10 μg Cas9 (Thermo Fisher, A36499), and 4 μM ssODN donor template (IDT) were prepared in electroporation buffer to a final volume of 50 μL per sample. Following electroporation, the transfection mix was equilibrated for 20 min at 37°C. After equilibration, 1 μM HDR Enhancer V2 (IDT, 10007910) was added, and cells were subjected to a cold shock incubation for 48 h at 32°C. At 24 h post-electroporation, media containing 1 μM HDR Enhancer V2 was replaced with fresh media and cells were returned to 32°C until transfer to 37 °C at 48 h post-electroporation. At 72 h post-electroporation (time initial), cell pools were mixed thoroughly and split into 2 equivalent cell pools maintained in culture for 14 days (time final), with or without 1,000 U/mL numan IFN-Alpha2b (pbl Assay Science, 11105-1). Media was replaced 2x per week and cells were split as needed. At time initial and time final, an aliquot of 100,000 cells were pelleted and genomic DNA was extracted (Qiagen, 69506) for PCR and Sanger sequencing. Editing efficiency for time initial, time final -IFN, and time final +IFN groups were evaluated using the ICE analysis tool (Synthego).
C64S KI:
[1] sgRNA: ACACCTGAATCAAGGAGTTA
[2] ssODN template: CTTATAGGCCTGGTTGGTTTACACAACATTGGACAGACCAGCTGTCTTAACTCCTTGATTCAGGTGTTCGTAATGAATGTGGACTTCACC
[3] PCR primers: (Forward, 5′- CGGCTGATTTTTGGATTTTTTTTGTAGAGACAT-3’; Reverse, 5′- ATAATACAAATGCTCATCAGCCTGAA-3′)
Recombinant protein expression and purification
The open reading frame DNA sequences of hUSP18(AA16-372) (Uniprot Q9UMW8), mUSP18(AA46-368) (Uniprot Q9WTV6), and pro-hISG15(AA1-165)C78S (Uniprot P05161) were synthesized by GENEWIZ. C78S was substituted to stabilize hISG15 for purification.65 hUSP18 and mUSP18 were cloned into pFastBac1 between BamHI and XhoI sites with an N-terminal His tag and hISG15 was inserted into pGEX6p1 (Cytiva, 28-9546-48) between BamHI and XhoI sites with an N-terminal GST tag.
We noted a discrepancy between the UniProt hUSP18 annotation (https://www.uniprot.org/uniprotkb/Q9UMW8/entry, accessed on December 23, 2023) and the experimentally determined open reading frame.66 Burkhart et al. demonstrated that the cryptic start codon CUG (amino acid 16 in UniProt) is the correct translation start site for full length USP18 and the canonical start codon AUG (amino acid 36 in UniProt) is the translation start site for the N-terminal truncated USP18 isoform (USP18-sf). Guided by the experimental evidence, we synthesized hUSP18(AA16-372) to include both isoforms.
C64S, C64S/C65S, and I60N hUSP18 mutations were introduced by quick-change mutagenesis and verified by Sanger sequencing. hUSP18 variants and mUSP18 were expressed in Spodoptera frugiperda (Sf9, ATCC CRL-1711) insect cells using the Bac-to-Bac Baculovirus Expression System (Invitrogen, 10359016). Sf9 cells were infected at a cell density of 4 × 106 cells/mL with a multiplicity of infection of 5.0. At 48 h after infection, cells were collected and resuspended in the lysis buffer containing 25 mM HEPES pH 8.0, 200 mM NaCl, 1 mM TCEP, 40 mM imidazole, EDTA-free complete protease inhibitor cocktail (Sigma, 56619600) and 50 U/mL Pierce universal nuclease (Thermo Fisher, 88702). Sf9 cells were lysed by sonication and His-USP18 protein was purified by Ni-affinity chromatography and His-tag cleavage by PreScission protease. Size exclusion chromatography was performed with t SEC buffer (25 mM HEPES pH 8.0, 200 mM NaCl, 1 mM TCEP). Intact mass spectrometry was performed using 6545 LC/Q-TOF (Agilent technologies). The intact mass analysis was conducted in positive ion mode. The nozzle voltage was 2 kV, capillary voltage was 4 kV, source temperature was 325°C, and the dry gas flow rate was 10 L/min. The protein (10 μL) was loaded by an LC system (Agilent 1290 Infinity II, Agilent Technologies, Santa Clara, CA) on an HPLC column (PLRP-S 1000 Å, 5 mm, 50 × 2.1 mm, Agilent Technologies, Santa Clara, CA). The following solvents were used: solvent A, water with 0.1% formic acid; solvent B, acetonitrile with 0.1% formic acid. The protein was eluted at 400 μL/min with the following gradient: 2% B in 1 min, 85% B in 9 min, 95% B in 0.1 min, 95% B from 10.1–12.0 min then went back to 2% in 0.1 min. For data analysis, spectra were coadded and deconvoluted using Agilent MassHunter software with a 0.5 Da mass step and mass range of 10–150 kDa.
GST-hISG15 was expressed in Escherichia coli BL21(DE3) (Thermo Fisher, EC0114) at OD600 0.6–0.8 with 0.1 mM IPTG and 20°C and lysed in the lysis buffer by sonication. After GST-affinity chromatography and tag cleavage by PreScission protease, hISG15 was further purified by size exclusion chromatographywith SEC buffer. The codon-optimized gene corresponding to SARS-CoV-2 PLpro domain (Nsp3 residues 1562–1878; Uniprot P0DTD1) was synthesized as a gBlock gene fragment by Integrated DNA Technologies and was subcloned into a pET28a vector using NcoI and XhoI restriction enzymes (New England Biolabs). The protein was expressed with uncleavable N-terminal Avi and His6x tags in BL21(DE3) E. coli cells (Novagen SinglesTM). The protein was isolated from the lysate using Ni-NTA Agarose resin (Qiagen) and was purified to homogeneity and >95% purity by size exclusion chromatography using a HiLoad Superdex 75 pg 16/600 column (Cytiva) in a buffer containing 50 mM HEPES pH 8, 200 mM NaCl, 1 mM TCEP, and 5% glycerol.
Genetically modified cell lines
To generate hUSP18 C64S and I60N KI cells, 1 × 106 HAP1 cells were electroporated (Maxcyte STx) in 50 × 3 processing assemblies using electroporation buffer (Maxcyte, EPB-1) and cell-line specific protocols recommended by the manufacturer. Ribonucleoprotein complexes consisting of 200 pmol sgRNA (Synthego, CRISPRevolution sgRNA EZ kit), 10 μg Cas9 (Thermo Fisher, A36499), 4 μM ssODN donor template (IDT) were prepared in electroporation buffer to a final volume of 50 μL per sample. Following electroporation, the transfection mix was equilibrated for 20 min at 37°C. After equilibration, 1 μM HDR Enhancer V2 (IDT, 10007910) was added, and cells were subjected to a cold shock incubation for 48 h at 32°C. At 24 h post-electroporation, media containing 1 μM HDR Enhancer V2 was replaced with fresh culture media and cells were returned to 32°C until transfer to 37°C at 48 h post-electroporation. Cells were subjected to single-cell cloning on the CellCelector (Sartorius) into 96-well plates. Single-cell clones were confirmed by Sanger sequencing.
C64S:
[1] sgRNA: ACACCTGAATCAAGGAGTTA
[2] ssODN template: CTTATAGGCCTGGTTGGTTTACACAACATTGGACAGACCAGCTGTCTTAACTCCTTGATTCAGGTGTTCGTAATGAATGTGGACTTCACC
[3] PCR primers: (Forward, 5′-CGGCTGATTTTTGGATTTTTTTTGTAGAGACAT-3’; Reverse, 5′-ATAATACAAATGCTCATCAGCCTGAA-3′)
I60N:
[1] sgRNA: CTGGTTGGTTTACACAACAT
[2] ssODN template: CTTGCCCTCAGCATTTTTTTCTCTTCCCCTTATAGGCCTGGTTGGTTTACACAATAACGGACAGACCTGCTGCCTTAACTCCTTGATTCAGGTGTTCGT
[3] PCR primers: (Forward, 5′-CGGCTGATTTTTGGATTTTTTTTGTAGAGACAT-3’; Reverse, 5′-AAAGTACCAATACTTGGGTCGCACCC-3′)
To generate HCT116 hUSP18 KO cells inFigures S2B and 2C, 1 × 106 WT HCT116 cells (ATCC; CCL-247) were electroporated (Maxcyte STx) in OC100 processing assemblies using electroporation buffer (Maxcyte, EPB-1) and optimization protocol HCT116. Ribonucleoprotein complexes consisting of 200 pmol sgRNA (Synthego, CRISPRevolution sgRNA EZ kit) and 10 μg Cas9 (Thermo Fisher, A36499) were prepared in electroporation buffer to a final volume of 100 μL per sample. Following electroporation, the transfection mix was equilibrated for 20 min at 37°C before addition of fresh media for cell recovery. Knockout efficiency was confirmed by Sanger sequencing and ICE analysis tool (Synthego).
[1] sgRNA 1 for USP18 KO cells: CAGGCCCTGGGACGCTCTCT
[2] sgRNA 2 for USP18 KO cells: GCAGCCCAGAGAGCGTCCCA
[3] PCR primers: (Forward, 5′-TCTAAGACCTGCTCTTTGGCATCAGAA-3′, Reverse, 5′-AAAGCTCTGCGGCTTCAGAGCGGAGG -3′)
To generate HAP1 UBA7 KO and UBA7/USP18 double KO cells in Figure 5, 1 × 106 WT HAP1 cells (Horizon Discovery, 631) or USP18 KO HAP1 cells (Horizon Discovery, HZGHC000492c006) were electroporated (Maxcyte STx) in OC25 × 3 processing assemblies using electroporation buffer (Maxcyte, EPB-1) and optimization protocol OP9. Ribonucleoprotein complexes consisting of 200 pmol sgRNA (Synthego, CRISPRevolution sgRNA EZ kit) and 10 μg Cas9 (Thermo Fisher, A36499) were prepared in electroporation buffer to a final volume of 25 μL per sample. Following electroporation, the transfection mix was equilibrated for 20 min at 37°C before addition of fresh media for cell recovery. Knockout efficiency was confirmed by Sanger sequencing and ICE analysis tool (Synthego).
[1] sgRNA for UBA7 KO cells: CCCATCCACAGGTATGTGCT
[2] sgRNA for UBA7/USP18 double KO cells: CCCTGAATCCTCTGCATGGC
[3] PCR primers: (Forward, 5′-GACCTGGACAGCTCTGTTGA-3′, Reverse, 5′-GCCACCTACACAAAGACCCT-3′)
ISG15-PA activity-based probe assay
hISG15(79-C)-propargylamide(PA) (hISG15-PA) and mISG15(77-C)-PA (mISG15-PA) probes were prepared with Intein-MESNa (sodium 2-mercaptoethanesulfonate) methodology similar to the previous reports.52 Briefly, ISG15-Intein-CBS proteins were expressed in BL21(DE3) and purified by Chitin-Binding beads (NEB, S6651) affinity purification followed by eluting with SEC buffer (25 mM HEPES pH 8.0, 200 mM NaCl, 1 mM TCEP) containing 50mM MESNa (Sigma,1392807). ISG15-MESNa conjugates were further purified by size exclusion chromatography with the SEC buffer followed by converting it to ISG15-PA by incubating with 5 times molar PA (Sigma, P50900) at RT. Excess PA was removed by desalting using PD10 desalting column (Cytiva, 17085101).
50 nM hUSP18 WT and 5000 nM hISG15-PA or 1 μM mUSP18 WT and 10 μM mISG15-PA were incubated at 37°C for 30 min to monitor the conjugation of hUSP18-hISG15 or mUSP18-mISG15, respectively. hUSP18-hISG15 conjugation was detected by western blot with the following primary antibody: rabbit anti-USP18 (Cell Signaling, 4813S). mUSP18-mISG15 conjugation was visualized by trihalo compound-based, stain-free SDS-PAGE (BioRad, 4568086).
hUSP18-hISG15 complex formation assay
50 μM hUSP18 WT was incubated with or without 75 μM hISG15 WT (molar ratio 1:1.5) at RT for 5 min, followed by running SEC with SEC buffer (25 mM HEPES pH 8.0, 200 mM NaCl, 1 mM TCEP). Complex formation was determined by comparing the SEC profiles of hUSP18, hISG15, and hUSP18 incubated with hISG15.
Cell viability and qRT-PCR assays
In Figures 1A and 1B, CT26 cells were plated in 96-well plates (Corning, 3585) at 10,000 cells per well and were subjected to a 5-point dose response of murine IFN-β (pbl Assay Sciences, 12410-1) starting at 1,000 U/mL with 3-fold dilution series. Cell confluence was measured every 4 h during IFN treatment (Sartorius, Incucyte S3). In Figure 1A, cells were plated as described above and after 72 h, lysed for gene expression analysis. Cell lysis, RNA extraction, and cDNA synthesis were performed with the Cells-to-CT gene expression kit (Invitrogen, AM1729). qRT-PCR was performed on the Applied Biosystems QuantStudio 7 Real-Time PCR System according to the manufacturer’s instructions with the following TaqMan gene expression assays: IFIT1 (Mm00515153_m1), ISG15 (Mm01705338_s1), and GAPDH (Mm03302249_g1). Results shown as expression 2−ΔCt relative to GAPDH.
In Figures 2B and 2C, HCT116 cells were plated in 96-well plates (Corning, 3585) at 5,000 cells/well in 100 μL media containing 0 or 1000 U/mL IFN-alpha2b (pbl Assay Science, 11105-1). Cell confluence was measured every 4 h during IFN treatment (Sartorius, Incucyte S3).
In Figures S2B and S2C, CT26 cells were plated in 96-well plates (Corning, 3585) at 5,000 cells per well and were treated with 111 U/mL murine IFN-alpha A (pbl Assay Science, 12100-1) for 24 h, prior to addition of Annexin V green dye (Sartorius, 4642). Individual Annexin V vials were resuspended in 425 μL of media and added at 1:100 dilution to cells. Cell confluence and Annexin V signal were measured every 4 h during IFN treatment (Sartorius, Incucyte S3). Annexin V signal was calculated by total integrated intensity per well to confluence.
In Figure 4C, HAP1 cells were subjected to an 11-point dose response of human IFN-alpha2b (pbl Assay Science, 11105-1) starting at 9,000 U/mL with 3-fold dilution series. Cells were plated in 384-well white microplates (Corning, 3570) at 3,000 cells/well in 50 μL media containing IFN-α for 72 h prior to cell lysis with 50 μL CellTiter-Glo 2.0 (Promega, G9243). The reaction was incubated in the dark for 20 min at RT. Luminescence was measured on the GloMax Discover (Promega, GM3000) and viability was normalized to 0 U/mL IFN control for each cell line.
In Figures 5C and 5D, HAP1 cells were plated in 96-well plates (Corning, 3585) at 10,000 cells per well and were subjected to a 9-point dose response of human Interferon Beta 1a (pbl Assay Science, 11410-2) starting at 27,000 U/mL with 3-fold dilution series. Cell confluence was measured every 4 h during IFN treatment (Sartorius, Incucyte S3). After 48 h image collection, cells were lysed for gene expression analysis. Cell lysis, RNA extraction, and cDNA synthesis were performed with the Cells-to-CT gene expression kit (Invitrogen, AM1729). qRT-PCR was performed on the Applied Biosystems QuantStudio 7 Real-Time PCR System according to the manufacturer’s instructions with the following TaqMan gene expression assays: IFIT1 (Hs0035663_g1), ISG15 (Hs00192713_m1), and GAPDH (Hs99999905_m1). Results shown as expression 2−ΔCt relative to GAPDH. In Figures 7B, S5B, and S5C, CT26 cells were subjected to a 9-point dose response of murine IFN-alpha A (pbl Assay Science, 12100-1) starting at 9,500 U/mL with 3.33 dilution series. Cells were plated in 96-well plates (Corning, 3598) at 5,000 cells/well in 100 μL media containing IFN-α for 72 h. Cell confluence was measured every 6 h during IFN treatment (Sartorius, Incucyte SX5). In Figure 7B, cells were lysed for viability analysis with 80 μL CellTiter-Glo 2.0 (Promega, G9243) after 72 h of image acquisition. The reaction was incubated in the dark for 20 min at RT. Luminescence was measured on the GloMax Discover (Promega, GM3000) and viability was normalized to 0 U/mL IFN control for each cell line.
Protein expression assays
For human HAP1 cell lines in Figures 4A, 4B, and 5A, 1 × 105 cells in 24-well plates were treated for 24 h in media containing 1,000 U/mL human IFN-beta 1a (pbl Assay Science, 11410-2). For mouse CT26 cell lines in Figures 7A and S5A, 1 × 106 cells in 6-well plates were treated for 24 h in media containing 1,000 U/mL murine IFN-alpha A (pbl Assay Science, 12100-1). Lysates were prepared in RIPA buffer (Thermo Scientific, 89900) containing 1× protease and phosphatase inhibitors (Thermo Scientific, 78440) and 250 U/mL benzonase (Sigma, E1014). Concentrations were measured using the Pierce BCA Protein Assay Kit (Thermo Fisher, 23228). Protein analysis was performed on a Wes system (ProteinSimple, 004–600) according to manufacturer’s instructions using a 12–230 kDa Separation Module (ProteinSimple, PS-PP03) and an anti-rabbit Detection Module (ProteinSimple, DM-001). The following primary antibodies against human proteins were used at the indicated dilution: rabbit anti-ISG15 (7H29L24) 1:50 (Thermo Fisher, 703131), rabbit anti-USP18 (D4E7) 1:25 (Cell Signaling, 4813S), and rabbit anti-Vinculin 1:300 (Abcam, Ab129002). The following primary antibodies against mouse proteins were used at the indicated dilution: rabbit anti-ISG15 (E5B3U) 1:50 (Cell Signaling, 89771), rabbit FAB anti-USP18 (#5080) 50 μg/mL (BioRad, AbD55080sco-1), and rabbit anti-Vinculin 1:300 (Abcam, Ab129002).
ISG15-Rho110 biochemical assay
USP18 enzymes and ISG15 substrates were diluted 2x in 20 mM Tris-HCl pH 7.5 (Thermo Fisher 15567027), 150 mM NaCl, 0.05% CHAPS (Millipore Sigma C5070), 0.05% BSA (Thermo Fisher 15260037), and 0.5 mM TCEP (UBP Bio CHT#P1021-100). Recombinant hUSP18 (WT, C64S, I60N) or mUSP18 were incubated with recombinant human ISG15-Rho110 (South Bay Bio, SBB-PS0002) or mouse ISG15-Rho110 (Ac-ISG15prox-Rho110MP, UbiQ Bio BV, UBIQ-127). Enzymes and substrates were combined at the indicated concentrations in a black 384-well plate (Costar, 3820), briefly centrifuged, and immediately read at an interval of 2 min on an EnVision 2104 reader with excitation filter 485 nm and emission filter 531 nm. For progression curves, enzymes and substrates were mixed at the indicated concentration and fluorescence from the cleaved substrate was monitored and recorded in the plate reader every two minutes for the indicated time. For KM determinations, KM(apparent) was calculated in accordance with standard methods. Briefly, a matrix of enzyme concentration ranges was titrated across a range of fluorescent human or mouse ISG15-Rho110 substrate concentrations, and the resultant reactions generated liberated fluorophore from the substrate. The resultant fluorescence was monitored and quantified in a plate reader every two minutes for at least 30 min. From these data, RFU versus time progression curves were generated and initial linearity was determined by non-linear regression in Prism for each enzyme and substrate combination. Enzyme and substrate combinations that resulted in high and linear initial velocities of the reaction were then included in a plot of the velocity (RFUs/time) versus substrate concentration. The velocity versus substrate concentration curve was curve fit in Prism by Michaelis-Menten method for substrate versus velocity to determine KM(apparent)and VMAX(apparent). The reported values are an average of two determinations at different enzyme concentrations rounded to two significant digits.
Biolayer interferometry assay
Binding assays for hUSP18 and hISG15 complex formation were performed on an Octet Red384 biolayer interferometry instrument (ForteBio, Inc.) at 25°C in running buffer (20 mM HEPES pH7.5, 150 mM NaCl, 1 mM TCEP, 0.2 mg/mL BSA, and 0.02% Tween-20). 1000 rpm plate shake speed was maintained throughout the experiment. Biotinylated hUSP18 was prepared at 50 nM and captured on streptavidin coated sensors (ForteBio, 18–5019) yielding approximately 1 nm response, followed by a wash step. Sensors were then dipped for 600 s of association phase in wells containing 7-point serial dilution of hISG15 starting at 10 mM with 3.33 dilution series, followed by 600 s dissociation in blank wells. The nm response after association was plotted as a function of the hISG15 concentration and fit to a standard binding isotherm in GraphPad Prism to obtain the KD.
pSTAT1/total STAT1 HTRF assay
20,000 HAP1 cells were seeded in 96-well plates for 16–24 h with IMDM phenol-red free media (Gibco, 21056023) + 10% HI-FBS (Thermo Fisher, 16140071) prior to adding indicated concentration of human IFN-alpha 2b (pbl Assay Science, 11105-1) for 4 h IFN prime. Cells were washed 1x with PBS and fresh media without IFN was added to allow cells to rest. After 16–24 h rest, the indicated concentration of human IFN-alpha 2b (pbl Assay Science, 11105-1) was added for a 15 min pulse prior to cell lysis. pSTAT1 and total STAT1 levels were measured according to manufacturer’s protocol for pSTAT1 Tyr701 (Perkin Elmer, 63ADK026PEG) and total STAT1 (Perkin Elmer, 63ADK096PEG) HTRF kits in HTRF 96-well low volume plates (Perkin Elmer, 66PL96025). HTRF signal was read on an EnVision 2105 reader with excitation filter 320 nm and emission filters 665 nm and 615 nm. pSTAT1/total STAT1 levels were normalized as ratio to account for baseline total STAT1 levels after the initial IFN prime.
In vitro deISGylation of endogenous substrates
To obtain endogenously ISGylated human or mouse proteins, HAP1 hUSP18 KO cells (Horizon Discovery, HZGHC000492c006) were treated for 24 h with 1,000 U/mL human IFN-alpha 2b (pbl Assay Science, 11105-1) or CT26 mUsp18 KO cells were treated for 24 h with 1,000 U/mL murine IFN-alpha A (pbl Assay Science, 12100-1) prior to cell lysis in 20 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% NP40, 0.05% BSA, 0.05% CHAPS, and 0.5 mM TCEP. Lysate concentration was measured using Pierce BCA Protein Assay Kit (Thermo Fisher, 23228), adjusted to 1 μg/μL, and incubated with indicated concentrations of recombinant hUSP18 (WT, C64S, I60N), mUSP18, or SARS-CoV-2 PLpro for 1 h at RT. Western blot analysis was performed on a Wes system (ProteinSimple, 004–600) according to manufacturer’s instructions using a 12–230 kDa Separation Module (ProteinSimple, PS-PP03) and anti-rabbit Detection Module (ProteinSimple, DM-001). The following primary antibodies against human proteins were used at the indicated dilution: rabbit anti-ISG15 (7H29L24) 1:50 (Thermo Fisher, 703131), and rabbit anti-Vinculin 1:500 (Abcam, Ab129002). The following primary antibodies against mouse proteins were used at the indicated dilution: rabbit anti-ISG15 (E5B3U) 1:50 (Cell Signaling Technology, 89771), and rabbit anti-Vinculin 1:300 (Abcam, Ab129002).
Pro-ISG15 cleavage assay
1 μM recombinant hUSP18 (WT, C64S, C64S/C65S, I60N) and 5 μM recombinant pro-hISG15 were incubated for 3 or 10 min at 37°C in the buffer of 25 mM HEPES pH 8.0, 200 mM NaCl, 1 mM TCEP. Cleavage of pro-hISG15(AA1-165) to mature hISG15(AA1-157) was monitored by SDS-PAGE as well as mass spectrometry using 6545 LC/Q-TOF (Agilent technologies) as described above in “recombinant protein expression and purification”.
Competitive ISG15-Rh110 inhibition assay
Assays were run at RT in 50 mM HEPES (pH 7.5), 150 mM NaCl, 1 mM TCEP, 0.2 mg/mL BSA, 0.02% Tween 20. mISG15-Rho110 (Ac-ISG15prox-Rh110MP, UbiQ Bio BV, UBIQ-127) was prepared in 1x buffer at 2 μM prior to addition of mUSP18 or hUSP18 and immediately mixed 1:1 with non-fluorogenic hISG15 or mISG15 in the assay plate (Corning 3820; 20 μL per well) for a final concentration of 1 μM mISG15-Rho110 and 1 nM hUSP18 or 2 nM mUSP18. For non-fluorogenic hISG15 and mISG15, a serial dilution of eight concentrations were tested at the following final concentrations: for hUSP18, 2 to 250 nM hISG15 or 0.4 to 50 μM mISG15 and for mUSP18, 0.1 to 50 μM hISG15 or mISG15. The plate was briefly centrifuged to remove air bubbles and transferred to a Tecan M1000 plate reader. Fluorescence was monitored at 492/525 nm with 8 nm bandwidth filters for 1 h. The change of fluorescence was fit to a linear time dependence. The slopes were then fit to a standard, noncooperative EC50 equation: Y = Y0/(1+(X/EC50)). Here, Y0 is the maximal rate observed in the absence of any competitive ligand, X is the concentration of competitive ligand, and EC50 is the concentration of competitive ligand which yields one-half the Y0 value.
Syngeneic mouse models
Two rounds of electroporation were performed to generate mUsp18 KO CT26 cells. First, WT CT26 cells were electroporated with sgRNA-mUsp18#1 (TGTACAGCCCACGCAAATC) and Cas9 and expanded for 4 days prior to a second round of electroporation with sgRNA-mUsp18#2 (AAAGTCAGCCATCCCAACGT) and Cas9. Rosa26 KO cells were generated by a single round of electroporation with sgRNA-Rosa26 (GAAGATGGGCGGGAGTCTTC) and Cas9. PCR amplicons surrounding mUsp18 sgRNA cut sites were sequenced (MiSeq, Illumina) and frameshift mutation frequency for triploid CT26 cells was estimated to be ∼65% by the following equation: [1-((1 – FSsg1) ∗ (1 – FSsg2))]3 where FS = % frameshift mutation for a given sgRNA. Loss-of-function KO frequency may be higher given that large in-frame mutations may also result in loss-of-function. mUSP18 protein reduction was confirmed by proteomic analysis of expanded mUsp18−/− pools, mRosa26 −/− pools, and parental WT CT26 prior to implantation. For the proteomics experiment, cells were treated with 333 U/mL murine IFN-β for 48 h to induce mUSP18 expression prior to cell lysis.
Bottom-up mass-spectrometry-based proteomics analysis was performed, with relative quantitation. CT26 cells were lysed in 2% SDS (sodium dodecyl sulfate), 150 mM NaCl, 50 mM Tris-HCl pH 8.5 and protein concentration was measured by microBCA assay. Lysate was reduced by 5 mM DTT (dithiothreitol) for 25 min at 56°C, then alkylated by 15 mM IAA (iodoacetamide) for 30 min in the dark. Proteins were purified by chloroform methanol precipitation. Protein pellets were redissolved in 8 M urea, 20 mM EPPS (4-(2-Hydroxyethyl)-1-piperazinepropanesulfonic acid) pH 8.5. Samples were diluted to 4 M urea, 20 mM EPPS pH 8.5 and Lys-C (75:1 protein to enzyme mass ratio) was added with overnight incubation at RT. Samples were further diluted to 1.6 M urea, 20 mM EPPS pH 8.5 and trypsin (75:1 protein to enzyme mass ratio) was added with overnight incubation at 37°C. Samples were labeled with TMTpro-16plex, pooled together and desalted. Offline high pH fractionation was performed on the combined sample to generate 24 fractions. The fractions were desalted, redissolved in 1% formic acid and 2% acetonitrile, and analyzed on Orbitrap Fusion Lumos with 3 h LC gradient and MS3 quantification. Data were searched with Proteome Discoverer 2.4 with dynamic modifications (methionine oxidation, protein N-terminal acetylation, protein N-terminal methionine loss), static modifications (TMTpro on lysines and peptide N-terminus, cysteine carbamidomethylation) and trypsin digestion with up to 2 missed cleavages.
All procedures performed on animals were in accordance with regulations and established guidelines and were reviewed and approved by an Institutional Animal Care and Use Committee or through an ethical review process. BALB/c mice (strain code – 028), 6–8 weeks old, were procured from Charles River Laboratories and housed under standard 12:12 light:dark cycle in ventilated racks at a room temperature of 72°F and Relative Humidity between 30 and 70%. After acclimatization, 1 × 106 Rosa26−/− or Usp18−/− CT26 pools or 5 × 105 WT, KO, or C61S KI mUSP18 CT26 clones were implanted subcutaneously in the right flank of each mouse (Parental CT26 line prior to genetic modification: ATCC, CRL-2638TM). Tumor volume was measured at least twice weekly with a caliper using the longest dimension (length) and the longest perpendicular dimension (width) once tumors became palpable. Tumor volume was estimated with the formula: (L×W2)/2.
Sequence alignments
Sequence alignments for hUSP18 vs. mUSP18 and hISG15 vs. mISG15 were generated using CLUSTAL W.67 Sequence differences between hUSP18 and mUSP18 or hISG15 and mISG15 were mapped onto the hUSP18-hISG15 structure in the AlphaFold ternary complex model using PyMOL (The PyMOL Molecular Graphics System, Version 2.5.2 Schrödinger, LLC).
AlphaFold modeling
Prior to building the hUSP18-hISG15-hSTAT2 tertiary model in AlphaFold, the predicted disordered region of USP18 (N-terminal 15 amino acid residues) were removed. The tertiary model structure was generated using AlphaFold-Multimer68,69 with default settings. The top PDB file was further analyzed to elucidate USP18 catalytic and scaffolding functions. PyMOL was used to generate tertiary structural models.
Quantification and statistical analysis
All statistical analysis was performed using Graphpad Prism Version 9. Data collected as raw values are shown as mean ± SEM. Details of statistical methods are reported in the figure legends and Table S1.
Acknowledgments
We thank Jon Oyer and Rubina Tuladhar for generously sharing the CRISPR LAPSE method ahead of publication; Ellene Mashalidis for the purification of recombinant SARS-CoV-2 PLpro; Kim F. Fennell for recombinant protein efforts; Brooke Conti Trousdale, Jennie Altman, and Drew Nager for comments on the article; Pfizer’s Worldwide Research, Development and Medical Postdoctoral Program for their support; Pfizer colleagues in Emerging Science and Innovation for stimulating discussion; and David Shields for his leadership and thoughtful feedback.
Author contributions
Experiments were performed by the following authors: HW and VJ (CRISPR LAPSE); VJ (HTRF); HW, CWL, KL, VJ, and JS (cell line generation and characterization); DRH, CWL, MY, and ECR (recombinant protein assays); ZKJ, EC, BY, and AG (in vivo models); YX (proteomics). FW, PDW, VF, VJ, PL, ACG, ECR, BMK, and APF contributed to the conception, design, and/or interpretation of the work. VJ, VF, and PDW wrote the article with input from all authors.
Declaration of interests
VJ, HW, CWL, DRH, KL, ECR, MY, ZKJ, EC, YX, JS, BY, AG, PL, ACG, VF, PDW, and FW are current or former employees of Pfizer and may own Pfizer stock. BMK and APF receive research funding from Pfizer.
This work was supported by Pfizer, and APF and BMK are funded by the Chinese Academy of Medical Sciences (CAMS) Innovation Fund for Medical Science (CIFMS), China (grant nr - 2018-I2M-2-002).
Published: March 27, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2024.109593.
Supplemental information
References
- 1.McNab F., Mayer-Barber K., Sher A., Wack A., O'Garra A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015;15:87–103. doi: 10.1038/nri3787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Snell L.M., McGaha T.L., Brooks D.G. Type I Interferon in Chronic Virus Infection and Cancer. Trends Immunol. 2017;38:542–557. doi: 10.1016/j.it.2017.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schneider W.M., Chevillotte M.D., Rice C.M. Interferon-stimulated genes: a complex web of host defenses. Annu. Rev. Immunol. 2014;32:513–545. doi: 10.1146/annurev-immunol-032713-120231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bekisz J., Baron S., Balinsky C., Morrow A., Zoon K.C. Antiproliferative Properties of Type I and Type II Interferon. Pharmaceuticals. 2010;3:994–1015. doi: 10.3390/ph3040994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gessani S., Conti L., Del Cornò M., Belardelli F. Type I interferons as regulators of human antigen presenting cell functions. Toxins. 2014;6:1696–1723. doi: 10.3390/toxins6061696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zitvogel L., Galluzzi L., Kepp O., Smyth M.J., Kroemer G. Type I interferons in anticancer immunity. Nat. Rev. Immunol. 2015;15:405–414. doi: 10.1038/nri3845. [DOI] [PubMed] [Google Scholar]
- 7.Boukhaled G.M., Harding S., Brooks D.G. Opposing Roles of Type I Interferons in Cancer Immunity. Annu. Rev. Pathol. 2021;16:167–198. doi: 10.1146/annurev-pathol-031920-093932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cheon H., Borden E.C., Stark G.R. Interferons and their stimulated genes in the tumor microenvironment. Semin. Oncol. 2014;41:156–173. doi: 10.1053/j.seminoncol.2014.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Crow Y.J., Stetson D.B. The type I interferonopathies: 10 years on. Nat. Rev. Immunol. 2022;22:471–483. doi: 10.1038/s41577-021-00633-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Taft J., Bogunovic D. The Goldilocks Zone of Type I IFNs: Lessons from Human Genetics. J. Immunol. 2018;201:3479–3485. doi: 10.4049/jimmunol.1800764. [DOI] [PubMed] [Google Scholar]
- 11.Barber D.L., Wherry E.J., Masopust D., Zhu B., Allison J.P., Sharpe A.H., Freeman G.J., Ahmed R. Restoring function in exhausted CD8 T cells during chronic viral infection. Nature. 2006;439:682–687. doi: 10.1038/nature04444. [DOI] [PubMed] [Google Scholar]
- 12.François-Newton V., Magno de Freitas Almeida G., Payelle-Brogard B., Monneron D., Pichard-Garcia L., Piehler J., Pellegrini S., Uzé G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS One. 2011;6 doi: 10.1371/journal.pone.0022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kang D., Jiang H., Wu Q., Pestka S., Fisher P.B. Cloning and characterization of human ubiquitin-processing protease-43 from terminally differentiated human melanoma cells using a rapid subtraction hybridization protocol RaSH. Gene. 2001;267:233–242. doi: 10.1016/s0378-1119(01)00384-5. [DOI] [PubMed] [Google Scholar]
- 14.Zhang X., Bogunovic D., Payelle-Brogard B., Francois-Newton V., Speer S.D., Yuan C., Volpi S., Li Z., Sanal O., Mansouri D., et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature. 2015;517:89–93. doi: 10.1038/nature13801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Meuwissen M.E.C., Schot R., Buta S., Oudesluijs G., Tinschert S., Speer S.D., Li Z., van Unen L., Heijsman D., Goldmann T., et al. Human USP18 deficiency underlies type 1 interferonopathy leading to severe pseudo-TORCH syndrome. J. Exp. Med. 2016;213:1163–1174. doi: 10.1084/jem.20151529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alsohime F., Martin-Fernandez M., Temsah M.H., Alabdulhafid M., Le Voyer T., Alghamdi M., Qiu X., Alotaibi N., Alkahtani A., Buta S., et al. JAK Inhibitor Therapy in a Child with Inherited USP18 Deficiency. N. Engl. J. Med. 2020;382:256–265. doi: 10.1056/NEJMoa1905633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martin-Fernandez M., Buta S., Le Voyer T., Li Z., Dynesen L.T., Vuillier F., Franklin L., Ailal F., Muglia Amancio A., Malle L., et al. A partial form of inherited human USP18 deficiency underlies infection and inflammation. J. Exp. Med. 2022;219 doi: 10.1084/jem.20211273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malakhov M.P., Kim K.I., Malakhova O.A., Jacobs B.S., Borden E.C., Zhang D.E. High-throughput immunoblotting. Ubiquitiin-like protein ISG15 modifies key regulators of signal transduction. J. Biol. Chem. 2003;278:16608–16613. doi: 10.1074/jbc.M208435200. [DOI] [PubMed] [Google Scholar]
- 19.Malakhov M.P., Malakhova O.A., Kim K.I., Ritchie K.J., Zhang D.E. UBP43 (USP18) specifically removes ISG15 from conjugated proteins. J. Biol. Chem. 2002;277:9976–9981. doi: 10.1074/jbc.M109078200. [DOI] [PubMed] [Google Scholar]
- 20.Knobeloch K.P., Utermöhlen O., Kisser A., Prinz M., Horak I. Reexamination of the role of ubiquitin-like modifier ISG15 in the phenotype of UBP43-deficient mice. Mol. Cell Biol. 2005;25:11030–11034. doi: 10.1128/MCB.25.24.11030-11034.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Malakhova O.A., Yan M., Malakhov M.P., Yuan Y., Ritchie K.J., Kim K.I., Peterson L.F., Shuai K., Zhang D.E. Protein ISGylation modulates the JAK-STAT signaling pathway. Genes Dev. 2003;17:455–460. doi: 10.1101/gad.1056303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Arimoto K.I., Löchte S., Stoner S.A., Burkart C., Zhang Y., Miyauchi S., Wilmes S., Fan J.B., Heinisch J.J., Li Z., et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat. Struct. Mol. Biol. 2017;24:279–289. doi: 10.1038/nsmb.3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gruber C., Martin-Fernandez M., Ailal F., Qiu X., Taft J., Altman J., Rosain J., Buta S., Bousfiha A., Casanova J.L., et al. Homozygous STAT2 gain-of-function mutation by loss of USP18 activity in a patient with type I interferonopathy. J. Exp. Med. 2020;217 doi: 10.1084/jem.20192319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Duncan C.J.A., Thompson B.J., Chen R., Rice G.I., Gothe F., Young D.F., Lovell S.C., Shuttleworth V.G., Brocklebank V., Corner B., et al. Severe type I interferonopathy and unrestrained interferon signaling due to a homozygous germline mutation in STAT2. Sci. Immunol. 2019;4 doi: 10.1126/sciimmunol.aav7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Basters A., Geurink P.P., El Oualid F., Ketscher L., Casutt M.S., Krause E., Ovaa H., Knobeloch K.P., Fritz G. Molecular characterization of ubiquitin-specific protease 18 reveals substrate specificity for interferon-stimulated gene 15. FEBS J. 2014;281:1918–1928. doi: 10.1111/febs.12754. [DOI] [PubMed] [Google Scholar]
- 26.Jiménez Fernández D., Hess S., Knobeloch K.P. Strategies to Target ISG15 and USP18 Toward Therapeutic Applications. Front. Chem. 2019;7:923. doi: 10.3389/fchem.2019.00923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Durfee L.A., Lyon N., Seo K., Huibregtse J.M. The ISG15 conjugation system broadly targets newly synthesized proteins: implications for the antiviral function of ISG15. Mol. Cell. 2010;38:722–732. doi: 10.1016/j.molcel.2010.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang Y., Thery F., Wu N.C., Luhmann E.K., Dussurget O., Foecke M., Bredow C., Jiménez-Fernández D., Leandro K., Beling A., et al. The in vivo ISGylome links ISG15 to metabolic pathways and autophagy upon Listeria monocytogenes infection. Nat. Commun. 2019;10:5383. doi: 10.1038/s41467-019-13393-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao C., Hsiang T.Y., Kuo R.L., Krug R.M. ISG15 conjugation system targets the viral NS1 protein in influenza A virus-infected cells. Proc. Natl. Acad. Sci. USA. 2010;107:2253–2258. doi: 10.1073/pnas.0909144107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang Y., Zhong G., Zhu L., Liu X., Shan Y., Feng H., Bu Z., Chen H., Wang C. Herc5 attenuates influenza A virus by catalyzing ISGylation of viral NS1 protein. J. Immunol. 2010;184:5777–5790. doi: 10.4049/jimmunol.0903588. [DOI] [PubMed] [Google Scholar]
- 31.Perng Y.C., Lenschow D.J. ISG15 in antiviral immunity and beyond. Nat. Rev. Microbiol. 2018;16:423–439. doi: 10.1038/s41579-018-0020-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ketscher L., Hannß R., Morales D.J., Basters A., Guerra S., Goldmann T., Hausmann A., Prinz M., Naumann R., Pekosz A., et al. Selective inactivation of USP18 isopeptidase activity in vivo enhances ISG15 conjugation and viral resistance. Proc. Natl. Acad. Sci. USA. 2015;112:1577–1582. doi: 10.1073/pnas.1412881112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lai C., Struckhoff J.J., Schneider J., Martinez-Sobrido L., Wolff T., García-Sastre A., Zhang D.E., Lenschow D.J. Mice lacking the ISG15 E1 enzyme UbE1L demonstrate increased susceptibility to both mouse-adapted and non-mouse-adapted influenza B virus infection. J. Virol. 2009;83:1147–1151. doi: 10.1128/JVI.00105-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Giannakopoulos N.V., Arutyunova E., Lai C., Lenschow D.J., Haas A.L., Virgin H.W. ISG15 Arg151 and the ISG15-conjugating enzyme UbE1L are important for innate immune control of Sindbis virus. J. Virol. 2009;83:1602–1610. doi: 10.1128/JVI.01590-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Freitas B.T., Durie I.A., Murray J., Longo J.E., Miller H.C., Crich D., Hogan R.J., Tripp R.A., Pegan S.D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020;6:2099–2109. doi: 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- 36.Munnur D., Teo Q., Eggermont D., Lee H.H.Y., Thery F., Ho J., van Leur S.W., Ng W.W.S., Siu L.Y.L., Beling A., et al. Altered ISGylation drives aberrant macrophage-dependent immune responses during SARS-CoV-2 infection. Nat. Immunol. 2021;22:1416–1427. doi: 10.1038/s41590-021-01035-8. [DOI] [PubMed] [Google Scholar]
- 37.Desai S.D., Haas A.L., Wood L.M., Tsai Y.C., Pestka S., Rubin E.H., Saleem A., Nur-E-Kamal A., Liu L.F. Elevated expression of ISG15 in tumor cells interferes with the ubiquitin/26S proteasome pathway. Cancer Res. 2006;66:921–928. doi: 10.1158/0008-5472.CAN-05-1123. [DOI] [PubMed] [Google Scholar]
- 38.Fan J.B., Arimoto K.i., Motamedchaboki K., Yan M., Wolf D.A., Zhang D.E. Identification and characterization of a novel ISG15-ubiquitin mixed chain and its role in regulating protein homeostasis. Sci. Rep. 2015;5 doi: 10.1038/srep12704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Pinto-Fernandez A., Salio M., Partridge T., Chen J., Vere G., Greenwood H., Olie C.S., Damianou A., Scott H.C., Pegg H.J., et al. Deletion of the deISGylating enzyme USP18 enhances tumour cell antigenicity and radiosensitivity. Br. J. Cancer. 2021;124:817–830. doi: 10.1038/s41416-020-01167-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Okumura F., Okumura A.J., Uematsu K., Hatakeyama S., Zhang D.E., Kamura T. Activation of double-stranded RNA-activated protein kinase (PKR) by interferon-stimulated gene 15 (ISG15) modification down-regulates protein translation. J. Biol. Chem. 2013;288:2839–2847. doi: 10.1074/jbc.M112.401851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arimoto K.I., Miyauchi S., Troutman T.D., Zhang Y., Liu M., Stoner S.A., Davis A.G., Fan J.B., Huang Y.J., Yan M., et al. Expansion of interferon inducible gene pool via USP18 inhibition promotes cancer cell pyroptosis. Nat. Commun. 2023;14:251. doi: 10.1038/s41467-022-35348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Park J.H., Yang S.W., Park J.M., Ka S.H., Kim J.H., Kong Y.Y., Jeon Y.J., Seol J.H., Chung C.H. Positive feedback regulation of p53 transactivity by DNA damage-induced ISG15 modification. Nat. Commun. 2016;7 doi: 10.1038/ncomms12513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mustachio L.M., Lu Y., Tafe L.J., Memoli V., Rodriguez-Canales J., Mino B., Villalobos P.A., Wistuba I., Katayama H., Hanash S.M., et al. Deubiquitinase USP18 Loss Mislocalizes and Destabilizes KRAS in Lung Cancer. Mol. Cancer Res. 2017;15:905–914. doi: 10.1158/1541-7786.MCR-16-0369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guo Y., Chinyengetere F., Dolinko A.V., Lopez-Aguiar A., Lu Y., Galimberti F., Ma T., Feng Q., Sekula D., Freemantle S.J., et al. Evidence for the ubiquitin protease UBP43 as an antineoplastic target. Mol. Cancer Ther. 2012;11:1968–1977. doi: 10.1158/1535-7163.MCT-12-0248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mustachio L.M., Kawakami M., lu Y., Rodriguez-Canales J., Mino B., Behrens C., Wistuba I., Bota-Rabassedas N., Yu J., Lee J.J., et al. The ISG15-specific protease USP18 regulates stability of PTEN. Oncotarget. 2017;8:3–14. doi: 10.18632/oncotarget.13914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ferreira J.C., Fadl S., Rabeh W.M. Key dimer interface residues impact the catalytic activity of 3CLpro, the main protease of SARS-CoV-2. J. Biol. Chem. 2022;298 doi: 10.1016/j.jbc.2022.102023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Corey D.R., Craik C.S. An Investigation into the Minimum Requirements for Peptide Hydrolysis by Mutation of the Catalytic Triad of Trypsin. J. Am. Chem. Soc. 1992;114:1784–1790. [Google Scholar]
- 48.Craik C.S., Roczniak S., Largman C., Rutter W.J. The Catalytic Role of the Active Site Aspartic Acid in Serine Proteases. Science. 1987;237:909–913. doi: 10.1126/science.3303334. [DOI] [PubMed] [Google Scholar]
- 49.Sprang S., Standing T., Fletterick R.J., Stroud R.M., Finer-Moore J., Xuong N.-H., Hamlin R., Rutter W.J., Craik C.S. The Three-Dimensional Structure of Asn102 Mutant of Trypsin: Role of Asp102 in Serine Protease Catalysis. Science. 1987;237:905–909. doi: 10.1126/science.3112942. [DOI] [PubMed] [Google Scholar]
- 50.Vasou A., Nightingale K., Cetkovská V., Bamford C.G., Andrejeva J., Randall R.E., McLauchlan J., Weekes M.P., Hughes D.J. A co-opted ISG15-USP18 binding mechanism normally reserved for deISGylation controls type I IFN signalling. bioRxiv. 2021 doi: 10.1101/2021.06.01.446527. Preprint at. [DOI] [Google Scholar]
- 51.Vuillier F., Li Z., Commere P.H., Dynesen L.T., Pellegrini S. USP18 and ISG15 coordinately impact on SKP2 and cell cycle progression. Sci. Rep. 2019;9:4066. doi: 10.1038/s41598-019-39343-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Basters A., Geurink P.P., Röcker A., Witting K.F., Tadayon R., Hess S., Semrau M.S., Storici P., Ovaa H., Knobeloch K.P., Fritz G. Structural basis of the specificity of USP18 toward ISG15. Nat. Struct. Mol. Biol. 2017;24:270–278. doi: 10.1038/nsmb.3371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Speer S.D., Li Z., Buta S., Payelle-Brogard B., Qian L., Vigant F., Rubino E., Gardner T.J., Wedeking T., Hermann M., et al. ISG15 deficiency and increased viral resistance in humans but not mice. Nat. Commun. 2016;7 doi: 10.1038/ncomms11496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Qiu X., Taft J., Bogunovic D. Developing Broad-Spectrum Antivirals Using Porcine and Rhesus Macaque Models. J. Infect. Dis. 2020;221:890–894. doi: 10.1093/infdis/jiz549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Clancy A., Rusilowicz-Jones E.V., Wallace I., Swatek K.N., Urbé S., Clague M.J. ISGylation-independent protection of cell growth by USP18 following interferon stimulation. Biochem. J. 2023;480:1571–1581. doi: 10.1042/BCJ20230301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Potu H., Sgorbissa A., Brancolini C. Identification of USP18 as an important regulator of the susceptibility to IFN-alpha and drug-induced apoptosis. Cancer Res. 2010;70:655–665. doi: 10.1158/0008-5472.CAN-09-1942. [DOI] [PubMed] [Google Scholar]
- 57.Magor K.E., Miranzo Navarro D., Barber M.R.W., Petkau K., Fleming-Canepa X., Blyth G.A.D., Blaine A.H. Defense genes missing from the flight division. Dev. Comp. Immunol. 2013;41:377–388. doi: 10.1016/j.dci.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Qian W., Wei X., Zhou H., Jin M. Molecular cloning and functional analysis of duck ubiquitin-specific protease 18 (USP18) gene. Dev. Comp. Immunol. 2016;62:39–47. doi: 10.1016/j.dci.2016.04.008. [DOI] [PubMed] [Google Scholar]
- 59.Hong B., Li H., Lu Y., Zhang M., Zheng Y., Qian J., Yi Q. USP18 is crucial for IFN-γ-mediated inhibition of B16 melanoma tumorigenesis and antitumor immunity. Mol. Cancer. 2014;13:132. doi: 10.1186/1476-4598-13-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Li G., Shi W., Xu Y., Li K., Chen Z., Lv M., Lv J., Qiu T., Qian Q., Ji J., et al. The USP18-FBXO6 axis maintains the malignancy of ovarian cancer. Biochem. Biophys. Res. Commun. 2022;593:101–107. doi: 10.1016/j.bbrc.2022.01.020. [DOI] [PubMed] [Google Scholar]
- 61.Bogunovic D., Byun M., Durfee L.A., Abhyankar A., Sanal O., Mansouri D., Salem S., Radovanovic I., Grant A.V., Adimi P., et al. Mycobacterial disease and impaired IFN-gamma immunity in humans with inherited ISG15 deficiency. Science. 2012;337:1684–1688. doi: 10.1126/science.1224026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Blaszczyk K., Nowicka H., Kostyrko K., Antonczyk A., Wesoly J., Bluyssen H.A.R. The unique role of STAT2 in constitutive and IFN-induced transcription and antiviral responses. Cytokine Growth Factor Rev. 2016;29:71–81. doi: 10.1016/j.cytogfr.2016.02.010. [DOI] [PubMed] [Google Scholar]
- 63.Singh A., Padariya M., Faktor J., Kote S., Mikac S., Dziadosz A., Lam T.W., Brydon J., Wear M.A., Ball K.L., et al. Identification of novel interferon responsive protein partners of human leukocyte antigen A (HLA-A) using cross-linking mass spectrometry (CLMS) approach. Sci. Rep. 2022;12 doi: 10.1038/s41598-022-21393-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Yang Z., Xian H., Hu J., Tian S., Qin Y., Wang R.F., Cui J. USP18 negatively regulates NF-kappaB signaling by targeting TAK1 and NEMO for deubiquitination through distinct mechanisms. Sci. Rep. 2015;5 doi: 10.1038/srep12738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Narasimhan J., Wang M., Fu Z., Klein J.M., Haas A.L., Kim J.J.P. Crystal structure of the interferon-induced ubiquitin-like protein ISG15. J. Biol. Chem. 2005;280:27356–27365. doi: 10.1074/jbc.M502814200. [DOI] [PubMed] [Google Scholar]
- 66.Burkart C., Fan J.B., Zhang D.E. Two independent mechanisms promote expression of an N-terminal truncated USP18 isoform with higher DeISGylation activity in the nucleus. J. Biol. Chem. 2012;287:4883–4893. doi: 10.1074/jbc.M111.255570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Thompson J.D., Higgins D.G., Gibson T.J. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Evans R., O’Neill M., Pritzel A., Antropova N., Senior A., Green T., Žídek A., Bates R., Blackwell S., Yim J., et al. Protein Complex Prediction with AlphaFold-Multimer. bioRxiv. 2022 doi: 10.1101/2021.10.04.463034. Preprint at. [DOI] [Google Scholar]
- 69.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Data reported in this paper will be shared by the lead contact upon request.
-
•
This paper does not report original code.