

Abstract
Small molecules that modulate the 14-3-3 protein-protein interaction (PPI) network represent valuable therapeutics and tool compounds. However, access has been lost to 14-3-3 PPI molecular glues of the cotylenin class, leading to investigations into practical chemical syntheses of congeners and analogues. Here we report a concise synthesis of (−)-cotylenol via a 10-step asymmetric entry into a diversifiable 5-8-5 core. This route features a mild Liebeskind-Srogl fragment coupling that tolerates unprecedented steric hindrance to produce a highly congested ketone, and a tandem Claisen-ene cascade that establishes the 8-membered ring. Late-stage control of stereochemistry and functionality leads to (−)-cotylenol and sets the stage for focused library synthesis.
Graphical Abstract
INTRODUCTION
Cotylenin A (1) is a member of the fusicoccanes, a family of 5-8-5 tricyclic diterpenoids produced by phytopathogenic fungi (Figure 1).1,2 Initially isolated as a plant growth regulator, 1 has since been shown to induce differentiation of human acute myeloid leukemia (AML) in primary culture,3 sensitize human cancer types to existing drugs,4a,5 and significantly decrease levels of the tumorigenic transcription factor c-Myc.6,7 Its aglycon cotylenol8 (3) is also bioactive, inducing differentiation in murine leukemia cells at a modestly lower (~10×) potency than 1.9 The activity of these compounds is believed to result from their function as “molecular glues” that can selectively stabilize (or disrupt10) complexes between the 14-3-3 signaling hub and its numerous client proteins.11 This property has attracted attention in both academia and industry,12 since 14-3-3 clients include cancer-relevant proteins such as C-RAF, p53, and BAD,4a,13 and 14-3-3 PPIs have been proposed to underlie resistance to standard-of-care drugs (e.g., cisplatin, etoposide, doxorubicin).14 As such, researchers have sought to broadly understand how 1 and 3 modulate different PPIs within the 14-3-3 interactome, as well as determine structure-activity relationships (SAR) to optimize potency and selectivity.4,9,15,16 Despite important progress, realization of these objectives has been hampered for at least 12 years because the producer organism of 1 and 3, a Cladosporium species, has lost the ability to proliferate in culture.17 Both the supply and diversification of the cotylenol scaffold are thus critical to advance the cotylenin chemotype towards therapeutic applications, analogous to immunomodulatory imide drug (IMiD) molecular glues.18
Figure 1.
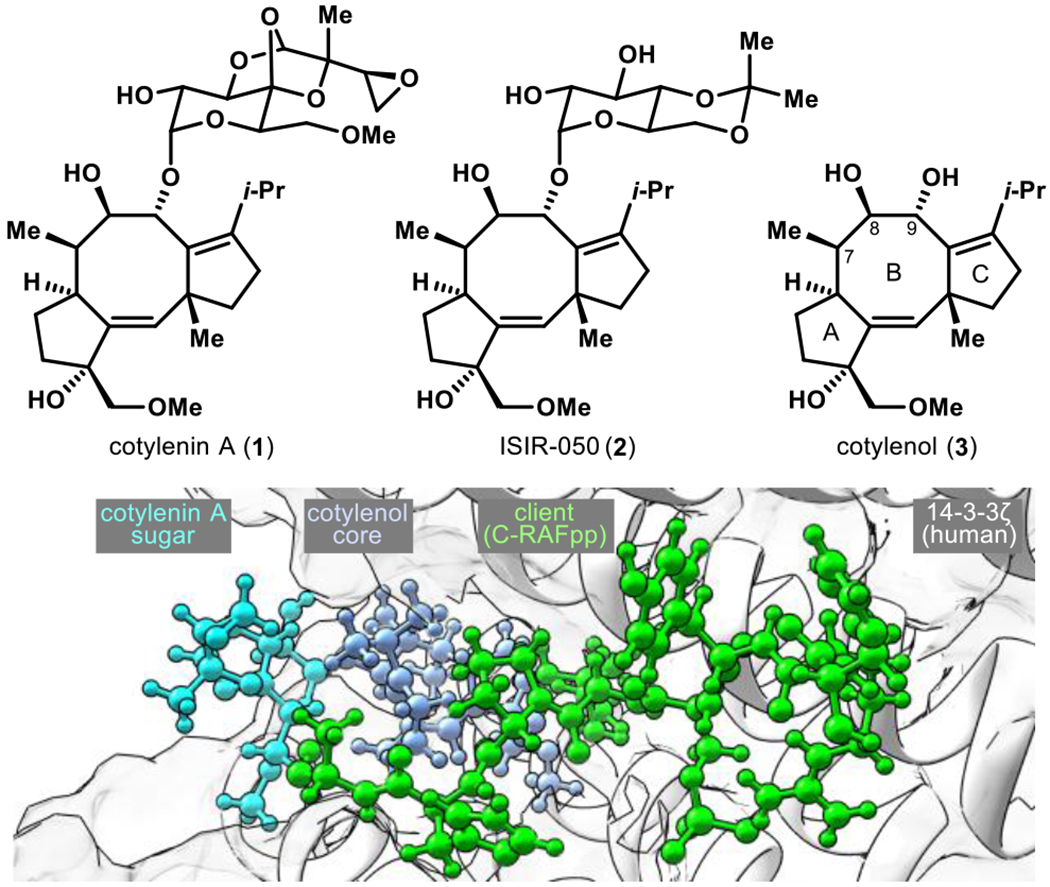
Fusicoccanes such as cotylenin A function as molecular glues between 14-3-3 proteins and phosphoprotein clients (from PDB: 4IHL, ref. 4a). C-RAFpp = diphosphorylated C-RAF peptide.
Currently, access to material requires use of mimics prepared through multistep semisynthesis (e.g., 2 in 14 steps from fusicoccin A)19 or total synthesis. One total synthesis of cotylenin A has been reported to date (25 steps, 0.15%),20 along with two syntheses of its aglycon, cotylenol (3) (21–32 steps, <1–3.9% yield).20,21 Here we report an alternative synthesis of 3 that provides expedient access to material and rapidly reaches a scaffold amenable to diversification.
RESULTS AND DISCUSSION
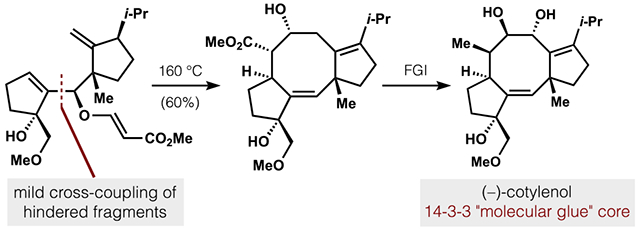
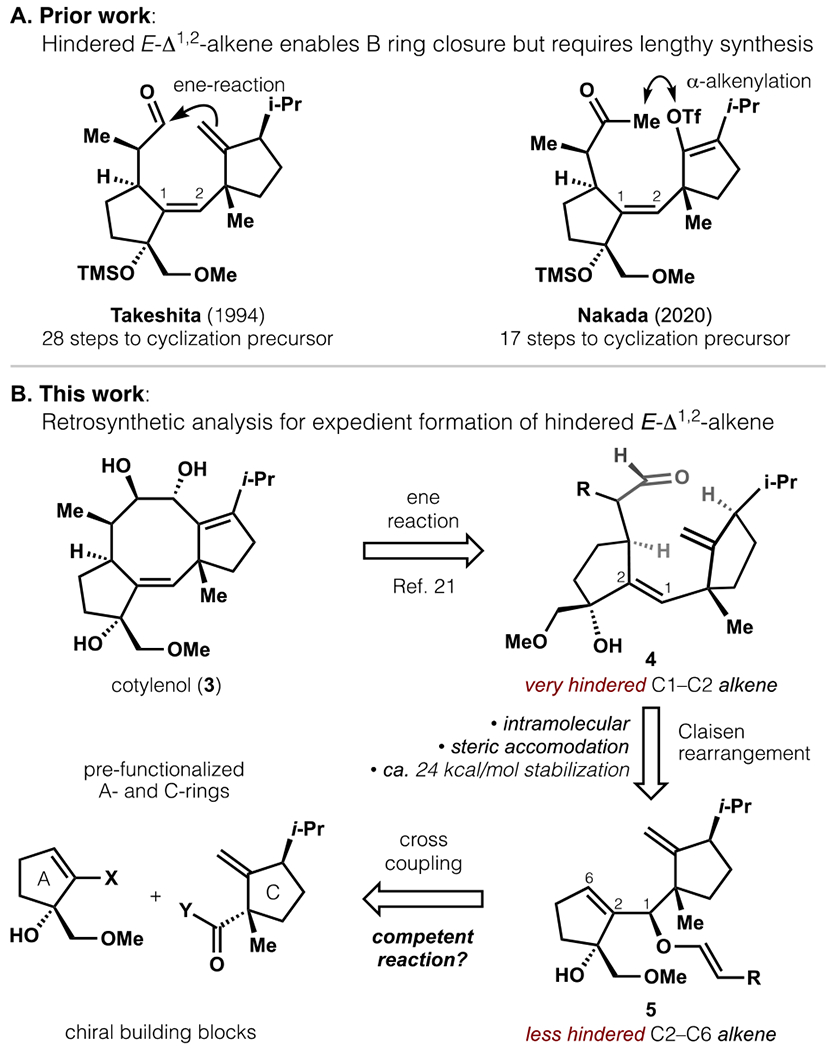
Prior syntheses by Takeshita and Nakada revealed that assembly of the Δ1,2-alkene with an E-configuration enabled efficient cyclooctene closure via ene or α-alkenylation reactions (Figure 2).20,21 However, synthesis of the cyclization precursors required 28 and 17 steps, respectively, due to the extreme steric congestion that flanked the alkene. As an important step towards broad studies of the cotylenin chemotype, we aimed to access 3 via expedient synthesis of substrate 4, which incorporated the E-Δ1,2-alkene with all native A- and C-ring functionality. As shown by X-ray crystallography, these rings form extensive contacts with 14-3-3 and its client in a deep cleft,22 whereas the C7–9 bridge and sugar motif point towards solvent-exposed regions. To forge the encumbered Δ1,2-alkene, we reasoned that the severe steric demands would be most readily accommodated in an intra-molecular rearrangement coupled to a strong driving force. A Claisen rearrangement appeared well-suited due to 1) exothermicity of C=O bond formation to offset steric repulsion, 2) established models to understand and control product stereochemistry,23 and 3) chemoselectivity.24 Analysis of Claisen transition states and experimental feedback (see SI and Scheme 7) eventually suggested allyl vinyl ether 5 as the required starting material, which could arrive in convergent fashion from prefunctionalized A- and C-rings.25
Figure 2.
Synthetic strategies to access cotylenol.
Scheme 7.
Incremental progress towards identification of a suitable Claisen rearrangement substrate.
First-generation synthesis of A- and C-ring fragments and their cross-coupling.
Our first-generation synthesis aimed to couple the A- and C-ring fragments through an addition of an A-ring vinyllithium to a C-ring electrophile. We therefore targeted an A-ring hydrazone and a C-ring aldehyde, which could be united under Shapiro reaction conditions.
Preparation of the A-ring began with acyloin cyclization of dimethyl glutarate,26 followed by a Zn(OTf)2-catalyzed Mukaiyama aldol reaction with dimethoxymethane (Scheme 1a). The trimethylsilyl ether was cleanly deprotected with Montmorillonite K10 in MeOH to afford ketone rac-6. Chiral preparative supercritical fluid chromatography (SFC) provided access to pure enantiomers ((R)- and (S)-6), where the absolute configuration was assigned by derivatization and X-ray crystallography (see SI). Condensation of (S)-6 with TrisNHNH2 produced hydrazone 7, and the tertiary alcohol was silylated to produce 8.
Scheme 1.
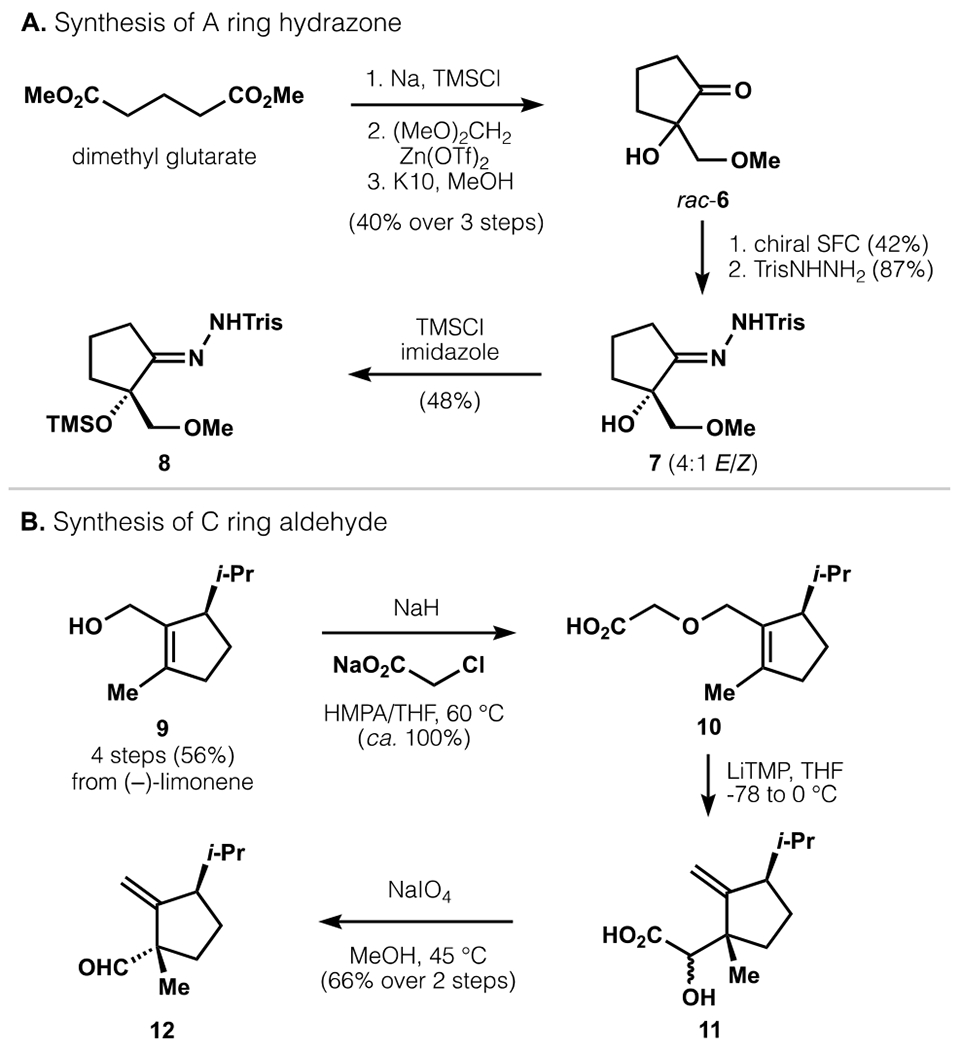
Initial synthesis of A- and C-ring fragments, revised in Schemes 3, 5 and 6.
Synthesis of the C-ring began with known alcohol 9, prepared in 50 mmol quantities from (−)-limonene in 4 steps and 56% yield (Scheme 1b).27 Alkylation with sodium chloroacetate formed 10 in quantitative yield. To generate the C11 quaternary center, 10 was treated with LiTMP to effect a [2,3]-Wittig rearrangement. Oxidative cleavage of the resulting α-hydroxyacid 11 afforded aldehyde 12 in 66% yield over two steps.
With sulfonylhydrazone 8 and aldehyde 12 in hand, we pursued their union via a Shapiro reaction that would convert 8 to its corresponding alkenyllithium. However, subjection of silyl-protected hydrazone 8 to standard Shapiro conditions (2 equiv. n-BuLi, THF, −78 °C) consistently resulted in retro-[1,4]-Brook rearrangement to generate a vinyl silane (protonated form of 13, Scheme 2a). Productive reaction with aldehyde 12 required an excess of n-BuLi (3 equiv) to generate either a nucleophilic silicate anion or organolithium (14). Under such conditions, allylic alcohol 15 was produced with high diastereoselectivity, possibly from Felkin-Anh control or coordination of the C-ring aldehyde to the A-ring lithium alkoxide. Unfortunately, this diastereomer proved unproductive in the synthesis according to classic Claisen rearrangement models combined with experimental validation (vide infra, also see SI). Use of free alcohol-containing substrate 7 averted the retro-[1,4]-Brook rearrangement, but still resulted in addition to 12 with the incorrect facial selectivity (Scheme 2b). Attempts to reverse stereoselectivity with additives or alternative organometallics proved unsuccessful.
Scheme 2.
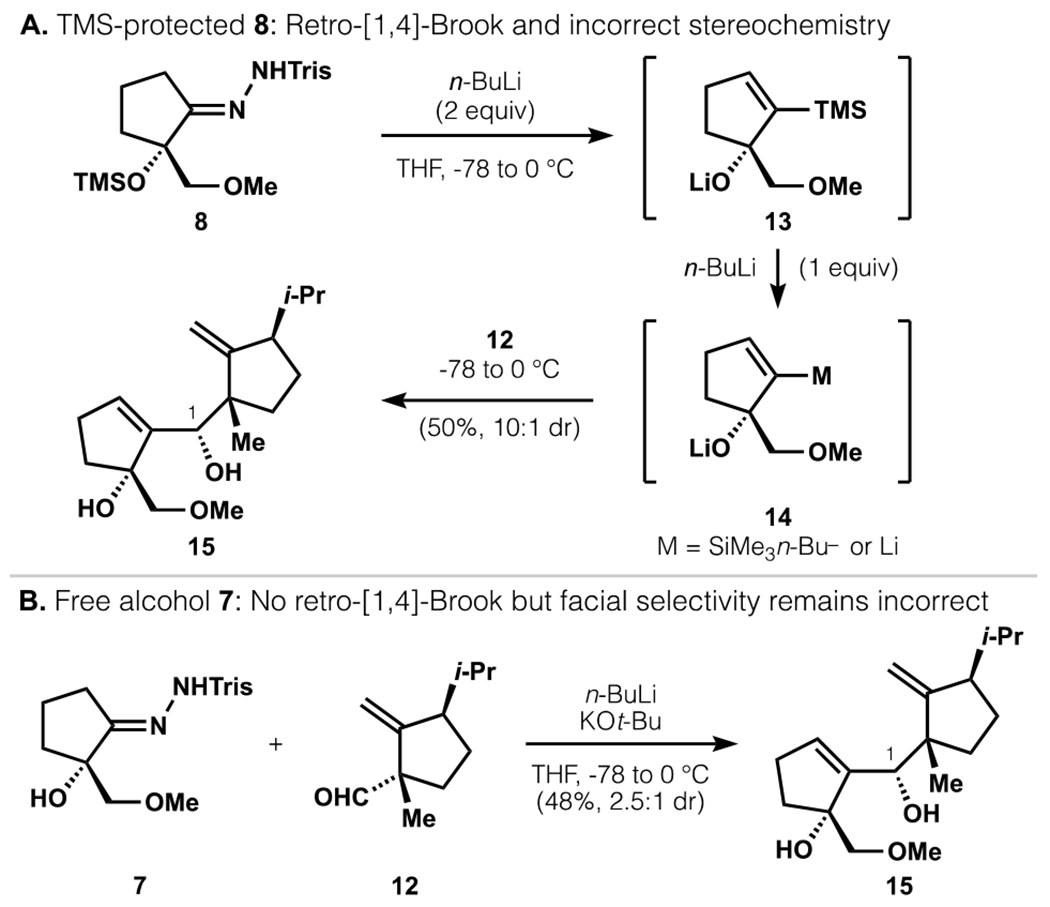
Addition to C-ring aldehyde yields incorrect diastereomer 15.
Because addition to the C-ring aldehyde afforded the undesired alcohol epimer, we sought a higher oxidation state electrophile to enable formation of a ketone that could be stereoselectively reduced. We thus targeted a C-ring thioester as both an electrophile and a gateway to alternative carboxylic acid derivatives (Scheme 3a).
Scheme 3.
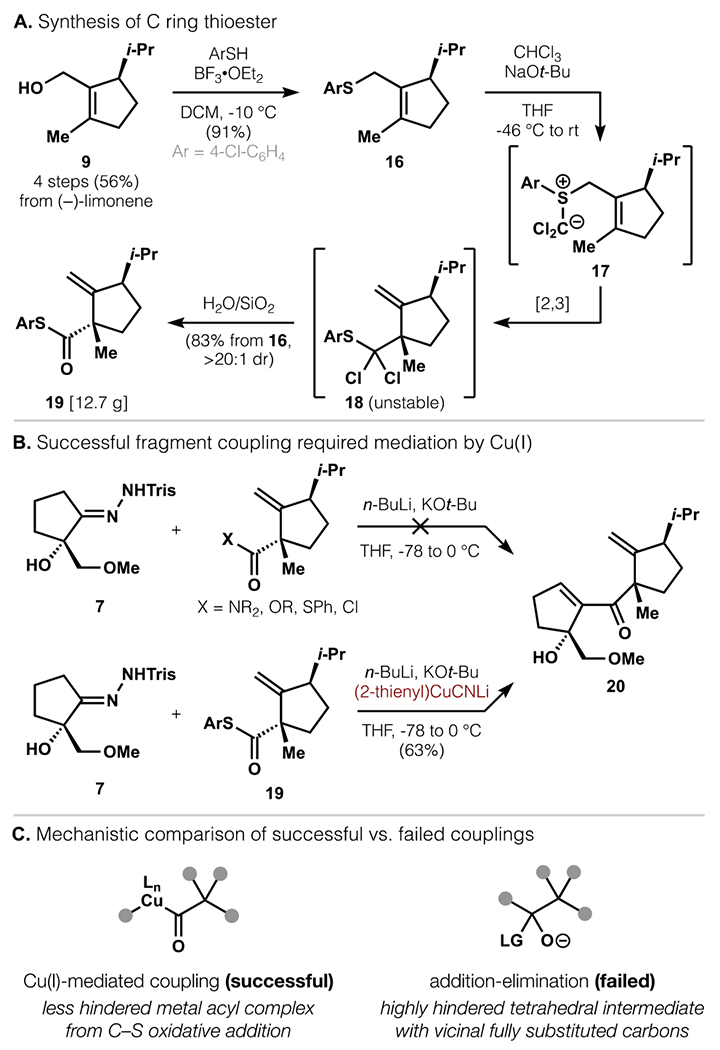
Realization of fragment coupling with a higher oxidation state C-ring electrophile.
Alcohol 9 was converted to thioether 16 through BF3•OEt2-promoted substitution with 4-chlorothiophenol. This thioether was then reacted with dichlorocarbene to form sulfonium ylide 17, which underwent facile [2,3]-Wittig rearrangement to deliver intermediate 18.28 Chromatography on hydrated silica gel converted this dichlorothioether to thioester 19 as a single diastereomer in 83% yield from 16. The use of NaOt-Bu in place of KOt-Bu enabled higher conversions and yields, due to either a lower rate of alkoxide addition to :CCl2 or slower α-elimination that limits :CCl2 concentration and thus homodimerization.29,30 This preparation of thioester 19 scaled easily to produce >10 grams in a single pass.
To merge the A- and C-rings while avoiding the retro-Brook rearrangement encountered with 8, we returned to free alcohol 7 (Scheme 3b). Treatment of 7 with n-BuLi (>3 equiv) and KOt-Bu at 0 °C, however, did not result in coupling with numerous acyl electrophiles, including acid chlorides. Given that reactivity with aldehyde 12 was achieved (vide supra), it was evident that the A-ring could be converted to a competent nucleophile, but that the steric hindrance of poorly electrophilic carboxylic acid derivatives prevented reaction. This challenge was ultimately overcome with a Cu(I)-mediated coupling.31 In this protocol, 7 was treated with n-BuLi and KOt-Bu, followed by the (2-thienyl)CuCNLi complex developed by the Lipshutz group,32 and lastly 19 to yield 63% of product 20. 4-Chloro substitution on the thioester was necessary to achieve good yield, whereas the parent phenyl thioester led to a low yield and low conversion of the C-ring electrophile. Whereas Cu(I)-mediated couplings of thioesters with hard organometallic nucleophiles have seen significant development, their use in the convergent coupling of highly functionalized or hindered fragments is scarce.33
The success of this Cu(I)-promoted reaction likely resulted from its mechanistic deviation from typical acyl substitution reactions via carbonyl addition-elimination (Scheme 3c). For the reaction of interest (7 + 19 to 20), an addition-elimination mechanism would require formation of an hindered tetrahedral intermediate with vicinal, fully substituted carbons. In contrast to 1,2-addition, the Cu(I)-mediated process has been proposed to involve C–S oxidative addition to form a considerably less hindered metal acyl complex, which then undergoes C–C reductive elimination.34 An analogous consideration of mechanism and steric effects would later guide reaction selection in our second-generation route (vide infra).
With access to 20 secured, an alcohol-directed Luche reduction (NaBH4, CeCl3•7H2O, −78 °C) advanced the material towards a Claisen rearrangement substrate (Scheme 4). The unstable diol 21 was obtained with the desired C1 stereochemistry as a single desired diastereomer in ca. 80% yield.35 The high reactivity and stereochemical outcome are consistent with transition state TS1, in which the C3 tertiary alcohol directs reduction and the C-ring occupies a pseudo-equatorial position. Alternative scenarios of ketone-cerium coordination or chelation are disfavored by the following data: C3 silyl ether 22 did not undergo any reduction under identical Luche conditions, and the C3 epimer of 20 delivered the C1-epimeric alcohol (20:1 dr).
Scheme 4.
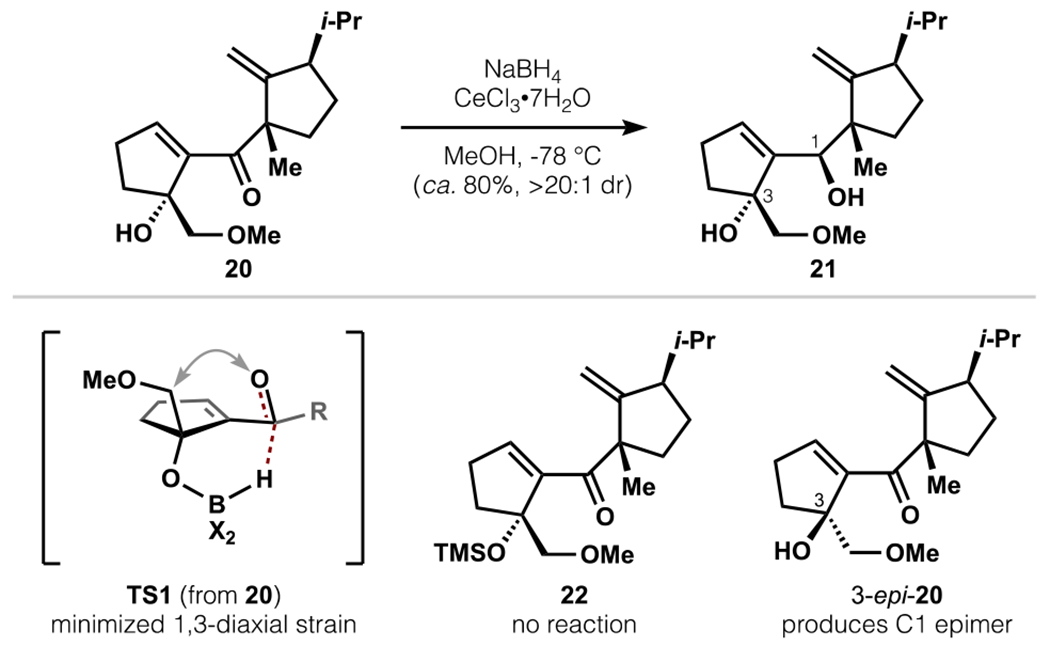
Stereoselective Luche reduction to establish desired C1 configuration.
Whereas this route established convergent access to targeted intermediate 21, several issues noted by the Referees significantly limited its practicality and use in medicinal chemistry. First, the use of preparative chiral SFC constrained material throughput due to instrument time (13 h per gram of (S)-6), cost ($90/h), and reliance on specialized facilities (Waters 150 AP). Second, the convergent coupling proved difficult to scale and was most reliably carried out with 40 mg 7. Operationally, it required tedious and carefully timed preparation of four discrete organometallic species ((2-thienyl)Li, (2-thienyl)CuCNLi, A-ring vinyllithium, A-ring cuprate). Moreover, yields were difficult to reproduce because of the reaction’s extreme sensitivity to adventitious water and oxygen.36 With the objective to broadly enable studies of the cotylenin chemotype, we sought improved material throughput via an asymmetric synthesis of the A-ring and a more practical convergent coupling.
Enantioselective synthesis of A-ring fragment and second-generation cross-coupling with C-ring.
Our initial efforts towards an asymmetric A-ring synthesis targeted ketone 6 in enantiomerically enriched form. Despite its structural simplicity, its enantioselective synthesis was repeatedly thwarted by poor enantioselectivity or an inability to productively elaborate synthetic intermediates. Consequently, we explored the synthesis of a cyclic vinylboron coupling partner, encouraged by precedent for the formation of cyclic vinylboronic esters by ring-closing metathesis37 and the versatility of the C–B bond as a precursor to C–X or C–[M] species. This sequence commenced with ketone 23, which is known in one step from methoxyacetonitrile38 or accessible from methoxyacetic acid in a highly scalable 2-step protocol (Scheme 5).39 To establish the chiral C3 tertiary alcohol, 23 was subjected to addition of trimethylsilylacetylene in the presence of Et2Zn and a chiral amino alcohol ligand, inspired by foundational work by Noyori.40
Scheme 5.
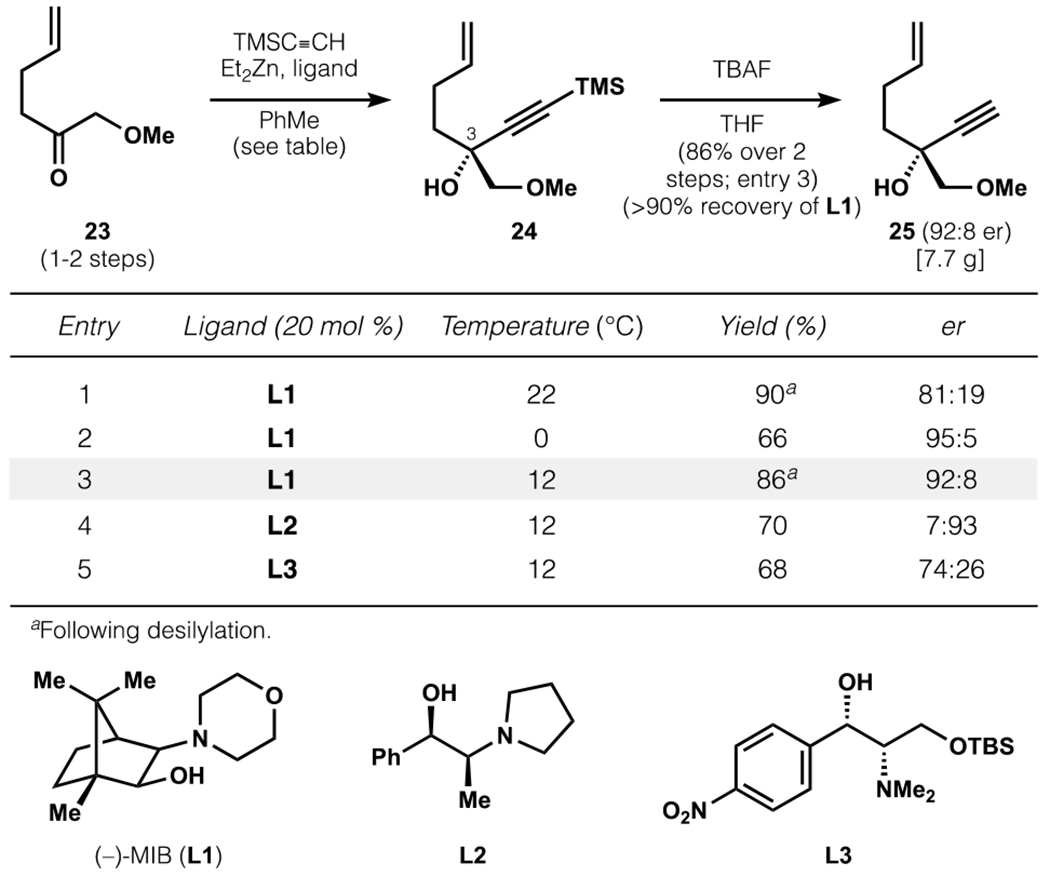
Asymmetric synthesis of C3 tertiary alcohol for second-generation A-ring synthesis.
The use of 20 mol% (−)-MIB41 (L1), derived from (+)-camphor in 3 steps, was found to give the desired product in 81:19 er at room temperature (90% over 2 steps, following desilylation). Cooling to 0 °C improved enantioselectivity (95:5 er), but at the expense of yield (66%). An intermediate temperature (12 °C, dioxane/dry ice) provided both high yields and enantioselectivity, and L1 was found to be superior to other chiral amino alcohols used for asymmetric alkynylation of ketones with proximal coordinating groups.42 Using the optimized conditions, alkynylation and subsequent desilylation produced propargyl alcohol 25 (92:8 er) in 86% yield over two steps. This protocol delivered >7 grams of 25 in a single pass, and L1 could be recovered in >90% yield through extractive workup.
Scalemic propargyl alcohol 25 was subjected to an efficient α-selective hydroboration based on a protocol from the Carretero group, introducing a handle for subsequent cross-coupling while setting the stage for A-ring closure (Scheme 6).43 Cyclization of 26 was accomplished through ring-closing metathesis with the Hoveyda-Grubbs II catalyst (5 mol %), which provided the boronic ester coupling partner 27 in 74% yield. This material was used without chromatography to avoid substantial losses on silica gel (74% vs. 53%). This robust sequence enabled straightforward access to >2 grams of 27 in a single pass and 5.2 grams over 3 runs.
Scheme 6.
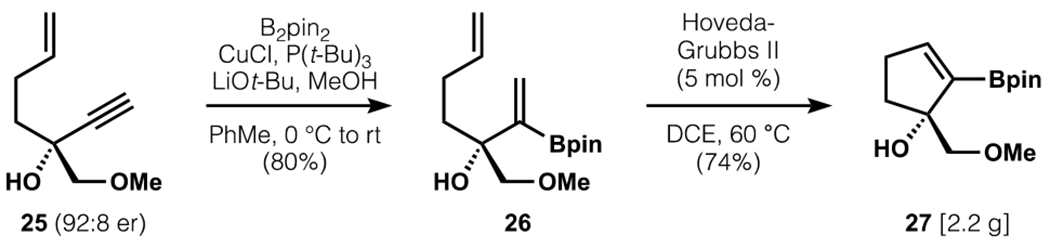
Elaboration to an A-ring cyclic vinylboronic ester.
Access to gram amounts of pinacolboronic ester 27 allowed us to explore cross-coupling methods more practical than the cuprate-thioester coupling (Scheme 3). In keeping with lessons from the first-generation cuprate coupling, we first investigated transition metal-catalyzed cross-couplings that proceeded through metal acyl complexes to avoid hindered tetrahedral intermediates. We considered a Liebeskind-Srogl coupling (Figure 3) to be ideal, as no change to either substrate would be necessary.44 However, no literature precedent supported use of a hindered, cyclic boronic ester nucleophile and model studies were discouraging. For example, PhBpin failed to couple with 19. PhB(OH)2 showed improved reactivity (12%, Figure 3F) but 27 could not be cleanly converted to its boronic acid due to facile 1,3-transposition of the allylic alcohol. In light of these unfavorable results, we were surprised to find that boronic ester 27 engaged thioester 19 in a Liebeskind-Srogl coupling under typical conditions (Pd2(dba)3, P(2-furyl)3, CuTC, THF), albeit in only 30% yield at 50 °C. Whereas variation of phosphine ligands, Pd pre-catalysts, and Cu reagents failed to improve reaction efficiency, boric acid (B(OH)3) exhibited positive effects, possibly as a Lewis acid, pinacol scavenger,45 hydroxide donor to Pd,46 or precursor to borate esters involving the C3 tertiary alcohol.47 Its inclusion nearly doubled the yield to 54% (1.4 g of 20 in one pass, Figure 3a) and allowed efficient coupling to occur without heating. This represents a remarkably facile synthesis of a hindered ketone under neutral conditions at room temperature. The successful coupling of a hindered vinylboronic ester contrasts with the current state-of-the-art, which requires the use of vinylstannanes48 to form challenging C–C bonds. Vinylborons, on the other hand, have been restricted to unhindered cases (trans-1-alkenyl) and almost always require the use of boronic acids.49
Figure 3.
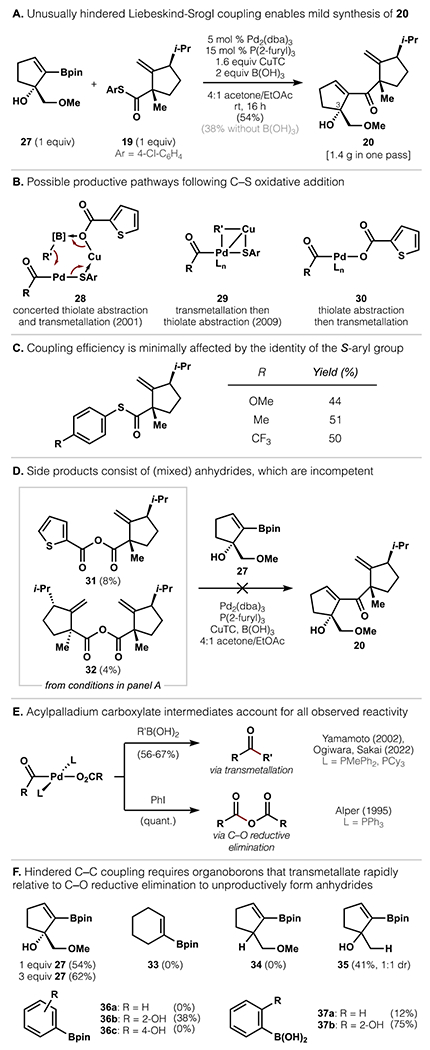
Discovery and investigation of an unusually hindered Liebeskind-Srogl fragment coupling.
Intrigued by the mild formation of hindered ketone 20 from a nontraditional Liebeskind-Srogl partner, we sought a better understanding of this reaction and the determinants of its success. The generally restricted scope of vinylborons in hindered Liebeskind-Srogl couplings suggested transmetallation to be a limiting factor (see below and Figure 3F). In the canonical coupling of boronic acids, Liebeskind proposed simultaneous thiolate abstraction and transmetallation, both mediated by CuTC (28, Figure 3b).50 A computational study subsequently proposed stepwise transfer of the organoboron to an acyl-Pd2+-thiolate, followed by thiolate departure (29).51 A third possibility involved thiolate abstraction by CuTC to form an acylpalladium carboxylate intermediate (30), followed by transmetallation. Although such an intermediate has not been invoked in the Liebeskind-Srogl coupling, it is a proposed intermediate in Pd-catalyzed couplings of arylcarboxylic acid anhydrides with boronic acids.52
In the course of improving the cross-coupling, we evaluated the reaction dependence on the S-aryl group, which would directly participate in the pathways 28 and 29 but not 30. Electronically differentiated S-aryl groups led to similar yields, suggesting that 28 and 29 (Figure 3c) may not be major contributors to productive transmetallation of the boronic ester onto Pd2+. Although 28 and 29 could not be rigorously excluded, they did not account for all observed reactivity, as revealed by analysis of unpurified reaction mixtures. Identifiable side products included 3-epi-20 (4%, from the minor enantiomer of 27) and mixed anhydride 31 (8%), derived from 19 and CuTC (Figure 3d). The symmetrical anhydride 32 was also observed (4%), possibly resulting from hydrolysis of 31 under the reaction conditions, followed by acylation by a second molecule of 31. The structure of both anhydrides was confirmed by independent synthesis, and their stability to chromatographic purification testifies to the exceedingly encumbered environment of the C-ring. When subjected to the cross-coupling conditions, both 31 and 32 failed to yield product (20), establishing that anhydride formation is nonproductive.
These findings are most consistent with the intermediacy of an acylpalladium carboxylate complex (e.g. 30), which has been shown through prior stoichiometric studies to account for all observed reactivity (Figure 3e). Yamamoto, Ogiwara and Sakai demonstrated that C–C coupling occurs in good yield upon treatment of related acylpalladium carboxylate complexes with arylboronic acids.52b,53 Alper has shown that a complex with the less donating PPh3 ligand spontaneously undergoes C–O reductive elimination to generate anhydrides.54 This process occurs following dissociation of a phosphine ligand, and is of relevance since our experiments employ P(2-furyl)3, which is both thermodynamically and kinetically more labile than PPh3.55 Notably, numerous studies have provided evidence against C–O bond formation via outer-sphere nucleophilic attack on the acyl group, which would not require complex 30.56 Interestingly, in the coupling of less hindered systems, Liebeskind and Srogl have only observed decarbonylation as a side reaction, suggesting that the steric demands of the substrate may alter relevant reaction pathways.44
The likely involvement of intermediate 30 suggested that extremely hindered Liebeskind-Srogl couplings require efficient transmetallation to a congested acylpalladium carboxylate complex in order to outcompete C–O reductive elimination. Consistent with this mechanistic picture, the use of excess boronic ester 27 (3 equiv) improved the yield of 20 (62% vs. 54% under identical conditions, Figure 3f), and not because the reaction was limited by protodeboronation.57 To investigate whether proximal functional groups influenced the transmetallating ability of 27, closely related boronic esters were reacted with thioester 19. Unsubstituted boronic ester 33 and C3-deoxy boronic ester 34 failed to couple with 19, whereas hydroxyl-substituted substrate 35 proved competent (36%). These observations were mirrored in arylboronic esters and acids, where the presence of a 2-hydroxy group significantly enhanced reactivity relative to the parent compound (36a–c, 37a–b). The pendant hydroxy group may serve to lower the barrier to transmetallation via coordination to Pd2+ as a directing group (as has been demonstrated for alkylboronic esters58), or hydrogen bonding with a Pd-bound acyl group or carboxylate; the coordination of boric acid may strengthen these interactions. While the experiments above demonstrated the beneficial effect of the proximal alcohol, it was not found to be broadly sufficient to enable cross-coupling with hindered thioester 19 (see SI). These data emphasize the general difficulty of hindered Liebeskind-Srogl reactions and the unique efficiency of the coupling to form 20.
These insights into the reactivity of boronic ester 27 led us to evaluate the extent to which hindered Liebeskind-Srogl couplings depended on the thioester partner. In contrast to the boronic ester, numerous sterically encumbered thioesters coupled successfully (38a–h, Figure 4). These experiments revealed several insights. First, the transformation did not rely on the exocyclic alkene in 19 to stabilize acylpalladium species.59 Second, the reaction yield increased for less encumbered thioesters, possibly due to faster transmetallation of less-hindered acylpalladium carboxylates relative to C–O reductive elimination. Finally, under no circumstances did we observe decarbonylation, possibly since decarbonylation to form tert-alkylpalladium complexes incurs prohibitive steric penalties. Overall, this investigation has shown that the Liebeskind-Srogl coupling can enable facile synthesis of hindered ketones if transmetallation is accelerated, and we have outlined key considerations for the success of this demanding transformation.
Figure 4.
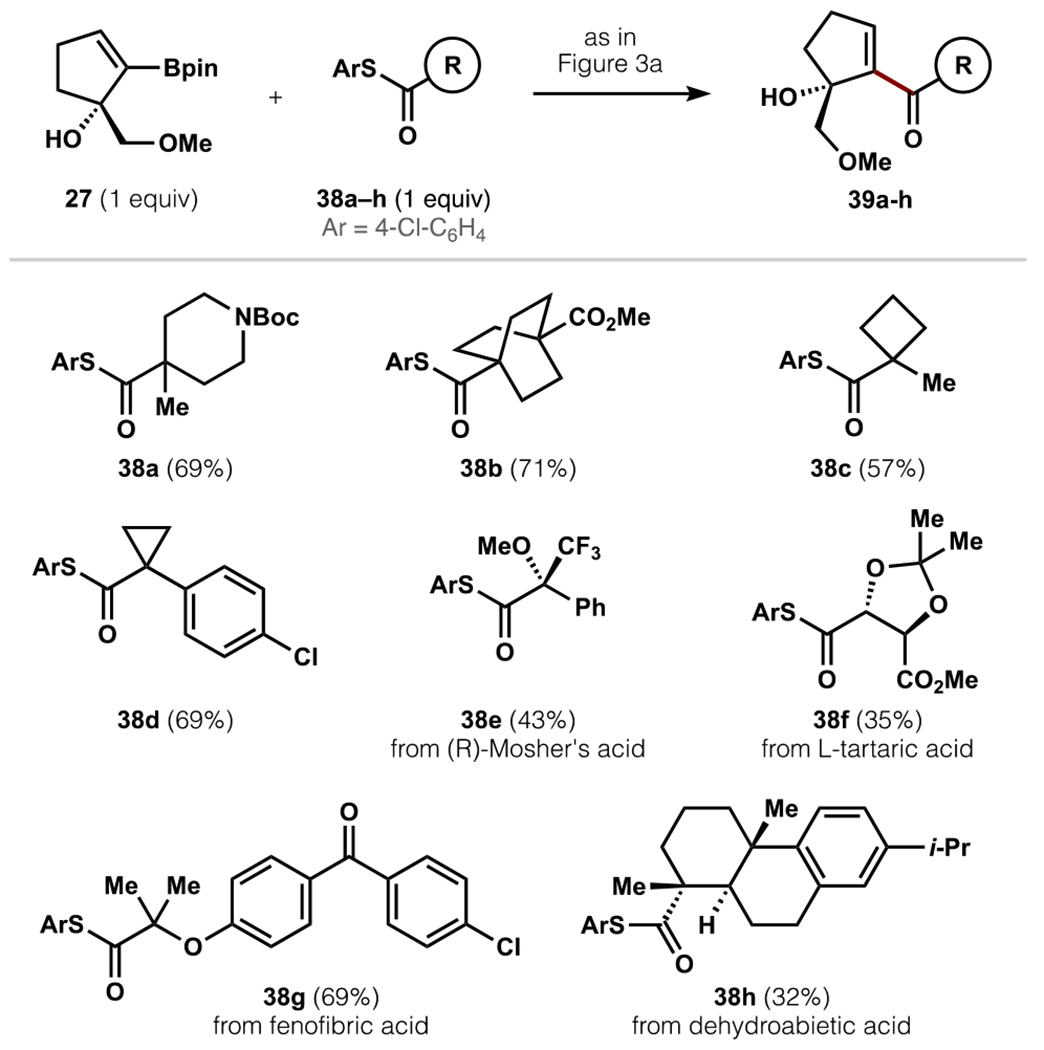
Scope of hindered ketones. Reaction conditions: 27 (0.12 mmol), 38a–h (1 equiv), Pd2(dba)3•CHCl3 (5 mol %), P(2-furyl)3 (15 mol %), CuTC (1.6 equiv), B(OH)3 (2 equiv), 4:1 acetone/EtOAc, rt, 16 h.
More importantly, this second-generation route to 20 accomplished the two goals outlined during its conception. The first goal of developing an asymmetric A-ring synthesis was made possible by an enantioselective ketone alkynylation, which enabled rapid elaboration to a vinylboronic ester. The second goal of developing a robust, operationally simple and scalable cross-coupling was achieved through a B(OH)3-enhanced Liebeskind-Srogl coupling. This transformation produced hindered ketone 20 under mild conditions and obviated the laborious and sensitive organocuprate coupling from the first-generation synthesis. The coupling has been scaled to produce >1 gram of 20 per run, intercepting the end of the first-generation route with far greater material throughput.
Claisen rearrangement and completion of cotylenol.
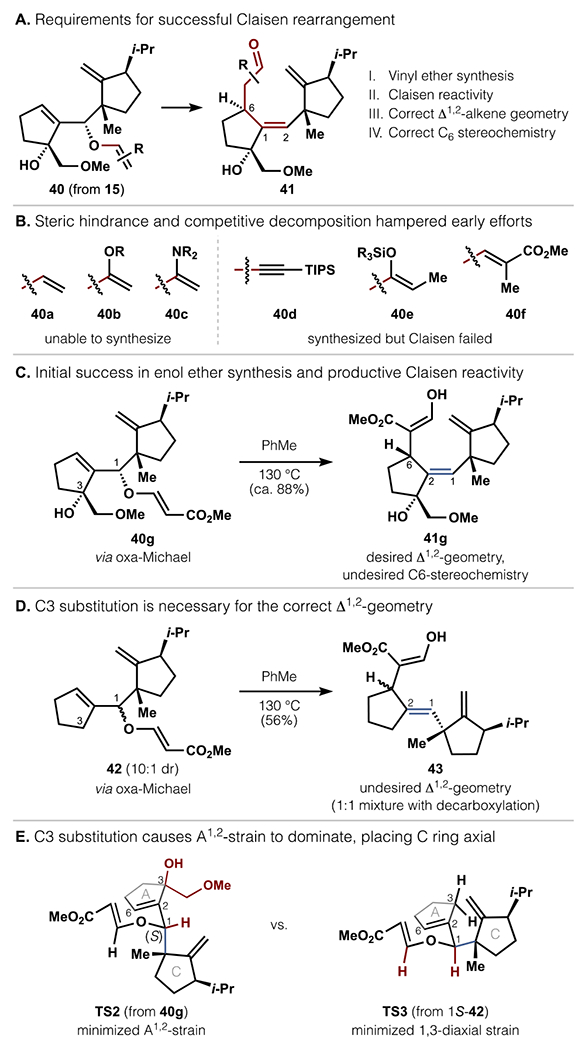
Access to the 5-8-5 scaffold and completion of cotylenol (3) required identification of a suitable enol ether substrate for Claisen rearrangement to construct the highly congested Δ1,2-alkene (Scheme 7a). A successful substrate would need to: (1) be accessible from the hindered C1 alcohol, (2) undergo Claisen rearrangement faster than decomposition (especially elimination of the C3 tertiary allylic alcohol), (3) afford the correct E-geometry at the formed Δ1,2-alkene, and (4) afford the correct R configuration at C6.
With these criteria in mind, we initially attempted to surmount these challenges from allylic alcohol 15, which was more readily accessed than its epimer 21 during initial route development (Scheme 2). The first task of forging the enol ether was met with resistance. Steric hinderance prevented synthesis of vinyl ether 40a using alkenyl electrophiles with Pd2+ or Hg2+ catalysis, while extensive decomposition thwarted access to silyl ketene acetal/hemiaminal 40b and 40c under forcing Johnson- or Eschenmoser-Claisen conditions (Scheme 7b). Enol ether synthesis was eventually accomplished to form ynol ether 40d (from Waser’s reagent60) and enol ethers 40e,f (from esterification/silylation or oxa-Michael addition). However, these compounds failed to undergo productive Claisen rearrangements, likely due to barriers exceeding those of decomposition pathways.
Further experimentation revealed that vinylogous ester 40g was accessible from oxa-Michael addition (see SI) and reactive in the Claisen rearrangement (Scheme 7c). Upon heating in PhMe at 130 °C, 40g converted into 41g in 56% yield, furnishing the hindered Δ1,2-alkene with the necessary E-geometry for elaboration to 3. The alkene geometry was rationalized as shown in TS2: the C3 substitution on the A-ring caused A1,2-strain to dominate over 1,3-diaxial strain, forcing the C-ring to occupy a pseudoaxial position in the chair-like transition state (Scheme 7e).23 The importance of C3-substitution was verified with model substrate 42 (Scheme 7d).61 This compound also underwent Claisen rearrangement, but the absence of C3-substitution rendered A1,2-strain insufficient to overcome 1,3-diaxial strain. Thus, the C-ring instead occupied a pseudoequatorial position in TS3, producing 43 with the undesired alkene geometry.
Although enol ether 40g satisfied 3 out of 4 requirements, its Claisen rearrangement yielded the incorrect C6 stereochemistry, a result of the 1S-configuration. Our collective findings led us to posit that all four requirements could be met with the C1-epimer of 40g.
Realization of a successful stereoselective Claisen rearrangement (Scheme 8) began with C1-epimeric alcohol 21, which was accessed via Liebeskind-Srogl coupling and reduction (Scheme 4). Oxa-Michael addition of the hindered C1 alcohol to methyl propiolate was mediated by N-methylmorpholine (NMM) at 0 °C with exceptional ease, likely through an alkoxide/enammonium caged pair.62 The resulting vinyl ether 44 was heated in silylated glass to provide 5-8-5 tricycle 45 via a stereoselective Claisen rearrangement/ene reaction cascade.21,63 In accordance with the insights gained from Scheme 7, the A-ring substitution at C3 and the R-configuration at C1 translated cleanly to the requisite hindered E-alkene and the 6R-configuration, respectively (TS4). The E-alkene placed the C8 aldehyde proximal to the Δ9,10-alkene, such that the thermal conditions also effected an ene reaction (TS5) analogous to that developed by Takeshita.21b,c This protocol readily afforded 750 mg of the advanced tricycle 45 in a single pass.
Scheme 8.
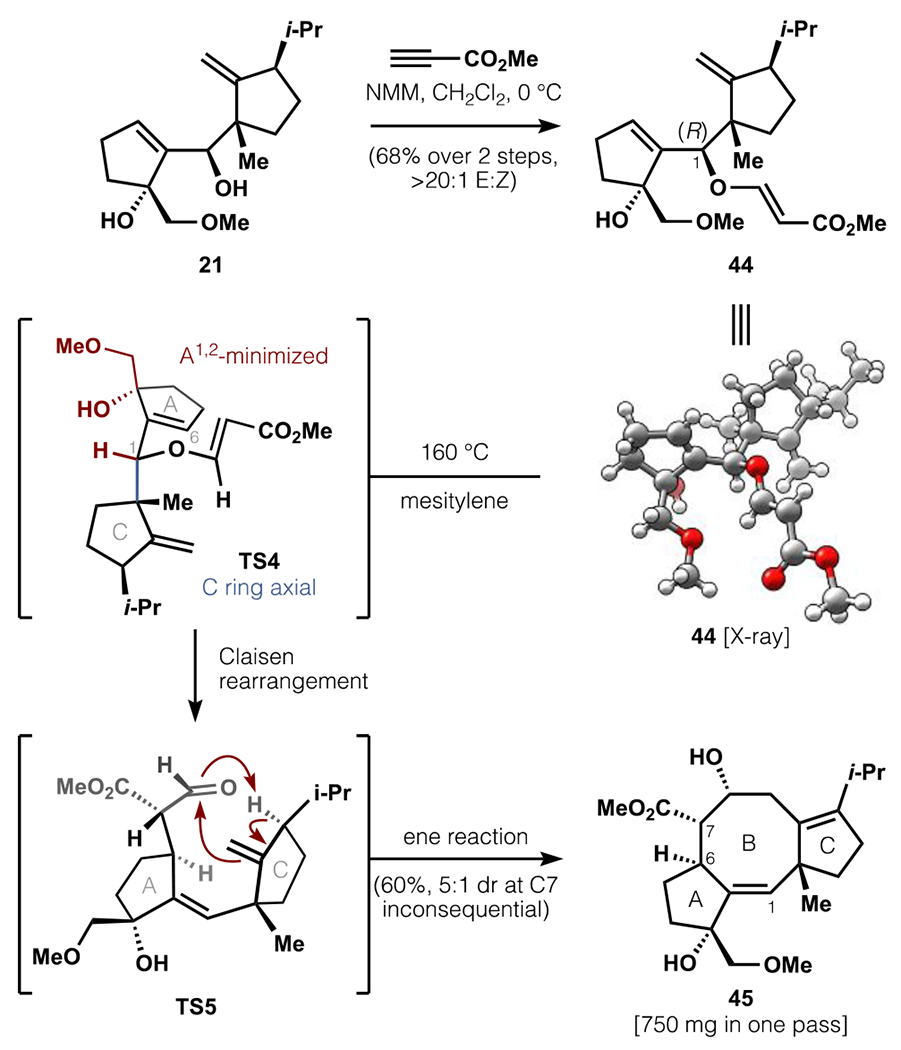
Tandem Claisen-ene cascade.
A simple sequence then converted 45 to cotylenol (3). First, β-hydroxyketone 46 was obtained following uneventful oxidation of the β-hydroxyester and decarboxylative aldol reaction with formaldehyde (Scheme 9a). Elimination of the β-hydroxyl group yielded enone 47, which was subjected to a series of stereoselective transformations to establish the C7–9 stereotriad. α-Hydroxylation of 47 at C9 proved efficient (94%) and highly stereoselective (>20:1 dr), contrasting prior syntheses wherein oxidations of related intermediates delivered mixtures of C9 epimers (2.7–1.5:1).20,21 The rigid conformation enforced by the all-sp2 C7–9 bridge of 50 (the potassium enolate of 47) may allow reagents to avoid the i-Pr substituent but not the C18 methyl on the opposite face (Scheme 9b).64 Late-stage intermediates of prior syntheses possessed pseudo-equatorial methyl groups at C7, which likely twisted the enolate (relative to 50) to expose the internal face. α-Hydroxyketone 48 was then subjected to a diimide reduction, which also occurred from the exterior face with high dr (7:1). Finally, Nakada’s protocol for directed reduction furnished 3 in 83% yield and >20:1 dr without recourse to C3 alcohol protection/deprotection as used previously.20
Scheme 9.
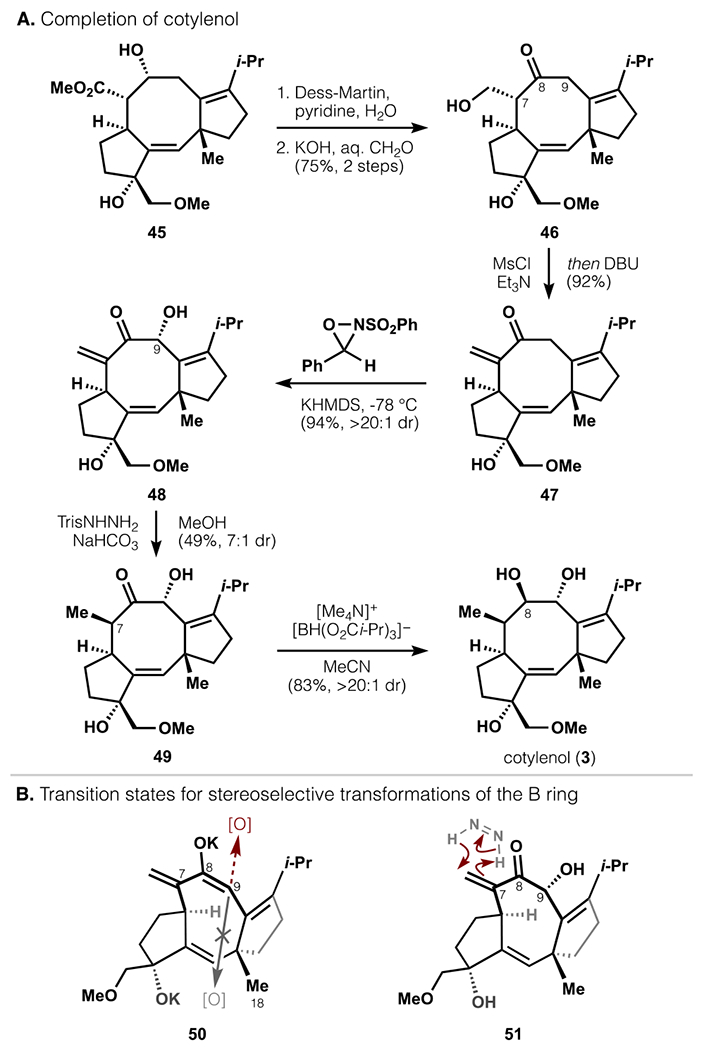
Completion of cotylenol via stereoselective B-ring manipulations.
CONCLUSION
In summary, we have developed a short synthesis of (−)-cotylenol (16 linear steps, 9 steps from convergence of A and C-rings) via mild Liebeskind-Srogl coupling of hindered and fully functionalized A- and C-rings, and a Claisen-ene cascade reaction (Scheme 10). The synthetic sequence scales well, expediently affording 750 mg of 5-8-5 scaffold 45 in 10 linear steps (3.8%, 1st generation versus 4.2–6.8%, 2nd generation route, depending on preparation of A-ring starting material). Furthermore, the route is highly amenable to diversification at numerous positions: the A and C rings through cross-coupling, and the B-ring through manipulation of the Claisen-ene product 45. We plan to leverage this synthesis to prepare a focused library of analogues and explore the selective engagement of 14-3-3 protein/client complexes.65 These efforts will benefit from available crystal structures for 14-3-3 protein/client/cotylenin A complexes and existing SAR data for cotylenin congeners and semisynthetic fusicoccin analogues. We aim to identify novel natural product-based lead compounds for future development, and efforts toward this goal are underway.66
Scheme 10.
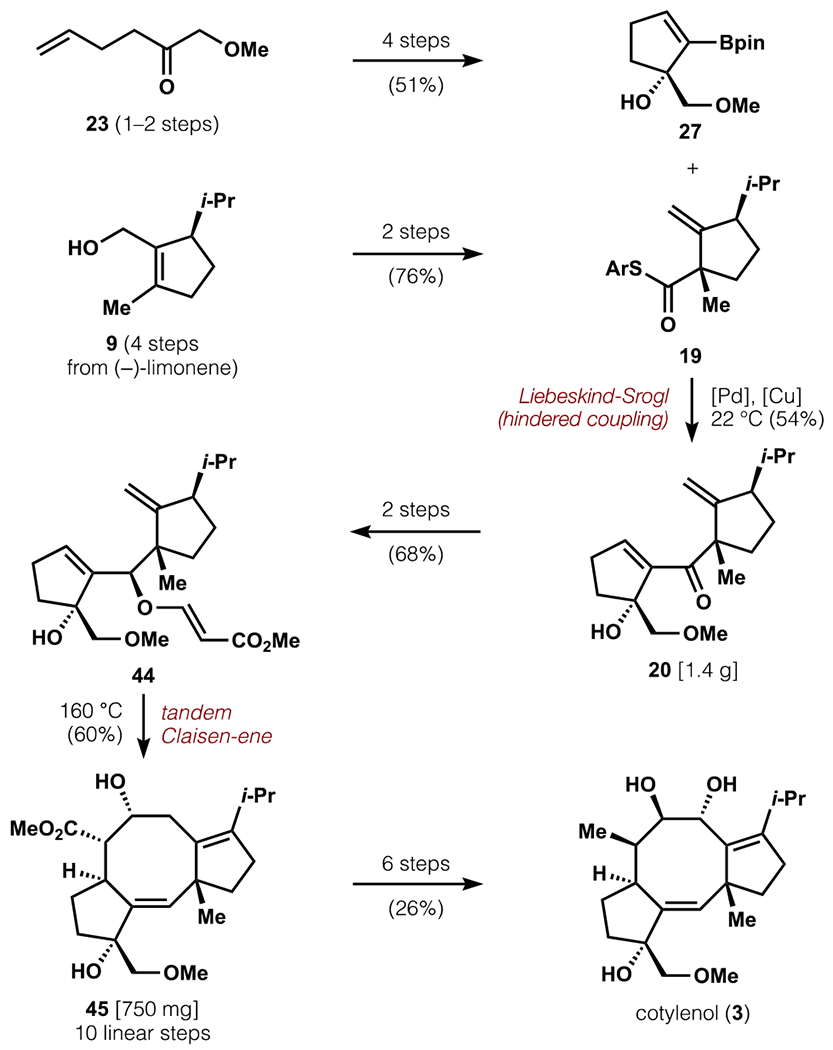
Summary of completed synthesis.
Supplementary Material
ACKNOWLEDGMENT
Arnold Rheingold, Milan Gembicky, Jack Bailey and Erika Samolova are acknowledged for X-ray crystallographic analysis. We thank Dr. L. Pasternack and Dr. D.-H. Huang for NMR assistance. The Scripps Automated Synthesis Facility directed by Jason Chen performed separations and analysis of key intermediates, including preparative chiral SFC separation of rac-6. Support was provided by the National Institutes of Health (R35 GM122606 to R. A. S.; F32 CA278405 to S.I.T.), the Kellogg Graduate School (D.W.S.), the NSF (GRF to T.R.H.), the JSPS (postdoctoral fellowship to A.K.) and the Uehara Memorial Foundation (postdoctoral fellowship to R.S.).
Footnotes
A provisional patent has been filed: U.S. Ser. No. 63/408,740.
Supporting Information. Experimental procedures, characterization data and structural assignments. This material is available free of charge via the Internet at http://pubs.acs.org.
REFERENCES
- (1).(a) de Boer AH; Leeuwen IJ “Fusicoccanes: diterpenes with surprising biological functions” Trends Plant Sci. 2012, 17, 360. [DOI] [PubMed] [Google Scholar]; (b) Marra M; Camoni L; Visconti S; Fiorillo A; Evidente A “The Surprising Story of Fusicoccin: A Wilt-Inducing Phytotoxin, a Tool in Plant Physiology and a 14-3-3-Targeted Drug. Biomolecules” Biomolecules 2021, 11, 1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).(a) Sassa T; Tojyo T; Munakata K Isolation of a New Plant Growth Substance with Cytokinin-like Activity. Nature 1970, 227, 379. [DOI] [PubMed] [Google Scholar]; (b) Sassa T Cotylenins, Leaf Growth Substances Produces by a Fungus. Part I. Isolation and Characterization of Cotylenins A and B. Agr. Biol. Chem 1971, 35, 1415. [Google Scholar]; (c) Sassa T; Togashi M; Kitaguchi T The structures of cotylenins A,B,C,D and E. Agr. Biol. Chem 1975, 39, 1735. [Google Scholar]
- (3).Yamada K; Honma Y; Asahi KI; Sassa T; Hino KI Tomoyasu S Differentiation of Human Acute Myeloid Leukae-mia Cells in Primary Culture in Response of Cotylenin A, a Plant Growth Regulator. Br. J. Haematol 2001, 114, 814. [DOI] [PubMed] [Google Scholar]
- (4).(a) Molzan M; Kasper S; Röglin L; Skwarczynska M; Sassa T; Inoue T; Breitenbuecher F; Ohkanda J; Kato N; Schuler M; Ottmann C Stabilization of Physical RAF/14-3-3 Interaction by Cotylenin A as Treatment Strategy for RAS Mutant Cancers. ACS Chem. Biol 2013, 8, 1869. [DOI] [PubMed] [Google Scholar]; (b) Ottmann C; Weyand M; Sassa T; Inoue T; Kato N; Wittinghofer A; Oecking C A Structural Rationale for Selective Stabilization of Anti-Tumor Interactions of 14-3-3 Proteins by Cotylenin A. J. Mol. Biol 2009, 386, 913. [DOI] [PubMed] [Google Scholar]
- (5).(a) Honma Y; Ishii Y; Yamamoto-Yamaguchi Y; Sassa T; Asahi K-I Cotylenin A, a Differentiation-Inducing Agent, and IFN-Cooperatively Induce Apoptosis and Have an Antitumor Effect on Human Non-Small Cell Lung Carcinoma Cells in Nude Mice. Cancer Res. 2003, 63, 3659. [PubMed] [Google Scholar]; (b) Kasukabe T; Okabe-Kado J; Kato N; Sassa T; Honma Y Effects of Combined Treatment with Rapamycin and Cotylenin A, a Novel Differentiation-Inducing Agent, on Human reast Carcinoma MCF-7 Cells and Xenografts. Breast Cancer Res. 2005, 7, R1097. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Honma Y; Kasukabe T; Yamori T; Kato N; Sassa T Antitumor Effect of Cotylenin A plus Interferon-α: Possible Therapeutic Agents against Ovary Carcinoma. Gynecol. Oncol 2005, 99, 680. [DOI] [PubMed] [Google Scholar]
- (6).Ikejiri F; Honma Y; Okada T; Urano T; Suzumiya J Cotylenin A and tyrosine kinase inhibitors synergistically inhibit the growth of chronic myeloid leukemia cells. Int. J. Oncol, 2018, 52, 2061. [DOI] [PubMed] [Google Scholar]
- (7).Madden SK; de Araujos AD; Gerhardt M; Mason JM Taking the Myc out of cancer: toward therapeutic strategies to directly inhibit c-Myc. Mol. Cancer, 2021, 20, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).(a) Sassa T; Negoro T; Ueki H Production and Characterization of a New Fungal Metabolite, Cotylenol. Agr. Biol. Chem 1972, 36, 2281. [Google Scholar]; (b) Sassa T Structure of Cotylenol, the Aglycone of the Cotylenins Leaf Growth Substances. Agr. Biol. Chem 1972, 36, 2037. [Google Scholar]; (c) Sassa T; Takahama A; Shindo T The Stereostructure of Cotylenol, the Aglycone of Cotylenins Leaf Growth Substances. Agr. Biol. Chem 1975, 39, 1729. [Google Scholar]
- (9).Cotylenin A and cotylenol bind analogously at the 14-3-3/client interface, as demonstrated through X ray crystal structures (the sugar moiety of cotylenin A is partly solvent-exposed):; Anders C; Higuchi Y; Koschinsky K; Bartel M; Schumacher B; Thiel P; Nitta H; Preisig-Müller R; Schlichthörl G; Renigunta V; Ohkanda J; Daut J; Kato N; Ottmann C A Semisynthetic Fusicoccane Stabilizes a Protein-Protein Interaction and Enhances the Expression of K+ Channels at the Cell Surface. Chem. Biol 2013, 20, 583. [DOI] [PubMed] [Google Scholar]
- (10).Kaplan A; Andrei SA; van Regteren Altena A; Simas T; Banerjee SL; Kato N; Bisson N; Higuchi Y; Ottmann C; Fournier AE “Polypharmacological perturbation of the 14-3-3 adaptor protein interactome stimulates neurite outgrowth” Cell Chem. Biol 2020, 27, 657. [DOI] [PubMed] [Google Scholar]
- (11).(a) Stevers LM; Sijbesma E; Botta M; MacKintosh C; Obsil T; Landrieu I; Cau Y; Wilson AJ; Karawajczyk A; Eickhoff J; Davis J Modulators of 14-3-3 protein–protein interactions. J. Med. Chem 2017, 61, 3755. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Pennington K; Chan T; Torres M; Andersen JL The dynamic and stress-adaptive signaling hub of 14-3-3: emerging mechanisms of regulation and context-dependent protein–protein interactions. Oncogene 2018, 37, 5587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Ambagon Therapeutics Home Page. https://www.ambagontx.com (accessed 2023-07-19)
- (13).(a) Doveston RG; Kuusk A; Andrei SA; Leysen S; Cao Q; Castaldi MP; Hendricks A; Brunsveld L; Chen H; Boyd H; Ottmann C Small-Molecule Stabilization of the P53 – 14-3-3 Protein-Protein Interaction. FEBS Lett. 2017, 591, 2449. [DOI] [PubMed] [Google Scholar]; (b) Hermeking H The 14-3-3 Cancer Connection. Nat. Rev. Cancer 2003, 931. [DOI] [PubMed] [Google Scholar]
- (14).(a) Sinha P; Kohl S; Fischer J; Hütter G; Kern M; Köttgen E; Dietel M; Lage H; Schnölzer M; Schadendorf D Identification of Novel Proteins Associated with the Development of Chemoresistance in Malignant Melanoma Using Two-Dimensional Electrophoresis. Electrophoresis 2000, 21, 3048. [DOI] [PubMed] [Google Scholar]; (b) Mori M; Vignaroli G; Cau Y; Dinic̈ J; Hill R; Rossi M; Colecchia D; Pešić M; Link W; Chiariello M; Ottmann C; Botta M Discovery of 14-3-3 Protein-Protein Interaction Inhibitors that Sensitize Multidrug-Resistant Cancer Cells to Doxorubicin and the Akt Inhibitor GSK690693. ChemMedChem 2014, 9, 973. [DOI] [PubMed] [Google Scholar]
- (15).Andlovic B; Heilmann G; Ninck S; Andrei SA; Centorrino F; Higuchi Y; Kato N; Brunsveld L; Arkin M; Menninger S; Choidas A; Wolf A; Klebl B; Kaschani F; Kaiser M; Eickhoff J; Ottmann C IFNα Primes Cancer Cells for Fusicoccin-Induced Cell Death via 14-3-3 PPI Stabilization. Cell Chem. Biol 2023, 30, 573. [DOI] [PubMed] [Google Scholar]
- (16).(a) Wolter M; de Vink P; Filipe Neves J; Srdanović S; Higuchi Y; Kato N; Wilson A; Landrieu I; Brunsveld L; Ottman C Selectivity via Cooperativity: Preferential Stabilization of the p65/14-3-3 Interaction with Semisynthetic Natural Products. J. Am. Chem. Soc 2020, 142, 11772. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Andrei SA; de Vink P; Sijbesma E; Han L; Brunsveld L; Kato N; Ottmann C, Higuchi Y Rationally Designed Semisynthetic Natural Product Analogues for Stabilization of 14-3-3 Protein–Protein Interactions. Angew. Chem. Int. Ed 2018, 57, 13470. [DOI] [PubMed] [Google Scholar]; (c) Kawakami K; Hattori M; Inoue T; Maruyama Y; Ohkanda J; Kato N; Tongu M; Yamada T; Akimoto M; Takenaga K; Sassa T; Suzumiy J; Honma Y A Novel Fusicoccin Derivative Preferentially Targets Hypoxic Tumor Cells and Inhibits Tumor Growth in Xenografts. Anticancer Agents Med. Chem 2012, 12, 791. [DOI] [PubMed] [Google Scholar]
- (17).Ono Y; Minami A; Noike M; Higuchi Y; Toyomasu T; Sassa T; Kato N; Dairi T Dioxygenases, Key Enzymes to Determine the Aglycon Structures of Fusicoccin and Brassici-cene, Diterpene Compounds Produced by Fungi, J. Am. Chem. Soc 2011, 133, 2548. [DOI] [PubMed] [Google Scholar]
- (18).(a) Hansen JD; Correa H; Nagy MA; Alexander M; Plantevin V; Grant V; Whitefield B; Huang D; Kercher T; Harris R; Narla RK; Leisten J; Tang Y; Moghaddam M; Ebinger K; Piccotti J; Havens CG; Cathers B; Carmichael J; Daniel T; Vessey R; Hamann LG; Leftheris K; Mendy D; Baculi F; LeBrun LA; Khambatta G; Lopez-Girona A Discovery of CRBN E3 Ligase Modulator CC-92480 for the Treatment of Relapsed and Refractory Multiple Myeloma. J. Med. Chem 2020, 63, 6648. [DOI] [PubMed] [Google Scholar]; (b) Guirguis AA; Ebert BL Lenalidomide: Deciphering Mechanisms of Action in Myeloma, Myelodysplastic Syndrome and Beyond. Curr. Opin. Cell Biol 2015, 37, 61. [DOI] [PubMed] [Google Scholar]
- (19).Inoue T; Higuchi Y; Yoneyama T; Lin B; Nunomura K; Honma Y; Kato N Semisynthesis and biological evaluation of a cotylenin A mimic derived from fusicoccin A. Bioorg. Med. Chem. Lett 2018, 28, 646. [DOI] [PubMed] [Google Scholar]
- (20).(a) Uwamori M; Osada R; Sugiyama R; Nagatani K; Nakada M Enantioselective Total Synthesis of Cotylenin A. J. Am. Chem. Soc 2020, 142, 5556. [DOI] [PubMed] [Google Scholar]; For preparation of starting material 11 of ref. 20a in 5 steps and 67% yield see:; (b) Yama-no Y; Nishiyama Y; Aoki A; Maoka T; Wada A Total synthesis of lycopene-5,6-diol and γ-carotene-5′,6′-diol stereoisomers and their HPLC separation. Tetrahedron 2017, 73, 2043 [Google Scholar]; (c) Maruoka K; Ooi T; Nagahara S; Yamamoto H Organoaluminum-catalyzed rearrangement of epoxides a facile route to the synthesis of optically active β-siloxy aldehydes. Tetrahedron 1991, 47, 6983. [Google Scholar]
- (21).(a) Kato N; Okamoto H; Arita H; Imaoka T; Miyagawa H; Takeshita H Stereoselective Construction of Functionalized Fusicoccane Framework. Synlett, 1994, 5, 337. [Google Scholar]; (b) Okamoto H; Arita H; Kato N; Takeshita H Total Synthesis of (−)-Cotylenol, a Fungal Metabolite Having a Leaf Growth Activity. Chem. Lett 1994, 23, 2335. [Google Scholar]; (c) Kato N; Okamoto H; Takeshita H Total Synthesis of Optically Active Cotylenol, a Fungal Metabolite Having a Leaf Growth Activity. Intramolecular Ene Reaction for an Eight-Membered Ring Formation. Tetrahedron, 1996, 52, 3921. [Google Scholar]; The ene reaction has also been used to assemble 5-8-5 skeletons that lack the Δ1,2-alkene:; (d) Kato N; Wu X; Tanaka S; Takeshita H Structure Elucidation of “Hy-droxycycloaraneosene” by Unambiguous Total Synthesis. An Eight-Membered Ring formation via a Lewis Acid-Catalyzed Ene-Reaction. Chem. Lett 1989, 18, 91. [Google Scholar]
- (22).PDB: 3SP5, see Ref. 9.
- (23).Castro AMM “Claisen Rearrangement over the Past Nine Decades” Chem. Rev 2004, 104, 2939. [DOI] [PubMed] [Google Scholar]
- (24).Snyder SA; Tang Z-Y; Gupta R “Enantioselective Total Synthesis of (−)-Napyradiomycin A1 via Asymmetric Chlorination of an Isolated Olefin” J. Am. Chem. Soc 2009, 131, 5744. [DOI] [PubMed] [Google Scholar]
- (25).For discussions and examples of convergent fragment couplings, see:; (a) Dibrell SE; Tao Y; Reisman SE “Synthesis of Complex Diterpenes: Strategies Guided by Oxidation Pattern Analysis” Acc. Chem. Res 2021, 54, 1360. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jamison CR; Overman LE “Fragment Coupling with Tertiary Radicals Generated by Visible-Light Photocatalysis” Acc. Chem. Res 2016, 49, 1578. [DOI] [PubMed] [Google Scholar]
- (26).Jarkas N; Voll RJ; Williams L; Camp VM; Good-man MM (R,S)-anti-1-amino-2-[18F]fluorocyclopentyl-1-carboxylic acid: synthesis from racemic 2-benzyloxycyclopentanone and biological evaluation for brain tumor imaging with positron emission tomography. J. Med. Chem 2010, 53, 6603. [DOI] [PubMed] [Google Scholar]
- (27).(a) Wender PA; Bi FC; Brodney MA; Gosselin F Asymmetric Synthesis of the Tricyclic Core of Cyathane Diterpenes via a Transition Metal Catalyzed [5+2] Cycloaddition. Org. Lett 2001. ¸3, 2105. [DOI] [PubMed] [Google Scholar]; (b) Daeppen C; Kaiser M; Neuburger M; Gademann K Preparation of Antimalarial Endoperoxides by a Formal [2+2+2] Cycloaddition. Org. Lett 2015, 17, 5420. [DOI] [PubMed] [Google Scholar]
- (28).Andrews G; Evans DA The stereochemistry of the rearrangement of allylic sulfonium ylids: A new method for the stereoselective formation of asymmetry at quaternary carbon. Tet. Lett 1972, 50, 5121. [Google Scholar]
- (29).Martel B; Hiriart JM Nouveaux intermediaires α-halomethylmetalliques stables: formation et stabilite remarquable D’α-halomethylsodium et D’α-halomethylpotassium. Tet. Lett 1971, 12, 2737. [Google Scholar]
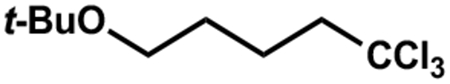
-
(30).We note that the reaction produces the following side product, apparently derived from NaOt-Bu, THF and CHCl3. The mechanism of its formation is not known.
- (31).Anderson RJ; Henrick CA; Rosenblum LD A General Ketone Synthesis: Reaction of Organocopper Reagents with S-Alkyl and S-Aryl Thioester. J. Am. Chem. Soc 1974, 96, 3654. [Google Scholar]
- (32).Lipshutz BH; Koerner M; Parker DA 2-Thienyl(cyano)copper Lithium. A Lower Order, Stable “Cuprate in a Bottle” Precursor to Higher Order Reagents. Tet. Lett 1987, 28, 945. [Google Scholar]
- (33).For rare examples of convergent couplings, see:; (a) Collum DB; McDonald III JH; Still WC Synthesis of the Polyether Antibiotic Monensin. 3. Coupling of Precursors and Transformation to Monensin. J. Am. Chem. Soc 1980, 102, 2120. [Google Scholar]; (b) Kato D; Murase T; Talode J; Nagae H; Tsurugi H; Seki M; Mashima K Diarylcuprates for the Selective Syntheses of Mulifunctionalized Ketones from Thioesters under Mild Conditions. Chem. Eur. J 2022, 28, e202200474. [DOI] [PubMed] [Google Scholar]
- (34).Yoshikai N; Iida R; Nakamura E Mechanism of the Nucleophilic Substitution of Acyl Electrophiles using Lithium Organocuprates. Adv. Synth. Catal 2008, 350, 1063. [Google Scholar]
- (35).The instability of alcohol 21 led to higher yields if purification occurred after the next step.
- (36).On occasion, α-hydroxylation of 7 has been observed.
- (37).For selected examples, see:; (a) Kleban I; Krokhmaliuk Y; Reut S; Shuvakin S; Pendyukh VV; Khyzhan OI; Yarmoliuk DS; Tymtsunik AV; Rassukana YV; Grygorenko OO Multigram Synthesis of Heterabicyclo[n.1.0]alkan-1-yl Trifluoroborates. Eur. J. Org. Chem 2021, 6551. [Google Scholar]; (b) Heinrich CF; Durand D; Starck J; Michelet V Ruthenium Metathesis: A Key Step to Access a New Cyclic Tetrasubstituted Olefin Platform. Org. Lett 2020, 22, 7064. [DOI] [PubMed] [Google Scholar]
- (38).(a) Reznichenko AL; Hampel F; Hultzsch KC Kinetic Resolution of Aminoalkenes by Asymmetric Hydroamination: A Mechanistic Study. Chem. Eur. J 2009, 15, 12819. [DOI] [PubMed] [Google Scholar]; (b) Clemens JJ; Bechara WS; Bookser BC; Cleveland T; Coon T; Gallant M; Grootenhuis PDJ; Hadida Ruah SS; Laterreur J; Miller MT; Paraselli P; Ramtohul YK; Reddy TJ; Sturino C; Valdez L; Zhou J Macrocycles Containing a 1,3,4-Oxadiazole Ring for Use as Modulators of Cystic Fibrosis Transmembrane Conductance Regulator. US WO2022109573 A1, 2022. [Google Scholar]
- (39).The 2-step procedure involves Weinreb amide synthesis and addition of 3-butenylmagesium bromide. See SI for details.
- (40).(a) Kitamura M; Kawai SK; Noyori R Catalytic Asymmetric Induction. Highly Enantioselective Addition of Dialkylzincs to Aldehydes. J. Am. Chem. Soc 1986, 108, 6071. [DOI] [PubMed] [Google Scholar]; (b) Pu L; Yu H-B Catalytic Asymmetric Organozinc Additions to Carbonyl Compounds. Chem. Rev 2001, 101, 757. [DOI] [PubMed] [Google Scholar]
- (41).Nugent WA MIB: An Advantageous Alternative to DIAB for the Addition of Organozinc Reagents to Aldehydes. Chem. Comun 1999, 1369. [Google Scholar]
- (42).(a) Patel NR; Nawrat CC; McLaughlin M; Xu Y; Huffman MA; Yang H; Li H; Whittaker AM; Andreani T; Lévesque F; Fryszkowka A; Brunskill A; Tschaen DM; Maloney KM Synthesis of Islatravir Enabled by a Catalytic, Enantioselective Alkynylation of a Ketone. Org. Lett 2020, 22, 4659. [DOI] [PubMed] [Google Scholar]; (b) Jiang B; Chen Z; Tang X Highly Enantioselective Alkynylation of α-Keto Ester: An Efficient Method for Constructing a Chiral Tertiary Carbon Center. Org. Lett 2002, 4, 3451. [DOI] [PubMed] [Google Scholar]
- (43).Moure AL; Mauleón P; Gómez Arrayás R; Carretero JC Formal Regiocontrolled Hydroboration of Unbiased Internal Alkynes via Borylation/Allylic Alkylation of Terminal Alkynes. Org. Lett 2013, 15, 2054. [DOI] [PubMed] [Google Scholar]
- (44).Liebeskind LS; Srogl J Thiol Ester-Boronic Acid Coupling. A Mechanistically Unprecedented and General Ketone Synthesis. J. Am. Chem. Soc 2000, 122, 11260. [Google Scholar]
- (45).For roles of B(OH)3 in the Chan-Lam coupling, see:; Vantourout JC; Miras HN; Isidro-Llobet A; Sproules S; Watson AJB Spectroscopic Studies of the Chan-Lam Amination: A Mechanism Inspired Solution to Boronic Ester Reactivity. J. Am. Chem. Soc 2017, 139, 4769. [DOI] [PubMed] [Google Scholar]
- (46).Thomas AA; Zahrt AF; Delaney CP; Denmark SE Elucidating the Role of Boronic Esters in the Suzuki-Miyaura Reaction: Structural, Kinetic, and Computational Investigations. J. Am. Chem. Soc 2018, 140, 4401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).1H NMR spectra of 27 and either B(OH)3 or B(OMe)3 in acetone-d6 show no association in the ground state, although transient adducts remain a possibility.
- (48).(a) Cheng H-G; Chen H; Liu Y; Zhou Q The Liebeskind–Srogl Cross-Coupling Reaction and its Synthetic Applications. Asian J. Org. Chem 2018, 7, 490. [Google Scholar]; (b) Serrano R; Boyko YD; Hernandez LW; Lotuzas A; Sarlah D Total Syntheses of Scabrolide A and Yonarolide. J. Am. Chem. Soc 2023, 145, 8805. [DOI] [PubMed] [Google Scholar]
- (49).(a) Yang H; Li H; Wittenberg R; Egi M; Huang W; Liebeskind LS Ambient Temperature Synthesis of High Enantiopurity N-Protected Peptidyl Ketones by Peptidyl Thiol Ester–Boronic Acid Cross-Coupling. J. Am. Chem. Soc 2007, 129, 1132. [DOI] [PMC free article] [PubMed] [Google Scholar]; For two examples of coupling unhindered catecholboronic esters see:; (b) Fenneteau J; Vallerotto S; Ferrie L; Figadere B Liebeskind–Srogl Cross-Coupling on γ-Carboxyl-γ-Butyrolactone Derivatives: Application to the Side Chain of Amphidinolides C and F. Tet. Lett 2015, 56, 3758. [Google Scholar]; For the sole example of a Z-substituted vinylboronic acid, see:; (c) Lovell KM; Vasiljevik T; Araya JJ; Lozama A; Prevatt-Smith KM; Day VC; Dersch CM; Rothman RB; Butelman ER; Kreek MJ; Prisinzano TE Semisynthetic Neoclerodanes and Kappa Opioid Receptor Probes. Bioorg. Med. Chem 2012, 20, 3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Savarin C; Liebeskind LS Nonbasic, Room Temperature, Palladium-Catalyzed Coupling of Aryl and Alkenyl Iodides with Boronic Acids Mediated by Copper(I) Thiophene-2-carboxylate (CuTC). Org. Lett 2001, 3, 2149. [DOI] [PubMed] [Google Scholar]
- (51).Musaev DG; Liebeskind LS On the Mechanism of Palladium(0) Catalyzed, Copper(I) Carboxylate Mediated Thioorganic–Boronic Acid Desulfitative Coupling. A Noninnocent Role for the Carboxylate Ligand. Organometallics 2009, 28, 4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).(a) Gooßen LJ; Ghosh K Palladium-Catalyzed Synthesis of Aryl Ketones from Boronic Acids and Carboxylic Acids or Anhydrides. Angew. Chem. Int. Ed 2001, 40, 3458. [DOI] [PubMed] [Google Scholar]; (b) Kakino R; Yasumi S; Shimizu I; Yamamoto A Synthesis of Unsymmet-rical Ketones by Palladium-Catalyzed Cross-Coupling Reaction of Carboxylic Anhydrides with Organoboron Compounds. Bull. Chem. Soc. Jpn 2002, 75, 137. [Google Scholar]; (c) Gooßen LJ; Koley D; Her-mann HL; Thiel W The Palladium-Catalyzed Cross-Coupling Reaction of Carboxylic Anhydrides with Arylboronic Acids: A DFT Study. J. Am. Chem. Soc 2005, 127, 11102. [DOI] [PubMed] [Google Scholar]
- (53).Hattori H; Ogiwara Y; Sakai N Formation, Characterization, and Reactivity of Acyl Palladium Complexes in Pd(OAc)2/PCy3-Catalyzed Transformation of Acyl Fluorides. Organometallics 2022, 41, 1509. [Google Scholar]
- (54).Grushin VV; Alper H Indirect Formation of Carboxylic Acids via Anhydrides in the Palladium-Catalyzed Hydroxycarbonylation of Aromatic Halides. J. Am. Chem. Soc 1995, 117, 4305. [Google Scholar]
- (55).Farina V; Krishnan B Large Rate Accelerations in the Stille Reaction with Tri-2-furylphosphine and Triphenylarsine as Palladium Ligands: Mechanistic and Synthetic Implications. J. Am. Chem. Soc 1991, 113, 9585. [Google Scholar]
- (56).(a) van Leeuwen PWNM; Zuideveld MA; Swennenhuis BHG; Freixa Z; Kramer PCJ; Goubitz K; Fraanje J; Lutz M; Spek AL Alcoholysis of Palladium(II) Complexes Relevant to the Alternating Copolymerization of Ethene and Carbon Monoxide and the Alkoxycarbonylation of Alkenes: The Importance of Cis-Coordinating Phosphines. J. Am. Chem. Soc 2003, 125, 5523. [DOI] [PubMed] [Google Scholar]; (b) Ozawa F; Kawasaki N; Okamoto H; Yamamoto T; Yamamoto A Mechanisms of Double and Single Carbonylation Reactions of Aryl Iodides Catalyzed by Palladium Complexes to Give α-Keto Esters and Esters. Organometallics 1987, 6, 1640. [Google Scholar]; (c) Komiya S; Akai Y; Tanaka K; Yamamoto T; Yamamoto A Reductive Elimination of Aryl Carboxylates from Acyl(aryloxy)nickel(II) and -palladium(II) Complexes. Organometallics 1985, 4, 1130. [Google Scholar]
- (57).When 1 equiv of 27 is used, ~20% unreacted 27 remains when the reaction begins to stall. The reaction therefore does not stall due to an absence of 27 to participate in the coupling.
- (58).(a) Blaisdell TP; Morken JP Hydroxyl-Directed Cross-Coupling: A Scalable Synthesis of Debromohamigeran E and Other Targets of Interest. J. Am. Chem. Soc 2015, 137, 8712. [DOI] [PMC free article] [PubMed] [Google Scholar]; For an example of OH-containing functionality directing other elementary steps at Pd, see:; (b) Houpis IN; Huang C; Nettekoven U; Chen JG; Liu R; Canters M Carboxylate-Directed Cross-Coupling Reactions in the Synthesis of Trisubstituted Benzoic Acids. Org. Lett 2008, 10, 5601. [DOI] [PubMed] [Google Scholar]
- (59).Willis MC Transition Metal Catalyzed Alkene and Alkyne Hydroacylation. Chem. Rev 2010, 110, 725. [DOI] [PubMed] [Google Scholar]
- (60).Hari DP; Caramenti P; Waser J “Cyclic Hypervalent Iodine Reagents: Enabling Tools for Bond Disconnection via Reactivity Umpolung” Acc. Chem. Res 2018, 51, 3212. [DOI] [PubMed] [Google Scholar]
- (61).Stereochemistry was not assigned to C1 of 18 or C6 of 19 due to multiple rotatable bonds, but since 19 could not be advanced, the issue was considered irrelevent.
- (62).Winterfeldt E; Preuss H “Der sterische Verlauf von Additionen an die Dreifachbindung” Chem. Ber 1966, 99, 450. [Google Scholar]
- (63).For alternative approaches to related 5-8-5 ring systems, see:; (a) Wang Y-Q; Xu K; Min L; Li C-C “Asymmetric Total Syntheses of Hypoestin A, Albolic Acid, and Ceroplastol II” J. Am. Chem. Soc 2022, 23, 10162. [DOI] [PubMed] [Google Scholar]; (b) Chen B; Wu Q; Xu D; Zhang X; Ding Y; Bao S; Zhang X; Wang L; Chen Y A Two-Phase Approach to Fusicoccane Synthesis to Uncover a Com-pound that Reduces Tumourigenesis in Pancreatic Cancer Cells. Angew. Chem. Int. Ed 2022, 61, e202117476. [DOI] [PubMed] [Google Scholar]; (c) Thach DQ; Brill ZG; Grover HK; Esguerra KV; Thompson JK; Maimone TJ “Total Synthesis of (+)-6-epi-Ophiobolin A” Angew. Chem. Int. Ed 2020, 59, 1532. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Brill ZG; Grover HK; Maimone TJ “Enantioselective synthesis of an ophiobolin sesterterpene via a programmed radical cascade” Science 2016, 352, 1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).This simple model does not account for the role of aggregation state in stereoselectivity. For example, see:; Romesberg FE; Collum DB “Mechanism of Lithium Dialkylamide-Mediated Ketone and Imine Deprotonations: An MNDO Study of Monomer and Open Dimer Pathways” J. Am. Chem. Soc 1995, 117, 2166. [Google Scholar]
- (65).Woo S; Shenvi RA Natural Product Synthesis through the Lens of Informatics. Acc. Chem. Res 2021, 54, 1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).For an outstanding chemoenzymatic approach to cotylenol and the fusicoccins by Jiang and Renata, archived simultaneously with this work, see:; Jiang Y; Renata H Modular Chemoenzymatic Synthesis of Ten Fusicoccane Diterpenoids. ChemRxiv. 2023-May-21. DOI: 10.26434/chemrxiv-2023-gfh8l. (accessed 2023-08-21). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.