

Abstract
Background
In the absence of antiretroviral therapy (ART), over 50% of HIV‐infected infants progress to AIDS and death by 2 years of age. However, there are challenges to initiation of ART in early life, including the possibility of drug resistance in the context of prevention of mother‐to‐child transmission (PMTCT) programs, a paucity of drug choices , uncertain dosing for some medications and long‐term toxicities. Key management decisions include when to start ART, what regimen to start, and whether and when to substitute drugs or interrupt therapy. This review, an update of a previous review, aims to summarize the currently available evidence on this topic and inform the ART management in HIV‐infected children less than 3 years of age.
Objectives
To evaluate 1) when to start ART in young children (less than 3 years); 2) what ART to start with, comparing first‐line non‐nucleoside reverse transcriptase inhibitor (NNRTI) and protease inhibitor (PI)‐based regimens; and 3) whether alternative strategies should be used to optimize antiretroviral treatment in this population: induction (initiation with 4 drugs rather than 3 drugs) followed by maintenance ART, interruption of ART and substitution of PI with NNRTI drugs once virological suppression is achieved on a PI‐based regimen.
Search methods
Search methods
We searched for published studies in the Cochrane HIV/AIDS Review Group Trials Register, The Cochrane Library, Pubmed, EMBASE and CENTRAL. We screened abstracts from relevant conference proceedings and searched for unpublished and ongoing trials in clinical trial registries (ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform).
Selection criteria
We identified RCTs that recruited perinatally HIV‐infected children under 3 years of age without restriction of setting. We rejected trials that did not include children less than 3 years of age, did not provide stratified outcomes for those less than 3 years or did not evaluate either timing of ART initiation, choice of drug regimen or treatment switch/interruption strategy.
Data collection and analysis
Two reviewers independently applied study selection criteria, assessed study quality and extracted data. Effects were assessed using the hazard ratio (HR) for time‐to‐event outcomes, relative risk for dichotomous outcomes and weighted mean difference for continuous outcomes.
Main results
A search of the databases identified a total of 735 unique, previously unreviewed studies, of which 731 were excluded to leave 4 new studies to incorporate into the review. Four additional studies were identified in conference proceedings, for a total of 8 studies addressing when to start treatment (n=2), what to start (n=3), whether to substitute lopinavir/ritonavir (LPV/r) with nevirapine (NVP) (n=1), whether to use an induction‐maintenance ART strategy (n=1) and whether to interrupt treatment (n=1).
Treatment initiation in asymptomatic infants with good immunological status was associated with a 75% reduction (HR=0.25; 95%CI 0.12‐0.51; p=0.0002) in mortality or disease progression in the one trial with sufficient power to address this question. In a smaller pilot trial, median CD4 cell count was not significantly different between early and deferred treatment groups 12 months after ART.
Regardless of previous exposure to nevirapine for PMTCT, the hazard for treatment failure at 24 weeks was 1.79 (95%CI 1.33, 2.41) times higher in children starting ART with a NVP‐based regimen compared to those starting with a LPV/r‐based regimen (p=0.0001) with no clear difference in the effect observed for children younger or older than 1 year. The hazard for virological failure at 24 weeks was overall 1.84 (95%CI 1.29, 2.63) times higher for children starting ART with a NVP‐based regimen compared to those starting with a LPV/r‐based regimen (p=0.0008) with a larger difference in time to virological failure (or death) between the NVP and LPV/r‐based regimens when ART was initiated in the first year of life.
Infants starting a LPV/r regimen and achieving sustained virological suppression who then substituted LPV/r with NVP after median 9 months on LPV/r were less likely to develop virological failure (defined as at least one VL greater than 50 copies/mL) compared with infants who started and stayed on LPV/r (HR=0.62, 95%CI 0.41, 0.92, p=0.02). However the hazard for confirmed failure at a higher viral load (>1000 copies/mL) was greater among children who switched to NVP compared to those who remained on LPV/r (HR=10.19, 95% CI 2.36, 43.94, p=0.002).
Children undergoing an induction‐maintenance ART approach with a 4‐drug NNRTI‐based regimen for 36 weeks, followed by 3‐drug ART, had significantly greater CD4 rise than children receiving a standard 3‐drug NNRTI‐based ART at 36 weeks (mean difference 1.70 [95%CI 0.61, 2.79] p=0.002) and significantly better viral load response at 24 weeks (OR 1.99 [95%CI 1.09, 3.62] p=0.02). However, the immunological and virological benefits were short‐term.
The one trial of treatment interruption that compared children initiating continuous ART from infancy with children interrupting ART was terminated early because the duration of treatment interruption was less than 3 months in most infants. Children interrupting treatment had similar growth and occurrence of serious adverse events as those in the continuous arm.
Authors' conclusions
ART initiation in asymptomatic children under 1 year of age reduces morbidity and mortality, but it remains unclear whether there are clinical benefits to starting ART in asymptomatic children diagnosed with HIV infection between 1‐3 years.
The available evidence shows that a LPV/r‐based first‐line regimen is more efficacious than a NVP‐based regimen, regardless of PMTCT exposure status. New formulations of LPV/r are urgently required to enable new WHO recommendations to be implemented. An alternative approach to long‐term LPV/r is substituting LPV/r with NVP once virological suppression is achieved. This strategy looked promising in the one trial undertaken, but may be difficult to implement in the absence of routine viral load testing.
A 4‐drug induction‐maintenance approach showed short‐term virological and immunological benefits during the induction phase but, in the absence of sustained benefits, is not recommended as a routine treatment strategy. Treatment interruption following early ART initiation in infancy was challenging for children who were severely immunocompromised in the context of poor clinical immunological condition at ART initiation due to the short duration of interruption, and is therefore not practical in ART treatment programmes where close monitoring is not feasible.
Keywords: Humans; Infant; Anti‐HIV Agents; Anti‐HIV Agents/administration & dosage; Anti‐HIV Agents/therapeutic use; CD4 Lymphocyte Count; Disease Progression; Drug Administration Schedule; Drug Combinations; HIV Infections; HIV Infections/drug therapy; HIV Infections/mortality; HIV Infections/transmission; Induction Chemotherapy; Induction Chemotherapy/methods; Infectious Disease Transmission, Vertical; Infectious Disease Transmission, Vertical/prevention & control; Lopinavir; Lopinavir/administration & dosage; Lopinavir/therapeutic use; Maintenance Chemotherapy; Maintenance Chemotherapy/methods; Nevirapine; Nevirapine/administration & dosage; Nevirapine/therapeutic use; Ritonavir; Ritonavir/administration & dosage; Ritonavir/therapeutic use
Plain language summary
Using antiretroviral drugs to treat children under 3 years old who have HIV infection
Children under 3 years of age who have HIV infection have a high risk of dying without antiretroviral therapy (ART). However, treatment in this age group is challenging because there are high levels of virus in the blood and few suitable drug choices. Results from this systematic review show that ART soon after birth is preferable to delaying treatment, because infants are less likely to die or become sick. Starting a first‐line treatment regimen that includes lopinavir/ritonavir rather than nevirapine is preferable, because infants and young children are less likely to have to stop treatment, whether or not they had previously been exposed to nevirapine. However, lopinavir/ritonavir is more expensive than nevirapine. It is also currently only available as an inconvenient liquid, which tastes bitter and has to be refrigerated, making it challenging to implement in all parts of the world. While waiting for better formulations to become available, it may be possible to switch from lopinavir/ritonavir to nevirapine once the HIV virus levels become undetectable. However, based on the evidence currently available, a viral load test would be required to identify those children who could safely substitute lopinavir/ritonavir with nevirapine. Viral loads are expensive and not widely available in most countries in sub‐Saharan Africa. An alternative treatment approach is to give a stronger drug combination (four different drugs together) when treatment is first started, then reduce down to three drugs after a short while. However, this strategy did not appear to have long‐term benefits. A 'treatment interruption' strategy, in which infants start ART soon after birth but then stop medication after 1‐2 years, is difficult to implement. Children stopping ART need to restart it very quickly to prevent them becoming sick, and monitoring a child off treatment is challenging in settings with few resources.
Background
The number of children under the age of 15 years living with HIV increased from 1.6 million (range, 1.4 million‐2.1 million) in 2001 to 3.3 million (range 3.1 million‐3.8 million] in 2011. Despite a 43% decline in mother‐to‐child transmission (MTCT) between 2009‐2011, around 900 newly infected infants continue to be born daily (UNAIDS 2012). In the absence of antiretroviral therapy (ART), over 50% of HIV‐infected infants progress to AIDS and death by 2 years of age (Newell 2004). The introduction of ART has dramatically changed the natural history of HIV infection in children (Gortmaker 2001, Gibb 2000) and now, in well‐resourced countries, over 90% of HIV‐infected children reach the age of 10 years (Walker 2004). However, at the end of 2011, only an estimated 28% of children eligible for ART in low‐ and middle‐income countries were receiving it (UNAIDS 2012).
The natural history of perinatal HIV infection differs from that of primary infection in adults. Rapid disease progression is a hallmark of HIV infection during the first 2 years of life, especially in resource‐limited settings. Perinatal infection occurs either in utero (mostly during the third trimester), during delivery, or after birth through breastfeeding. The pattern of viraemia in vertically infected children differs from that in infected adults, with HIV RNA levels remaining high throughout infancy (first 12 months of life) and, in the absence of treatment, decreasing only slowly to adult set‐point levels over the next few years in those with slow disease progression (McIntosh 1996). This difference most likely reflects the immaturity of the paediatric immune system in controlling viral replication. Viral load is generally a poor predictive marker of disease progression during infancy (Dunn 2008).
CD4 counts in healthy young children are naturally higher than in older children or adults and slowly decline to adult levels by approximately 5 years of age (Wade 1994). Age is therefore an important consideration in assessing by CD4 count the risk of progression for HIV‐infected children (Dunn 2008). Percentage CD4 count tends to vary less with age and is preferred in the first 5 years of life as a marker of HIV disease progression; however, the predictive value of the CD4 cell percentage for progression to AIDS or death is lower in children under two years of age, compared with older children. Infants have a higher short‐term risk of clinical progression compared to older children and are at greater risk of acquiring opportunistic infections, such as Pneumocystis jiroveci pneumonia (PcP) and cytomegalovirus (CMV), even at high CD4 counts (HPPMCS 2003).
The Cross Continents Collaboration for Kids (3Cs4kids) study, which combined longitudinal data from approximately 2500 untreated HIV‐infected children mostly enrolled in African studies (one study from Brazil), evaluated the prognostic value of selected laboratory and growth markers for 12‐month risk of mortality (3Cs4kids 2008). There were insufficient studies focusing on sub‐Saharan African infants to include children <12 months of age in this analysis. Nevertheless, prognosis for children with a given CD4 count or percentage was shown to be poorer at younger ages, and the predictive value of both markers improved with age, similar to findings for European/US children (HPPMCS 2003). Moreover, both CD4 percentage and count were shown to be less effective in discriminating between low and high mortality risk for children in resource‐limited, compared to well‐resourced, settings.
Because there are no good markers to predict disease progression during infancy, criteria for initiation of ART in HIV‐infected infants have varied over time. Until 2007, there was no consensus between settings: US guidelines generally recommended consideration of ART for infants more strongly than did European guidelines, which only strongly recommended treatment for those with symptomatic disease or immunosuppression, but included the option to initiate ART in all infants. World Health Organization (WHO) recommendations for resource‐limited settings gave CD4 threshold criteria, but these were based on analyses of CD4 percentage and viral load from European/US cohorts (Dunn 2008). Over time, there has been a general shift in paediatric guidelines towards earlier initiation of ART, recognizing that the goals of treatment are not only to reduce opportunistic infections and AIDS‐related mortality, but also to maximize long‐term growth, neurodevelopment and immunological health (WHO 2010, WHO 2013, Laughton 2012)
Although early initiation of ART may be beneficial for young children, lifelong treatment is problematic, given the limited availability of appropriate drugs, long‐term ART toxicity, difficulties with adherence, risk of viral resistance, and cost of such a strategy. Although over 20 antiretroviral drugs are licensed worldwide for the treatment of HIV‐infected adults, many are unlicensed or do not have appropriate formulations for very young children. Ideal antiretroviral drugs for young children are crushable or dispersible fixed‐dose combination tablets that are convenient for healthcare providers to dose, easy for caregivers to administer and palatable for infants. However, although these are available for nevirapine‐based first‐line regimens, drugs such as lopinavir/ritonavir (LPV/r) cannot easily be combined with other antiretroviral drugs and are currently only available as poorly palatable liquid suspensions, or as tablets that cannot be crushed. Even where appropriate formulations are available, drug metabolism varies with age and optimal dosing is uncertain for many drugs in young children. Treatment options are therefore limited for infants and young children.
Efficacy of ART in young children may be affected by maternal transmission of drug‐resistant virus, arising either from multi‐drug exposure in high‐income countries or exposure to prevention of mother‐to‐child transmission (PMTCT) interventions, which usually include non‐nucleoside reverse transcriptase inhibitor (NNRTI) drugs in low‐ and middle‐income countries. The prevalence of transmitted primary drug resistance in perinatal HIV infection increased by 58% between 1998 and 2002 in resource‐rich countries (Karchava 2006, Persaud 2007). Scale‐up of ART is expected to increase the prevalence of resistant transmitted virus in resource‐limited settings over time (Gupta 2012). Infants who have HIV infection and are exposed to nevirapine through infant prophylaxis and maternal treatment or prophylaxis have demonstrable viral resistance (Martinson 2007, Arrive 2007, Church 2009), potentially compromising the response to nevirapine‐containing first‐line treatment regimens (Lockman 2007, Musiime 2009). Since NNRTI‐based PMTCT regimens are standard of care in most settings, the occurrence of drug resistance needs to be carefully considered in recommendations for first‐line infant and young child treatment regimens.
ART started in early childhood therefore presents considerable challenges in terms of drug choice, particularly when considering the need for effective lifelong therapy with minimal toxicity. Immature renal function, altered hepatic enzyme activity and differences in drug absorption lead to variation in systemic exposure to antiretrovirals among infants. Administration of some nucleoside reverse transcriptase inhibitors (NRTIs) is associated with increased rates of lactic acidosis, pancreatitis and hepatitis, due to mitochondrial toxicity (McComsey 2002). Similar to HIV‐infected adults, disorders of lipid metabolism and fat redistribution (the lipodystrophy syndrome) have been described in children receiving NRTIs and protease inhibitors (PIs) (Arpadi 2013,Jaquet 2000 , Vigano 2003, Piloya 2012, Kinabo 2013)
An alternative approach to the currently recommended lifelong treatment could be to start ART in infancy, followed by a period of treatment interruption. This strategy may allow the child to be protected during the period of greatest risk for HIV disease progression and mortality, but enable time off therapy beyond 1‐2 years of age to reduce toxicity, cost and risk of resistance. Definitive, long‐term ART would be restarted when the child met standard age‐related treatment criteria.
This review in its previous version (Penazzato 2012) focused on ART for HIV‐infected children under 2 years of age because of the unique issues relevant to this age group. HIV infection during the first 2 years of life is characterized by high mortality and significant morbidity during a period of rapid growth and neurodevelopment, yet prognostic markers to guide management decisions are poorly predictive and treatment is much more challenging. New treatment approaches in early life require a systematic appraisal of the available evidence in order to inform clinical decisions as well as national policies.
This current revision of the review extends the upper age limit to children under 3 years of age which, despite being an arbitrary age limit, broadly reflects the major differences which make the management of younger children distinct from older children, such as: the need to successfully control high viral loads, rapid disease progression and the limited drug options (eg. limited data and experience with efavirenz and tenofovir). New trial data on management of children <3 years of age have also emerged since the previous version of this review was published, and the authors decided to update the systematic review to help inform revision of international treatment guidelines.
Objectives
The first objective of this review is to assess when to start ART in young children. The efficacy of early ART initiation in HIV‐infected children less than 3 years of age will be compared to deferred ART, started according to clinical or immunological criteria. The second objective is to address the question of what type of ART regimen to start with, comparing efficacy and toxicity of non‐nucleoside reverse transcriptase inhibitor (NNRTI) and protease inhibitor (PI)‐based regimens. If data are available, the third objective of this review is to address the efficacy and safety of alternative strategies to optimize antiretroviral treatment: either an induction‐maintenance approach to ART initiation, treatment interruption or ART switch strategies.
Methods
Criteria for considering studies for this review
Types of studies
Randomized controlled trials.
Types of participants
Perinatally HIV‐infected children under 3 years of age were included from any setting.
Types of interventions
The following types of intervention were assessed:
‐ Timing of treatment: use of early compared to deferred ART
‐ Choice of treatment: use of NNRTI‐ versus PI‐based regimens, in combination with any NRTI backbone
‐ Substitution of LPV/r with NVP following initiation with PI‐based regimens.
‐ Interruption of treatment, compared to continuous early ART
‐ Induction‐maintenance treatment: Initiating ART with more than 3 antiretroviral drugs for an induction period, then moving to maintenance treatment with a standard 3‐drug regimen
Types of outcome measures
The planned primary outcome measure was mortality and disease progression (defined as occurrence of new AIDS events ‐ CDC Class C, or WHO stage 4 disease‐ and other serious HIV‐related events ‐ CDC Class B or WHO stage 3 disease). The planned secondary outcome measures were: Increase of CD4 percentage from ART initiation (as defined by each study); virological suppression (HIV RNA viral load below the level of assay detectability, typically 400 or 50 copies/mL plasma); virological failure (as defined by each study, typically over 1000 copies/mL plasma); change in growth from baseline values following ART initiation (absolute weight and height percentiles or Z‐scores); neurodevelopmental outcome (as defined by each study, for example Griffiths Mental Development Scales Scores: mean locomotor quotient and mean general quotient (Griffiths 1976; Luiz 2001); serious adverse events (SAE) and drug‐related adverse events according to the NIAID SAE grade 1 to 4 rating criteria (NIAID 2009).
Depending on the data available in the study report or provided by investigators the outcome definitions described above were modified for analysis and pooling. When data on only one trial were available for a question, individual trial results were described.
Search methods for identification of studies
The search was performed in consultation with the HIV/AIDS Trials Search Co‐ordinator. The original search was conducted on 1st November 2010 and was subsequently repeated on the 1st August 2012 for the purpose of this updated version of the review . We sought to identify all relevant studies, from 1997 to the search date, regardless of language or publication status, by searching the Cochrane HIV/AIDS Review Group Trials Register, The Cochrane Library, PubMed, EMBASE and the Cochrane Central Register of Controlled Trials (CENTRAL). In addition, the following specific search terms were used: infant, child, p(a)ediatric, highly active antiretroviral therapy, anti‐retroviral agents, early antiretroviral therapy, deferred antiretroviral therapy, HIV infection, human immunodeficiency virus, acquired immunodeficiency syndrome, NNRTI, non‐nucleoside reverse transcriptase inhibitors, NRTI, nucleoside reverse transcriptase inhibitors, PI, protease inhibitors, randomised controlled trial, and controlled clinical trial.
Also, abstracts from the following relevant conference proceedings were screened for potentially eligible trials: World AIDS Conference; International AIDS Society conference (IAS) and Conference on Retroviruses and Opportunistic Infections (CROI), for abstracts presented through 2012. We also searched for unpublished and ongoing studies by considering prospective clinical trial registries (ClinicalTrials.gov and the WHO International Clinical Trials Registry Platform), and by contacting research organizations and experts in the field.
Data collection and analysis
Selection of studies
Two of the authors (MP and AJP) independently screened records identified by the search. Studies were identified if they met the eligibility criteria and were only rejected on initial screening if they did not include children less than 3 years of age or did not contain at least one comparison of ART approach in terms of initiation, drug regimen choice or optimisation strategy. Due to the minor revision of the inclusion criteria for this current update, the screening of the original version of the review was revised to ensure application of the new age inclusion criteria.
Data extraction and management
MP and AJP independently extracted data from the studies on patient characteristics, interventions and outcomes onto pre‐designed forms. They independently cross‐checked and assessed these data, with any disagreement resolved by consensus with a third review author (EJA), when necessary. When a review author had authored an eligible study, an independent party assisted in both the extraction of data and assessment of methodological quality of that study.
If insufficient data were available in the study report, further information was sought from the publication authors.
Assessment of risk of bias in included studies
Various aspects of the methodological quality of included studies were assessed independently by MP and AJP using the risk of bias tool (Higgins 2011). Any discrepancies were resolved by consensus with a third review author (EJA).
Measures of treatment effect
For meta‐analysis of time‐to‐event outcomes, such as death and disease progression, the most appropriate statistic is the hazard ratio (HR). Where available, the HR and associated statistics were extracted directly from the trial report. When a HR was not provided in the trial report and only Kaplan‐Meier curves were available, trial investigators were contacted to obtain the relevant HRs and related statistics.
For dichotomous outcomes, such as decline by 10% in CD4%, a risk ratio (RR) of the rate of the occurrence was calculated from events and number of patients. For continuous outcomes, such as change in CD4 percentage or weight and height Z‐score, the mean difference was calculated.
Dealing with missing data
Where the publication did not provide enough data, and this could preclude reliable estimation of treatment effects, we contacted the trial authors for further information. If insufficient data were still available for any particular outcome, this was described qualitatively rather than analysed quantitatively.
Assessment of heterogeneity
Any qualitative or quantitative heterogeneity indicated by a Chi‐squared test (P<0.2) was investigated, where possible, by considering trial‐level explanatory factors and the subgroup analyses described below.
Assessment of reporting biases
Formal methods (as described by Egger in the Cochrane Handbook, (Higgins 2011) were planned to investigate the presence of reporting bias, but where few trials existed, the likelihood of reporting bias was instead described.
Data synthesis
Where more than one trial was identified for the questions being addressed, the HRs or RRs for each outcome for each trial were combined in meta‐analysis to give a pooled HR or RR, using the fixed‐effect model (FEM). A random‐effect model (REM) was also used to test the robustness of the results to the choice of model.
Subgroup analysis and investigation of heterogeneity
Pre‐specified trial subgroup analyses were carried out according to age, previous antiretroviral NNRTI exposure or documented NNRTI resistance, if sufficient data were available.
Sensitivity analysis
If heterogeneity was detected and could not be explained by subgroup analyses then sensitivity analyses were conducted.
Results
Description of studies
See: Characteristics of included studies; Characteristics of ongoing studies
Result of search
Overall, Medline/PubMed yielded 864 records, CENTRAL 952, EMBASE 986 for a total of 2802 records. Of the 2043 records remaining after duplicates were removed, only 4 referred to studies that were eligible for inclusion in the review as the remainder were not randomized control trials, did not include the population of interest or assessed efficacy of obsolete drug regimens. One additional study was identified in conference proceedings (Figure 1). From the search update Medline/PubMed yielded 697 records, CENTRAL 870 and EMBASE 354 to give a total of 881 records. Of the 735 records remaining after duplicates were removed, 4 referred to studies that were eligible for inclusion in the review. Four additional studies were identified in conference proceedings.
1.
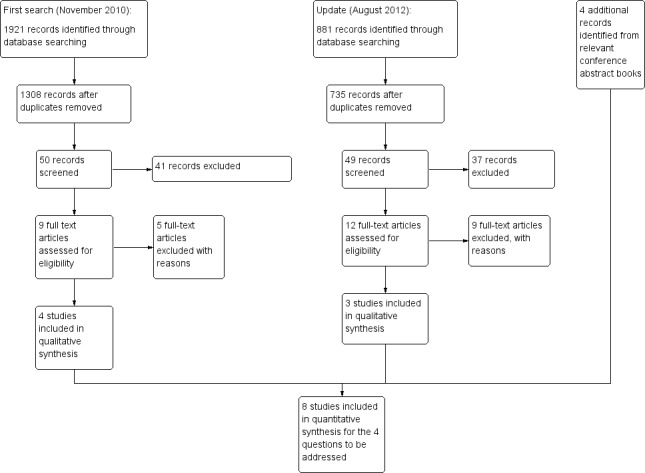
Study flow diagram.
In total, two of these studies were eligible for the question of when to start treatment, three for what to start, one for the strategy of substituting LPV/r with NVP, one for planned treatment interruption and one for the induction‐maintenance strategy.
Two additional trials on substitution of LPV/r with EFV were found but enrolment was only recently completed and the trials are still ongoing.
Included trials
1. When to start
The CHER study (Violari 2008) was a 6 year randomised controlled trial conducted in South Africa. Infants (n=377) from 6 to 12 weeks of age who had asymptomatic HIV infection and a CD4 percentage of 25% or more were randomised to immediate or deferred antiretroviral treatment with a LPV/r‐based regimen containing lamivudine and zidovudine. In two of three study arms infants were started on immediate antiretroviral treatment and remained on it for either one or two years before treatment discontinuation; in the other arm infants started deferred ART according to WHO 2006 clinical or immunological criteria. The primary outcome was mortality or disease progression. Immunological response, growth and toxicity were also assessed in the three study arms. After the second review by the Data and Safety Monitoring Board in June 2007, the decision was taken to stop recruitment into the deferred therapy group and infants in this group were reassessed for initiation of antiretroviral therapy. Results of the early vs deferred groups were analysed and published as soon as possible after this decision. The trial has continued its planned 4.5‐6 year follow‐up and reported final results.
A second study, Paediatric Early HAART STI Study (PEHSS), conducted in Durban, South Africa (Prendergast 2008 (a)) was also included to address the timing of treatment initiation. This was a randomised controlled trial of 63 infants, designed as a feasibility pilot study to evaluate three approaches to antiretroviral treatment of HIV‐infected infants. Infants were randomised at diagnosis to one of three study arms: deferred ART, started once clinical or immunological criteria were reached; immediate ART given for 1 year, then stopped; or immediate ART given with up to three structured treatment interruptions to 18 months of age, then stopped. Four‐drug antiretroviral therapy (zidovudine/lamivudine/nelfinavir/nevirapine) was used as the first‐line antiretroviral regimen. The primary endpoint was the proportion of infants progressing to AIDS by 3 years. However, mortality, morbidity, virological and immunological response were also measured at one year from treatment initiation and compared between study arms.
2. What to start with
P1060 was a randomised trial in six African countries of initial therapy with zidovudine and lamivudine plus either NVP or LPV/r in HIV‐infected children 6 to 36 months of age, who qualified for treatment according to WHO criteria. Children enrolled in cohort 1 (Palumbo 2010) (N=164) had all been exposed to single‐dose nevirapine (sd‐NVP) prophylaxis as part of PMTCT interventions. The primary endpoint was treatment failure (a composite of virological failure or discontinuation of the study drugs for any reason, including death) by study week 24. Secondary endpoints were virological failure (defined as a confirmed plasma HIV‐1 RNA level of less than 1 log10 copies per millilitre below the study entry level at 12 to 24 weeks after the initiation of treatment or a confirmed plasma HIV‐1 RNA level of more than 400 copies per millilitre at 24 weeks) or death by study week 24; time to virological failure or discontinuation over the follow‐up period and time to virological failure or death over the follow‐up period. Immunological response, growth and toxicity were also reported. Enrollment in this cohort was terminated early on the recommendation of the Data and Safety Monitoring Board.
In P1060 cohort 2 (Violari 2012), a parallel randomised trial to P1060 cohort 1, 288 children between 2 and 36 months of age who had not been exposed to single‐dose nevirapine prophylaxis (or to any antiretroviral drug taken by the mother) as part of PMTCT interventions were randomised to NVP‐based or LPV/r‐based first line antiretroviral therapy. The primary endpoint, similar to cohort 1, was treatment failure (a composite of virological failure or discontinuation of the study drugs for any reason, including death) by study week 24. The secondary endpoints were: virological failure (defined as a confirmed plasma HIV‐1 RNA level of less than 1 log10 copies per millilitre below the study entry level at 12 to 24 weeks after the initiation of treatment or a confirmed plasma HIV‐1 RNA level of more than 400 copies per millilitre at 24 weeks) or death by study week 24; time to virological failure or discontinuation over the follow‐up period and time to virological failure or death over the follow‐up period. Immunological response, growth and toxicity were also reported. This study was also terminated early as the Data Safety Monitoring Board recommended unblinding the study results in October 2010 when 24 week endpoints had been reached by all participants.
In the PROMOTE‐pediatric trial (Achan 2012), Ugandan children aged 2 months to 5 years who were either ART‐naive, or who were currently suppressed on NNRTI‐based ART, were randomized to either an NNRTI‐based or PI‐based regimen and followed for 6 months to 2 years. The study was designed to investigate the impact of ART regimen on incidence of malaria, in an area of high‐intensity malaria transmission. 185 children were enrolled, and randomized to NNRTI‐based (N=93) or PI‐based (N=92) ART at median age 3.1 years (range 0.3‐6.0 years). The NNRTI was nevirapine in children <3 years of age, and efavirenz in children >3 years of age, together with an NRTI backbone of zidovudine and lamivudine (with substitution of either stavudine or abacavir for zidovudine in cases of anaemia). The primary endpoint (reported for 170 children in the published trial) was malaria incidence; secondary endpoints included incidence of complicated malaria, efficacy and safety of antimalarial therapy and pharmacokinetic characteristics of lumefantrine. However, protocol‐defined analyses included evaluating the non‐inferiority of LPV/r compared to NNRTI‐based ART, with virological (proportion of children with viral load <400 copies/mL at 48 weeks) and immunological (mean CD4 change at 48 weeks) endpoints. Of the 185 randomized children, 131 were ART‐naive and 54 had been on previous NNRTI‐based ART with viral load <400 copies/mL at study entry; after randomization, 176 reached week 48, and 163 of these had viral load data available. After contacting the study authors, non‐prespecified secondary analyses were undertaken using the same endpoints as the P1060 trials.
3. Substitute LPV/r with NVP
The NEVEREST study (Coovadia 2010) was a randomised controlled trial that enrolled 323 infants under two years of age who had previously been exposed to NVP. After initiating treatment with LPV/r plus lamivudine and stavudine, 195 infants who had maintained a viral load of <400 HIV‐1 RNA copies/mL for at least 3 months were randomised either to remain on LPV/r or to substitute with NVP and were followed for a further 52 weeks, maintaining the NRTI backbone. Mortality, virological suppression, immunological response, growth and toxicity were assessed in the two study groups. Follow‐up data to week 156 were also collected and analysed (Kuhn 2012).
4.Induction‐Maintanance
The ARROW trial (ARROW trial team 2013)was an open randomised parallel‐group trial of 1206 ART‐naive (except for PMTCT exposure) children aged 3 months ‐ 17 years (31% below 3 years of age), meeting 2006 WHO criteria to start ART in Uganda and Zimbabwe. The trial had a factorial design , with randomisation 1:1 at ART initiation to either clinically driven monitoring (CDM) or laboratory (CD4 count/percent and toxicity including haematology and biochemistry) plus clinical monitoring (LCM), and open‐ label randomisation to one of three ART regimens. Arm A started 3‐drug ART (abacavir, lamivudine and NNRTI), whilst Arms B and C started 4‐drug, 2‐class ART (abacavir, lamivudine, zidovudine and NNRTI). After a 36 week induction period, Arm B children stopped zidovudine (and continued abacavir, lamivudine and NNRTI) and Arm C children stopped the NNRTI (continuing a triple NRTI regimen of abacavir, lamivudine and zidovudine). The specific NNRTI (nevirapine/efavirenz) was chosen by clinicians according to local availability and age. The primary efficacy endpoint for the induction‐maintenance randomisation was change in CD4 count at weeks 72 and 144. Secondary endpoints included new WHO 3/4 event or death; new or recurrent WHO 3/4 event or death; new WHO stage 4 event or death; new or recurrent WHO stage 4 event or death; mortality; viral load at weeks 72 and 144; weight, height and BMI; and grade 3/4 adverse events or SAEs.
5. Planned treatment interruption
The Optimizing Pediatric HIV‐1 Therapy 03 Study (OPH03 ‐NCT00428116 ) was an open, randomised trial of continuous vs interrupted treatment, recruiting Kenyan infants who started ART below 13 months of age, had been on ART >24 months and had CD4 >25% and normalized growth. Follow‐up was monthly for growth parameters and 3‐monthly for CD4 counts, for a planned total of 18 months. Infants were randomised to continue (n=21) or interrupt (n=21) treatment at a median (IQR) age of 29 (29, 34) and 30 (29,35) months, respectively. The primary endpoints were weight‐for‐height Z‐scores (WHZ) and serious adverse events. Infants restarted ART if their CD4% was ≤25%, or dropped >1/3 below peak, or if they developed an opportunistic infection or had poor growth. In July 2011, the Data Safety and Monitoring Board recommended stopping the study prematurely because of the high proportion of early restarts in those interrupting ART. At the time of trial cessation, one child had withdrawn from each arm; 5 infants in the continuous arm and 6 in the interruption arm were followed for the planned 18 months.
The question concerning treatment interruption 1 or 2 years after ART initiation has also been addressed by both CHER (Cotton 2012) and PEHSS (Prendergast 2008 (a)). However, both studies were designed prior to the WHO recommendation of immediate ART in perinatally infected infants, so the standard of care arm in each was based on prior WHO guidelines, with initiation of treatment at immunological or clinical thresholds. Neither trial therefore includes a continuous immediate treatment arm, which is now the standard of care against which an interruption strategy would be evaluated. We therefore excluded from the systematic review the subset of results that specifically referred to planned treatment interruption in these two trials.
Ongoing trials
To our knowledge no other randomised controlled trials are underway for this specific age group to assess time to treatment initiation, optimal first line regimen or optimisation strategies similar to those described above, but two additional ongoing studies (ANRS 12206‐ MONOD, NCT01146873‐NEVEREST 3) are assessing safety and efficacy of substituting LPV/r with EFV in children who started treatment early on a LPV/r‐based regimen.
Risk of bias in included studies
The sequence generation was computerized and performed centrally by the trial statistician in all the studies included. Allocation was adequately concealed by using opaque envelopes or electronic interfaces, opened at the time of randomisation in all studies (Figure 2).
2.
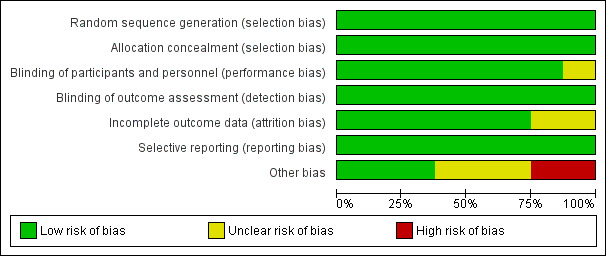
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Participants were not blinded to arm allocation, but the study endpoints were unlikely to be affected by unmasking. Blinding of outcome assessment was only reported for the CHER trial (Violari 2008); however, endpoint assessments for P1060 (Palumbo 2010, Violari 2012), PROMOTE (Achan 2012, Ruel 2013), NEVEREST (Coovadia 2010, Kuhn 2012), ARROW (ARROW trial team 2013) and OPH03 (Wamalwa 2012) relied mainly on laboratory measurements, which are unlikely to be affected by unmasking.
Incomplete outcome data were reported in detail for NEVEREST, where a modified intent to treat analysis was conducted, but few patients were excluded. An intent to treat analysis of all patients was performed for the CHER trial, P1060 cohort 1 and 2, ARROW and OPH03 such that attrition bias is unlikely to significantly affect the results. No loss to follow‐up was reported for the PEHSS trial (Prendergast 2008 (b)).
The primary outcome was pre‐specified in the study protocols and provided in study reports or by the investigators, so selective reporting at least of this outcome is unlikely. Virological and immunological outcomes were also reported as expected given the nature of the questions under investigation.
Given the small number of studies, it was not possible to formally assess other reporting biases. However, given the extensive searches of the standard and grey literature and trial registers, and also contact with experts in the field, it is unlikely that publication bias is an issue in this review (Figure 3).
3.
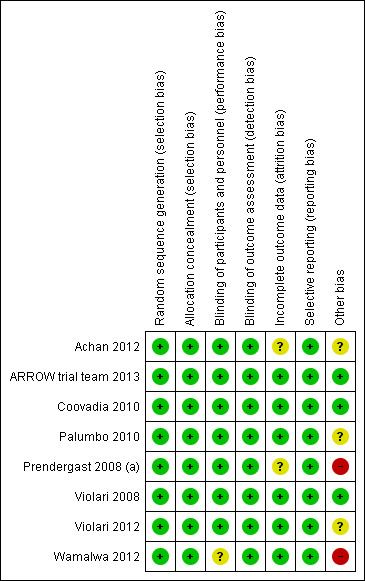
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
P1060 cohort 1 and cohort 2 were terminated early as recommended by the DSMB on the basis of the pre‐specified criteria, and the CHER trial was modified to recall and evaluate all deferred arm infants for initiation of immediate ART. Despite pre‐specified criteria applied for termination of P1060, potential biases due to early termination of the studies should be considered. Similarly, early termination was recommended by the DSMB in OPH03 due to the high rate of re‐start in the interruption arm; the short time spent off ART in the interruption arm may have reduced the ability to detect differences in growth (the primary endpoint) between groups.
The PEHSS trial was originally designed as a feasibility study and was not powered to assess differences in mortality between arms. Similarly, the PROMOTE trial was designed to assess malaria outcomes rather than virological and immunological responses across arms and was therefore not powered to compare virological response with LPV/r vs NVP‐based regimens. The NEVEREST trial enrolled patients who had already achieved virological suppression and it may therefore limit the generalizability of the findings.
Effects of interventions
1. When to start
Two studies assessed when to start treatment. The pooled HR for time to death for these two trials of 0.36 (95%CI 0.18‐0.74) suggests a significant 64% relative reduction in mortality among infants starting ART early compared to those starting deferred ART once clinical and immunological criteria had been met (Figure 4). However, there is strong evidence (p=0.005, I²=87%) that the effect of early ART on mortality differs between these two studies, such that the fixed effect model is not entirely appropriate. While the random‐effects model suggests the effect of ART on mortality is much smaller with very large confidence intervals (HR 0.89, 95%CI 0.05‐15) this gives considerable weight to the small Durban pilot study which may not be appropriate, as it was not designed or powered to address this question. Certainly, the weight of the current evidence, as provided by the CHER study, is in favour of early ART (HR=0.24, 95%CI 0.11, 0.51, p<0.001).
4.
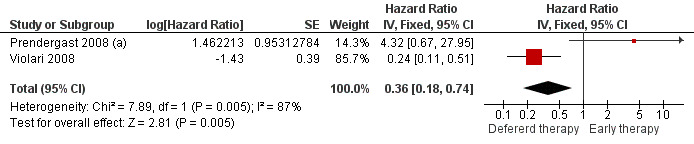
Forest plot of comparison: 1 Early vs Deferred antiretroviral treatment, outcome: 1.1 Mortality.
The combined outcome of mortality or disease progression could only be assessed for the CHER trial as the PEHSS study only reported hospitalizations. Early treatment in asymptomatic infants with good immunological status was strongly associated with a 75% reduction (HR=0.25, 95%CI 0.12‐0.51, p=0.0002, Figure 5) in time to mortality or disease progression as compared to deferring treatment until clinical and immunological criteria were met.
5.
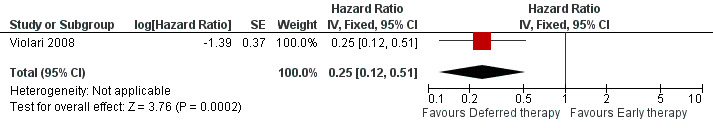
Forest plot of comparison: 1 Early vs Deferred antiretroviral treatment, outcome: 1.2 Mortality or disease progression.
Immunological response was not combined in meta‐analysis as the two studies provided different immunological endpoints. In the PEHSS study median CD4 cell count was not significantly different between groups 12 months after ART initiation (immediate group 33% vs deferred group 32%; P=0.70; Mann–Whitney test). In the CHER study the mean changes from baseline in the CD4 percentage were reported and absolute difference between the early‐therapy group and the deferred‐therapy group was 12.3% (p<0.001) at 12 weeks, 11.5% (p<0.001) at 32 weeks, 9.3% (p<0.001) at 24 weeks and 6.7% by week 40.
2. What to start
Three studies assessed what antiretroviral regimen to start in children <3 years of age (for the purpose of this analysis data from the PROMOTE trial were restricted to ART‐naive children in this age group). No study was powered to assess mortality or disease progression as independent endpoints. Overall, the hazard for treatment failure (a composite of virological failure or discontinuation of the study drugs for any reason, including death) was 1.79 (95%CI 1.33‐2.41) times higher in children starting ART with a NVP‐based regimen compared to those starting with a LPV/r‐based regimen (p<0.0001, Figure 6). However, there is some evidence (p=0.08, I²=60%) that the effect of starting LPV/r or NVP on treatment failure differs between these three studies, such that the fixed effect model is not entirely appropriate. While the random‐effects model suggests the effect of NVP‐based regimen on treatment failure is much smaller with wide confidence intervals (HR 1.58, 95%CI 0.93‐2.67, p=0.09) this gives considerable weight to the smaller PROMOTE study which may not be appropriate, as it was not designed or powered to address this question. Certainly, the weight of the current evidence, as provided by the P1060 studies, is for a benefit in the use of LPV/r‐based regimens (HR=0.24, 95%CI 0.11, 0.51, p<0.001; Figure 7). When considering only the two cohorts of P1060, the findings are consistent across studies (p=0.88, I²=0%), suggesting that results are similar for NNRTI‐exposed and unexposed children. There was no clear difference in effect by age group (subgroup heterogeneity: p=0.97; I²=0%) with a similar increase in the risk of treatment failure in children starting NVP‐based regimens regardless of whether they were older (HR=2.00 95%CI 1.32, 3.03, p=0.001) or younger (HR=2.03, 95%CI=1.24‐3.32, p=0.005) than 12 months. The results were similar when a random‐effects model was used.
6.
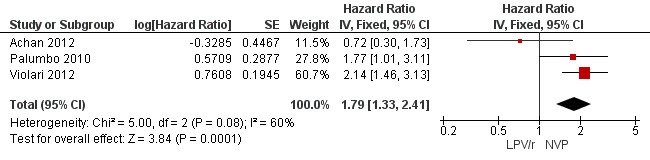
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.1 Treatment failure (virological failure or treatment discontinuation).
7.
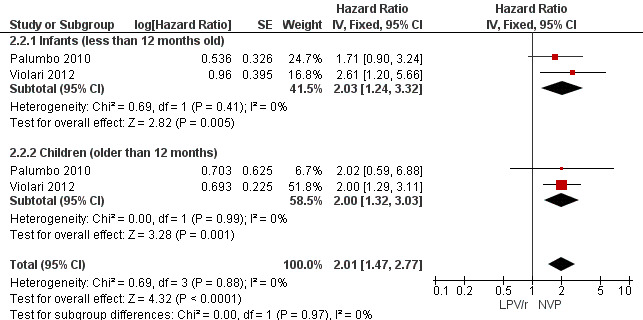
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.2 Treatment failure (virological failure or treatment discontinuation).
The hazard for virological failure (defined as a confirmed plasma HIV‐1 RNA level of less than 1 log10 copies per millilitre below the study entry level at 12 to 24 weeks after the initiation of treatment or a confirmed plasma HIV‐1 RNA level of more than 400 copies per millilitre at 24 weeks) was overall 1.84 (95%CI 1.29‐2.63) times higher for children starting ART with a NVP‐based regimen compared to those starting with a LPV/r‐based regimen (p=0.0008, Figure 8). However, there is good evidence (p=0.04, I²=70%) that the effect of starting LPV/r or NVP on virological failure differs between these three studies, such that the fixed effect model is not entirely appropriate. While the random‐effects model suggests the effect of NVP‐based regimen on virological failure is much smaller with wide confidence intervals (HR 1.71, 95%CI 0.83‐3.52, p=0.15) this gives considerable weight to the small PROMOTE study which may not be appropriate, as it was not designed or powered to address this question. When considering only the two cohorts of P1060 some modest heterogeneity was still present (P=0.23, I²=31%, Figure 9) across studies suggesting that a difference may exist between NNRTI‐exposed and unexposed children. However, the random‐effects model gives a similar result (HR=2.46 95%CI 1.48‐4.08) and the evidence of association (p<0.0001) between a NVP‐based regimen and a shorter time to virological failure (or death) remains very strong. The heterogeneity seems to be partly explained by a difference in effect by age group (subgroup heterogeneity: p=0.04). The HR for virological failure (or death) in the NVP‐based regimen group compared to the LPV/r‐based regimen group was larger in infants below 12 months (HR 3.88, 95%CI 2.06‐7.30, p<0.0001), compared to those above 12 months (HR 1.67, 95%CI 1.03‐2.70, p=0.04) with no differences between trials within these age‐groups. This suggests a larger difference in time to virological failure (or death) between the NVP and LPV/r‐based regimens when ART is initiated in the first year of life.
8.
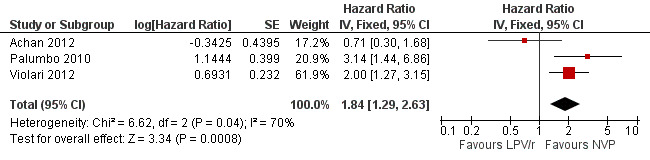
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.3 Virological failure or death.
9.
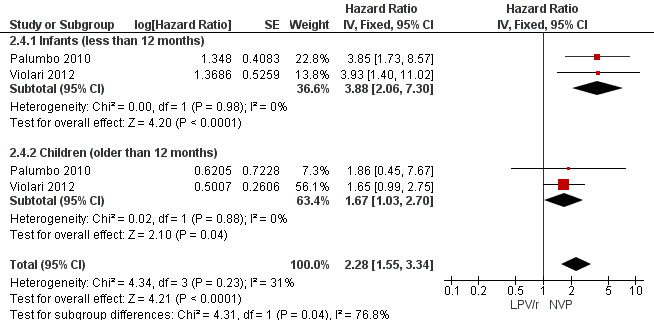
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.4 Virological failure or death.
Subgroup analysis accounting for the presence of documented baseline NNRTI resistance was not possible as an adequate measure of effect in the subgroups could not be obtained for both studies. However, in P1060 cohort 1 (Palumbo 2010) it was reported that among those children with documented NNRTI resistance, the difference between the proportion reaching the primary endpoint in the two study arms was much larger (83.3% vs 18.2%) than that observed (35.8% vs 20.3%) in those without resistance (p=0.02 for interaction).
There was weak evidence of an association between treatment arm and immunological response (mean difference (MD)=1.18, 95%CI ‐0.50, 2.86, p=0.17, Figure 10) and heterogeneity was limited (p=0.30; I² = 17%). The random‐effects model gave similar results (MD=1.22, 95%CI ‐0.72, 3.16, p=0.22). These results provide some indication that the increase in CD4% may be greater in the NVP arm compared to the LPV/r arm.
10.
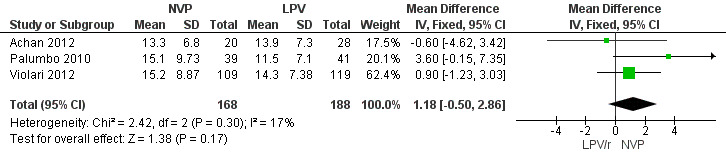
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.5 Change in CD4% from baseline.
In contrast to the findings for the primary endpoints, the increase in weight‐for‐age Z‐score was greater in the NVP arm compared to the LPV/r arm (MD=0.24, 95%CI ‐0.02, 0.50, p=0.07, Figure 11). These findings do not appear to be consistent across studies as a considerable degree of heterogeneity was found (p= 0.10; I² = 57%). A similar effect was observed in the random‐effects model (MD=0.19, 95%CI ‐0.23, 0.61, p=0.38) but the association appears to be much weaker.
11.
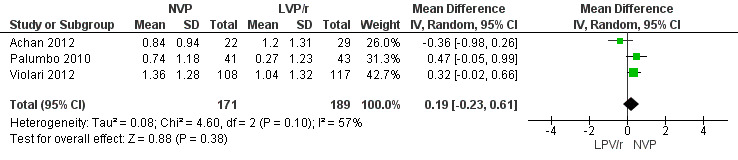
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.6 Change in weight z‐score from baseline.
Similarly, a weak association was found between treatment arm and change in mean height‐for‐age Z‐score when the fixed‐effect model was used (MD=0.16, 95%CI ‐0.01, 0.32, p=0.07, Figure 12), but disappeared when the random‐effects model was used (0.15, 95%CI ‐0.15, 0.46, p=0.32) , with the change being higher in the NVP group compared to the LPV/r group. Some heterogeneity was detected across studies (p=0.10, I²=57%).
12.
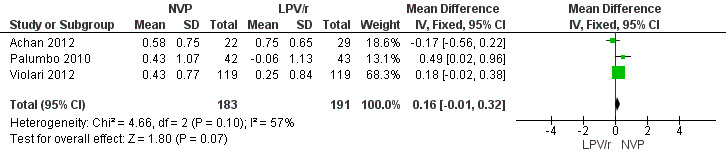
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.7 Change in height z‐score from baseline.
Adverse events associated with treatment were not significantly more frequent in the NVP arm with either the fixed ( RR=1.21, 95%CI 0.88, 1.65, p=0.23, Figure 13) or random effects model (RR=1.17, 95%CI 0.79, 1.73, p=0.44). Some difference in effect across studies was found (heterogeneity: p=0.22; I²=34%) .
13.
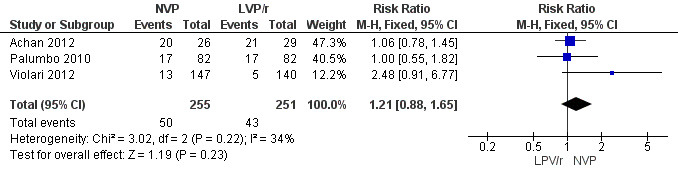
Forest plot of comparison: 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, outcome: 2.8 Adverse events.
3. Substituting LPV/r with NVP
Only one trial addressing the question of LPV/r substitution was included. In the NEVEREST trial (Coovadia 2010), investigating infants and young children who had previously been exposed to NVP, the risk of having at least one VL greater than 50 copies/mL was lower in children substituting NVP for LPV/r after a median of 9 months on LPV/r‐based regimen (having achieved virological suppression) compared to those remaining on a LVP/r‐based regimen (HR=0.62, 95%CI 0.41‐0.92, p=0.02, Figure 14).
14.
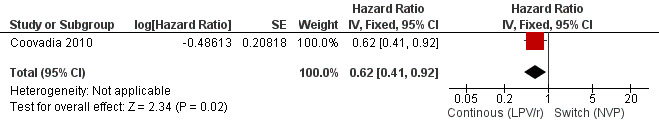
Forest plot of comparison: 3 Switch to NVP vs continue on LPV/r, outcome: 3.2 Virological failure (any VL>50 copies/mL).
However, the hazard for confirmed virological failure (>1000 copies/mL) was higher among children substituting LPV/r with NVP as compared to those remaining on LPV/r (HR=10.19, 95% CI 2.36, 43.94, p=0.002, Figure 15). CD4% increase was lower in the control (LPV/r) group compared to the NVP group (RR=0.22,95% CI 0.07‐0.74, p=0.01, Figure 16). Weight‐for‐age Z scores were similar on average, but fewer children in the NVP group experienced a decline in weight‐for‐age (RR=0.32, 95% CI 0.11‐0.94, p=0.04, Figure 17). Grade 3 or 4 elevation in ALT levels was more common in the NVP group but events were rare and no association was found with study arm (RR=1.80, 95% CI 0.55‐5.97, p=0.33, Figure 18). Similarly, grade 3 or 4 neutropenia was rare and similar across arms (RR=1.72, 95% CI 0.42‐6.99, p=0.45, Figure 19).
15.
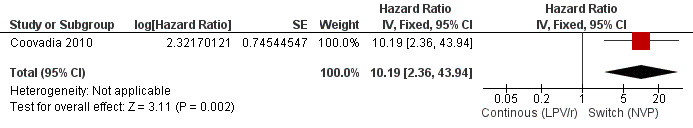
Forest plot of comparison: 3 Switch to NVP vs continue on LPV/r, outcome: 3.3 Virological failure (confirmed VL>1000 copies/mL).
16.
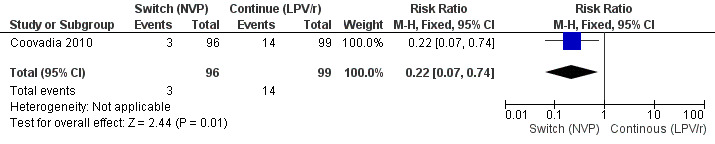
Forest plot of comparison: 3 Switch to NVP vs continue on LPV/r, outcome: 3.4 Decline by 10% in CD4% at week 52.
17.
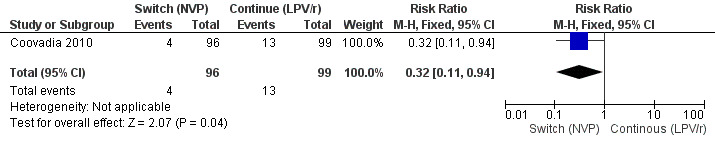
Forest plot of comparison: 3 Switch to NVP vs continue on LPV/r, outcome: 3.5 Decline by 1 z‐score in weight‐for‐age at week 52.
18.
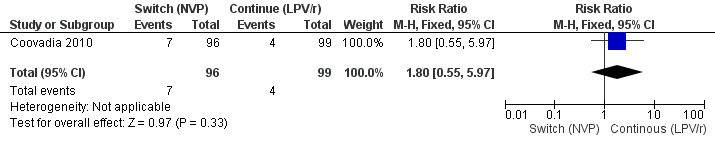
Forest plot of comparison: 4 Switch to NVP vs continue on LPV/r, outcome: 4.5 ALT increase (Grade 3/4).
19.
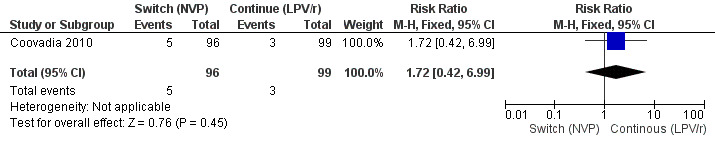
Forest plot of comparison: 4 Switch to NVP vs continue on LPV/r, outcome: 4.6 Neutropenia (grade 3/4).
4. Induction‐Maintance
One trial (ARROW trial team 2013) investigated an induction‐maintenance ART strategy in children and we report here results for the subset of 370 (31%) children below 3 years of age. This trial was also designed to assess the benefits of clinically driven monitoring or routine laboratory and clinical monitoring for toxicity and efficacy.
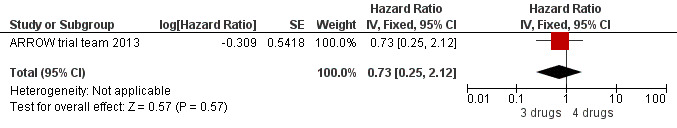
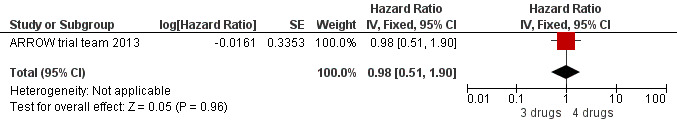
There was no significant difference in the primary endpoint (mean change in CD4 percentage from baseline) between children who received a standard 3‐drug regimen (Arm A) and those who received an induction‐maintenance regimen (Arms B and C) either at 72 weeks (mean difference 0.70 [95%CI ‐0.51, 1.91] p= 0.33, Figure 20) or 144 weeks (mean difference ‐0.20 [95%CI ‐1.48, 1.08] p= 0.69, Figure 21); however, at 36 weeks (not the primary endpoint, but the end of the induction period in Arms B and C) there was a significantly greater CD4 increase in the 4‐drug, compared to 3‐drug, arms (mean difference 1.70 [95%CI 0.61, 2.79] p=0.002, Figure 22). At 24 weeks virological response was better in those children who received an induction‐maintenance regimen (Arms B and C) than in those who received a standard 3‐drug regimen (Arm A) (OR 1.99 [95%CI 1.09, 3.62] p=0.02, Figure 23); however, this effect was not maintained at 48 weeks (OR 0.96 [95%CI 0.50, 1.83] p=0.90) and 144 weeks (OR 1.03 [95%CI 0.51, 2.07] p=0.94, Figure 24). There were no differences between groups in mortality (HR 0.71 [95%CI 0.16, 3.21] p=0.66, Figure 25) or mortality and disease progression (HR 0.73 [95%CI 0.25, 2.12] p=0.57, Figure 26, HR 0.98 [95%CI 0.51, 1.90] p=0.96, Figure 27), and no differential growth between children receiving initial 4‐drug or 3‐drug regimens .
20.
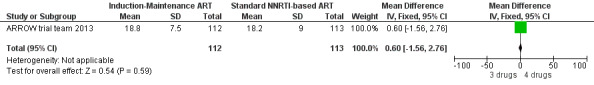
Forest plot of comparison: 6 Induction‐Maintance strategy, outcome: 6.2 Increase in CD4% from baseline at week 72.
21.
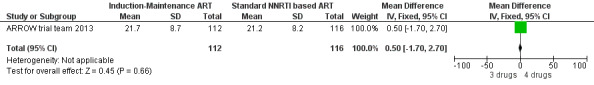
Forest plot of comparison: 6 Induction‐Maintance strategy, outcome: 6.3 Increase in CD4% from baseline at week 144.
22.
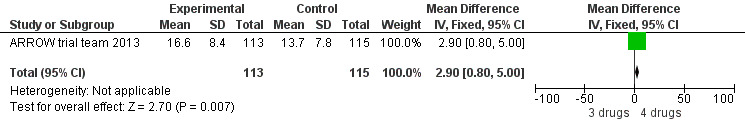
Forest plot of comparison: 6 Induction‐Maintance strategy, outcome: 6.1 Increase in CD4% from baseline at week 36.
23.
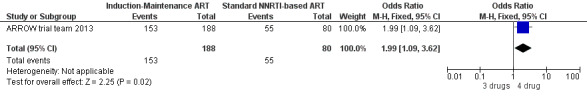
Forest plot of comparison: 5 Induction‐Maintance strategy, outcome: 5.4 Virologic response (VL<400 copies/ml) at 24 weeks.
24.
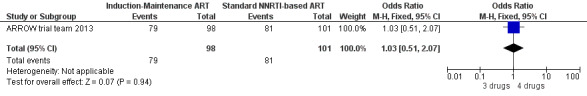
Forest plot of comparison: 5 Induction‐Maintance strategy, outcome: 5.7 Virologic response (VL<400 copies/ml) at 144 weeks.
25.
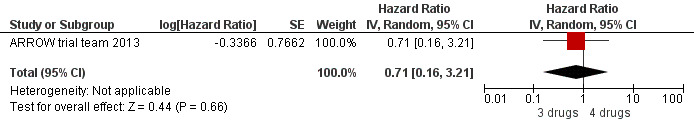
Forest plot of comparison: 6 Induction‐Maintance strategy, outcome: 6.5 Mortality.
26.
Forest plot of comparison: 6 Induction‐Maintance strategy, outcome: 6.6 New WHO 4/death event‐free survival.
27.
Forest plot of comparison: 6 Induction‐Maintance strategy, outcome: 6.7 New WHO 3/4/death event‐free survival.
5. Planned treatment interruption
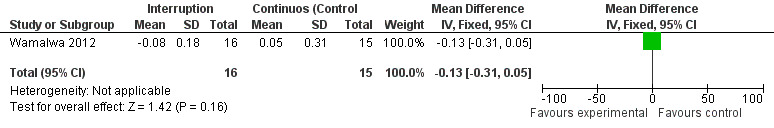
Although three trials explored the impact of planned treatment interruption after initiation of ART, only one study was designed to directly compare continuous early ART (now the international standard of care for infants) versus interrupting for a certain period of time. In the OPH‐03 trial, in which children who started ART in infancy were randomised either to treatment interruption or continuous ART, growth and serious adverse events were similar between arms: 6‐month mean change in weight‐for‐age Z‐score was 0.05 vs –0.08 (MD= ‐0.13, 95% CI ‐0.31 to 0.05, p=0.16, Figure 28), in height‐for‐age Z‐score was 0.06 vs –0.04 (MD= ‐0.10, 95% CI ‐0.33 to 0.13, p= 0.39, Figure 29), and in weight‐for‐height Z‐score was 0.07 vs –0.04 (MD= ‐0.11, 95% CI ‐0.32 to 0.10, p= 0.30, Figure 30) in the continue vs planned treatment interruption arms, respectively. One serious adverse event was observed in each arm (OR=1.00, 95% CI 0.006 to 17.12 p=1.00, Figure 31). However, the trial was stopped early because 14/21 (67%) infants in the interruption arm had to restart ART within 3 months of treatment interruption.
28.
Forest plot of comparison: 5 Planned treatment interruption strategy, outcome: 5.1 Change in WAZ z‐score.
29.
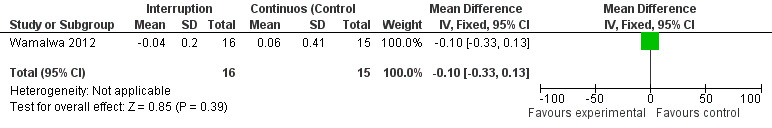
Forest plot of comparison: 5 Planned treatment interruption strategy, outcome: 5.2 Change in HAZ z‐score.
30.
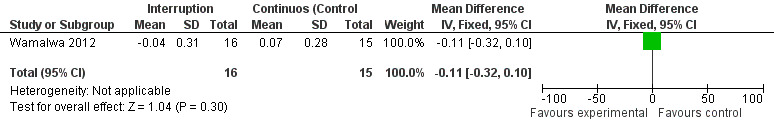
Forest plot of comparison: 5 Planned treatment interruption strategy, outcome: 5.3 Change in WHZ z‐score.
31.
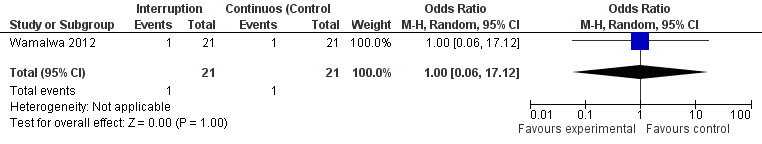
Forest plot of comparison: 5 Planned treatment interruption strategy, outcome: 5.4 Serious adverse events.
Discussion
Paediatric HIV infection remains an important public health problem, with an estimated 900 infants newly infected each day (UNAIDS 2012). Treating young children is challenging because of lack of appropriate antiretroviral formulations, adherence difficulties, concerns regarding pharmacokinetics and reliance on caregivers. Furthermore, there are concerns regarding long‐term drug toxicity, development of resistance and the cost of lifelong treatment when starting young children on ART. However, it has long been recognised that disease progression in infancy is rapid, and that the markers used to guide treatment decisions in older children and adults do not reliably identify those infants at highest risk of morbidity and mortality (HPPMCS 2003). Mindful of these competing factors, and in the absence of randomised evidence, clinicians have historically varied in their practice regarding when to start ART in young children. Furthermore, decisions around which ART regimen to start are complicated by the possibility of drug resistance in the context of nevirapine exposure, fewer drug choices for infants, uncertain dosing for some medications and long‐term toxicities. However, as reviewed here, there are now data from randomised trials to address some of these issues and guide clinicians in treatment choices and management strategies for young children.
The question of when to start ART has been clarified by results from the CHER trial. In this trial, which recruited asymptomatic South African infants between 6‐12 weeks of age with well‐preserved CD4 counts, the data were so clear at interim DSMB review that results were released early and infants randomised to deferred ART were recalled to assess the need to start immediate treatment. There was a significant difference in both morbidity and mortality between infants randomised to immediate versus deferred ART. Deaths were often sudden and were not classically AIDS‐defining; gastroenteritis and pneumonia predominated and mortality occurred even among infants with high CD4 counts. This highlights the difficulty of following up untreated infants, even in the setting of a trial, because of the rapidity of disease progression. Data from another trial included in this analysis, conducted in Durban, South Africa, showed a reduction in morbidity, but no significant difference in mortality between infants randomised to immediate versus deferred ART. Although there appears to be heterogeneity in these results, the Durban trial was a much smaller (n=63) feasibility study of different approaches to antiretroviral management in infancy; this trial was neither designed nor powered to address the question of ‘when to start’ ART. The results of the CHER trial were felt by policy makers to be generalisable to all settings, as reflected by changes in US (DHHS 2011), European (PENTA 2009) and WHO guidelines (WHO 2010) which all now recommend initiation of ART early in infancy. It should be noted, however, that most infants in CHER had been exposed to PMTCT interventions (as single dose NVP) and thus were infected despite prophylaxis, suggesting in utero acquisition of HIV infection, which may be associated with faster disease progression compared with those infected either intrapartum or postnatally through breast milk (Italian Register 1999, Mphatswe 2007). Infants in CHER were also identified early in life, as would be anticipated from a well functioning PMTCT programme. However, in many settings early infant diagnosis (EID) is delayed or even unavailable, and loss‐to‐follow‐up from PMTCT and EID programmes is high (Chatterjee 2010). Infants recruited to CHER had relatively well preserved CD4 counts (CD4>25% was an entry criterion), but given the speed of disease progression after birth, many HIV‐infected infants outside of a trial setting will have progressed to a CD4 count below this threshold by 6‐12 weeks. It may therefore be anticipated in ART programmes that mortality will be higher than that reported for immediate therapy arms in CHER. This highlights the critical nature of an integrated and effective PMTCT/early infant diagnosis programme so that infants can benefit from early diagnosis and immediate initiation of ART.
The trials analysed here do not address the question of whether ART should be initiated in infants diagnosed with HIV at birth. The most recent description of a “functional cure” in a US infant (Persaud 2013) has opened a widespread debate on the value of initiating treatment in the first 48 hours of life following a positive PCR result at birth. Current studies are investigating the impact of treatment started close to birth.
Whilst the majority of perinatally infected infants progress rapidly, a minority will be long‐term non‐progressors. An asymptomatic, HIV‐infected child who is diagnosed in late infancy or beyond the first year of life may not require immediate initiation of ART. WHO guidelines in 2010, in the absence of trial data, but recognising that disease progression remains high in children from 1‐2 years of age, made a pragmatic recommendation for universal ART below 2 years. Revised guidelines in 2013 recommend initiation of ART regardless of clinical and immunological criteria for all children less than 5 years, with the goal of simplifying the initation of paediatric treatment (particularly in the absence of CD4 count or percent measurements) and providing programmatic advantages to facilitate rapid scaling up of paediatric ART in resource limited settings.
Immediate ART, at least in young infants, reduces morbidity and mortality and may improve neurodevelopmental outcomes (Laughton 2012). Data from the trials presented here show that good virological and immunological outcomes are achievable in early life. However, there are concerns regarding cost, long‐term toxicity and viral resistance, in addition to the feasibility of early ART in terms of availability, palatability and adherence.
The availability of paediatric formulations is also an important consideration for fragile health systems, so that there are advantages in harmonising drugs and formulations with those needed for adults. The choice of first‐ line therapy is therefore critical, particularly in the context of lifelong treatment.
The P1060 trials addressed the question of what ART to start, comparing a NVP‐based regimen with a LPV/r‐based regimen in young children below 3 years of age. Two parallel trials recruited both sd‐NVP‐exposed children (cohort 1) and sd‐NVP‐unexposed children (cohort 2). It is well recognised that a substantial proportion of infants and young children exposed to sd‐NVP at birth develop NNRTI resistance (Arrive 2007), and previous data suggested that this may compromise the efficacy of NVP‐based regimens (Lockman 2007). This concern was further consolidated by the P1060 cohort 1 data, which showed a significantly higher treatment failure rate among sd‐NVP‐exposed children randomized to a NVP‐ compared to LPV/r‐based regimen. Unexpectedly, sd‐NVP‐unexposed children in cohort 2 also showed a higher treatment failure rate if randomized to a NVP, compared to LPV/r, regimen. These trials were not powered to evaluate mortality or disease progression as independent endpoints. The primary outcome, treatment failure, was a composite endpoint defined as virological failure or discontinuation of the study treatment for any reason, including death; the endpoint was fairly short‐term, at 24 weeks. P1060 trial investigators were as rigorous as possible in ensuring correct allocation of NVP exposure status, although it is difficult to completely rule out the possibility that some infants in cohort 2 were NVP‐exposed. Children in cohort 2 were older than children in cohort 1. After stratifying by age, the combined analysis of cohort 1 and 2 did not show any difference between infants and children older than 12 months for the primary endpoint of treatment failure (which included virological failure, toxicity leading to discontinuation and death); however, a considerable difference was detected for the secondary endpoint comprising virological failure or death. This inconsistency in the subgroup analyses could be explained by a higher number of interruptions due to toxicity in children older than 12 months in the NVP arm. The strength of association between starting ART with NVP and having a higher risk of virological failure in infants (less than 12 months) was much larger than the risk observed in older children, suggesting that NNRTI resistance may have played a role. It is plausible that there is an attenuated effect in older children, who benefit from a "wash out" period during which NNRTI mutations fade and only re‐emerge once selective pressure of the drug is re‐established. These findings are consistent with the NEVEREST cohort, where detection of NNRTI‐resistant strains was inversely correlated with increasing age at treatment initiation (Hunt 2011). By contrast, a post‐hoc analysis of data from the PROMOTE‐PEDS trial in Uganda, in which young children (2mo‐5yrs) were randomized to a LPV/r‐ or NNRTI‐based regimen, showed no significant difference between arms for children <3 years of age, using the same endpoints as the P1060 trial. However, the finding of no overall difference between regimens needs to be interpreted with caution, because the study was neither designed nor powered to assess these the superiority of one of the two regimens.
Overall, the inclusion of PROMOTE‐PEDS led to considerable heterogeneity between the three trials that explored first‐line PI vs NNRTI regimens in young children; however, there were no clear differences in effect between the two P1060 studies. It remains unclear in P1060 why LPV/r should be superior to NVP even in children not previously exposed to sd‐NVP for PMTCT, as this has not been observed in adults nor in the PENPACT 1 trial (PENPACT1 study team). There are several theoretical explanations. First, infants may be disadvantaged by a NVP‐based regimen because of the low genetic barrier to resistance in the context of high viral loads during early life. Of note, the recommended nevirapine dose was increased during the course of the P1060 trial following release of new WHO guidelines (from 4 mg/kg for 14 days and 7 mg/kg twice a day thereafter, to the currently FDA recommended dose of 160‐200 mg/m2/dose once daily for 14 days followed by 160‐200 mg/m2/dose twice‐daily) . However, the authors did not find evidence for a dose effect to explain the observed failure rate among NVP‐treated children. Second, it has been proposed that the use of lead‐in dosing, whereby NVP is given only once‐daily for the first 2 weeks, may lead to under‐dosing and thereby facilitate the development of NNRTI resistance and virological failure. In the CHAPAS 1 trial where children were randomised to initiate ART with full dose versus ‘lead‐in’ for 2 weeks with half dose NVP, the viral load responses at 48 and 96 weeks were the same in the two groups (Mulenga 2010); however, subsequent analyses showed that even using the fixed dose combination baby pills in which the NVP dose is at the upper end of recommendations, NVP levels at 4 weeks were low in young infants (Fillekes 2013). Further investigation of early pharmacokinetics and viral load with ‘lead‐in' NVP dosing is planned in the IMPAACT P1103 trial. Third, it is possible that infants without documented NNRTI exposure were actually carrying NNRTI‐resistant virus, as reported by observational studies and surveillance data from South Africa and Zimbabwe (Kuhn 2013, Apollo 2013). Studies in South Africa have indicated that levels of transmitted resistance remain low (Pillay 2008) despite treatment scale‐up, so acquisition of NNRTI resistance mutations as a consequence of multiresistant strains circulating at a population level seems unlikely. Although population sequencing was conducted in a subset of the P1060 study population, more sensitive allele‐specific resistance assays of baseline samples from the 1060 cohort 2 trial are ongoing to rule out the possibility of undocumented NVP exposure.
Although the P1060 trials suggest that LPV/r‐based regimens are preferable for all infants, regardless of PMTCT exposure status, these data are in contrast to those from the PENPACT‐1 study (PENPACT1 study team), a multinational trial of first‐line NNRTI versus PI therapy in children . Over 4 years of follow up, PENPACT‐1 found no significant difference between children starting NNRTI‐ or PI‐based regimens in primary (change in log10 HIV‐1 RNA VL from baseline) or secondary endpoints (regimen switch, change in CD4% from baseline, VL <400 copies/ml at week 24 on first‐line ART, VL <400 copies/ml at 4 years, continued VL suppression on first‐line ART, failure of second‐line ART, grade 3/4 adverse events, new CDC stage C events and resistance). Children in this trial were older (median age 6.5 (IQR: 2.8‐12.9) years), and predominantly living in Europe and USA. PENPACT‐1 was not included in the current systematic review because subgroup data were not available for children <3 years of age, and first‐line ART in the PI arm included un‐boosted PIs (nelfinavir and ritonavir), which are known to be sub‐optimal and are no longer used. A large observational study of 437 infants followed in European national cohorts for a median of 5.9 years (EPPICC 2011) did not confirm the P1060 trial findings. In this study, no difference was observed in viral load suppression and CD4 response between infants initiating ART with a PI versus NNRTI, but only a limited number of infants were started on a boosted‐PI containing regimens. It is interesting to speculate whether the NRTI backbone or the particular NNRTI agent used had an important influence on outcomes. Although the first‐line NRTIs in P1060 and PROMOTE‐PEDS were zidovudine and lamivudine, a variety of backbones were used in EPPICC and PENPACT‐1; almost one‐quarter of children in PENPACT‐1 received abacavir and lamivudine, which were shown to be superior to zidovudine and lamivudine in the PENTA 5 trial (PENTA 2002). Whether there is a difference in efficacy between nevirapine and efavirenz or whether acquired mutations to these drugs fade or re‐emerge differently is still unclear, particularly in light of the limited experience with EFV in young children. In PENPACT‐1, almost two‐thirds of children in the NNRTI arm received efavirenz; by contrast, all children in P1060 received nevirapine, because efavirenz is not used below 3 years at the time of this trial.
Overall, despite differences between the results of the P1060 trials and previous published data in European populations, there is good evidence for superiority of LPV/r over NVP, at least for the short‐term composite outcome assessed by these trials.
To our knowledge, no other trial is addressing the question of PI versus NNRTI 3‐drug ART regimens or versus regimens containing new classes of drugs (eg. integrase inhibitors) started in early life. The one trial of sd‐NVP‐exposed infants underway in Kenya(ClinicalTrials.gov reference NCT00427297 trial) was terminated early following the results of the P1060 trial. Data presented here have therefore provided a dilemma to policy‐makers: high‐quality data from two randomised controlled trials demonstrate a benefit to LPV/r, but there are currently practical disadvantages to LPV/r over NVP, including cost, palatability, cold chain requirements and available formulations for infants (Prendergast 2012). However, as effective PMTCT interventions are rapidly scaled up the number of new perinatal infections will decrease and those who are infected are increasingly likely to have been exposed to PMTCT drugs. Since these infants are eligible for PI‐based ART on the grounds of prior NNRTI exposure, the additional impact of a change in policy (to recommend PI‐based ART for all infants and young children) is expected to be limited and benefits may result from recommending a single preferred drug within the same age group. Furthermore, there is emerging evidence of detectable NNRTI resistance among children without any history of exposure to ARV drugs, suggesting that prior PMTCT exposure may not be a reliable marker for identifying children at higher risk of HIV resistance to NNRTI (Apollo 2013, Kuhn 2013). Revised WHO 2013 guidelines therefore recommend LPV/r‐based ART for infants and young children, regardless of prior PMTCT exposure; however, where use of LPV/r is not feasible, ART should be initiated without any delay with NVP (WHO 2013).
An alternative approach to long‐term LPV/r, which was investigated in the NEVEREST trial, would be to start ART with a LPV/r‐based regimen and switch to NVP (maintaining the NRTI backbone) once virological suppression is achieved. Interpretation of the NEVEREST results for the two virological endpoints would suggest that, at least for selected patients (those without pre‐treatment NVP resistance and with documented sustained virological suppression), switching to NVP could be a successful strategy. Children who switched to NVP were more likely to maintain viraemia below 50 copies/ml compared to those continuing LPV/r. By contrast, virological failure (>1000 copies/mL) was more common in those who switched to NVP and was strongly related to presence of pre‐treatment NNRTI mutations. The authors suggest that the apparent inconsistency between the two virological endpoints may be explained by the sub‐optimal adherence profile of infants continuing unpalatable LPV/r, leading to occasional blips of HIV viraemia, whereas virological failure (VL>1000 copies/mL) occurred more commonly in those switching to NVP, because of tits low genetic barrier to resistance. Although the NEVEREST strategy appears promising and results are now confirmed by the week 156 follow‐up data (Kuhn 2012), the findings may not be easily generalisable since enrolled children had all achieved and sustained viral load <400 copies/mL for at least 3 months within the first 12 months of treatment. This selected population is likely to have better adherence, fewer problems with ART tolerability and an improved prognosis, compared to an unselected population. Furthermore, this strategy require virological monitoring, both to identify eligible children and to detect early virological failure on NVP. Nevertheless, the NEVEREST strategy has been included as a potential approach to management of young children in the revised WHO guidelines (WHO 2013), in settings where virological monitoring is feasible. A simplified approach, using a fixed duration of LPV/r‐based ART, followed by a switch to EFV‐based ART once the child is 3 years or older, would be more practical in settings without the capacity for viral load monitoring, but would benefit from better formulations of LPV/r to be more easily implemented . Additional data to further inform these approaches are awaited: similar PI‐sparing strategies are under investigation in the MONOD trial (ANRS 12206‐ MONOD) in children under two years of age and in the NEVEREST‐3 trial (NCT01146873‐NEVEREST 3) in children between 3 and 5 years of age.
ARROW is the only randomised trial to have compared an induction‐maintenance approach to the management of ART in children; approximately one‐third of children in this trial were below 3 years of age. There was evidence of benefit during the induction phase with 4 drugs, with greater virological suppression (at week 24) and CD4 increase (at week 36), particularly among children with low baseline CD4 counts. However, there was no long‐term sustained benefit from an induction‐maintenance approach, since change in CD4 count by 144 weeks was not significantly different between arms. Previous data on an induction‐maintenance strategy came from a European cohort study(EPPICC 2011), which reported both immunological and virological benefit of a 4 drug approach in infants starting treatment in the first year of life most were still on the 4 drugs regimen at their 12 months outcome measure. Whether sustained benefits would have been seen in the ARROW trial with a longer induction period (beyond 36 weeks) is unclear. However, 4‐drug regimens represent an additional burden on ART costs and currently a long‐term induction‐maintenance strategy is not recommended in light of the limited, short‐term gains observed in this trial.
An alternative approach to the currently recommended lifelong treatment would be to start early ART in infancy, followed by a period of treatment interruption. Theoretically, this strategy may allow the child to be protected during the period of greatest risk for HIV disease progression and mortality, but enable time off therapy beyond 1‐2 years of age to reduce toxicity, cost and risk of resistance. Definitive, long‐term ART would be restarted when the child meets standard age‐related treatment criteria. Only one trial was assessed in this review, because it was the only trial to compare the current standard approach (early lifelong ART for infants) with an interruption approach. . The OPH‐03 trial found that disease progression was rapid after ART interruption, with two‐thirds of children restarting ART within 3 months. Two South African trials (PEHSS and CHER), which started treatment in healthier babies and had different designs but similar rationale to OPH03, also found that young children stopping ART needed to restart sooner than would be feasible in a public health approach, where close follow‐up and monitoring of children off ART is challenging. Whether children could safely interrupt therapy at a later time‐point, when disease progression is less rapid, may be worth investigating further. The PENTA 11 trial (PENTA 11), conducted predominantly in Europe, suggested that such a strategy was safe and well accepted in older children (median age 9 years) but also highlighted the importance of selecting patients with good immunological characteristics (particularly high nadir CD4 count), as being those most likely to benefit from treatment interruption .
Authors' conclusions
Implications for practice.
Recommendations for treatment initiation in children <2 years of age have already been updated in all settings since 2010, because of the unequivocal benefit of early treatment initiation in infancy. New 2013 WHO guidelines now recommend that children between 2‐5 years should also initiate ART irrespective of clinical and immunological status in order to simplify treatment programmes in high‐burden settings and allow rapid and wide scale up of paediatric ART. However, given the uncertainty of the clinical benefits offered by early ART in asymptomatic children, countries with high ART coverage, good access to CD4 testing and adequate retention, in treatment programmes may continue to use clinical and immunological thresholds to determine treatment initiation in children 2‐5 years of age. Findings from the P1060 trial have provided the scientific basis for a change in the first line recommended ART for children less than 3 years, which is now LPV/r‐based where feasible. Few high burden countries are currently using LPV/r as first‐line treatment in NVP‐exposed infants and adopting these new recommendation for children under 3 years will require a significant expansion of LPV/r provision. Recognising the challenges in LPV/r formulation and supply chain management, where LPV/r is not feasible WHO guidelines continue to recommend first‐line NVP‐based regimens, which have the benefit of being available as fixed‐dose scored dispersible tablets. A multi‐unit particle preparation of LPV/r, stored in a capsule designed to be opened and sprinkled, has been evaluated in the CHAPAS 2 trial in Uganda and Zambia (ISRCTN01946535), and provides promise that more suitable LPV/r formulations for infants and young children will become available. The Drugs for Neglected Diseases Initiative (DNDi) has identified 4‐in‐1 first‐line sprinkle formulations for young children as a priority area for development by 2015. Given the findings of the ARROW induction‐maintenance and the OPH‐03 treatment interruption trials, these strategies are not currently recommended within treatment programmes.
Implications for research.
There is little evidence to guide whether all children diagnosed with HIV infection between 1‐5 years of age should start ART, as has been recommended on practical and operational grounds by WHO. Although the risk of disease progression remains high at this age, a considerable proportion of children surviving beyond the first year of life will be slower progressors, who may not fulfil clinical or immunological criteria to start ART until late childhood or adolescence (Ferrand 2009). The PREDICT trial (Puthanakit 2012)
showed no survival benefit to earlier treatment of older children. However, there may still be a benefit to starting early ART, in terms of neurodevelopmental outcome, amelioration of the inflammatory consequences of HIV (so‐called 'non‐AIDS' morbidity) and long‐term immune reconstitution. Future trials may provide clarity on this issue.
The role of newer drug classes, such as integrase inhibitors, has yet to be evaluated in young children, but these could provide an alternative to NNRTI‐ or PI‐based first‐line regimens. The integrase inhibitor dolutegravir is appealing for use in infancy because of its once‐daily dosing, high potency at a low milligram dose and no requirement for boosting. In the meantime, assuming that PI‐based regimens become standard‐of‐care first‐line ART, further evidence is urgently required to better inform policy on the most appropriate sequencing following initiation with a LPV/r‐based regimen. Further data are needed on the possibility of substituting LPV/r with NVP or EFV, how best to identify those children who may benefit from such a strategy and the mid‐ and long‐term impact on treatment sequencing and virological control.
There is also a need for operational research on the programmatic and economic consequences, as well as long term toxicities (in particular metabolic and lipids) and options for second line ART of initiating children <3 years on LPV/r in resource‐limited settings.
What's new
| Date | Event | Description |
|---|---|---|
| 23 February 2014 | New citation required and conclusions have changed | New studies were available to inform the questions addressed by this review. |
Acknowledgements
We are particularly grateful to: Jane Lindsey, Paul Palumbo, Avy Violari and the P1060 trial team investigators who provided supplementary data for these analyses; Louise Kuhn and the NEVEREST team; Grace John‐Stewart and the OPH03 team; Theodore Ruel, Jane Achan and the PROMOTE‐PEDS team. Diana Gibb, Mark Cotton and Jayne Thierny contributed to the original version of this review and Steve Welch provided independent study review in the original version . We are grateful to Amberle Durano, Tara Horvath, Gavrilah Wells and Joy Oliver for their support in the search and update process.
Data and analyses
Comparison 1. Early vs Deferred antiretroviral treatment.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Mortality | 2 | Hazard Ratio (Fixed, 95% CI) | 0.36 [0.18, 0.74] | |
| 2 Mortality or disease progression | 1 | Hazard Ratio (Fixed, 95% CI) | 0.25 [0.12, 0.51] |
1.1. Analysis.
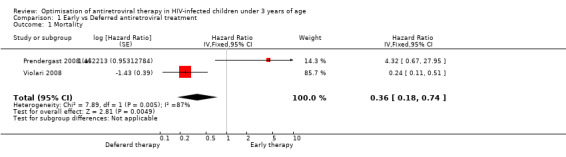
Comparison 1 Early vs Deferred antiretroviral treatment, Outcome 1 Mortality.
1.2. Analysis.
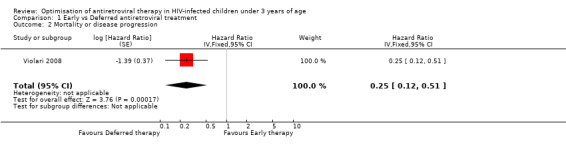
Comparison 1 Early vs Deferred antiretroviral treatment, Outcome 2 Mortality or disease progression.
Comparison 2. NVP‐based vs LPV/r‐based first line antiretroviral therapy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Treatment failure (virological failure or treatment discontinuation) | 3 | Hazard Ratio (Fixed, 95% CI) | 1.79 [1.33, 2.41] | |
| 2 Treatment failure (virological failure or treatment discontinuation) | 2 | Hazard Ratio (Fixed, 95% CI) | 2.01 [1.47, 2.77] | |
| 2.1 Infants (less than 12 months old) | 2 | Hazard Ratio (Fixed, 95% CI) | 2.03 [1.24, 3.32] | |
| 2.2 Children (older than 12 months) | 2 | Hazard Ratio (Fixed, 95% CI) | 2.00 [1.32, 3.03] | |
| 3 Virological failure or death | 3 | Hazard Ratio (Fixed, 95% CI) | 1.84 [1.29, 2.63] | |
| 4 Virological failure or death | 2 | Hazard Ratio (Fixed, 95% CI) | 2.28 [1.55, 3.34] | |
| 4.1 Infants (less than 12 months) | 2 | Hazard Ratio (Fixed, 95% CI) | 3.88 [2.06, 7.30] | |
| 4.2 Children (older than 12 months) | 2 | Hazard Ratio (Fixed, 95% CI) | 1.67 [1.03, 2.70] | |
| 5 Change in CD4% from baseline | 3 | 356 | Mean Difference (IV, Fixed, 95% CI) | 1.18 [‐0.50, 2.86] |
| 6 Change in weight z‐score from baseline | 3 | 360 | Mean Difference (IV, Random, 95% CI) | 0.19 [‐0.23, 0.61] |
| 7 Change in height z‐score from baseline | 3 | 374 | Mean Difference (IV, Fixed, 95% CI) | 0.16 [‐0.01, 0.32] |
| 8 Adverse events | 3 | 506 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.21 [0.88, 1.65] |
2.1. Analysis.
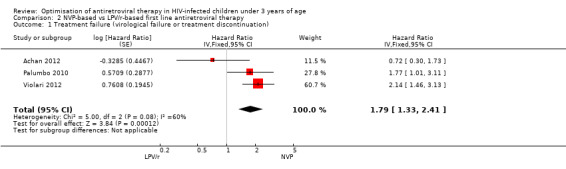
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 1 Treatment failure (virological failure or treatment discontinuation).
2.2. Analysis.
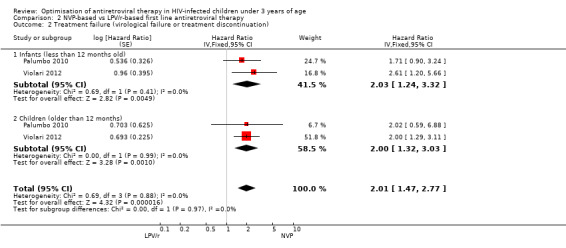
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 2 Treatment failure (virological failure or treatment discontinuation).
2.3. Analysis.
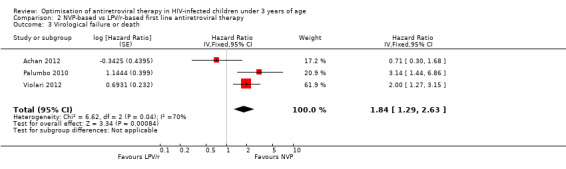
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 3 Virological failure or death.
2.4. Analysis.
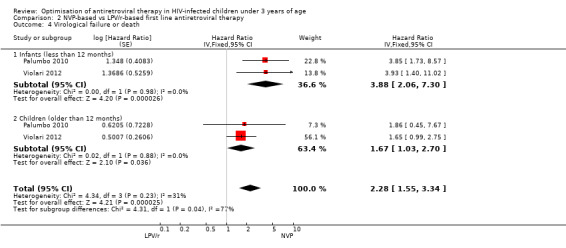
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 4 Virological failure or death.
2.5. Analysis.
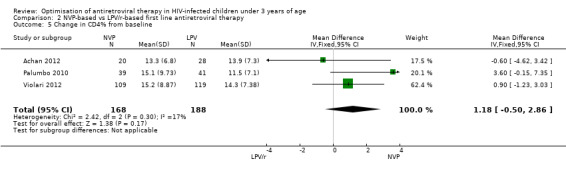
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 5 Change in CD4% from baseline.
2.6. Analysis.
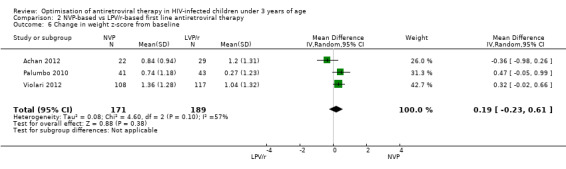
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 6 Change in weight z‐score from baseline.
2.7. Analysis.
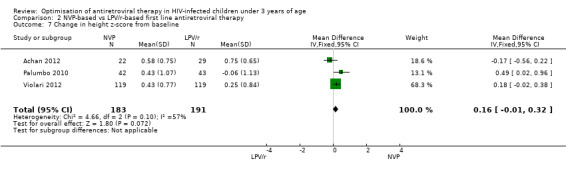
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 7 Change in height z‐score from baseline.
2.8. Analysis.
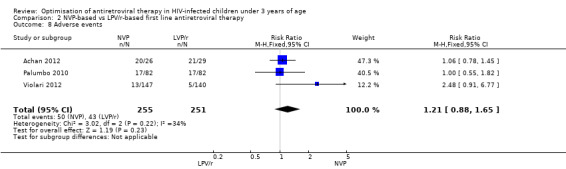
Comparison 2 NVP‐based vs LPV/r‐based first line antiretroviral therapy, Outcome 8 Adverse events.
Comparison 3. Switch to NVP vs continue on LPV/r.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Virological failure (any VL>50 copie/ml) | 1 | Hazard Ratio (Fixed, 95% CI) | 0.62 [0.41, 0.92] | |
| 2 Virological failure (confirmed VL>1000 copies/ml) | 1 | Hazard Ratio (Fixed, 95% CI) | 10.19 [2.36, 43.94] | |
| 3 Decline by 10% in CD4% at week 52 | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.22 [0.07, 0.74] |
| 4 Decline by 1 z‐score in weight‐for‐age at week 52 | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.11, 0.94] |
| 5 ALT increase (Grade 3/4) | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.80 [0.55, 5.97] |
| 6 Neutropenia (grade 3/4) | 1 | 195 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.72 [0.42, 6.99] |
3.1. Analysis.
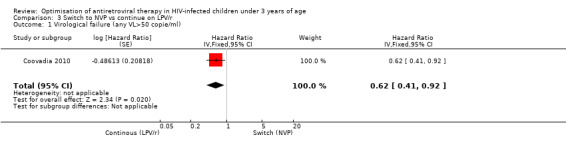
Comparison 3 Switch to NVP vs continue on LPV/r, Outcome 1 Virological failure (any VL>50 copie/ml).
3.2. Analysis.
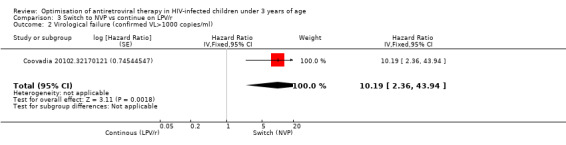
Comparison 3 Switch to NVP vs continue on LPV/r, Outcome 2 Virological failure (confirmed VL>1000 copies/ml).
3.3. Analysis.
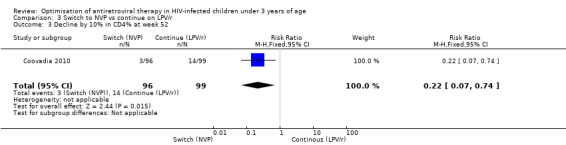
Comparison 3 Switch to NVP vs continue on LPV/r, Outcome 3 Decline by 10% in CD4% at week 52.
3.4. Analysis.
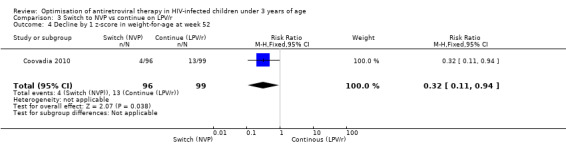
Comparison 3 Switch to NVP vs continue on LPV/r, Outcome 4 Decline by 1 z‐score in weight‐for‐age at week 52.
3.5. Analysis.
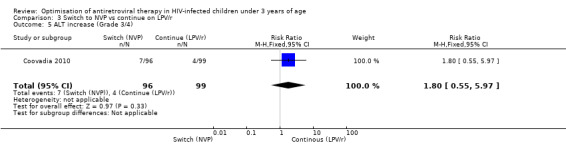
Comparison 3 Switch to NVP vs continue on LPV/r, Outcome 5 ALT increase (Grade 3/4).
3.6. Analysis.
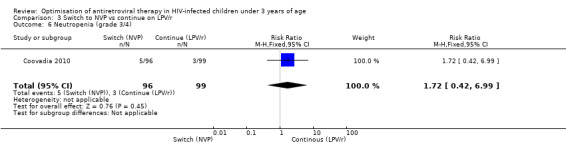
Comparison 3 Switch to NVP vs continue on LPV/r, Outcome 6 Neutropenia (grade 3/4).
Comparison 4. Induction‐Maintance strategy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Increase in CD4% from baseline at week 36 | 1 | 228 | Mean Difference (IV, Fixed, 95% CI) | 2.90 [0.80, 5.00] |
| 2 Increase in CD4% from baseline at week 72 | 1 | 225 | Mean Difference (IV, Fixed, 95% CI) | 0.60 [‐1.56, 2.76] |
| 3 Increase in CD4% from baseline at week 144 | 1 | 228 | Mean Difference (IV, Fixed, 95% CI) | 0.5 [‐1.70, 2.70] |
| 4 Virologic response (VL<400 copies/ml) at 24 weeks | 1 | 268 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.99 [1.09, 3.62] |
| 5 Virologic response (VL<400 copies/ml) at 36 weeks | 1 | 312 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.28 [0.73, 2.23] |
| 6 Virologic response (VL<400 copies/ml) at 48 weeks | 1 | 195 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.96 [0.50, 1.83] |
| 7 Virologic response (VL<400 copies/ml) at 144 weeks | 1 | 199 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.03 [0.51, 2.07] |
| 8 Mortality | 1 | Hazard Ratio (Random, 95% CI) | 0.71 [0.16, 3.21] | |
| 9 New WHO 4/death event‐free survival | 1 | Hazard Ratio (Fixed, 95% CI) | 0.73 [0.25, 2.12] | |
| 10 New WHO 3/4/death event‐free survival | 1 | Hazard Ratio (Fixed, 95% CI) | 0.98 [0.51, 1.90] |
4.1. Analysis.
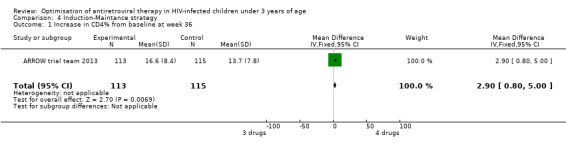
Comparison 4 Induction‐Maintance strategy, Outcome 1 Increase in CD4% from baseline at week 36.
4.2. Analysis.
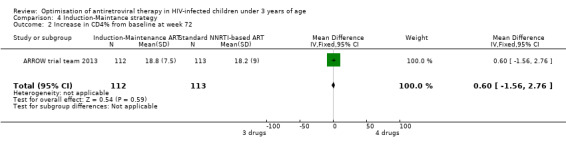
Comparison 4 Induction‐Maintance strategy, Outcome 2 Increase in CD4% from baseline at week 72.
4.3. Analysis.
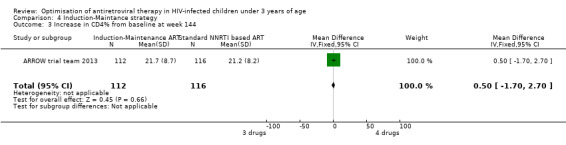
Comparison 4 Induction‐Maintance strategy, Outcome 3 Increase in CD4% from baseline at week 144.
4.4. Analysis.
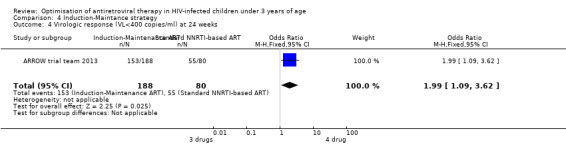
Comparison 4 Induction‐Maintance strategy, Outcome 4 Virologic response (VL<400 copies/ml) at 24 weeks.
4.5. Analysis.
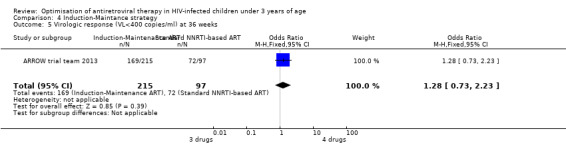
Comparison 4 Induction‐Maintance strategy, Outcome 5 Virologic response (VL<400 copies/ml) at 36 weeks.
4.6. Analysis.
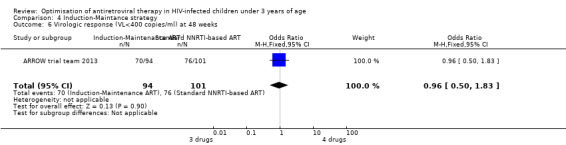
Comparison 4 Induction‐Maintance strategy, Outcome 6 Virologic response (VL<400 copies/ml) at 48 weeks.
4.7. Analysis.
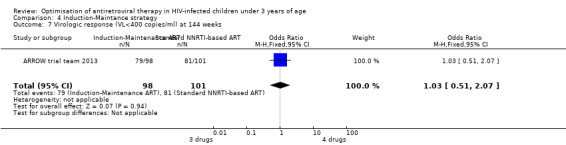
Comparison 4 Induction‐Maintance strategy, Outcome 7 Virologic response (VL<400 copies/ml) at 144 weeks.
4.8. Analysis.
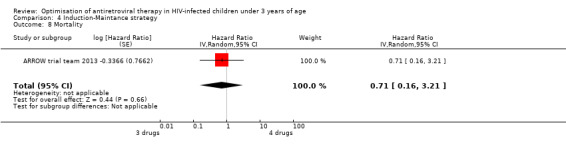
Comparison 4 Induction‐Maintance strategy, Outcome 8 Mortality.
4.9. Analysis.
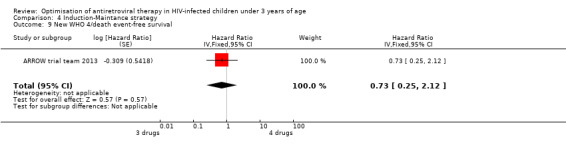
Comparison 4 Induction‐Maintance strategy, Outcome 9 New WHO 4/death event‐free survival.
4.10. Analysis.
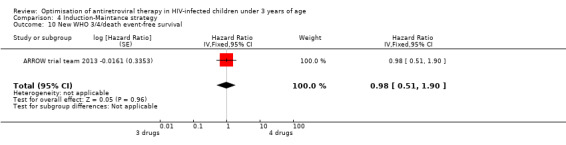
Comparison 4 Induction‐Maintance strategy, Outcome 10 New WHO 3/4/death event‐free survival.
Comparison 5. Planned treatment interruption strategy.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in WAZ z‐score | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.13 [‐0.31, 0.05] |
| 2 Change in HAZ z‐score | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.1 [‐0.33, 0.13] |
| 3 Change in WHZ z‐score | 1 | 31 | Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.32, 0.10] |
| 4 Serious adverse events | 1 | 42 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 17.12] |
5.1. Analysis.
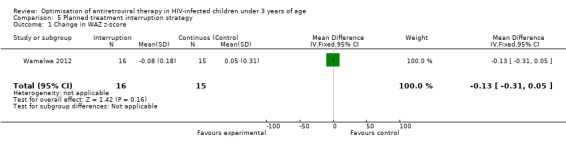
Comparison 5 Planned treatment interruption strategy, Outcome 1 Change in WAZ z‐score.
5.2. Analysis.
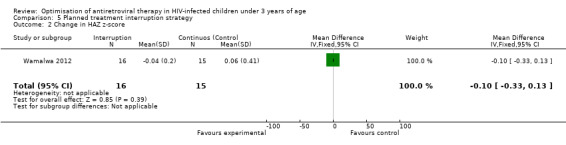
Comparison 5 Planned treatment interruption strategy, Outcome 2 Change in HAZ z‐score.
5.3. Analysis.
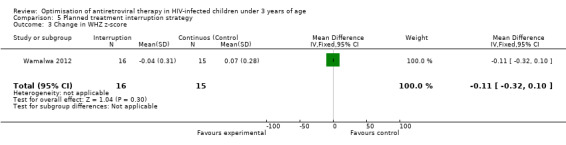
Comparison 5 Planned treatment interruption strategy, Outcome 3 Change in WHZ z‐score.
5.4. Analysis.
Comparison 5 Planned treatment interruption strategy, Outcome 4 Serious adverse events.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Achan 2012.
| Methods | Open label, single site, randomized clinical trial | |
| Participants | Participants were HIV infected children aged 2 months to 5 years eligible for ART or currently receiving NNRTI‐ based ART with virological suppression (HIV RNA <400 copies/mL). | |
| Interventions | Participants were randomized to receive either LPV/r‐based or NNRTI‐based ART and followed for 2 years. Control: NVP or EFV‐based regimen Intervention: LPV/r‐based regimen |
|
| Outcomes | Primary endpoint: Incidence‐density of malaria, defined as the number of incident episodes of malaria per time at risk. Secondary endpoints: ‐ Incidence of any adverse events, defined as severity grade 2 or higher that were possibly, probably or definitely related to study drugs ‐ Virologic efficacy of LPV/r versus NNRTI‐based ART in HIV‐infected children, with test for non‐inferiority in the proportion of children who achieve HIV viral RNA suppression at 48 weeks ‐ Immunologic efficacy of LPV/r versus NNRTI‐based ART, with test for non‐inferiority in the change from baseline CD4 cell count and % at 2 time points; 48 and 96 weeks. ‐ Association between nutritional status and HIV‐related outcomes, including ART levels. |
|
| Notes | Virologic and immunologic efficacy were predefined sub‐analyses but the trial was not design to compare the two ART regimens in terms of HIV disease outcomes. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Children were stratified by HIV treatment status (naive vs. on ART) to ensure balance between the intervention and control arms within these groups. To protect against temporal changes in either subject referral or study procedures, randomization will was blocked with a randomly permuted block size of 2 or 4 (from study protocol) |
| Allocation concealment (selection bias) | Low risk | Two sets of sealed sequentially numbered envelopes contained the treatment assignment and the assignment key was held in the central administrative data offices by a staff member not in contact with study subjects (from study protocol) |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None, but outcomes are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | None, but outcome measurements are unlikely to be biased by unmasking |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 2 children withdrew after randomisation/enrolment but before initiating study drug; 4 children prematurely stopped study arm drug (3 on NNRTI and 1 on PI). Noteworthy that 10 of the 12 premature withdrawals, including 3 of the 4 deaths, were from the NNRTI arm. While in no cases were withdrawals attributed to ART/NNRTI related reasons, this disproportionate loss from the NNRTI arm represents at least a potential for bias towards no significant differences in AE’s between the arms. ITT analysis. |
| Selective reporting (reporting bias) | Low risk | Study protocol available in the public domain, risk of selective reporting is very unlikely |
| Other bias | Unclear risk | The study was not designed or powered to compare treatment efficacy or to establish superiority of one of the two regimens. |
ARROW trial team 2013.
| Methods | Open‐label randomised trial | |
| Participants | 1206 symptomatic HIV infected infants and children in Uganda and Zimbabwe. Inclusion criteria: ART naive Children between 6 months to 17 years with confirmed, documented diagnosis of HIV‐1 infection and eligible for ART Parents or guardians, and children where appropriate according to age and knowledge of HIV status, must be willing and able to give informed consent for randomisation to clinically driven monitoring (CDM) or laboratory and clinical monitoring (LCM) and to first‐line ART strategy Exclusion criteria Children who were:unlikely to attend regularly; likely to have poor adherence; with acute infection; receiving chemotherapy for malignancy or receiving medication contraindicated by ART; with laboratory abnormalities, which are a contraindication for the patient to start ART; pregnant or breastfeeding. |
|
| Interventions | Two strategic approaches for management of antiretroviral therapy (ART): 1. Clinically driven monitoring (CDM) or laboratory plus clinical monitoring (LCM). 2. Continuous first‐line ART with three drug, two class regimen, comprising two NRTIs plus one NNRTI, or induction with four drugs (two classes) followed by maintenance with three drugs. |
|
| Outcomes | The primary endpoints were: 1. Monitoring practice (n = 1206): a. Efficacy: progression to a new WHO stage 4 event or death b. Safety: any adverse events of grade 3 or 4, which are not HIV‐related only 2. ART strategies for first‐line therapy (n=1206): a. Efficacy: Change in CD4 percentage at 72 and 144 weeks b. Safety: any adverse events of grade 3 or 4, which are not HIV‐related only |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation was stratified by centre, and by factorial randomisations within the overarching ARROW trial (CDM vs LCM, and first‐line ART induction/maintenance strategy). The computer‐generated sequentially numbered randomisation list (variable block sizes) was pre‐prepared by the Trial Statistician and incorporated within the secure database at each trial centre, connected to but not located within each clinical centre. |
| Allocation concealment (selection bias) | Low risk | Allocation was undertaken by clinicians phoning the local trials centre. Trial managers could access the next number but not the whole list. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None, but outcomes are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | None, but outcome measurements are unlikely to be biased by unmasking |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | "Of 1206 randomised, 28 withdrew or were lost to follow up during the whole follow up period (approximately 2.5% LTFU)" (from correspondence). ITT analysis. |
| Selective reporting (reporting bias) | Low risk | Study protocol available in the public domain, risk of selective reporting is very unlikely |
| Other bias | Low risk | |
Coovadia 2010.
| Methods | Randomized open label trial | |
| Participants | 323 children less than 2 years old previously exposed to NNRTI, median age 20 months (IQR 10‐36) in the control and 19 (IQR 9‐43) months in the switch group. Inclusion criteria: met clinical and immunological criteria for treatment initiation (WHO 2006), exposed to NVP and started on PI‐based regimen; achieved and sustained less than 400 copies/ml for at least 3 months within the first 12 months of treatment Exclusion criteria: children receiving tuberculosis treatment, having abnormalities in ALT greater than grade 2 (grading from Division of AIDS guidelines) and needing acute treatment for opportunistic infections (except TB) or tumours |
|
| Interventions | Control: continuous PI based ART Intervention: switch to NVP based ART |
|
| Outcomes | Mortality; Any viral load >50 copies/ml; Confirmed viral load >1000 copies/ml; Change in CD4%; Change in weight and height Z scores; Adverse events | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | quote:" Randomization was done in cohort blocks of variable size between 8 and 12. Allocations were generated by the study statistician" |
| Allocation concealment (selection bias) | Low risk | Quote from trial manuscript: "Allocations were concealed in opaque envelopes opened on site at the time of randomisation" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None, but outcomes are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | None, but outcome measurements are unlikely to be biased by unmasking |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | quote: "Modified intent‐to‐treat analyses were conducted excluding those children (n = 3) missing virologic outcome data" Missing data were reported in detail |
| Selective reporting (reporting bias) | Low risk | Study protocol available in the public domain, risk of selective reporting is very unlikely |
| Other bias | Low risk | |
Palumbo 2010.
| Methods | Phase II randomised clinical trial | |
| Participants | 123 children less than 36 months old Age:> 6 months to < 36 months (up to but not including the 3rd birthday) Inclusion criteria: If either the mother or the child had received NVP; subsequently amended to require specifically that the child had received nevirapine. Had to have diagnosis or AIDS by 60 days from birth and be formula fed from birth. Treatment eligible as defined by the WHO paediatric algorithm. HIV‐1 RNA >5,000 copies/mL within 60 days prior to study entry/randomisation Exclusion criteria: TB treatment or not met inclusion criteria |
|
| Interventions | NVP‐based vs LPV/r‐based first line antiretroviral therapy | |
| Outcomes | Treatment failure (virological failure or discontinuation for any cause included death); virological failure; change in CD4%; change in weight and height z scores; adverse events. | |
| Notes | Enrollment was terminated early on the recommendation of the data and safety monitoring board. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from the protocol: "dynamic permuted block system [stratified (<12months>)] " |
| Allocation concealment (selection bias) | Low risk | Quote from correspondence: "The Subject Registration and Randomization System provides a web‐based interface that leads site personnel through the checklist. If the subject satisfies the entry criteria, the system stores the subject's information in the central database and assigns a treatment based on a permuted block algorithm" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None (from correspondence), but outcomes are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | None (from correspondence), but outcome measurements are unlikely to be biased by unmasking |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT was performed and attrition bias is unlikely |
| Selective reporting (reporting bias) | Low risk | Protocol available, selective outcome reporting is unlikely |
| Other bias | Unclear risk | Early termination of the study: "Enrollment in this cohort was terminated early on the recommendation of the data and safety monitoring board." |
Prendergast 2008 (a).
| Methods | Pilot randomised controlled trial | |
| Participants | Age: 63 HIV infected infants from birth Inclusion criteria: born in one of the study hospitals and had confirmed intrauterine/intrapartum HIV infection and caregivers gave consent for enrolment. Exclusion criteria: gestational age less than 37 weeks, birth weight less than 2 kg, evidence of other congenital infections or severe abnormalities. |
|
| Interventions | Arm A: deferred ART, according to WHO 2003 guidelines Arm B: immediate ART given for 1 year, then stopped Arm C: immediate ART given with up to three structured treatment interruptions to 18 months of age, then stopped. |
|
| Outcomes | Mortality; morbidity; virological suppression; change in CD4% | |
| Notes | Data regarding stopping or interrupting strategy not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer generated sequence by trial statistician |
| Allocation concealment (selection bias) | Low risk | Quote from correspondence: " Opaque envelope at study sites were used for concealment allocation" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None, but outcomes are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | None, but outcome measurements are unlikely to be biased by unmasking |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No loss to follow up was reported. |
| Selective reporting (reporting bias) | Low risk | Major outcomes to address efficacy and safety of antiretroviral therapy were all reported, therefore risk of bias is unlikely. |
| Other bias | High risk | The trial was a feasibility study and was not designed or powered to assess difference in mortality between the two arms. |
Violari 2008.
| Methods | Phase 3 open‐label randomised control trial | |
| Participants | 377 infants 6 to 12 weeks of age who had HIV infection (DNA PCR positive; RNA above 1000 copies/ml) Inclusion criteria: CD4 percentage of 25% or more. Exclusion criteria: Presence of a severe CDC Stage B or Stage C disease, Grade 3 or 4 laboratory values for alanine aminotransferase (ALT) and aspartate aminotransferase (AST), absolute neutrophil count, haemoglobin, electrolytes, creatinine or clinical toxicity at screening, as defined by age appropriate toxicity tables; presence of any major congenital abnormalities that were life‐threatening and any acute and clinically significant medical event at randomisation; inability to tolerate oral medication; birth weight <2 kilograms; use of investigational drugs or any medications that are disallowed with protease inhibitors or non‐nucleoside reverse transcriptase inhibitors and; inability of parent or legal guardian to attend regularly scheduled study visits. |
|
| Interventions | Infants were randomly assigned to receive one of three treatments: early limited antiretroviral therapy for 96 weeks, early limited antiretroviral therapy for 40 weeks, or deferred therapy | |
| Outcomes | By week 24: Time to death or failure of the first‐line antiretroviral therapy; HIV disease progression; change in CD4% during follow up; grade 3 and 4 drug‐related events. | |
| Notes | After review by the data and safety monitoring board, the deferred‐therapy group was modified, and infants in this group were all reassessed for initiation of antiretroviral therapy. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The randomisation schedule was prepared centrally by the trial statistician and faxed to the study sites." |
| Allocation concealment (selection bias) | Low risk | Quote from correspondence: " prepared by the trial statistician and communicated by fax to the sites" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None, but outcomes are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "An independent endpoint review committee reviewed all deaths and CDC stage C and severe stage B events without knowledge of CD4 values, status of antiretroviral therapy, or randomised treatment assignments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Loss to follow up was 6% in the early treatment arm and 3% in deferred treatment arm. Intent to treat TT analysis was performed by censoring LTFU. |
| Selective reporting (reporting bias) | Low risk | Protocol available, selective outcome reporting is unlikely. |
| Other bias | Low risk | Early stop: "After a review by the data and safety monitoring board, the deferred‐therapy group was modified, and infants in this group were all reassessed for initiation of antiretroviral therapy". The guiding statistical criterion for “proof beyond reasonable doubt” was based on a difference of at least 3 SD in the log relative hazard (or nominal P<0.001) in any interim analysis (according to the Haybittle–Peto rule). |
Violari 2012.
| Methods | Phase II randomised clinical trial | |
| Participants | 288 children less than 36 months old Age: > 2 months to < 36 months (up to but not including the 3rd birthday) Inclusion criteria: HIV‐1 RNA >5,000 copies/mL within 60 days prior to study entry/randomisation. This can also be considered as the confirmatory HIV test in patients in whom only one positive HIV test is available at the time of screening, at sites where only one HIV PCR is normally performed for diagnosis of HIV infection. ARV naïve except for ART used in attempts to prevent intrapartum MTCT. (Infant ART use for < 1 week postpartum for prevention of MTCT is allowed.) Treatment eligible as defined by the WHO paediatric algorithm. Parent or legal guardian able and willing to provide signed informed consent and to have the subject followed at the clinical site. Maternal use of ARVs during pregnancy and/or during labor is permitted with the exception of NNRTIs. Documentation of lack of NVP exposure. Exclusion criteria: Grade >2 AST or ALT at screening. Any Grade >3 laboratory toxicity at screening. Receipt of ART other than for prevention of intrapartum MTCT. Infants who received ART past the first week of life (e.g. for prevention of breast milk transmission) were excluded from study entry. Acute, serious infections requiring active treatment (prophylaxis allowed [e.g. PCP, cryptococcal meningitis]). Subjects could be receiving treatment for active TB if this did not include rifamycin drugs, and with approval by the study chairs. Chemotherapy for active malignancy. History of cardiac conduction abnormality and underlying structural heart disease.Report of any maternal NVP or other NNRTI exposure prior to or during the pregnancy and during breastfeeding with this child, including single dose NVP, documented by either verbal report or through the clinic or hospital record (use of ART from the NRTI or PI classes was allowed). Report of infant NVP exposure at any time, including during the first week of life, documented by either verbal report or through the clinic or hospital record. |
|
| Interventions | NVP‐based vs LPV/r‐based first line antiretroviral therapy | |
| Outcomes | Treatment failure (virological failure or discontinuation for any cause included death) at 24 weeks; virological failure or death at 24 weeks; time to treatment failure (virological failure or discontinuation for any cause included death); time to virological failure or death; change in CD4%; change in weight and height z scores; adverse events | |
| Notes | In October 2010, the Data Safety Monitoring Board recommended un‐blinding the study results. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from the protocol:"dynamic permuted block system [stratified (<12months>)] " |
| Allocation concealment (selection bias) | Low risk | Quote from correspondence: " The Subject Registration and Randomization System provides a web‐based interface that leads site personnel through the checklist. If the subject satisfies the entry criteria, the system stores the subject's information in the central database and assigns a treatment based on a permuted block algorithm" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | None (from correspondence), but outcome measurements are unlikely to be biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | None (from correspondence), but outcome measurements are unlikely to be biased by unmasking |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT was performed and attrition bias is unlikely |
| Selective reporting (reporting bias) | Low risk | Protocol available, selective outcome reporting is unlikely |
| Other bias | Unclear risk | Early termination of the study:"In October 2010, the Data Safety Monitoring Board recommended un‐blinding the study results. " |
Wamalwa 2012.
| Methods | Open label randomised clinical trial. | |
| Participants | 150 infants who initiated HAART at <13 months of age Inclusion Criteria: A. Infants newly initiating HAART ‐ Less than 13 months of age ‐ HIV‐1 DNA detection with confirmation (positive on two HIV‐1 DNA filter paper tests) ‐ Caregiver of infant plans to reside in Nairobi for at least 3 years (reported by caregiver) ‐ Caregiver is able to provide sufficient location information B. Infants already receiving HAART ‐ Initiated HAART at <13 months of age ‐ Records confirming HIV positive status ‐ Documentation of CD4% and weight prior to HAART initiation ‐ Must be on 1st line drug regimen Eligibility for randomisation: ‐ Completed 24 months of treatment with HAART ‐ Normalized growth: weight for height z‐score > ‐0.5; Child's weight must be above the 5th weight‐for‐age percentile and the weight curve must not be flat or falling (i.e. cross 2 major percentile lines or more over the past 3 months) ‐ CD4% >= 25 ‐ Children who recently initiated or who require anti‐tuberculosis treatment at the time of randomisation were ineligible for randomisation. |
|
| Interventions | Infants will be treated with HAART regimen for 24 months after which those who have immune reconstitution and adequate growth (˜100) will be randomised to continuous versus deferred therapy. Clinical outcomes will be compared in these children to determine if interruption is a safe and beneficial strategy. | |
| Outcomes | Primary outcome: growth and severe adverse events Secondary outcome: Incidence of morbidities, specifically pneumonia, diarrhoea, and hospitalisation. CD4% and VL post‐randomisation |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote from the protocol: "Block randomization scheme with variable block sizes was generated using STATA 8.0 ralloc.ado v2.2.1. Treatments were allocated in 1:1 ratio. Blocks of variable sizes were generated in equal proportions. All the study investigators and staff will be blinded to the block number, block size and sequence in the block." |
| Allocation concealment (selection bias) | Low risk | Quote from the protocol: "The treatments will be assigned via pre‐prepared sealed, opaque envelopes and the envelopes will be ordered in the sequence of treatment assignments generated by the STATA code." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | None (by study design). Outcome measurements could be potentially biased by unmasking |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | (from correspondence) " Outcomes (virologic, immunologic) and adverse events (laboratory assays for signs of toxicity) were assessed by the lab without reference to treatment allocation.Growth measurements and morbidity assessments were performed in an unblinded fashion, standard protocols for measurement of growth parameters and for documentation of adverse events were employed for each arm. Analysis was blinded to arm allocation." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Attrition was similar in each arm and was due to similar reasons (2 participants withdrew post‐randomization,1 in each arm). ITT analysis was performed by censoring LTFU. |
| Selective reporting (reporting bias) | Low risk | Protocol available, endpoints have been reported as pre‐specified and selective outcome reporting is unlikely |
| Other bias | High risk | The sample size calculation did not account for either early restart or unplanned interruption. In addition, the sample size calculation was performed before the change in treatment guidelines to recommend initiation of ART for children with a CD4% less than 25% (increased from 20%). This change in treatment guidelines prompted a revision in criteria for restart of ART in the interrupted arm from 20% to 25%. DSMB recommended early termination of the study due to high proportion of early restart. |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Ananworanich 2008 | Children younger than 1 year were excluded from the study. |
| Cotton 2012 | The trial did not include an arm that received continuous ART from birth |
| King 2005 | Children included in the study were not treatment naive. |
| Krogstad 2002 | Children included in the study were not treatment naive. |
| Lockman 2007 | No comparison between NNRTI‐based and PI‐based was provided. |
| Luzuriaga 2004 | This trial was not randomised. |
| NCT00427297 | This trial was interrupted for lack of equipoise. |
| PENPACT1 study team | Children less than 2 years old were poorly represented and no stratification by age was provided for the measure of effect. |
| Prendergast 2008 (b) | The trial did not include an arm that received continuous ART from birth |
| Puthanakit 2012 | Definition of "early" and "deferred" treatment did not match predefined inclusion criteria. |
| Wiznia 2000 | Children included in the study were not treatment naive. |
| Wongsawat 2010 | Addressing a different comparison: CD4 count versus CD4% as criteria to start treatment |
Characteristics of ongoing studies [ordered by study ID]
ANRS 12206‐ MONOD.
| Trial name or title | ANRS 12206 – MONOD: International phase 2b‐3 randomised clinical trial to assess two once‐daily simplified antiretroviral triple therapies among HIV‐infected children early treated by a 12‐month twice daily triple therapy between 6 weeks and 24 months of age and in virological success in Africa: the MONOD Project (Burkina Faso, Ivory Coast, Rwanda) |
| Methods | Open label randomised clinical trial |
| Participants | Inclusion Criteria for antiretroviral treatment initiation:
‐ infant follow‐up in one of the trial sites
‐ HIV‐1 infection diagnosed by RT PCR after 6 weeks of life
‐ age between 3 and 12 month at antiretroviral treatment initiation
‐ antiretroviral‐naive except if received for the prevention of mother to child HIV transmission
‐ HB>=7 g/dl, neutrophils>750/mm3, creatinine<3xULN, TGO and TGP<3xULN
‐ signed informed consent
Exclusion Criteria for antiretroviral treatment initiation:
‐ HIV‐2 infection or HIV‐1/HIV‐2 co‐infection
‐ Known intolerance to one of the trial treatments
‐ HB<7 g/dl, neutrophils<750/mm3, creatinine>3xULN, TGO or TGP>3xULN Inclusion Criteria for randomisation at 12 months in the simplification phase: ‐ age 24 months at most ‐ virological success, defined as 2 consecutive undetectable HIV RNA measured by RT PCR at least 3 months apart. Exclusion Criteria for randomisation at 12 months in the simplification phase: ‐ virological failure after the first 12 months of antiretroviral treatment |
| Interventions | Drug: AZT‐3TC‐LPV/r twice a day Drug: ABC‐3TC‐EFV once a day Drug: ABC‐3TC‐LPV/r once a day |
| Outcomes | Primary Outcome Measures: ‐ Virological success at 25 months (HIV RNA < 50 copies / mL) Secondary Outcome Measures: ‐ Virological success at 12 months (HIV RNA < 400 copies / mL) ‐ Immunological response at 12 and 25 months (CD4+ lymphocyte absolute count and percentage) ‐ Antiretroviral and cotrimoxazole pharmacokinetic parameters at 6, 19 and 25 months. ‐ Tolerance at 12 and 25 month (occurrence of grade 3 and 4 adverse events related to the trial treatment, particularly occurrence of immune reconstitution inflammatory syndrome) ‐ Adherence at 12 and 25 months ‐ Resistance to antiretroviral at 12 and 25 months |
| Starting date | June 2011 |
| Contact information | Dr. Valeriane Leroy Valeriane.leroy@isped.u‐bordeaux2.fr |
| Notes |
NCT01146873‐NEVEREST 3.
| Trial name or title | Treatment Options for Protease Inhibitor‐exposed Children (NEVEREST‐III) |
| Methods | Phase 3 Open label Randomized Efficacy Study (Factorial Assignment) |
| Participants | 400 HIV‐infected children Inclusion criteria: ‐3 to 5 years of age at time of screening for this trial if enrolled from outside or any age if enrolled from control arm of Neverest II. ‐Reliable history or documented exposure to NVP used as part of PMTCT ‐Initiated antiretroviral therapy with LPV/r at age less than 36 months ‐Receiving LPV/r‐based ART for at least 12 months ‐At least one viral load measurement less than 50 copies/ml conducted as part of screening for the study ‐ALT measurement grade I or less (DAIDS Toxicity Tables 2004) (Appendix A). These may be repeated until ALTs normalize if necessary. Exclusion criteria: ‐Prior treatment with any NNRTI drug as part of a therapeutic regimen ‐Substitution of other NRTI drugs (instead of 3TC and D4T which are the standard first line regimen) will be allowed. |
| Interventions | ‐ Active Comparator A: Group 1: Lopinavir/ritonavir (LPV/r)
Participants are assigned to remain on their current LPV/r‐based antiretroviral regimen
‐ Experimental A: Group 2: Efavirenz (EFV)
Participants are assigned to switch to an EFV‐based antiretroviral regimen ‐ Active Comparator B: Group D: D4T Children are assigned to remain on their current antiretroviral regimen, which includes D4T ‐ Experimental B: Group A: Abacavir (ABC) Children stop taking D4T and switch to ABC. |
| Outcomes | Primary Outcome Measures: ‐ Maintenance of viral suppression through 24 and 48 weeks post randomization (HIV RNA<50 copies/ml) ‐ Confirmed viral rebound through 24 weeks and 48 weeks post randomization (HIV RNA>1000 copies/ml) Secondary Outcome Measures: ‐ Magnitude of CD4 response, hospital admissions, new stage II or greater clinical conditions through 48 weeks ‐ ALT elevations, HDL, LDL, CRP, fat distribution through 48 weeks ‐ Drug related toxicities, including fat distribution and metabolic parameters, including when co‐treated for tuberculosis ‐ Adherence to medication through 48 weeks (assessed by pharmacy reconciliation of medications brought back) |
| Starting date | June 2010 (expected completion September 2014) |
| Contact information | Louise Kuhn lk24@columbia.edu |
| Notes |
Differences between protocol and review
Depending on the data available in the study report or provided by investigators the outcome definitions described in the protocol were slightly modified for analysis and pooling. When data on only one trial were available for a question, individual trial results were described. Update of this review required expansion of the inclusion criteria from 2 to 3 years old.
Contributions of authors
MP and AP were the lead authors. MP and AP scrutinised identified studies for eligibility, extracted data and assessed the methodological quality of included studies. MP performed the analysis. All authors critically reviewed the manuscript before submission.
Declarations of interest
AJP is a co‐investigator on the PEHSS and ARROW trials. EJA is a co‐investigator on the P1060 and NEVEREST trials.
Edited (conclusions changed)
References
References to studies included in this review
Achan 2012 {published and unpublished data}
- Achan j, Kahuru A, Ikilezi G, Ruel T, Clark T, Charlebois E, Rosenthal P, Dorsey G, Havlir D, Kamya M. Significant Reduction in Risk of Malaria among HIV+ Children Receiving Lopinavir/ritonavir‐based ART Compared to NNRTI‐based ART, a Randomized Open‐label Trial. March 2012; Vol. CROI.
ARROW trial team 2013 {published and unpublished data}
- ARROW Trial team. Routine versus clinically driven laboratory monitoring and first‐line antiretroviral therapy strategies in Africanchildren with HIV (ARROW): a 5‐year open‐label randomised factorial trial [Routine versus clinically driven laboratory monitoring and first‐line antiretroviral therapy strategies in Africanchildren with HIV (ARROW): a 5‐year open‐label randomised factorial trial]. Lancet. 2013 Mar 6;370(12):62198‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coovadia 2010 {published data only}
- Coovadia A, Abrams EJ, Stehlau R, Meyers T, Martens L, Sherman G, Hunt G, Hu CC, Tsai WY, Morris L, Kuhn L. Reuse of nevirapine in exposed HIV‐infected children after protease inhibitor‐based viral suppression: a randomised controlled trial. JAMA 2010;304(10):1082‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Palumbo 2010 {published and unpublished data}
- Palumbo P, Lindsey JC, Hughes MD, Cotton MF, Bobat R, Meyers T, Bwakura‐Dangarembizi M, Chi BH, Musoke P, Kamthunzi P, Schimana W, Purdue L, Eshleman SH, Abrams EJ, Millar L, Petzold E, Mofenson LM, Jean‐Philippe P, Violari A. Antiretroviral treatment for children with peripartum nevirapine exposure. N Engl J Med 2010;363(16):1510‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prendergast 2008 (a) {published and unpublished data}
- Prendergast A, Mphatswe W, Tudor‐Williams G, Rakgotho M, Pillay V, Thobakgale C, McCarthy N, Morris L, Walker BD, Goulder P. Early virological suppression with three‐class antiretroviral therapy in HIV‐infected AFrican infants. AIDS 2008;22(11):1333‐43. [DOI] [PubMed] [Google Scholar]
Violari 2008 {published data only}
- Violari A, Cotton MF, Gibb DM, Babiker AG, Steyn J, Madhi SA, Jean‐Philippe P, McIntyre JA. Early Antiretroviral Therapy and Mortality among HIV‐Infected Infants. N Engl J Med 2008;359:2233‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Violari 2012 {published and unpublished data}
- Violari A, Lindsey JC, Hughes MD, Mujuru HA, Barlow‐Mosha L, Kamthunzi P, Chi BH, Cotton MF, Moultrie H, Khadse S, Schimana W, Bobat R, Purdue L, Eshleman SH, Abrams EJ, Millar L, Petzold E, Mofenson LM, Jean‐Philippe P, Palumbo P. NVP‐ vs LPV/r‐based ART among HIV+ Infants in Resource‐limited Settings: The IMPAACT P1060 Trial [Nevirapine versus ritonavir‐boosted lopinavir for HIV‐infected children.]. N Engl J Med. 2012 Jun 21; Vol. 366, issue 25:2380‐9. [DOI] [PMC free article] [PubMed]
Wamalwa 2012 {published data only (unpublished sought but not used)}
- Wamalwa D, Benki‐Nugent S, Langat A, Tapia K, Ngugi E, Moraa H, Otieno V, Richardson B, Overbaugh J, John‐Stewart G. [Treatment Interruption in Infants following 24 Months of Empiric ART: Kenya]. 19th Conference on Retroviruses and Opportunistic Infections‐ Seattle, Washington State (US). March, 2012.
References to studies excluded from this review
Ananworanich 2008 {published data only}
- Ananworanich J, Kosalaraksa P, Siangphoe U, Engchanil C, Pancharoen C, Lumbiganon P, Intasan J, Apateerapong W, Chuenyam T, Ubolyam S, Bunupuradah T, Lange J, Cooper DA, Phanuphak P, HIV‐NAT 010 Study Team. A feasibility study of immediate versus deferred antiretroviral therapy in children with HIV infection. AIDS Res Ther 2008;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cotton 2012 {published data only}
- Cotton M, Violari A, Gibb G, Otwombe K, Josipovic D, Panchia R, Jean‐Philippe P, Handelsman E, McIntyre J, Babiker A and the CHER Team. Early ART followed by Interruption Is Safe and Is Associated with Better Outcomes than Deferred ART in HIV+ Infants: Final Results from the 6‐Year Randomized CHER Trial, South Africa. CROI 2012. March 2012; Vol. 28 LB.
King 2005 {published data only}
- King JR, Nachman S, Yogev R, Hodge J, Aldrovandi G, Hughes MD, Chen J, Wiznia A, Damle B, Acosta EP. Efficacy, tolerability and pharmacokinetics of two nelfinavir‐based regimens in human immunodeficiency virus‐infected children and adolescents: pediatric AIDS clinical trials group protocol 403. Pediatr Infect Dis J 2005;24(10):880‐5. [DOI] [PubMed] [Google Scholar]
Krogstad 2002 {published data only}
- Krogstad P, Lee S, Johnson G, Stanley K, McNamara J, Moye J, Jackson JB, Aguayo R, Dieudonne A, Khoury M, Mendez H, Nachman S, Wiznia A, Pediatric AIDS Clinical Trials Group 377 Study Team. Nucleoside‐analogue reverse‐transcriptase inhibitors plus nevirapine, nelfinavir, or ritonavir for pretreated children infected with human immunodeficiency virus type 1. Clin Infect Dis 2002;34(7):991‐1001. [DOI] [PubMed] [Google Scholar]
Lockman 2007 {published data only}
- Lockman S, Shapiro RL, Smeaton LM, Wester C, Thior I, Stevens L, Chand F, Makhema J, Moffat C, Asmelash A, Ndase P, Arimi P, Widenfelt E, Mazhani L, Novitsky V, Lagakos S, Essex M. Response to antiretroviral therapy after a single, peripartum dose of nevirapine. N Engl J Med 2007;356(2):135‐47. [DOI] [PubMed] [Google Scholar]
Luzuriaga 2004 {published data only}
- Luzuriaga K, McManus M, Mofenson L, Britto P, Graham B, Sullivan JL, PACTG 356 Investigators. A trial of three antiretroviral regimens in HIV‐1‐infected children. N Engl J Med 2004;350(24):2471‐80. [DOI] [PubMed] [Google Scholar]
NCT00427297 {unpublished data only}
- John‐Stewart G. Optimizing Pediatric HIV‐1 Treatment in Infants With Prophylactic Exposure to Nevirapine, Nairobi, Kenya. Clinicaltrials.gov NCT00427297.
PENPACT1 study team {published data only (unpublished sought but not used)}
- The PENPACT‐1 (PENTA 9/PACTG 390) Study Team. First‐line antiretroviral therapy with a protease inhibitor versus non‐nucleoside reverse transcriptase inhibitor and switch at higher versus low viral load in HIV‐infected children: an open‐label, randomised phase 2/3 trial. The Lancet Infectious Diseases 2011;11(4):273‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prendergast 2008 (b) {published data only}
- Prendergast A, Chonco F, Tudor‐Williams G, Mphatswe W, Cengimbo A, Thobakgale C, Dong C, Coovadia H, Walker B, Goulder P. Randomized controlled trial of 3 approaches to management of HIV‐infected infants [Randomized controlled trial of 3 approaches to management of HIV‐infected infants]. 16th Conferences on Retroviruses and Opportunistic Infections, Boston, Massachusetts, USA Canada 3‐6 February 2008;Abstract 77LB. [Google Scholar]
Puthanakit 2012 {published data only}
- Puthanakit T, Saphonn V, Ananworanich J, Kosalaraksa P, Hansudewechakul R, Vibol U, Kerr SJ, Kanjanavanit S, Ngampiyaskul C, Wongsawat J, Luesomboon W, Ngo‐Giang‐Huong N, Chettra K, Cheunyam T, Suwarnlerk T, Ubolyam S, Shearer WT, Paul R, Mofenson LM, Fox L, Law MG, Cooper DA, Phanuphak P, Chhi Vun M, Ruxrungtham K, on behalf of the PREDICT Study Group. Early versus deferred antiretroviral therapy for children older than 1 year infected with HIV (PREDICT): a multicentre,randomised, open‐label trial. The Lancet ID October 9, 2012;S1473‐3099(12):70242‐6. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiznia 2000 {published data only}
- Wiznia A, Stanley K, Krogstad P, Johnson G, Lee S, McNamara J, Moye J, Jackson JB, Mendez H, Aguayo R, Dieudonne A, Kovacs A, Bamji M, Abrams E, Rana S, Sever J, Nachman S. Combination nucleoside analogue reverse transcriptase inhibitor(s) plus nevirapine, nelfinavir, or ritonavir in stable antiretroviral therapy‐experienced HIV‐infected children: week 24 results of a randomized controlled trial‐‐PACTG 377. Pediatric AIDS Clinical Trials Group 377 Study Team. AIDS Res Hum Retroviruses 2000;16(12):1113‐21. [DOI] [PubMed] [Google Scholar]
Wongsawat 2010 {published data only}
- Wongsawat J, Puthanakit T, Kanjanavanit S, Hansudewechakul R, Ngampiyaskul C, Kerr SJ, Ubolyam S, Suwanlerk T, Kosalaraksa P, Luesomboon W, Ngo‐Giang‐Huong N, Chandara M, Saphonn V, Ruxrungtham K, Ananworanich J, PREDICT Study Group. CD4 cell count criteria to determine when to initiate antiretroviral therapy in human immunodeficiency virus‐infected children. Pediatr Infect Dis J 2010;29(10):966‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to ongoing studies
ANRS 12206‐ MONOD {published data only}
- Leroy V, Timite‐Konan M, Mutabazi V, Meda N. International phase 2b‐3 randomized clinical trial to assess two once‐daily simplified antiretroviraltriple therapies among HIV‐infected children early treated by a 12‐month twice daily triple therapybetween 6 weeks and 24 months of age and in virological success in Africa:the MONOD Project(Burkina Faso, Ivory Coast, Rwanda). clinicaltrial.gov NCT01127204. [Google Scholar]
NCT01146873‐NEVEREST 3 {unpublished data only}
- Treatment Options for Protease Inhibitor‐exposed Children (NEVEREST‐III). Ongoing study June 2010 (expected completion September 2014).
Additional references
3Cs4kids 2008
- Cross Continents Collaboration for Kids (3Cs4kids) Analysis and Writing Committee. Epidemiology and Social Markers for predicting mortality in untreated HIV‐infected children in resource‐limited settings: a meta‐analysis. AIDS 2 January 2008;22(1):97‐105. [http://journals.lww.com/aidsonline/Fulltext/2008/01020/Markers_for_predicting_mortality_in_untreated.12.aspx] [DOI] [PubMed] [Google Scholar]
Apollo 2013
- Apollo T, Zinyowera S, Dzangare J, Chakanyuka C, Mugurungi O, Penazzato M, Martin E, Jordan M, Perriens J, Bertagnolio S. [World Health Organization HIV Drug Resistance surveillance in children less than 18 months newly diagnosed with HIV in Zimbabwe]. 7th IAS Conference in HIV pathogenesis. treatment and prevention. Luala Lumpur, Malaysia. 30 June.
Arpadi 2013
- Arpadi S, Shiau S, Strehlau R, Martens L, Patel F, Coovadia A, Abrams EJ, Kuhn L. Metabolic abnormalities and body composition of HIV‐infected children on Lopinavir or Nevirapine‐based antiretroviral therapy. The Pediatric Infectious Disease Journal 2013;32(1):39‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
Arrive 2007
- Arrivé E, Newell ML, Ekouevi DK, Chaix ML, Thiebaut R, Masquelier B, Leroy V, Perre PV, Rouzioux C, Dabis F, Ghent Group on HIV in Women and Children. Prevalence of resistance to nevirapine in mothers and children after single‐dose exposure to prevent vertical transmission of HIV‐1: a meta‐analysis.. Int J Epidemiol 2007 Oct;36(5):1009‐21. [http://www.ncbi.nlm.nih.gov/pubmed/17533166?itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum&ordinalpos=4] [DOI] [PubMed] [Google Scholar]
Chatterjee 2010
- Chatterjee A, Tripathi S, Gass R, Hamunime N, Panha S, Kiyaga C, et al. Implementing services for Early Infant Diagnosis (EID) of HIV: a comparative descriptive analysis of national programs in four countries. BMC Public Health 2010;533(11):2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
Church 2009
- Church JD, Mwatha A, Bagenda D, Omer SB, Donnell D, Musoke P, Nakabiito C, Eure C, Bakaki P, Matovu F, Thigpen MC, Guay LA, McConnell M, Fowler MG, Jackson JB, Eshleman SH. In utero HIV infection is associated with an increased risk of nevirapine resistance in ugandan infants who were exposed to perinatal single dose nevirapine.. AIDS Res Hum Retroviruses 2009 Jul;25(7):673‐7.. [http://www.ncbi.nlm.nih.gov/pubmed/19552593?itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum&ordinalpos=3] [DOI] [PMC free article] [PubMed] [Google Scholar]
DHHS 2011
- Department of Health and Human Services. Antiretroviral Therapy and Medical Management of HIV‐ Infected Children. 2011.
Dunn 2008
- Dunn D, Woodburn P, Duong T, Peto J, Phillips A, Gibb D, Porter K, HIV Paediatric Prognostic Markers Collaborative Study (HPPMCS), Concerted Action on Sero‐Conversion to AIDS and Death in Europe (CASCADE) Collaboration. Current CD4 cell count and the short‐term risk of AIDS and death before the availability of effective antiretroviral therapy in HIV‐infected children and adults. [Current CD4 cell count and the short‐term risk of AIDS and death before the availability of effective antiretroviral therapy in HIV‐infected children and adults.]. J Infect Dis 2008 Feb 1;197(3):398‐404. [http://www.ncbi.nlm.nih.gov/pubmed/18248303?ordinalpos=19&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum] [DOI] [PubMed] [Google Scholar]
EPPICC 2011
- The European Pregnancy and Paediatric HIV Cohort Collaboration (EPPICC) study group in EuroCoord. Early antiretroviral therapy in HIV‐1‐infected infants, 1996‐2008: treatment response and duration of first‐line regimens. AIDS 2011 Nov 28;25(18):2279‐2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ferrand 2009
- Ferrand RA, Corbett EL, Wood R, et al. AIDS among older children and adolescents inSouthern Africa: projecting the time course and magnitude of the epidemic.. AIDS. 2009 Sep 24;23(15):2039‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fillekes 2013
Gortmaker 2001
- Gortmaker SL, Hughes M, Cervia J, Brady M, Johnson GM, Seage GR 3rd, Song LY, Dankner WM, Oleske JM, Pediatric AIDS Clinical Trials Group Protocol 219 Team. Effect of combination therapy including protease inhibitors on mortality among children and adolescents infected with HIV‐1. N Engl J Med. 2001 Nov 22;345(21):1522‐8. [DOI] [PubMed] [Google Scholar]
Griffiths 1976
- Griffiths R. The Abilities of Babies. University of London Press; A study in mental measurement 1976:pp. 5–44. [Google Scholar]
Gupta 2012
- Gupta RK, Jordan MR, Sultan BJ, Hill A, Davis DH, Gregson J, Sawyer AW, Hamers RL, Ndembi N, Pillay D, Bertagnolio S. Global trends in antiretroviral resistance in treatment‐naive individuals with HIV after rollout of antiretroviral treatment in resource‐limited settings: a global collaborative study and meta‐regression analysis. Lancet 2012 Oct 6;380(9849):1250‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins J, Green S. Cochrane handbook for systematic reviews of interventions, version 5.1.0. March 2011. [Google Scholar]
HPPMCS 2003
- Dunn D, HIV Paediatric Prognostic Markers Collaborative Study Group. Short‐term risk of disease progression in HIV‐1‐infected children receiving no antiretroviral therapy or zidovudine monotherapy: a meta‐analysis. [Short‐term risk of disease progression in HIV‐1‐infected children receiving no antiretroviral therapy or zidovudine monotherapy: a meta‐analysis.]. Lancet Nov 15;362(9396):1605‐11.. [http://www.ncbi.nlm.nih.gov/pubmed/14630440] [DOI] [PubMed] [Google Scholar]
Hunt 2011
- Hunt GM, Coovadia A, Abrams EJ, Sherman G, Meyers T, Morris L, Kuhn L. HIV‐1 drug resistance at antiretroviral treatment initiation in children previously exposed to single‐dose nevirapine. AIDS 2011 Jul 31;25(12):1461‐1469. [http://www.ncbi.nlm.nih.gov/pubmed/21633285] [DOI] [PMC free article] [PubMed] [Google Scholar]
Italian Register 1999
- The Italian Register for HIV Infection in Children. Rapid disease progression in HIV‐1 perinatally infected children born to mothers receiving zidovudine monotherapy during pregnancy.. AIDS 1999;13(8):927‐33. [PubMed] [Google Scholar]
Jaquet 2000
- Jaquet D, Lévine M, Ortega‐Rodriguez E, Faye A, Polak M, Vilmer E, Lévy‐Marchal C. Clinical and metabolic presentation of the lipodystrophic syndrome in HIV‐infected children. AIDS 2000 Sep 29;14(14):2123‐8.. [http://www.ncbi.nlm.nih.gov/pubmed?term=jaquet%202000%20HIV] [DOI] [PubMed] [Google Scholar]
Karchava 2006
- Karchava M, Pulver W, Smith L, Philpott S, Sullivan TJ, Wethers J, Parker MM.SourceWadsworth Center, New York State Department of Health, Albany, NY 12201‐2002, USA. Prevalence of drug‐resistance mutations and non‐subtype B strains among HIV‐infected infants from New York State. J Acquir Immune Defic Syndr 2006 Aug 15;42(5):614‐9. [http://www.ncbi.nlm.nih.gov/pubmed/16868498?ordinalpos=3&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum] [DOI] [PMC free article] [PubMed] [Google Scholar]
Kinabo 2013
- Kinabo GD, Sprengers M, Msuya LJ, et al. Prevalence of Lipodystrophy in HIV‐Infected Children in Tanzania on HAART. The Pediatric Infectious Disease Journal 2013;32:39‐44. [DOI] [PubMed] [Google Scholar]
Kuhn 2012
- Kuhn L, Coovadia A, Strehlau R, Meyers T, Martens L, Sherman G, Hunt G, Tsai W‐T, Morris L, Ambrams E. Long‐term Outcomes of Switching Children to NVP‐based Therapy after Initial Suppression with a PI‐based Regimen. [DOI] [PMC free article] [PubMed]
Kuhn 2013
- Kuhn L, Hunt G, Technau K, Coovadia A, Black V, Morris L, Abrams E. and Finding Infants with HIV Disease and Evaluating Resistance Study Group. [Pre‐treatment Drug Resistance Mutations among HIV+ Children <2 Years of Age Who Failed or Missed PMTCT: Johannesburg, South Africa]. 20th Conference on Retroviruses and Opportunistic Infections‐ Atlanta, Georgia. 3‐6 March 2013; Vol. Abstract 951.
Laughton 2012
- Laughton B, Cornell M, Grove D, Kidd M, Springer PE, Dobbels E, Rensburg AJ, Violari A, Babiker AG, Madhi SA, Jean‐Philippe P, Gibb DM, Cotton MF. Early antiretroviral therapy improves neurodevelopmental outcomes in infants. AIDS 2012 Aug 24;24(13):1685‐90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lewis 2012
- Lewis J, Walker AS, Castro H, Rossi A, Gibb DM, Giaquinto C, Klein N, Callard R. Age and CD4 count at initiation of antiretroviral therapy in HIV‐infected children: effects on long‐term T‐cell reconstitution.. J Infect Dis 2012 Feb 15;205(4):548‐56.. [DOI] [PubMed] [Google Scholar]
Lindsey 2012
- Lindsey J, Hughes M, A Violari, Eshleman S, Abrams E, Mofenson L, Jean‐Philippe P, Palumbo P and P1060 Study Team . Predictors of Virologic and Clinical Response to Nevirapine‐ vs Lopinavir/ritonavir‐based ART in Infants and Children with and without Prior Nevirapine Exposure for the PMTCT. CROI 2012. March 2012; Vol. paper 25.
Lockman 2007
- Lockman S, Shapiro RL, Smeaton LM, Wester C, Thior I, Stevens L, Chand F, Makhema J, Moffat C, Asmelash A, Ndase P, Arimi P, Widenfelt E, Mazhani L, Novitsky V, Lagakos S, Essex M. Response to Antiretroviral Therapy after a Single, Peripartum Dose of Nevirapine. N Engl J Med January 11, 2007, (356):135‐147. [http://www.nejm.org/doi/full/10.1056/NEJMoa062876] [DOI] [PubMed] [Google Scholar]
Luiz 2001
- Luiz D, Foxcroft C, Stewart R. The construct validity of the Griffiths Scales of Infant Development.. Child: care health and development. 2001;27:73–83. [DOI] [PubMed] [Google Scholar]
Martinson 2007
- Martinson NA, Morris L, Gray G, Moodley D, Pillay V, Cohen S, Dhlamini P, Puren A, Bhayroo S, Steyn J, McIntyre JA. J Acquir Immune Defic Syndr. 2007 Feb 1;44(2):148‐53.Selection and persistence of viral resistance in HIV‐infected children after exposure to single‐dose nevirapine.. J Acquir Immune Defic Syndr 2007 Feb 1;44(2):148‐53. [http://www.ncbi.nlm.nih.gov/pubmed/17117145?itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum&ordinalpos=16] [DOI] [PubMed] [Google Scholar]
McComsey 2002
- McComsey G. Update on mitochondrial toxicity of antiretrovirals and its link to lipodystrophy.. AIDS Rev 2002 Jul‐Sep;4(3):140‐7. [http://www.ncbi.nlm.nih.gov/pubmed/12416448] [PubMed] [Google Scholar]
McIntosh 1996
- McIntosh K, Shevitz A, Zaknun D, Kornegay J, Chatis P, Karthas N, Burchett SK. .Age‐ and time‐related changes in extracellular viral load in children vertically infected by human immunodeficiency virus. Pediatr Infect Dis J 1996 Dec;15(12):1087‐91. [http://www.ncbi.nlm.nih.gov/pubmed/8970217?itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum&ordinalpos=91] [DOI] [PubMed] [Google Scholar]
Mphatswe 2007
- Mphatswe W, Blanckenberg N, Tudor‐Williams G, Prendergast A, Thobakgale C, Mkhwanazi N, McCarthy N, Walker BD, Kiepiela P, Goulder P. High frequency of rapid immunological progression in African infants infected in the era of perinatal HIV prophylaxis.. AIDS 2007;21(10):1253‐61. [DOI] [PubMed] [Google Scholar]
Mulenga 2010
- Mulenga V, Cook A, Walker AS, Kabamba D, Chijoka C, Ferrier A, Kalengo C, Kityo C, Kankasa C, Burger D, Thomason M, Chintu C, Gibb DM. Strategies for nevirapine initiation in HIV‐infected children taking pediatric fixed‐dose combination "baby pills" in Zambia: a randomized controlled trial.. Clin Infect Dis. 2010 Nov 1;51(9):1081‐9. [DOI] [PubMed] [Google Scholar]
Musiime 2009
- Musiime V, Ssali F, Kayiwa J, Namala W, Kizito H, Kityo C, Mugyenyi P. Response to nonnucleoside reverse transcriptase inhibitor‐based therapy in HIV‐infected children with perinatal exposure to single‐dose nevirapine. AIDS Res Hum Retroviruses 2009 Oct;25(10):989‐96. [http://www.ncbi.nlm.nih.gov/pubmed/19778270] [DOI] [PubMed] [Google Scholar]
Newell 2004
- Newell ML, Coovadia H, Cortina‐Borja M, Rollins N, Gaillard P, Dabis F, Ghent International AIDS Society (IAS) Working Group on HIV Infection in Women and Children. Mortality of infected and uninfected infants born to HIV‐infected mothers in Africa: a pooled analysis.. Lancet 2004 Oct 2‐8;364(9441):1236‐43. [DOI] [PubMed] [Google Scholar]
NIAID 2009
- United States National Institute of Allergy and Infectious Diseases (NIAID). Division of AIDS, Table for Grading Adult and Pediatric Adverse Events (Toxicity Table),dated December 2004 (Clarification August 2009) [Available from: hiv.cochrane.org/sites/hiv.cochrane.org/files/uploads/Table_for_Grading_Severity_of_Adult_Pediatric_Adverse_Events.pdf].
PENTA 11
- Paediatric European Network for Treatment of AIDS. Response to planned treatment interruptions in HIV infection varies across childhood. AIDS 2010 Jan 16;24(2):231‐41. [DOI] [PubMed] [Google Scholar]
PENTA 2002
- Paediatric European Network for Treatment of AIDS (PENTA). Comparison of dual nucleoside‐analogue reverse‐transcriptase inhibitor regimens with and without nelfinavir in children with HIV‐1 who have not previously been treated: the PENTA 5 randomised trial [Comparison of dual nucleoside‐analogue reverse‐transcriptase inhibitor regimens with and without nelfinavir in children with HIV‐1 who have not previously been treated: the PENTA 5 randomised trial]. Lancet 2002 Mar;2(359(9308)):733‐40. [DOI] [PubMed] [Google Scholar]
PENTA 2009
- PENTA Steering Committee. PENTA 2009 guidelines for the use of antiretroviral therapy in paediatric HIV‐1 infection.. HIV Medicine 2009;10(10):591‐613. [DOI] [PubMed] [Google Scholar]
Persaud 2007
- Persaud D, Palumbo P, Ziemniak C, Chen J, Ray SC, Hughes M, Havens P, Purswani M, Gaur AH, Chadwick EG, Pediatric AIDS Clinical Trials Group P1030 Team. Early archiving and predominance of nonnucleoside reverse transcriptase inhibitor‐resistant HIV‐1 among recently infected infants born in the United States.. J Infect Dis 2007 May 15;195(10):1402‐10.. [http://www.ncbi.nlm.nih.gov/pubmed/17436219?ordinalpos=2&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum] [DOI] [PubMed] [Google Scholar]
Persaud 2013
- Persaud D, Gay H, Ziemniak C, Chen YH, Piatak M, Chun TW, Strain M, Richman D, Luzuriaga K. Absence of Detectable HIV‐1 Viremia after Treatment Cessation in an Infant.. N Engl J Med.. 2013 Oct 23.. [DOI] [PMC free article] [PubMed]
Pillay 2008
- Pillay V, Ledwaba J, Hunt G, Rakgotho M, Singh B, Makubalo L, Bennett DE, Puren A, Morris L. Antiretroviral drug resistance surveillance among drug‐naive HIV‐1‐infected individuals in Gauteng Province, South Africa in 2002 and 2004. Antivir Ther. ; 2008;13(Suppl 2):101‐7. [http://www.ncbi.nlm.nih.gov/pubmed/18575198] [PubMed] [Google Scholar]
Piloya 2012
- Piloya T, Bakeera‐Kitaka S, Kekitiinwa A, et al. Lipodystrophy among HIV‐infected children and adolescents on highly active antiretroviral therapy in Uganda: a cross sectional study.. Journal of the International AIDS Society 2012;15(2):17427. [DOI] [PMC free article] [PubMed] [Google Scholar]
Prendergast 2012
- Prendergast AJ, Penazzato M, Cotton M, Musoke P, Mulenga V, Abrams EJ, Gibb DM. Treatment of young children with HIV infection: using evidence to inform policymakers [Treatment of young children with HIV infection: using evidence to inform policymakers]. PLoS Med 2012;9(7):e1001273. [DOI] [PMC free article] [PubMed] [Google Scholar]
Ruel 2013
- Ruel T, Kakuru A, Ikilezi G, Mwangwa F, Dorsey G, Rosenthal P, Charlebois E, Havlir D, Kamya M, Achan J. [Comparison of Virologic and Immunologic Outcomes between HIV+ Ugandan Children Randomized to Ritonavir‐boosted Lopinavir or NNRTI‐ based ART]. 20th Conference on Retroviruses and Opportunistic Infections‐ Atlanta, Georgia, USA.. 3‐6 March 2013; Vol. Abstract 88 LB.
UNAIDS 2012
- UNAIDS. UNAIDS Report on the global AIDS epidemIc 2012. http://www.unaids.org/en/resources/campaigns/20121120_globalreport2012/globalreport/ 2012. [Google Scholar]
Vigano 2003
- Viganò A, Mora S, Testolin C, Beccio S, Schneider L, Bricalli D, Vanzulli A, Manzoni P, Brambilla P. Increased lipodystrophy is associated with increased exposure to highly active antiretroviral therapy in HIV‐infected children. J Acquir Immune Defic Syndr 2003 Apr 15;32(5):482‐9. [http://www.ncbi.nlm.nih.gov/pubmed/12679698] [DOI] [PubMed] [Google Scholar]
Wade 1994
- Wade AM, Ades AE. Age‐related reference ranges: significance tests for models and confidence intervals for centiles. Stat Med 1994 Nov 30;13(22):2359‐67.. [http://www.ncbi.nlm.nih.gov/pubmed/7855469?itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_RVDocSum&ordinalpos=2] [DOI] [PubMed] [Google Scholar]
Walker 2004
- Walker AS, Doerholt K, Sharland M, Gibb DM, Collaborative HIV Paediatric Study (CHIPS) Steering Committee. Response to highly active antiretroviral therapy varies with age: the UK and Ireland Collaborative HIV Paediatric Study. AIDS 2004 Sep 24;18(14):1915‐24. [http://www.ncbi.nlm.nih.gov/pubmed/15353977?ordinalpos=27&itool=EntrezSystem2.PEntrez.Pubmed.Pubmed_ResultsPanel.Pubmed_DefaultReportPanel.Pubmed_RVDocSum] [DOI] [PubMed] [Google Scholar]
WHO 2010
- WHO. Antiretroviral therapy for HIV infection in infants and children: Recommendations for a public health approach (2010 revision). 2010; Vol. http://www.who.int/hiv/pub/guidelines/paediatric020907.pdf. [PubMed]
WHO 2011
- WHO 2011. GLOBAL HIV/AIDS RESPONSE. Epidemic update and health sector progress towards Universal Access. Progress Report 2011. [http://whqlibdoc.who.int/publications/2011/9789241502986_eng.pdf]
WHO 2013
- World Health Organisation. Consolidated Guidelines on the use of Antiretroviral drugs for treating and preventing HIVinfection. June 2013.
References to other published versions of this review
Penazzato 2012
- Penazzato M, Prendergast A, Tierney J, Cotton M, Gibb D. Effectiveness of antiretroviral therapy in HIV‐infected children under 2 years of age. Cochrane Database of Systematic Reviews 2012 Jul 11, Issue 7. [PUBMED: 22786492] [DOI] [PubMed] [Google Scholar]