

Abstract
While autophagy genes are required for lifespan of long-lived animals, their tissue-specific roles in aging remain unclear. Here, we inhibited autophagy genes in Caenorhabditis elegans neurons, and found that knockdown of early-acting autophagy genes, except atg-16.2, increased lifespan, and decreased neuronal PolyQ aggregates, independently of autophagosomal degradation. Neurons can secrete protein aggregates via vesicles called exophers. Inhibiting neuronal early-acting autophagy genes, except atg-16.2, increased exopher formation and exopher events extended lifespan, suggesting exophers promote organismal fitness. Lifespan extension, reduction in PolyQ aggregates and increase in exophers were absent in atg-16.2 null mutants, and restored by full-length ATG-16.2 expression in neurons, but not by ATG-16.2 lacking its WD40 domain, which mediates noncanonical functions in mammalian systems. We discovered a neuronal role for C. elegans ATG-16.2 and its WD40 domain in lifespan, proteostasis and exopher biogenesis. Our findings suggest noncanonical functions for select autophagy genes in both exopher formation and in aging.
Macroautophagy (hereafter autophagy) is an intracellular recycling process by which cytosolic cargo is subjected to lysosomal degradation, referred to here as canonical autophagy. Autophagy plays important roles in numerous late-onset diseases including neurodegenerative disorders and has been directly linked to aging in multiple model organisms including the nematode C. elegans1. In this organism, RNA interference (RNAi) of multiple autophagy (Atg) genes during adulthood abrogates lifespan extension in long-lived mutants, which together with data from other organisms suggest that autophagy is required for longevity1. In contrast, RNAi inhibition of autophagy genes in wild-type (WT) C. elegans typically has limited effects on lifespan1, indicating that basal autophagy is not restricting normal aging. Still, the tissue-specific contributions of autophagy to organismal fitness and longevity remain unclear. The role of autophagy genes in neurons is of special interest because neuronal signaling plays key roles in several longevity paradigms2. Moreover, neuronal overexpression of Atg1 or Atg8 extends Drosophila lifespan3,4, whereas loss of either the autophagy gene Atg5 or Atg7 specifically in neurons causes neurodegeneration in mice5,6. While RNAi inhibition of several autophagy genes similarly increases neuronal protein aggregation in C. elegans7–9, neuronal autophagy gene functions remain to be systemically investigated in C. elegans, where neurons are generally refractory to RNAi10–13.
Multiple conserved autophagy (ATG) proteins function in the autophagy process, which comprises at least five steps: (i) initiation; (ii) nucleation and formation of a double-membrane structure (phagophore); (iii) phagophore elongation and ATG8 conjugation, followed by the enclosure to form an autophagosome that incorporates cargo including aggregated proteins; (iv) autolysosome formation by fusion of the autophagosome with single-membrane, acidic lysosomes; and, finally, (v) cargo degradation in the autolysosome14. ATG proteins acting in the initial steps of forming, elongating and closing the phagophore (i–iii) are encoded by early-acting autophagy genes, whereas proteins functioning in autolysosome formation and cargo degradation (iv and v) are encoded by late-acting autophagy genes15. During the early steps of canonical autophagy, phagophore elongation requires the conjugation of ATG8 to phosphatidylethanolamine (PE) by the ATG5–ATG12/ATG16 complex at both the inner and outer membranes of the growing double-membrane phagophore16, making ATG8 proteins common reporters for autophagosomes in model systems, including C. elegans17,18.
In recent years, emerging studies in mammalian tissue cultures have shown that, in addition to their roles in canonical autophagy, some early-acting autophagy proteins can also carry out functions in other cellular processes, such as phagocytosis and secretion. In these noncanonical activities, ATG8 is conjugated to single-membrane vesicles by the ATG5–ATG12/ATG16 complex19–21. While it remains underexplored how the ATG5–ATG12/ATG16 complex differentiates between double-membrane and single-membrane vesicles, the WD40 domain in the C terminus of ATG16L1 is specifically required for its noncanonical function in ATG8/LC3-associated phagocytosis in murine macrophages20, highlighting this domain of ATG16L1 as a key molecular entity that can separate its canonical and noncanonical functions, at least in phagocytosis. While a recent study has indicated a role for the WD40 domain of ATG16L1 in preventing Alzheimer’s disease in mice22, noncanonical functions for autophagy proteins have primarily been studied in mammalian cell culture23, and the relevance of noncanonical autophagy activities has yet to be studied in the context of organismal aging.
To comprehensively investigate the role of autophagy proteins in the neurons of a live animal, we used RNAi to inhibit autophagy genes functioning in each step of the autophagy process in a new C. elegans strain with increased specificity to neuronal RNAi. We found that neuronal inhibition of early-acting, but not late-acting, autophagy genes extended lifespan upon both whole-life and adult-only inhibition. This lifespan extension was accompanied by an unexpected decrease in neuronal aggregates of proteins with expanded polyglutamine (polyQ) stretches, a model for Huntington’s disease and an increase in the secretion of vesicles called exophers24, a recently identified extrusion event in neurons and muscle of C. elegans24,25, as well as in mammalian cells26,27. Notably, animals that formed exophers early in life in mechanosensory touch neurons, were significantly longer lived, suggesting a link of this extrusion event to organismal fitness. Importantly, the lifespan, polyQ aggregation and exopher phenotypes were all atg-16.2 dependent and could be rescued by pan-neuronal expression of ATG-16.2, an ortholog of mammalian ATG16L1, provided the ATG-16.2 protein included an intact WD40 domain. Our studies thus discovered that ATG-16.2 and its highly conserved WD40 domain are required for the induction of exophers in neurons experiencing proteotoxic stress and for the observed positive effects on polyQ aggregation, exopher formation and lifespan extension upon early-acting autophagy gene inhibition. Collectively, our findings highlight the possibility that noncanonical functions of autophagy proteins may be critical for tissue and organismal fitness via alternative secretory pathways, which could be relevant for disposal of misfolded proteins.
Results
Neuronal inhibition of early Atg genes extends lifespan
C. elegans neurons are generally refractory to RNAi because they lack the RNA channel/transporter protein SID-1 required for RNA uptake10–13. Previous studies investigating autophagy (Atg) genes by systemic or whole-body RNAi approaches have therefore not fully assessed the role of autophagy genes in neurons. To address this limitation, we constructed a new C. elegans strain capable of RNAi specifically in neurons by expressing SID-1 under the pan-neuronal rgef-1 promoter in sid-1(qt9) loss-of-function mutants, which avoids some complications of the commonly used unc-119 promoter28, which is not fully neuronal specific29,30. We validated our neuronal-specific RNAi strain in multiple ways, including by showing that the rgef-1 promoter remained expressed in neurons into late adulthood (Extended Data Fig. 1a). Feeding bacteria expressing double-stranded RNA (dsRNA) against genes with nonneuronal functions did not elicit phenotypes in this strain (Extended Data Fig. 1b–f), while knockdown of neuronal fluorescent markers and genes led to a 40–50% reduction of fluorescence (Extended Data Fig. 1g,h) and close to 100% phenotype penetration (Extended Data Fig. 1i), respectively. Moreover, the strain generally displayed a normal lifespan (in 14 of 16 experiments testing multiple independent lines; Supplementary Table 1).
Next, we tested the effect of neuronal-only knockdown of autophagy genes on C. elegans lifespan, using RNAi clones that have previously been shown to shorten the lifespan of longevity mutants upon whole-body RNAi treatment1 (all of the analyzed genes are expressed in C. elegans neurons29). Strikingly, we observed an extended lifespan for neuronal-only inhibition, initiated either from hatching (that is, whole-life) or from the first day of reproductive adulthood (that is, day 1, or adult-only), of genes with functions early in the pathway (that is, unc-51/ULK/Atg1, atg-13, bec-1/BECN1/Beclin1, atg-9, atg-7, atg-4.1 and lgg-1/ATG8; Fig. 1a), whereas knockdown of late-acting autophagy genes (that is, cup-5/MCOLN, epg-5, vha-13 and vha-15) were incapable of doing so, and vha-16 inhibition even shortened lifespan (Fig. 1b and Supplementary Tables 2 and 3). Interestingly, neuronal-only RNAi knockdown of early-acting gene atg-16.2 behaved differently with no lifespan extension in six experiments (Fig. 1b,c and Supplementary Table 3), making atg-16.2 inhibition an exception to other early-acting autophagy genes, which all showed beneficial effects following neuronal-only knockdown. These findings were particularly interesting considering a recent report in which neuronal inhibition of bec-1/BECN1 (using a sid-1(pk3321); unc-119p::sid-1 transgenic strain28) caused extended lifespan in aged, post-reproductive animals, whereas no lifespan extension was observed when young, reproductively active animals were subjected to neuronal bec-1/BECN1 RNAi31. This observation, along with the finding that systemic RNAi of several other early-acting autophagy genes could extend lifespan when initiated late in life, led the authors to hypothesize that autophagy gene function is subject to antagonistic pleiotropy in a neuronal-specific manner31. Our data contradict this hypothesis, because we show that neuronal-only inhibition of multiple early-acting autophagy genes, including bec-1/BECN1, extended lifespan even when inhibited from hatching (used throughout the rest of this study).
Fig. 1 |. With the exception of atg-16.2, neuronal inhibition of early-acting autophagy genes extends lifespan.

a, Schematic showing early-acting and late-acting genes in the autophagy process investigated in this study (see the introduction for additional information). Created using BioRender.com. b, Average mean lifespan change (% MLS change, indicated) in sid-1; rgef-1p::sid-1 animals after RNAi of the listed autophagy-related genes compared to control. All lifespan data were pooled irrespective of whole-life or adult-only RNAi initiation. Error bars indicate the s.d. P values: not significant (NS) P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, by a two-sided, one-sample t-test compared to hypothetical mean of 0. See Supplementary Table 2 for n, all P values and statistical details and Supplementary Table 3 for details of individual lifespan experiments. Shading of atg-16.2 emphasizes this RNAi treatment as an exception for lifespan extension by early-acting autophagy genes. c, Lifespan analyses of sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after whole-life atg-16.2, atg-7 or lgg-1/ATG8 RNAi compared to control (CTRL). Statistical significance was determined by two-sided log-rank test, NS, P = 0.07, ****P < 0.0001. See Supplementary Table 3 for details and repeats.
We next used atg-7 and lgg-1/ATG8 RNAi clones, two representative, early-acting autophagy gene RNAi clones with highly reproducible phenotypes, to test the effects of neuronal-only knockdown on healthspan measures, and found that these animals displayed unchanged swimming ability, pharyngeal pumping and progeny production (Extended Data Fig. 2a–c). Because defects in ciliated sensory neurons increase lifespan in C. elegans32, we also tested if neuronal-only RNAi against atg-7 or lgg-1/ATG8 affected the morphology of ciliated sensory neurons in dye-filling assays. Animals subjected to RNAi displayed normal dye-filling phenotypes compared to control animals on day 5 of adulthood (Extended Data Fig. 2d), indicating that defects in ciliated sensory neurons are not responsible for lifespan extension after neuronal-only knockdown of atg-7 or lgg-1/ATG8. To further test whether neuronal autophagy gene reduction affected neuronal physiology, we counted neuronal branches in mechanoreceptor neurons, such as the anterior and posterior lateral microtubule cells ALML/R, and PLML/R, the anterior and posterior ventral microtubule cells AVM and PVM, at an older age—a phenotype associated with neuronal dysfunction33,34. Animals treated with empty vector control showed branches originating from ALM and PLM mechanoreceptor neurons at old age (day 15)33,34, but not animals subjected to neuronal-only RNAi against atg-7 or lgg-1/ATG8 (Extended Data Fig. 2e), suggesting that inhibition of early-autophagy genes improves age-related decline in neuronal morphology. Finally, we tested the ability of animals to move toward a chemical attractant in chemotaxis assays and found that 5-day-old animals subjected to neuronal-only RNAi against atg-7 or lgg-1/ATG8 had improved chemotaxis to the attractant butanone (Extended Data Fig. 2f), suggesting improved neuronal function.
Taken together, we observed that neuronal-only knockdown of an extensive panel of early-acting autophagy genes, except atg-16.2, extended lifespan. The benefits induced by neuronal-only inhibition of at least atg-7 and lgg-1/ATG8 came with no notable effects on muscle-related healthspan measures, progeny production but improved neuronal morphology and function. These results demonstrate a differential role for early-acting versus late-acting autophagy genes in C. elegans neurons, and the differential effect indicates that the observed lifespan extension associated with their knockdown may not be a result of impairing lysosomal degradation per se.
Neuronal inhibition of Atg genes impairs autophagy
Our neuronal-only RNAi strain displayed unchanged autophagy dynamics in neurons (Extended Data Fig. 3a,b), intestine and body-wall muscle (Extended Data Fig. 3c,d). We next used these reagents to assess autophagy status after neuronal inhibition of autophagy genes. We used animals stably expressing GFP::LGG-1/ATG8, commonly used for the assessment of pre-autophagosomal phagophores and autophagosomes35, from the pan-neuronal rgef-1 promotor36. We subjected these animals to neuronal-only RNAi of the panel of autophagy genes and found that RNAi clones for all early-acting autophagy genes, with the exception of bec-1/BECN1 and atg-16.2, significantly decreased neuronal GFP::LGG-1/ATG8 punctae counts (Fig. 2a), consistent with early-acting autophagy genes being important for the formation of autophagosomes31. Late-acting autophagy genes, important for autophagosome–lysosome fusion and lysosomal degradation, either increased or did not change neuronal GFP::LGG-1/ATG8 punctae numbers, as expected (Fig. 2a). We repeated this experiment in neuronal-only RNAi strains expressing GFP::LGG-1(Gly116Ala) in neurons9. GFP::LGG-1(Gly116Ala) is lipidation deficient and lacks Gly116, which is important for conjugation to autophagosome membranes; as a consequence, GFP::LGG-1 (Gly116Ala) should be evenly distributed in the cytosol9,37,38. However, and as reported by us earlier9, a low level of GFP-positive punctae was observed in neurons of GFP::LGG-1(Gly116Ala)-expressing animals (Fig. 2b). While the nature of these GFP::LGG-1(Gly116Ala) structures remains to be fully characterized, they may represent protein aggregates39. Interestingly, GFP::LGG-1(Gly116Ala) expressing animals subjected to neuronal-only inhibition of all early-acting autophagy genes, including bec-1/BECN1 and atg-16.2, along with two late-acting genes, displayed a significant increase in the numbers of neuronal GFP::LGG-1(Gly116Ala) punctae (Fig. 2b), consistent with an accumulation of protein aggregates from autophagy block. To further corroborate this, we additionally performed autophagy flux assays40 by injecting the late-stage autophagy inhibitor bafilomycin A1 (BafA; a compound that blocks acidification of lysosomes41). As predicted, BafA did not lead to an increase in GFP-positive punctae in WT animals expressing the lipidation-deficient LGG-1(Gly116Ala) reporter, consistent with GFP::LGG-1(Gly116Ala) punctae not being competent autophagosomes (Fig. 2c). While BafA injection in animals subjected to bacteria expressing empty vector control increased the number of neuronal GFP::LGG-1/ATG8 punctae, as a reflection of active autophagy38,42, the number of neuronal GFP::LGG-1/ATG8 punctae was not increased when BafA was injected into animals after neuronal-only reduction of atg-7 and lgg-1/ATG8 (Fig. 2d), consistent with a block in neuronal autophagy. The alterations we observed in autophagy markers appeared specific to neurons because we did not observe any changes in GFP::LGG-1/ATG8 punctae in the intestine or in body-wall muscle after neuronal-only inhibition of early-acting autophagy genes atg-7 and lgg-1/ATG8 (Extended Data Fig. 3e,f).
Fig. 2 |. Neuronal inhibition of all early-acting autophagy genes impairs autophagy.

a, Representative images of nerve-ring neurons and mean number of GFP::LGG-1/ATG8 punctae of day 1 sid-1; rgef-1p::sid-1; rgef-1::gfp::lgg-1 animals after whole-life autophagy gene RNAi. Error bars are s.d. NS P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, by one-way analysis of variance (ANOVA) with Dunnett’s multiple-comparison test. See Supplementary Table 2 for n, all P values and statistical details. The brightness of the image displaying lgg-1/ATG8 RNAi was artificially enhanced. Scale bar, 10 μm. b, Representative images of nerve-ring neurons and mean number of non-lipidated GFP::LGG-1/ATG8(Gly116Ala) punctae in day 1 sid-1; rgef-1p::sid-1; rgef-1::gfp::lgg-1(Gly116Ala) animals after whole-life autophagy gene RNAi. Error bars are s.d. P values as in a. See Supplementary Table 2 for all P values and statistical details. The brightness of the nerve-ring image after lgg-1/ATG8 RNAi is artificially enhanced. Scale bar, 10 μm. c, Mean number of GFP::LGG-1/ATG8 punctae in nerve-ring neurons in day 1 rgef-1p::gfp::lgg-1 (WT)-expressing animals after injection of animals with vehicle (dimethylsulfoxide (DMSO); n = 27 animals) or BafA (n = 34 animals), and in animals expressing rgef-1p::gfp::lgg-1(Gly116Ala) (Gly116Ala; DMSO, n = 23; BafA, n = 28 animals), over three independent experiments. Error bars are s.d. NS P = 0.99, ****P < 0.0001, by two-way ANOVA with Tukey’s multiple-comparisons test. d, Mean number of GFP::LGG-1/ATG8 punctae in nerve-ring neurons after injection of animals with vehicle (DMSO) or BafA in day 1 sid-1; rgef-1p::sid-1; rgef-1::gfp::lgg-1 animals after whole-life atg-7 RNAi (DMSO, n = 30; BafA, n = 34 animals, NS P = 0.051) or lgg-1/ATG8 RNAi (DMSO, n = 33; BafA, n = 38 animals, NS P > 0.99) compared to control (CTRL) (DMSO, n = 27; BafA, n = 34 animals; ****P < 0.0001) over three independent experiments. Error bars are s.d. CTRL-DMSO versus atg-7-DMSO: NS P = 0.12; CTRL-DMSO versus lgg-1-DMSO: ****P < 0.0001, by two-way ANOVA with Tukey’s multiple-comparisons test. e, Representative images and mean fluorescence intensity in head regions of day 1 sid-1; rgef-1p::sid-1; sqst-1p::sqst-1::gfp animals after whole-life autophagy gene RNAi. Error bars are s.d. P values as in a. See Supplementary Table 2 for n, all P values and statistical details. Scale bar, 100 μm. Shading of atg-16.2 emphasizes this RNAi treatment as an exception for lifespan extension by early-acting autophagy genes (Fig. 1).
To further assess autophagy status in neurons, we used neuronal-only RNAi strains expressing a GFP-tagged version of the autophagy receptor p62/SQSTM1 (SQST-1 in C. elegans), which is a substrate for degradative autophagy, and mainly detected in neurons and pharynx, the feeding organ of C. elegans8. SQST-1::GFP significantly accumulated in the head after neuronal-only inhibition of atg-16.2 and most other early-acting genes (bec-1/BECN1, atg-7, atg-4.1 and lgg-1/ATG8) and two late-acting genes (cup-5/MCOLN and epg-5; Fig. 2e; RNAi of other atg genes were also increased, but did not reach statistical significance), indicating that these neuronal-only RNAi treatments generally resulted in an inhibition of canonical, degradative autophagy of at least SQST-1/p62 receptor-related substrates. Taken together, these autophagy reporter analyses illustrate that neuronal gene inhibition of early-acting autophagy genes impaired neuronal autophagy activity or cargo degradation.
Neuronal inhibition of early Atg genes reduces polyQ aggregation
Since neuronal deletion of Atg5 and Atg7 induces neurodegenerative phenotypes in mice5,6, we investigated protein aggregation models in C. elegans neurons. Consistent with previous work, we found that whole-body reduction (that is, primarily in nonneuronal tissues) of all tested autophagy genes increased the aggregation of YFP-tagged polyQ (that is, 40 glutamine (Q) repeats) in C. elegans neurons (Fig. 3a)7–9. We next asked if neuronal aggregate load was similarly affected when autophagy genes were reduced in neurons only. While no changes were observed in polyQ aggregation in sid-1(qt9) mutants or upon neuronal expression of SID-1 (Extended Data Fig. 4a), we unexpectedly observed that neuronal-only knockdown of all early-acting genes, except atg-16.2, decreased neuronal polyQ aggregate load. Knockdown of late-acting autophagy genes had no effect or increased the number of neuronal polyQ aggregates (Fig. 3b,c), paralleling the differential effect of early-versus-late autophagy gene knockdown we observed on lifespan extension (Fig. 1b). Moreover, whole-life, neuronal-only RNAi against atg-7 or lgg-1/ATG8 extended lifespan in short-lived animals expressing neuronal polyQ aggregates (Fig. 3d and Supplementary Table 4), indicating that the decreased aggregation load conferred by neuronal-only inhibition of autophagy genes correlates with consequential effects on longevity.
Fig. 3 |. Neuronal polyQ aggregation is increased by whole-body inhibition, but reduced by neuronal inhibition of early-acting autophagy genes, except atg-16.2.

a, Number of neuronal polyQ aggregates in day 5 rgef-1::Q40::yfp animals after whole-life autophagy gene RNAi, except for adult-only RNAi for vha-13, vha-15 and vha-16 to avoid larval arrest (unc-51/ULK and bec-1/BECN1 RNAi clones were previously tested by us with similar results9, and bec-1/BECN1 and atg-7 RNAi clones increase Htn-Q150 aggregation7). In the violin plots, the solid line indicates the median and dotted lines indicate quartiles. ***P < 0.001, ****P < 0.0001, by one-way ANOVA with Dunnett’s multiple-comparison test. See Supplementary Table 2 for n, all P values and statistical details. b, Number of neuronal polyQ aggregates in day 5 sid-1; rgef-1p::sid-1; rgef-1::Q40::yfp animals after whole-life autophagy gene RNAi. In the violin plots, the solid line indicates the median and dotted lines indicate quartiles. NS P > 0.05, *P < 0.05, ***P < 0.001, ****P < 0.0001, by one-way ANOVA with Dunnett’s multiple-comparison test. See Supplementary Table 2 for n, all P values and statistical details. Shading of atg-16.2 emphasizes this RNAi treatment as an exception for decreased polyQ aggregate number by early-acting autophagy genes. c, Representative images of nerve-ring neurons of day 5 sid-1; rgef-1p::sid-1; rgef-1::Q40::yfp animals after whole-life CTRL, atg-16.2, atg-7 or lgg-1/ATG8 RNAi with arrowheads indicating polyQ aggregates. Three experimental repeats. Scale bar, 20 μm. d, Lifespan analysis of sid-1; rgef-1p::sid-1 + rgef-1p::gfp; rgef-1::Q40::yfp animals after whole-life atg-7, or lgg-1/ATG8 RNAi compared to control (CTRL). Statistical significance was determined by two-sided log-rank test, ****P < 0.0001. See Supplementary Table 4 for details and repeats.
Neuronal inhibition of early Atg genes increases exopher formation
Similarly to human neurodegenerative diseases, neurons in C. elegans can rid themselves of toxic protein aggregates via biogenesis of large vesicles called exophers that jettison cytosolic material including polyQ aggregates from neurons into surrounding tissues24. Since whole-body RNAi in combination with RNAi in touch neurons (that is, using WT animals expressing mec-18p::sid-1) against early-acting autophagy genes bec-1/BECN1, atg-7 and lgg-1/lgg-2/ATG8 increases exopher generation in ALMR touch neurons, the neuronal subtype that most frequently produces exophers24, we tested the effect of neuronal-only knockdown of the panel of autophagy genes on exopher biogenesis in ALMR neurons using a transgenic mCherry reporter expressed from the mec-4 promoter (Fig. 4a)24,43. While baseline exopher numbers were unchanged in neuronal RNAi strains (Extended Data Fig. 4b), we found that neuronal-only knockdown of our panel of early-acting genes, but not late-acting genes, significantly increased exopher formation (Fig. 4b). In contrast, neuronal-only knockdown of atg-16.2, which did not extend lifespan (Fig. 1b) and increased neuronal aggregation of polyQ proteins (Fig. 2b), did not increase exopher generation (Fig. 2b), again highlighting atg-16.2 as an exception among the early-acting autophagy genes. Neuronal-only RNAi inhibition of atg-7 and lgg-1/ATG8 also extended lifespan in the mCherry touch neuron reporter background (Supplementary Table 4). Furthermore, adult-only neuronal inhibition of atg-7 and lgg-1/ATG8 was sufficient to induce exopher formation on day 2 of adulthood, while whole-body reduction (that is, primarily in nonneuronal tissues) of atg-7 and lgg-1/ATG8 did not increase exopher formation (Extended Data Fig. 4c,d). These findings further corroborate the correlation between exopher formation and lifespan extension caused by the neuronal reduction of early-acting autophagy genes. Q40::YFP was extruded in exophers in day 2 animals (Fig. 4c), and animals subjected to neuronal atg-7 and lgg-1/ATG8 RNAi produced significantly more exophers compared to control RNAi animals (Fig. 4d). These observations support exophers as a polyQ extrusion route, which may contribute to the reduced neuronal polyQ aggregate counts of day 5 animals subjected to neuronal-specific, early-autophagy gene RNAi (Fig. 3b).
Fig. 4 |. Neuronal inhibition of early-acting autophagy genes, except atg-16.2, induces exophers, which secrete Q40::yfp and extend lifespan.

a, Exophers originating from the ALMR neuron in sid-1; rgef-1p::sid-1; mec-4p::mCherry animals. Representative diagram showing two exophers (arrowheads) and the ALMR soma on day 2. Scale bar, 20 μm. b, Mean percentage of ALMR neurons with exophers in day 2 sid-1; rgef-1p::sid-1; mec-4p::mCherry animals after whole-life autophagy gene RNAi. Error bars are the s.d. NS P > 0.05, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, by two-sided Cochran–Mantel–Haenszel test to compare each RNAi to control (CTRL). See Supplementary Table 2 for n, all P values and statistical details. Shading of atg-16.2 emphasizes this RNAi treatment as an exception for increased exopher formation by early-acting autophagy genes. c, Representative images of an ALMR neuron of day 2 sid-1; rgef-1p::sid-1; mec-4p::mCherry; rgef-1p::Q40::yfp animals showing the expression of mCherry and Q40::yfp. polyQ collection in the soma (arrowhead)) and in the exopher is visible. Six experimental repeats. Scale bar, 20 μm. d, Mean percentage of ALMR neurons with exophers in day 2 sid-1; rgef-1p::sid-1; mec-4p::mCherry; rgef-1p::Q40::yfp animals after whole-life atg-7 RNAi, or lgg-1/ATG8 RNAi compared to control (CTRL). Error bars are the s.d. of n = 7 experiments with n = 317, 313 and 316 animals. *P = 0.016, **P = 0.0049, by two-sided Cochran–Mantel–Haenszel test. e, Lifespan analysis of mec-4p::mCherry animals sorted for presence of exophers on day 1–5 of adulthood. Statistical significance was determined by two-sided log-rank test, ****P < 0.0001. See Supplementary Table 5 for details and repeats.
To test if exopher formation could be directly linked to organismal lifespan, we analyzed mec-4p::mCherry expressing animals every day between day 1 and day 5 (refs. 24,43) for exophers from touch neurons, separating populations based on the presence or absence of exopher(s) and measured the lifespan of the two ‘binned’ populations. Remarkably, animals positive for exopher events in touch neurons were ~10–30% longer lived than those who did not (P < 0.009, log-rank, in four of five experiments; Fig. 4e and Supplementary Table 5). This suggests that exopher events in touch neurons (and likely other neurons not directly assayed) may contribute to improvements in organismal fitness. Moreover, when we plotted all our data obtained with the panel of autophagy RNAi clones and compared lifespan, polyQ and exopher data, we found a significant negative correlation between lifespan and polyQ aggregation (r = 0.9, P < 0.0001) between polyQ aggregation and exophers (r = 0.7, P = 0.008), and a positive correlation between lifespan and exophers (r = 0.06, P = 0.03; Extended Data Fig. 4g), consistent with links between these organismal phenotypes.
atg-16.2, but not atg-4.1, is required for exopher biogenesis
Considering that atg-16.2 inhibition in neurons did not extend lifespan, reduced protein aggregation, and increased exophers, we examined an atg-16.2(ok3224) null mutant alongside another early-acting autophagy null mutant, atg-4.1(bp501), for exopher formation. Exopher production on day 2 of adulthood in atg-16.2(ok3224) mutants was significantly decreased (Fig. 5a), unlike RNAi treated animals, which may reflect insufficient neuronal RNAi effects, or cell-nonautonomous contributions. In contrast, atg-4.1(bp501) mutants had increased exopher formation (Fig. 5a), similarly to neuronal-only RNAi inhibition of early-autophagy gene inhibitions (Fig. 4b)24. These findings highlight the requirement for atg-16.2 in exopher biogenesis.
Fig. 5 |. atg-16.2 is required for benefits of neuronal inhibition of early-autophagy genes.

a, Mean percentage of ALMR neurons with exophers in day 2 WT, atg-16.2(ok3224) and atg-4.1(bp501) animals expressing mec-4p::mCherry. Error bars are the s.d. WT (n = 261 animals) versus atg-16.2 (n = 274 animals) (n = 7 experiments, **P = 0.007), WT (n = 273 animals) versus atg-4.1 (n = 243 animals) (n = 6 experiments, ****P < 0.0001) by two-sided Cochran–Mantel–Haenszel test. b, Mean GFP fluorescence intensity in head region in day 1 WT, atg-4.1(bp501) and atg-16.2(ok3224) animals expressing sqst-1p::sqst-1::gfp. Error bars are the s.d. WT (n = 30) versus atg-16.2 (n = 35), ****P < 0.0001; WT (n = 38) versus atg-4.1 (n = 58), ****P < 0.0001) by two-sided t-test over three independent experiments. Representative images from one experiment. Scale bar, 200 μm. c, Mean neuronal GFP::LGG-1/ATG8-positive and GFP::LGG-1(Gly116Ala)-positive punctae in day 1 WT, atg-4.1(bp501) and atg-16.2(ok3224) animals. Error bars are the s.d. WT-GFP::LGG-1 (n = 57) versus WT-GFP::LGG-1(Gly116Ala) (n = 61), ****P < 0.0001; atg-16.2-GFP::LGG-1 (n = 31) versus atg-16.2-GFP::LGG-1(Gly116Ala) (n = 32), ****P < 0.0001; atg-4.1-GFP::LGG-1 (n = 30) versus atg-4.1-GFP::LGG-1(Gly116Ala) (n = 28), NS P = 0.15. Comparison between strains: WT (n = 27) versus atg-16.2 (n = 31), ****P < 0.0001, WT (n = 30) versus atg-4.1 (n = 30) NS P = 0.75 > 0.05, ****P < 0.0001, over three independent experiments by two-way ANOVA with Tukey’s multiple-comparisons test. d, Mean neuronal GFP::LGG-1/ATG8-positive punctae in day 1 WT, atg-4.1(bp501) and atg-16.2(ok3224) animals after vehicle (DMSO) or BafA injections to block autophagy. Error bars are the s.d. WT-DMSO (n = 27) versus WT-BafA (n = 34), ****P < 0.0001; atg-16.2-DMSO (n = 28) versus atg-16.2-BafA (n = 34), *P = 0.043; atg-4.1-DMSO (n = 24) versus atg-4.1-BafA (n = 31), **P = 0.0097. Comparison between strains: WT (n = 27) versus atg-16.2 (n = 28), ****P < 0.0001, WT (n = 27) versus atg-4.1 (n = 24) ****P < 0.0001, over three independent experiments by two-way ANOVA with Tukey’s multiple-comparisons test. e,f, Lifespan analyses in sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals carrying atg-16.2(ok3224) (e) or atg-4.1(bp501) mutations (f) after whole-life atg-7, or lgg-1/ATG8 RNAi compared to control (CTRL). Two-sided log-rank test, NS P = 0.5, P = 0.6 (e), ****P < 0.0001 (f). See Supplementary Table 6 for details and repeats. g,h, Number of neuronal polyQ aggregates in day 5 sid-1; rgef-1p::sid-1 + rgef-1p::gfp; rgef-1::Q40::yfp animals carrying atg-16.2(ok3224) (g) or atg-4.1(bp501) mutations (h) after RNAi treatments as in e. In the violin plots, the solid line indicates the median and dashed lines indicate quartiles. n = 45 animals each in three independent experiments (g) atg-16.2: NS P = 0.76, P = 0.46; (h) atg-4.1: ****P < 0.0001 by one-way ANOVA with Dunnett’s multiple-comparisons test. i, Mean percentage of ALMR with exophers of day 2 sid-1; rgef-1p::sid-1 + rgef-1p::gfp; mec-4p::mCherry (WT) animals and atg-16.2(ok3224) animals after RNAi treatments as in e–h. Error bars are the s.d. of n = 7 experiments with n = 273, 255 and 257 animals (left) and n = 407, 411 and 357 animals (right). WT: ****P < 0.0001; atg-16.2: P = 0.62, P = 0.77 by two-sided Cochran–Mantel–Haenszel test.
To confirm that the observed exopher phenotypes were independent of defects in neuronal degradative autophagy, we assayed autophagy status in neurons of atg-16.2 and atg-4.1 mutants. As previously reported in C. elegans embryos44,45, both mutants displayed increased SQST-1::GFP levels compared to WT animals (Fig. 5b), consistent with a block of autophagy. Moreover, the two autophagy mutants displayed pronounced neuronal GFP::LGG-1 phenotypes. In contrast to neuronal-only RNAi (Fig. 2a), atg-16.2(ok3224) and atg-4.1(bp501) mutants displayed no reduction of GFP::LGG-1 punctae in nerve-ring neurons, and a significant increase in neuronal, lipidation-independent GFP::LGG-1(Gly116Ala) (Fig. 5c) punctae was observed, similarly to neuronal-only RNAi (Fig. 2b). To corroborate the block in neuronal autophagy in these mutants, we again performed autophagy flux assays40 and found no increase in neuronal GFP::LGG-1 punctae when BafA was injected into atg-16.2(ok3224) and atg-4.1(bp501) mutants (Fig. 5d). Taken together, our results are consistent with neuronal autophagy being blocked in atg-16.2(ok3224) and atg-4.1(bp501) mutants. In conclusion, we identified a novel requirement for atg-16.2 in exopher biogenesis, which is seemingly independent of defects in neuronal degradative autophagy.
atg-16.2 is required for benefits of neuronal autophagy inhibition
We next tested whether atg-16.2 was required for the benefits of neuronal autophagy gene inhibition. Neuronal inhibition of atg-7 or lgg-1/ATG8 RNAi increased lifespan and decreased polyQ aggregates in atg-4.1(bp501) but not atg-16.2(ok3224) mutants (Fig. 5e–h and Supplementary Table 6). Consistently, atg-16.2 was required for the increased exopher formation upon neuronal-only atg-7 and lgg-1/ATG8 RNAi (Fig. 5i). The absence of phenotypes in atg-16.2(ok3224) mutants was not because these animals were refractory to RNAi, as they responded normally to RNAi clones for genes expressed outside neurons (Extended Data Fig. 5), and neuronal inhibition of the insulin/IGF-1-like receptor daf-2 increased lifespan in atg-16.2(ok3224) mutants (Supplementary Table 7), similarly to WT animals (Supplementary Table 3). Taken together with our autophagy analyses (Fig. 5b–d), these results indicate that neuronal-only RNAi of at least atg-7 and lgg-1/ATG8 can reduce neuronal polyQ aggregates, and induce exopher formation, as well as extend lifespan, irrespective of whether canonical autophagy is engaged, consistent with a cargo degradation-independent mechanism in neurons. Our data highlight that this underlying mechanism involves atg-16.2, which was required for all the abovementioned phenotypes.
ATG-16.2’s WD40 domain is required for exopher formation
To further address a mechanistic role for ATG-16.2 in exopher biogenesis, we next tested its cell-autonomous functions by rescuing ATG-16.2 expression in neurons of atg-16.2 mutants. ATG16 is a conserved protein (Extended Data Fig. 6a), containing an ATG5-interacting motif, a coiled-coil domain and a seven-bladed β-propeller WD40 domain that mediates protein interactions at its C terminus46 (Fig. 6a,b). The Saccharomyces cerevisiae ATG16 ortholog does not contain a WD40 domain (Extended Data Fig. 6a), and the WD40 domain seems dispensable for canonical, degradative autophagy in metazoans20. To test the function of the C. elegans ATG-16.2 WD40 domain, we created and analyzed a truncated protein devoid of the C-terminal WD40 domain (ATG-16.2ΔWD40)45 (Fig. 6a). In parallel, we also constructed and investigated a protein with mutations in two conserved residues (Extended Data Fig. 6a), i.e., phenylalanine 394 to alanine and isoleucine 509 to methionine (ATG-16.2(Phe394Ala, Ile509Met). Phe394, corresponding to Phe467 in human ATG16L1, is a surface-facing residue on Blade 4 of the WD40 domain (Fig. 6b) that creates a surface pocket with residues of Blade 5, and whose mutation to alanine in mammalian cells inhibits non-canonical autophagy functions of the WD40 domain20. We discovered the Ile509Met point mutation during the final stages of the publication process. Ile509, corresponding to Ile580 in human ATG16L1 and located between Blades 6–7 of the WD40 domain, is not surface exposed (Fig. 6b), and unlikely to play a functional role. However, future studies are needed for a comprehensive characterization and validation of any functional role of Ile509Met mutation in ATG16.2. We expressed each of these proteins, as well as a full-length ATG-16.2 from the rgef-1 promoter in neurons of atg-16.2(ok3224) mutants with similar gene expression levels to WT (Extended Data Fig. 6b). atg-16.2(ok3224) mutants displayed an increased number of GFP::LGG-1/ATG8 punctae (Fig. 5c,d) due to blocked autophagy (Fig. 5b,d), but the neuronal rescue of full-length ATG-16.2, ATG-16.2ΔWD40 and ATG-16.2(Phe394Ala) all significantly reduced the elevated number of neuronal GFP::LGG-1/ATG8 punctae observed in atg-16(ok3224) mutants to levels equal or close to those of WT (Fig. 6c). Thus, the three ATG-16.2 constructs make functional transgenes that are at least partially sufficient in rescuing autophagy deficiencies in atg-16(ok3224) mutants, and the ATG-16.2 WD40 domain is not required for neuronal autophagy in adult C. elegans, as in C. elegans embryos45 and mammalian cells20.
Fig. 6 |. The WD40 domain of ATG-16.2 is dispensable for autophagosome formation but required for exophergenesis.

a, Schematic of ATG-16.2 rescue constructs. Full-length ATG-16.2 protein includes ATG5-interacting motif, coiled-coil domain (CC) and WD40 domain. ATG-16.2ΔWD40 is a truncated ATG-16.2 protein containing amino acids 1–234. ATG-16.2(Phe394Ala, Ile509Met) protein contains a point mutation of phenylalanine to alanine in position 349 and in isoleucine to methionine in position 509 in the WD40 domain. See Extended Data Fig. 6a for the primary structure of ATG-16.2. b, AlphaFold model of ATG-16.2 (UniProt Q09406). The N-terminal region of ATG-16.2 is predicted to contain a helical structure with a conserved ATG5-interacting motif and a CC domain. The C terminus contains a seven-bladed WD40 beta-propeller domain. Phe394 (F) is located on blade 4 on the linker between β-sheets B-C, and Ile509 (I) is located on the linker between blade 6–7 before β-sheet A (enlarged). The enlarged surface model reveal indicates that Phe394 is surface exposed, whereas Iso509 is not. c, Mean GFP::LGG-1/ATG8-positive punctae in day 1 WT (n = 32), atg-16.2(ok3224) (n = 33) and atg-16.2(ok3224) mutants transgenically expressing full-length atg-16.2 (n = 32), atg-16.2 lacking the WD40 domain (ΔWD40) (n = 30) or atg-16.2(Phe394Ala, Ile509Met) (F394A, I509M) (n = 31) from the neuronal rgef-1 promoter. Error bars are the s.d. over three independent experiments, ****P < 0.0001, by one-way ANOVA by Dunnett’s multiple-comparisons test. d, Mean percentage of day 2 WT (n = 258 animals), atg-16.2(ok3224) (n = 271 animals) and atg-16.2(ok3224) animals expressing full-length atg-16.2 (n = 214 animals), atg-16.2 lacking the WD40 domain (ΔWD40) (n = 275 animals) or atg-16.2(Phe394Ala, Ile509Met) (F394A, I509M (n = 212 animals) from the neuronal rgef-1 promoter with ALMR exophers. Error bars are the s.d. of n = 6 experiments, ****P < 0.0001; ***P = 0.002, NS P = 0.30, P = 0.77 by two-sided Cochran–Mantel–Haenszel test.
Since atg-16.2 was required for exopher biogenesis (Fig. 5a,i), we next tested whether ATG-16.2 and its WD40 domain were cell-autonomously required for exopher formation. atg-16.2(ok3224) mutants expressing full-length ATG-16.2, but neither of the WD40 domain-defective ATG-16.2 constructs ΔWD40 and Phe394Ala/Ile609Met, significantly increased exopher numbers to WT levels (Fig. 6d). Collectively, these results indicate that a neuronal function of ATG-16.2 involving its WD40 domain is important for exopher formation.
ATG-16.2’s WD40 domain is required for benefits of neuronal autophagy inhibition
To analyze if neuronal ATG-16.2 was involved in the effects mediated by neuronal inhibition of early-acting autophagy genes, we created atg-16.2(ok3224) mutants capable of neuronal-only RNAi and expressing full-length neuronally expressed ATG-16.2, or WD40 domain-deficient ATG-16.2ΔWD40 and ATG-16.2(Phe394Ala/Ile509Met). We subjected these strains to neuronal-only RNAi of atg-7 or lgg-1/ATG8 and examined them for lifespan (Fig. 7a–c and Supplementary Table 7), neuronal polyQ aggregates (Fig. 7d–f) and exopher production (Fig. 7g–i). In the animals rescued with full-length ATG-16.2, neuronal knockdown of atg-7 or lgg-1/ATG8 extended lifespan (Fig. 7a), reduced neuronal polyQ (Fig. 7d) and increased exopher production (Fig. 7g), similarly to WT animals. In contrast, neuronal expression of ATG-16.2ΔWD40 or ATG-16.2(Phe394Ala/Ile509Met) failed to rescue lifespan extension (Fig. 7b,c), neuronal polyQ reduction (Fig. 7e,f) or exopher formation (Fig. 7h,i). These results demonstrate that the WD40 domain of ATG-16.2 is autonomously required for lifespan extension, neuronal polyQ aggregate reduction and increased exopher production in atg-16.2(ok3224) mutants following neuronal-only knockdown of early-acting autophagy genes.
Fig. 7 |. The WD40 domain of ATG-16.2 is for benefits of neuronal inhibition of early-autophagy genes.

a–c, Lifespan analysis of atg-16.2(ok3224); sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after whole-life atg-7 or lgg-1/ATG8 RNAi compared to control (CTRL). atg-16.2(ok3224) mutants were rescued by expressing full-length atg-16.2 (a), atg-16.2ΔWD40 (b) or atg-16.2(Phe394Ala, Ile509Met) (c) from the pan-neuronal promotor rgef-1. Statistical significance was determined by two-sided log-rank test, NS P > 0.05, ****P < 0.0001. See Supplementary Tables 3 and 6 for details and repeats. d–f, Number of neuronal polyQ aggregates in day 5 atg-16.2(ok3224); sid-1; rgef-1p::sid-1 + rgef-1p::gfp; rgef-1::Q40::yfp animals after whole-life atg-7 or lgg-1/ATG8 RNAi compared to control (CTRL). atg-16.2(ok3224) mutants were rescued by expressing full-length atg-16.2 (d), atg-16.2ΔWD40 (e) or atg-16.2(Phe394Ala, Ile509Met) (f) from the pan-neuronal promotor rgef-1. In the violin plots, solid lines indicate the median and dashed lines indicate quartiles. n = 30 animals each over three independent experiments. d, ***P = 0.0001, P = 0.0005; e, NS P = 0.14, P = 0.63; f, NS P = 0.63, P = 0.07, by one-way ANOVA with Dunnett’s multiple-comparisons test. g–i, Mean percentage of ALMR with exophers of day 2 atg-16.2(ok3224); sid-1; rgef-1p::sid-1 + rgef-1p::gfp; mec-4p::mCherry animals after whole-life atg-7, or lgg-1/ATG8 RNAi compared to control (CTRL). atg-16.2(ok3224) mutants expressed full-length atg-16.2 (g), atg-16.2ΔWD40 (h) or atg-16.2(Phe394Ala, Ile509Met) (i) from the pan-neuronal promotor rgef-1. Error bars are the s.d. g, atg-7 RNAi: n = 5, **P = 0.007; lgg-1//ATG8 RNAi: n = 7, **P = 0.003 (n = 304, 199 and 291 animals); h, atg-7 RNAi: n = 5, NS P = 0.49; lgg-1 RNAi: n = 6, NS P = 0.39; (n = 307, 186 and 330 animals) (i) atg-7 RNAi: n = 5, NS P = 0.43; lgg-1 RNAi: n = 6, NS P = 0.71 (n = 262, 188 and 240 animals); by two-sided Cochran–Mantel–Haenszel test.
Collectively, our results show a role for the WD40 domain of neuronal ATG-16.2 in exopher biogenesis and provide a potential mechanism by which pan-neuronal inhibition of early-acting autophagy genes decreases polyQ aggregation, possibly via increased exopher formation in at least some neurons. Increased exopher formation may also, at least in part, explain the lifespan extension induced by early-autophagy gene inhibition in C. elegans neurons.
Discussion
Here we demonstrated that RNAi of early-acting autophagy genes in C. elegans neurons significantly extended lifespan, autonomously improved neuron morphology and function in aged animals, reduced neuronal polyQ aggregates and increased the extrusion of neuronal exophers, which are large extracellular vesicles. These effects were independent of canonical, degradative autophagy, because downregulation of late-acting autophagy genes did not induce the same benefits. Moreover, atg-4.1 mutants, which have defects in autophagy, displayed the same phenotypes as WT animals following neuronal-only inhibition of early-acting autophagy genes. Instead, we found that extended lifespan, decreased polyQ aggregate number and increased exopher biogenesis after neuronal inhibition of early-acting autophagy genes required ATG-16.2, and particularly its WD40 domain-related functions. The ATG-16.2 WD40 domain is involved in the formation of exophers, which have been shown to secrete cytosolic material, including polyQ aggregates from C. elegans neurons, especially upon disruption of proteostatic pathways24,47. We thus propose a model in which the inhibition of early-acting autophagy genes leads to the ATG-16.2 WD40 domain-dependent formation of exophers, which may contribute to the decrease in neuronal protein aggregation and the observed lifespan extension in a direct or indirect fashion.
Mammalian ortholog ATG16L1 functions in the ATG12–ATG5/ATG16 complex to mediate ATG8 conjugation via PE to double-membrane autophagosomes, but can also via its highly conserved WD40 domain facilitate association of ATG8/LC3 to non-autophagosomal membranes, including endolysosomal membranes or single-membrane vesicles involved in LC3-associated phagocytosis in mouse macrophages20, by conjugation to both PE and phosphatidylserine (PS)48. We here found that the WD40 domain of C. elegans ATG-16.2 is required for basal exopher formation and for increased exopher formation upon neuronal inhibition of early-acting autophagy genes atg-7 and lgg-1/ATG8. In addition to C. elegans26,27, exophers have been observed in mammalian cardiomyocytes36 and neurons35, but exopher biogenesis is not well understood molecularly. Our observations raise interesting questions to address in future studies, including whether C. elegans ATG-16.2, via its WD40 domain and potential ATG16 WD40-specific binding partners like V-ATPase49, can get recruited to exopher membranes, and whether such potential noncanonical functions of ATG-16.2 would involve the recruitment and conjugation of ATG8 proteins to exopher membranes via both PE and PS in a manner dependent on the ATG12–ATG5 complex. To this end, neuronal-only reduction of lgg-1/ATG8 not only increased exopher formation but also reduced neuronal polyQ aggregation load, and increased lifespan similarly to other early-acting autophagy genes. While this could point toward an ATG8-independent function of ATG-16.2, the partial knockdown of neuronal lgg-1/ATG8 (~40–50% remaining after the RNAi knockdown treatment) could be sufficient for noncanonical autophagy but insufficient for canonical autophagy. Alternatively, ATG-16.2 could have an autophagy-independent function, that is, independent of its role in ATG8 lipidation, similarly to mammalian ATG16L1, which can facilitate hormone secretion via dense-core vesicles50 and exosome production51 in cellular models, as well as neuropeptide production in Drosophila52 in an autophagy-independent manner. Future experiments are needed to address the mechanism by which ATG-16.2 regulates exopher formation in touch neurons and possibly other neuronal subtypes after neuronal-only autophagy gene knockdown in C. elegans. We note that the C. elegans paralog ATG-16.1 is not expressed in touch neurons, but may play roles in other cell types, which will be important for future investigation.
Neuronal autophagy is important for protein homeostasis in the nervous system, as conditional knockdown of autophagy genes Atg5 and Atg7 in the central nervous system of mice leads to neurodegenerative phenotypes5,6 and loss-of-function mutations in the endolysosomal pathway frequently cause neurological disorders53,54. Autophagy has been shown to degrade aggregating proteins, including proteins with polyQ stretches55. In line with these observations, systemic/whole-body inhibition of all autophagy-related genes tested in WT C. elegans increased neuronal polyQ formation (this study and refs. 7–9). In contrast to nonneuronal knockdown, we report here that neuronal-only inhibition of early-acting autophagy genes reduced polyQ aggregate load in neurons. Our finding is consistent with recent studies demonstrating that polyQ proteins and aggregates can be transmitted from cell-to-cell in C. elegans56, in Drosophila57 and in induced pluripotent stem cells derived from patients with Huntington’s disease and transplanted into mouse brains58, possibly via extracellular vesicles59,60. Moreover, inhibition of autophagosome formation by silencing early-acting autophagy genes Atg5 or Vps34/Pik3c3 in neuronal cell culture models from mice and humans promotes exosomal secretion of aggregate-prone proteins, such as α-synuclein61, prions62 or an amyloid precursor63. Here, we observed neuronal polyQ-exopher extrusions in touch (ALM) neurons of young animals subjected to neuronal-only inhibition of early-acting autophagy genes. These observations raise several mechanistic questions, including what the biochemical properties and fate of the secreted polyQ proteins are and how they contribute to preventing polyQ aggregation at an older age. Likewise, it will be important to better understand the physiological role of potentially secreted, aggregated proteins as well as other exopher cargo in cell-to-cell communication, noting that our studies demonstrated a correlation between decreased neuronal aggregate load, increased exopher formation and lifespan extension upon neuronal inhibition of early-acting autophagy genes. One possibility may be that a general handoff of aggregates for remote degradation by their neighbors may maintain important neuronal functions that promote long life.
An especially surprising observation of this study was that neuronal-only inhibition of early-acting autophagy genes from hatching or the start of reproductive adulthood, using the same RNAi bacterial clones that shorten the lifespan of several long-lived C. elegans mutants (which would experience gene inhibition primarily in non-neuronal tissues)1, caused normal C. elegans to live ~20–30% longer. While we were not able to determine the extent of knockdown of autophagy-dependent genes in C. elegans neurons, neuronal-specific knockdown of fluorescent markers expressed in neurons led to 40–50% reduction in fluorescence. This reduction was sufficient to inhibit neuronal autophagy. The accompanying lifespan results are notable as autophagy gene RUBCN (Run domain Beclin-1-interacting and cysteine-rich domain-containing protein) negatively regulates autophagy, and whole-life, neuronal-only RNAi of rub-1/RUBCN using a sid-1(pk3321); unc-119p::sid-1 transgenic strain28 extends lifespan by increasing autophagy flux64. Thus, canonical autophagy induction in neurons can induce beneficial effects on lifespan in C. elegans. Our data, on the other hand, suggest that the neuronal-only inhibition of early-autophagy genes triggers a longevity mechanism independent of degradative autophagy and mediated by the WD40 domain of ATG-16.2. Considering the requirement of this highly conserved domain in noncanonical autophagy gene functions in mammals, we propose a similar role in adult C. elegans neurons involving possible secretion of lifespan-extending signals, either in exophers or in different types of atg-16.2-dependent secretory events. It will be interesting to address if and how the neuronal secretome may be affected in C. elegans atg-16.2 mutants.
In sum, we demonstrate that the inhibition of early-acting autophagy genes in neurons extended C. elegans lifespan, improved neuronal proteostasis and increased exopher formation mediated by the autonomous, WD40 domain-related function of ATG-16.2. If this mechanism is conserved in other organisms, modulating neuronal autophagy genes may provide a method to improve lifespan and neuronal proteostasis. In addition, as the WD40 domain of C. elegans ATG-16.2 was critical for lifespan determination, we speculate that noncanonical ATG16 functions may play a broader role in organismal aging than has been previously anticipated.
Methods
C. elegans strains and maintenance
C. elegans strains were maintained and cultured at 20 °C using Escherichia coli OP50 as a food source65 unless RNAi was initiated. All experiments were conducted using hermaphrodites. For RNAi experiments, animals were grown on HT115 bacteria from the time of RNAi initiation (see below). See Supplementary Table 8 for all strains used and created for this study. For maintenance and selection of animals expressing a hygromycin resistance marker, animals were grown on 6-cm NGM plates that were supplemented with 250 μl of 5 mg ml−1 of hygromycin B (GoldBio) in M9.
Construction of transgenic strains
The rgef-1p::sid-1 vector was made by Gateway cloning. The rgef-1 pan-neuronal promoter, sid-1 open reading frame and the unc-54 3′ untranslated region (UTR) were cloned into the Gateway destination vector pDEST_R3R4. The final product rgef-1p::sid-1 (pMH1141) was verified by sequencing. To construct the rgef-1p::atg-16.2 vector, cDNA of atg-16.2 was amplified by PCR from vector atg-16.2p::atg-16.2::gfp45 and ligated with pMH1307 (pSM vector66 containing rgef-1 promotor between NheI and SalI) by Gibson assembly. The final product and open reading frame of rgef-1p::atg-16.2 (pMH1387) was verified by sequencing. pMH1387 was used to change phenylalanine 394 (TTT) to alanine 394 (GCT) by site-directed mutagenesis to generate pMH1388. However, after acceptance of this manuscript, an additional mutation, Ile509Met, was discovered in pMH1388, and confirmed present in transgenic C. elegans strains MAH1038, ZB5329, and KUM48 (Supplementary Table 8), which all encode rgef-1p::atg-16.2(Phe394Ala, Ile509Met). To construct the rgef-1p::atg-16.2ΔWD40 vector, cDNA of atg-16.2delC, which encodes ATG-16.2(1–223), was amplified by PCR from atg-16.2p::atg-16.2 vector45 with added stop codon and ligated with pMH1307 (between NheI and SalI) by Gibson assembly. The final product rgef-1p::atg-16.2ΔWD40 (pMH1389) was verified by sequencing. Plasmid DNA was prepared using the Miniprep kit (Qiagen). Transgenic animals expressing an extrachromosomal array were created by gonadal microinjection of plasmids of interest with the indicated co-injection marker into indicated strains in Supplementary Table 8. A list of plasmids used and made in this study is provided in Supplementary Table 9. Primer information for plasmid construction is available in Supplementary Table 10.
RNA interference
RNAi was performed by feeding C. elegans with bacteria expressing dsRNA against the gene of interest. RNAi clones used in this study (Supplementary Table 9) were obtained from the Ahringer10 or the Vidal67 RNAi libraries. All RNAi clones were verified by sequencing. Two empty vector controls were used; the original L4440 plasmid, and L4440, digested with Eco RV and religated, which eliminated 114 bp (Eco RV restriction site was used to insert dsRNA; pMH1355). For RNAi experiments, HT115 bacteria were grown in liquid LB medium containing 0.1 mg ml−1 carbenicillin (Bio Pioneer), and 80-μl aliquots of bacteria were spotted onto 6-cm NGM plates with carbenicillin. Bacteria were allowed to grow for 1–2 days at room temperature. Before use, 80 μl 0.1 M IPTG (Promega) was added to the bacterial lawn to induce dsRNA expression before eggs (whole-life RNAi), larvae (L1 or L4 stage) or adults (adult-only RNAi) were transferred onto RNAi plates. Neuronal RNAi was performed in sid-1(qt9) mutants with sid-1 rescue under control of the pan-neuronal promoter rgef-1p. RNAi efficiency in mutants and neuronal RNAi strains was assessed with whole-life RNAi treatment on day 2 of adulthood. For RNAi specific against genes with muscle function, unc-112 and unc-22 RNAi penetrance was measured by paralysis and twitch-like movements, respectively. For RNAi specific against genes with hypodermal function, tsp-15 and bli-1 RNAi penetrance was determined by the appearance of blisters. For RNAi specific against genes with intestinal function, elt-2 RNAi penetrance was determined by a small phenotype and a clear and restricted appearing intestine. RNAi against the ubiquitously expressed rpl-2 led to larval arrest. RNAi specific against genes with neuronal function, snb-1 and unc-13, caused shrinker phenotype following gentle touch of the body with hair.
Fluorescence intensity measurements
Fluorescence intensity of GFP in nerve-ring neurons was measured in animals capable of neuronal RNAi to determine neuronal RNAi knockdown efficiency after feeding animals bacteria carrying empty vector control or expressing dsRNA against gfp. Fluorescence intensity of SQST-1::GFP was measured in either the head region or the whole body on the indicated days of adulthood either raised on OP50 bacteria or after animals were subjected to the indicated neuronal RNAi treatment. Animals were imaged on empty NGM plates after anesthetization with M9 medium containing 0.1% sodium azide. Images were acquired with a Leica DFC310 FX camera at 400 ms of exposure. The mean fluorescence intensity was measured in the nerve-ring neurons by outlining the same-sized circular region of interest using FIJI software (National Institutes of Health (NIH)) per animal and in whole body by outlining each worm, normalized to day 1 of adulthood or control RNAi.
Lifespan analysis
Lifespan was measured at 20 °C68. Synchronized animals were transferred onto 6-cm NGM plates seeded with E. coli OP50. Six plates were used for each strain with 20 animals per plate. The L4 larval stage was recorded as day 0 of lifespan, and animals were transferred every other day to a new NGM plate throughout the reproductive period. For lifespan experiments on RNAi bacteria, animals were fed dsRNA-expressing or control bacteria from hatching (whole-life), from larvae (L1 or L4 stage) or as adults (adult-only RNAi). For the exopher binning lifespans, strain ZB4065 (bzIs166[mec-4p::mCherry]) was used. Around 20–30 eggs were picked on a total of 60 P6 NGM plates with OP50 bacteria. From day 1 to day 5 of adulthood, animals were observed daily under a fluorescence microscope for exopher events, and animals with exophers (‘+ exopher’ group), assessed in a binary manner, were transferred onto P6 OP50 lifespan plates. Because exopher formation and subsequent clearance in the hypodermis takes ~3 days, our daily scoring assured that we would not miss exopher events43,69. On days 2 and 4, all animals were transferred away from their progeny. Animals that did not have exopher events by day 5 were transferred onto separate P6 OP50 lifespan plates for the ‘− exopher’ control group. Animals were scored as dead if they failed to respond to gentle prodding with a platinum-wire pick. All lifespan experiments performed for this study were numbered to indicate which RNAi clones were tested together in the same experiment (Supplementary Tables 1 and 3–7).
Healthspan measurements
Thrashing ability (that is, swimming), pharyngeal pumping and progeny production were assayed as measures of healthspan. For thrashing assays, animals on the indicated days of adulthood were transferred onto a 6-cm NGM media plate containing a drop of M9 medium, and body bends of 14–20 animals were counted for 20 s on a Leica stereoscope. Pharyngeal pumping was measured on the indicated days of adulthood by counting the grinder movements in the terminal pharyngeal bulb of 14–20 animals for 30 s on a Leica stereoscope. For the assessment of progeny production, 10 animals were singled on 6-cm NGM plates at the L4 larval stage and transferred daily onto fresh plates during the self-fertile reproductive span. The number of eggs/larvae produced by each animal per day was counted.
Dye-filling assay
Dye-filling experiments were performed on day 5 of adulthood. Animals were raised at 20 °C on corresponding RNAi plates for whole-life RNAi. On the day of experiment, 20 worms were transferred into 150 μl of a 10 ng μl−1 DiI (Invitrogen) solution diluted in M9 buffer. Animals were incubated for 2 h at 20 °C and then transferred to a fresh NGM plate seeded with OP50 bacteria to let them crawl for 1 h. Animals were mounted on a 2% agarose pad in M9 medium containing 0.1% sodium azide and imaged using a Zeiss Imager Z1 including apotome.2 with a Hamamatsu orca flash 4LT camera and Zen 2.3 software.
Neurite-branching analysis
Neurite branches were counted in sid-1; rgef-1p::sid-1 + rgef-1::gfp animals33. Eggs were raised on control HT115 bacteria or RNAi bacteria plates for whole-life RNAi. Branches from ALM and PLM mechanosensory neurons were counted on day 15 of adulthood. A branch was scored when a visible GFP-labeled branch was observed emanating from ALM or PLM mechanosensory neurons visualized using a Zeiss Imager Z1 including apotome at ×63 magnification.
Chemotaxis experiment
Chemotaxis assays were performed on day 5 of adulthood70,71. For each condition, ~250 animals were cultured at 20 °C on 6-cm semi-high growth media plates (25 g l−1 agar, 11.25 g l−1 Bacto Peptone, 3 g l−1 NaCl, 5 μg l−1 cholesterol in 100% ethanol, 1 mM CaCl2, 1 mM MgSO4, 25 mM KPO4 buffer pH 6 and 0.26 M carbenicillin in distilled water) and fed from hatching with bacteria expressing empty control vector or atg-7 and lgg-1/ATG8 dsRNA. At the L4 larval stage, animals were transferred onto plates coated with 50 μM 5-fluoro-2′-deoxyuridine. On day 5 of adulthood, animals were washed into 1.5-ml microcentrifuge tubes with M9 buffer and washed twice with M9 buffer. An empty 10-cm semi-high growth media carbenicillin plate for chemotaxis was prepared by pipetting 1 μl of 1 M sodium azide + 1 μl of 10% butanone in 95% ethanol (attractant), and 1 μl of 1 M sodium azide + 1 μl 95% ethanol (control) on either side of the plate. Animals were spotted in equal distance from the attractant and control at the origin and allowed to incubate for 1 h at room temperature. After incubation, animals within a 1.5-cm radius of the attractant, control and origin were counted. A chemotaxis index (CI) was then calculated: CI = [(Nattractant − Ncontrol)] / [(total − Norigin)].
Autophagy measurements
GFP::LGG-1 and lipidation-deficient GFP::LGG-1(Gly116Ala) punctae were counted in the nerve-ring neurons of animals expressing rgef-1p::gfp::lgg-1 (ref. 36) and rgef-1p::gfp::lgg-1(Gly116Ala)9, respectively, and in intestine and body-wall muscle of animals expressing lgg-1p::gfp::lgg-1 (ref. 35). For all experiments, animals were raised at 20 °C and assessed for autophagy status on day 1 of adulthood. Autophagy flux assays were performed by injecting BafA (BioViotica) or vehicle (DMSO, Sigma) into animals expressing rgef-1p::gfp::lgg-1 or rgef-1p::gfp::lgg-1(Gly116Ala)40. BafA was resuspended in DMSO to a stock concentration of 25 μM. The co-injection dye Texas Red dextran (3,000 MW; Molecular Probes) was resuspended in water to a stock concentration of 25 mg ml−1. The BafA or DMSO injection solution was injected close to the terminal pharyngeal bulb and animals were allowed to recover on 6-cm NGM plates with OP50 for 2 h. Surviving animals with intact nerve rings that scored positive for the red dye were used for GFP::LGG-1 punctae quantification. For imaging and punctae quantification, animals were mounted on a 2% agarose pad in M9 medium containing 0.1% sodium azide, and GFP-positive punctae were counted using a Zeiss Imager Z1 including apotome.2 with a Hamamatsu orca flash 4LT camera and Zen 2.3 software. The total number of GFP::LGG-1/Atg8-positive punctae was counted in body-wall muscle and the three to four most proximal intestinal cells at ×1,000 magnification. For imaging and punctae quantification of the nerve-ring neurons, z-stack images were acquired at a slice thickness of 1.0 μm.
PolyQ aggregation
The number of neuronal polyQ aggregates was counted in animals expressing rgef-1p::Q40::yfp in the nerve ring of individual animals on day 5 of adulthood8,9. Animals were raised at 20 °C on control HT115 bacteria or RNAi bacteria against the gene of interest. Animals were lined up on 6-cm NGM plates without food after anesthetization with M9 medium containing 0.1% sodium azide. Neuronal polyQ aggregates were counted under a Leica DFC310 FX camera at ×20 magnification.
Exopher measurements
Exophers were counted in animals expressing mec-4p::mCherry that were raised on OP50 or HT115 bacteria expressing empty vector or dsRNA from hatching or from the first day of adulthood43. Animals were scored on days 1–3, or just on day 2 of adulthood for exopher occurrence in a binary manner. Animals were either scored live on a Kramer dissecting scope with a ×20 objective or mounted on a microscope slide in a drop of M9 medium on an agarose pad, both containing 0.1% sodium azide as anesthesia using a Zeiss Imager Z1 including apotome at ×25 magnification. Each trial was graphed as a percentage of ALMR exophers. To determine colocalization of polyQ proteins in exophers, animals co-expressing mec-4p::mCherry and rgef-1p::Q40::yfp were assessed at day 2 of adulthood for exophers. mCherry-positive exophers in AMLR neurons were examined for polyQ colocalization, and z-stack images were obtained using a ×100 oil immersion objective, capturing stacks with a depth ranging from 4 to 8 μm at a slice thickness of 0.2 μm.
RT–qPCR
RT–qPCR was performed by isolating total RNA from a synchronized population of ∼2,000 day 1 animals raised on OP50 bacteria on 6-cm NGM plates, supplemented with hygromycin B where applicable9. After harvesting, the animal tissue was flash frozen in liquid nitrogen. RNA was extracted with TRIzol (Life Technologies), purified using a Qiagen RNeasy kit, and subjected to an additional DNA digestion step (Qiagen DNase I kit). Reverse transcription (1 μg RNA per sample) was performed using M-MuLV reverse transcriptase (Roche) and random 9-mer primers (New England Biolabs). qPCR was performed using the SYBR Green Master Mix (Roche) in a CFX384 machine (Bio-Rad). A standard curve was obtained for each primer set by serially diluting a mixture of different complementary DNAs, and the standard curves were used to convert the observed CT values to relative values. Three biological samples were analyzed, each with three technical replicates. The average and s.e.m. were calculated for each mRNA. mRNA levels of target genes were normalized to the mean of the housekeeping genes nhr-23 (nuclear hormone receptor), pmp-3 (putative ABC transporter) and cyn-1 (cyclophilin). Primer sequences are listed in Supplementary Table 10.
Statistics and reproducibility
Data were statistically analyzed using Excel, GraphPad Prism, STATA v.7 (Stata Corp) or Oasis.2 software72. No statistical methods were used to predetermine sample sizes, but our sample sizes are similar to those reported previously8,9,38,40,43,70. For all experiments, data distribution was assumed to be normal, but this was not formally tested. For two sample comparisons, an unpaired two-tailed t-test was used to determine significance. For three or more samples, a one-way ANOVA with Dunnett’s multiple-comparison test was used. For grouped comparisons, a two-way ANOVA with Tukey’s multiple comparisons was used to determine significance. Statistical significance of lifespan data was determined using a two-sided log-rank test, and statistics for lifespan data are provided in the Supplementary Information. The only data excluded from analyses were animals censored during lifespans. To compare the percentage mean lifespan change, we used a one-sample t-test and null-hypothesis testing. For the statistical comparison of the percentage of exophers formed, we used the Cochran–Mantel–Haenszel test. Blinding was performed where feasible, for most exopher measurements and for autophagy measurements assessing atg-16.2 rescue constructs. Animals for fluorescence measurements, autophagy measures and exopher measures were chosen under a bright-field microscope and therefore independent of their fluorescence intensity. No other randomization method was used.
Extended Data
Extended Data Fig. 1 |. Neuronal expression of SID-1, an RNA channel protein, leads to RNAi-competent neurons.

(a) Mean GFP fluorescence intensity in head region of rgef-1p::gfp animals on day 1 to day 12 of adulthood, relative to day 1. Error bars are s.d. of n = 3 experiments, with n = 29, 34, 32, 24, 28, 19 animals over 3 independent experiments. ns P = 0.78, P = 0.05, P = 0.116, ****P < 0.0001, **P = 0.005, by one-way ANOVA with Dunnett’s multiple comparisons test. (b-f) Wild-type animals (N2, WT), sid-1; rgef-1p::sid-1 + rgef-1p::gfp, and sid-1 mutants after whole-life RNAi against the indicated gene with tissue-specific functions, compared to control (CTRL). (b) Representative animals are shown with WT animals displaying paralysis (Prz) on unc-112 RNAi, larval arrest (Lva) on rpl-2 RNAi, blister formation (Bli) and larval arrest (Lva) on bli-1 RNAi, and clear (Clr) and larval arrest on elt-2 RNAi. Mean percent phenotypic penetrance after knockdown of genes with functions in (c) body-wall muscle; unc-22 – twitching and uncoordinated movement (Unc) (n = 8 experiments, ****P < 0.0001) and unc-112 – paralysis (n = 7 experiments, ****P < 0.0001), (d) a ubiquitous manner; rpl-2 – larval arrest (n = 8 experiments, ****P < 0.0001), (e) hypodermis; tsp-15 – blisters (n = 8 experiments, ****P < 0.0001) and bli-1 – blisters and larval arrest; (f) intestine; elt-2 – clear and larval arrest. Error bars are s.d. (g) Mean GFP fluorescence intensity in head region of day 1 sid-1; rgef-1p::sid-1 + rgef-1p::gfp rgef-1p::gfp animals after whole-life gfp RNAi compared to control (CTRL). Error bars are s.d. with n = 30 over 3 independent experiments. ****P < 0.0001, **P = 0.005, by two-tailed Student’s t-test. Scale bar: 100 μm. (h) Mean GFP fluorescence intensity in head region of day 1 sid-1; rgef-1p::sid-1 + rgef-1p::gfp rgef-1p::gfp animals after whole-life lgg-1 RNAi (n = 32) compared to control (CTRL) (n = 35). Error bars are s.d. over 3 independent experiments. ****P < 0.0001 by two-tailed Student’s t-test. Scale bar: 100 μm. (i) Mean percent of shrinker phenotype in day 2 WT, rde-1; unc-47p::rde-1::SL2::sid-1 (capable of GABA neuron-specific RNAi), sid-1; rgef-1p::sid-1 + rgef-1p::gfp, or sid-1 animals after two generations of whole-life snb-1 or unc-13 RNAi compared to control (CTRL). Error bars are s.d. with ****P < 0.0001 and ns P > 0.99 by one-way ANOVA with Dunnett’s multiple comparisons test.
Extended Data Fig. 2 |. Healthspan and neuronal phenotypes of animals after neuronal inhibition of atg-7 and lgg-1/ATG8.

(a) Mean body bends per 20 s of sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after whole-life atg-7, or lgg-1/ATG8 RNAi compared to control (CTRL). Error bars are s.e.m. of one representative experiment, each with n = 16 animals. Experiment was performed three times with similar results. Linear regression comparison versus CTRL: atg-7 RNAi: Pslope = 0.5; Py-intercept = 0.02; lgg-1/ATG8 RNAi: Pslope = 0.3; Py-intercept = 0.01. (b) Mean number of contractions in the terminal pharyngeal bulb per 30 s of sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after whole-life atg-7, or lgg-1/ATG8 RNAi compared to control (CTRL). Error bars are s.d. of n = 30 animals over 3 independent experiments. Linear regression comparison versus CTRL: atg-7 RNAi: Pslope = 0.4; Py-intercept = 0.4; lgg-1 RNAi: Pslope = 0.3; Py-intercept = 0.5. (c) Mean number of progeny produced per day in sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after atg-7 (n = 19 animals), or lgg-1/ATG8 RNAi (n = 18 animals) compared to control (CTRL) (n = 22 animals) over 2 independent experiments. Error bars are s.d. CTRL versus atg-7 RNAi: ns P = 0.72, 0.89, 0.91, 0.82, >0.99; CTRL versus lgg-1/ATG8 RNAi: ns P = 0.95, 0.96,0.60, 0.70, >0.99, by two-way ANOVA with Dunnett’s multiple comparisons test. (d) Analysis of integrity of sensory neurons in day 5 sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after atg-7, or lgg-1/ATG8 RNAi compared to control (CTRL). Sensory mutants daf-10(e1387) and osm-6(p811) are negative controls. Shown are representative images of n = 10 animals. Experiment was performed three times with similar results. Scale bar, 20 μm. (e) Representative image of neuronal branch (arrowhead) from ALM neuron in day 15 sid-1; rgef-1p::sid-1+ rgef-1p::gfp animals. Scale bar: 20 μm. Mean percent of animals with branches after whole-life atg-7, or lgg-1/ATG8 dsRNA compared to control (CTRL) of n = 4 experiments. Error bars are s.d. ***P = 0.0002, ****P < 0.0001 by Cochran-Mantel-Haenszel test. (f) Mean chemotaxis index of day 5 sid-1; rgef-1p::sid-1 + rgef-1p::gfp animals after whole-life atg-7, or lgg-1/ATG8 RNAi using the chemoattractant butanone. Error bars are 95% C.I. of n = 4 experiments. *P = 0.048, **P = 0.0094, by one-way ANOVA with Dunnett’s multiple comparisons test.
Extended Data Fig. 3 |. Autophagy status is unchanged in animals expressing sid-1 and in non-neuronal tissues after neuronal knockdown of early-acting autophagy genes.

(a) Mean neuronal GFP::LGG-1 and GFP::LGG-1(G116A) punctae in day 1 wild-type (sid-1 + /+) (n = 28, 29) and sid-1(qt9) (sid-1−/−) animals (n = 27, 27) with or without rgef-1p::sid-1 transgene (rgef-1p::sid-1 (+)) (n = 26, 29). Error bars are s.d. over 3 independent experiments. Comparison between strains: LGG-1: ns P = 0.75, P = 0.97, G116A: ns P = 0.98, P > 0.99. Comparison of lipidated and unlipidated structures: ****P < 0.0001, by two-way ANOVA with Tukey’s multiple comparisons test. (b) Mean sqst-1p::sqst-1::gfp fluorescence intensity in head region of day 1 wild-type (sid-1 + /+), and sid-1(qt9) (sid-1(−/−)) animals with or without rgef-1p::sid-1 transgene (rgef-1p::sid-1 (+)) on day 1 of adulthood. Error bars are s.d. of n = 31 animals over 3 independent experiments. ns P = 0.18 and P = 0.59 by one-way ANOVA with Dunnett’s multiple comparisons test. (c) GFP::LGG-1 punctae in intestinal cells of day 1 wild-type (sid-1 + /+) (n = 59) and sid-1(qt9) (sid-1−/−) animals (n = 62) with or without rgef-1p::sid-1 transgene (rgef-1p::sid-1 (+)) (n = 62). Violin plots with solid line indicating median and dashed lines indicating quartiles. ns P = 0.79, P = 0.99 by one-way ANOVA with Dunnett’s multiple comparisons test. (d) GFP::LGG-1 punctae in body-wall muscle areas of day 1 wild-type (sid-1 + /+) (n = 48) and sid-1(qt9) (sid-1−/−) animals (n = 45) with or without rgef-1p::sid-1 transgene (rgef-1p::sid-1 (+)) (n = 52). Violin plots with solid line indicating median and dashed lines indicating quartiles. ns P = 0.64, P = 0.70 by one-way ANOVA with Dunnett’s multiple comparisons test. (e) GFP::LGG-1 punctae in intestinal cells of day 1 sid-1; rgef-1p::sid-1 + rgef-1p::gfp; lgg-1p::gfp::lgg-1 animals after whole-life atg-7 (n = 65), or lgg-1/ATG8 (n = 48) RNAi compared to control (CTRL) (n = 81). Violin plots with solid line indicating median and dashed lines indicating quartiles. ns P = 0.98, P = 0.71 by one-way ANOVA with Dunnett’s multiple comparisons test. (f) GFP::LGG-1 punctae in body-wall muscle areas of day 1 sid-1; rgef-1p::sid-1 + rgef-1p::gfp; lgg-1p::gfp::lgg-1 animals after whole-life atg-7 (n = 54), or lgg-1/ATG8 (n = 53) RNAi compared to control (CTRL) (n = 49). Violin plots with solid line indicating median and dashed lines indicating quartiles. ns P = 0.34, P = 0.96 by one-way ANOVA with Dunnett’s multiple comparisons test.
Extended Data Fig. 4 |. Neuronal PolyQ aggregation, exopher formation and lifespan extension are correlated.

(a) Number of neuronal PolyQ aggregates in day 7 rgef-1::Q40::yfp wild-type (sid-1 + /+) (n = 41) and sid-1(qt9) (sid-1−/−) animals (n = 48 with or without rgef-1p::sid-1 transgene (rgef-1p::sid-1 (+)) (n = 42). Violin plots with solid line indicating median and dashed lines indicating quartiles. ns P = 0.79, P = 0.82 by one-way ANOVA with Dunnett’s multiple comparisons test. (b) Mean percent of ALMR neurons with exophers of day 2 mec-4p::mCherry wild-type (sid-1 + /+) (n = 228 animals) and sid-1(qt9) (sid-1−/−) (n = 262 animals) with or without rgef-1p::sid-1 transgene (rgef-1p::sid-1 (+)) (n = 295 animals). Error bars are s.d. of n = 7 experiments, ns P = 0.62, P = 0.54 by two-sided Cochran-Mantel-Haenszel test. (c) Mean percent of ALMR neurons with exophers of day 2 sid-1; rgef-1p::sid-1 + rgef-1p::gfp; mec-4p::mCherry animals (n = 247 animals) after adult-only atg-7 (n = 271 animals), or lgg-1/ATG8 (n = 285 animals) RNAi compared to control (CTRL). Error bars are s.d. of n = 6 experiments, *P = 0.028, P = 0.00006 by two-sided Cochran-Mantel-Haenszel test. (d) Mean percent of ALMR neurons with exophers of day 2 mec-4p::mCherry animals (n = 151 animals) after whole-life atg-7 (n = 157 animals), or lgg-1/ATG8 (n = 180 animals) RNAi compared to control (CTRL). Error bars are s.d. of n = 5 experiments, ns P = 0.39, P = 0.53 by two-sided Cochran-Mantel-Haenszel test. (e-g) Percent mean lifespan (LS) change (Fig. 1b), number of neuronal PolyQ aggregates (Fig. 3b), and mean percent of AMLR neurons with exophers (Fig. 4b) plotted against each other with simple linear regression (solid line with 95% C.I. as dashed lines). Numbers refer to specific RNAi treatment; 1unc-51/ATG1, 2atg-13, 3bec-1/BECN1, 4atg-9, 5atg-16.2, 6atg-7, 7atg-4.1, 8lgg-1/ATG8, 9cup-5, 10epg-5, 11vha-13, 12vha-15, 13vha-16. P values determined by two-sided Spearman correlation test.
Extended Data Fig. 5 |. atg-16.2 and atg-4.1 mutants display similar RNAi phenotypes.

(a-d) Phenotypes of day 2 WT, atg-16.2(ok3224), atg-4.1(bp501), and sid-1(qt9) animals after whole-life RNAi against a specific gene expressed in different tissues. Mean percent phenotypic penetrance after knockdown of genes with functions in (a) body-wall muscle; unc-22 – twitching and uncoordinated movement (Unc) and unc-112 – paralysis, (b) a ubiquitous manner; rpl-2 – larval arrest, (c) hypodermis; tsp-15 – blisters and bli-1 – blisters and larval arrest; (d) intestine; elt-2 – clear and larval arrest. Error bars are s.d. of n = 4 experiments. (a) unc-22 RNAi: ns P = 0.59, P = 0.87, ****P < 0.0001. unc-112 RNAi: ns P = 0.92, P = 0.96, ****P < 0.0001. (b) rpl-2 RNAi: ns P = 0.82, P = 0.96, ****P < 0.0001. (c) tsp-15 RNAi: ns P = 0.99, P = 0.64, ****P < 0.0001. bli-1 RNAi: ns P = 0.24, P = 0.99, ****P < 0.0001. (d) elt-2 RNAi: ns P = 0.76, P = 0.18, ****P < 0.0001 by one-way one-way ANOVA with Dunnett’s multiple comparisons test.
Extended Data Fig. 6 |. Multiple sequence alignment of ATG16 proteins, and atg-16.2 mRNA expression levels in C. elegans.
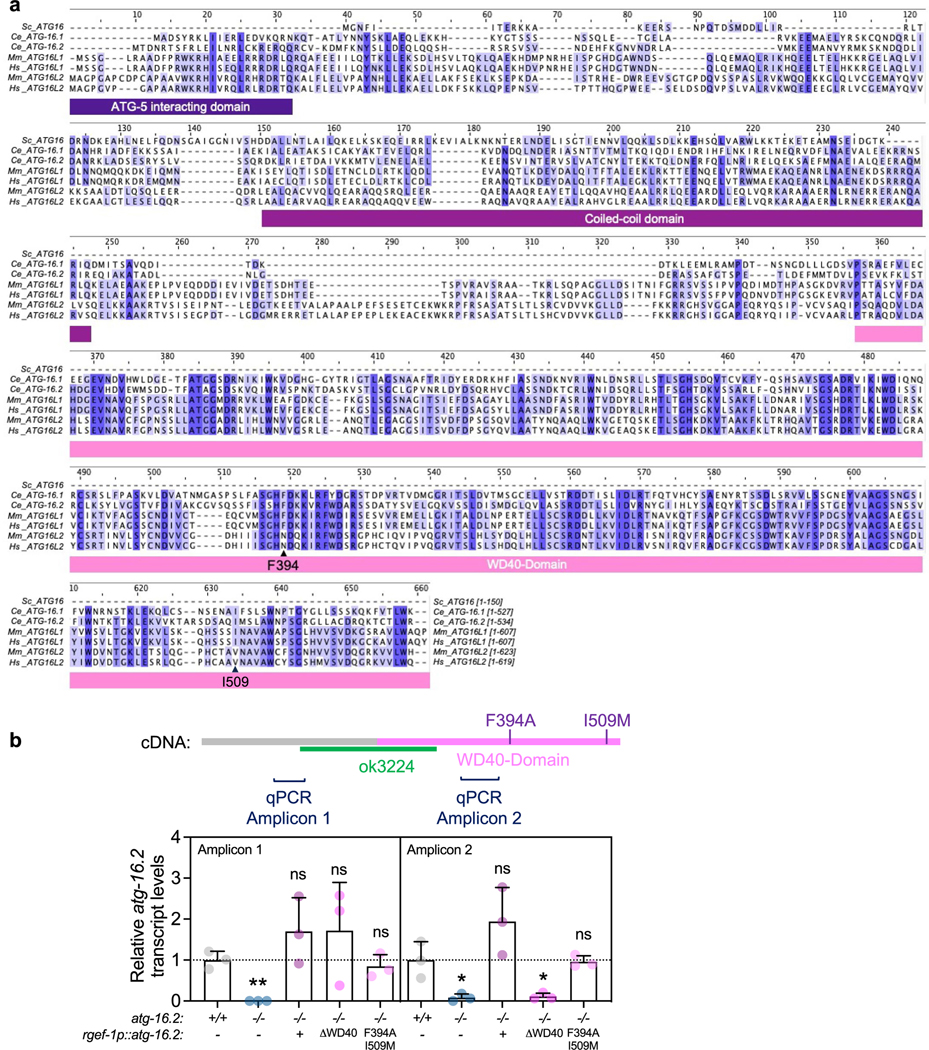
(a) Primary sequences of ATG16 proteins from S. cerevisiae (1 isoform, Sc_ATG16), C. elegans (2 isoforms, Ce_ATG-16.1, Ce_ATG-16.2), Mus musculus (2 isoforms, Mm_ATG16L1, MmATG16L2), and humans (2 isoforms, Hs_ATG16L1, Hs_ATG16L2) were aligned by the Clustal Omega and colored by conservation with darker shades indicated increased conservation ( Jalview). Protein elements of ATG16 proteins are drawn under the alignment and phenylalanine 349 and isoleucine 509 are indicated with an arrow. Sequences showed are ATG16 (NP_013882.1) in S. cerevisiae, ATG16.2 (NP_495299.2) in C. elegans, ATG16 (NP_001138124.2) in D. melanogaster, ATG16L1 (NP_001192320.1) in M. musculus, and ATG16L1 (NP_001350671.1) in H. sapiens. (b) Transcript levels of atg-16.2 in wild-type (WT), atg-16.2(ok3224), and atg-16.2(ok3224) animals expressing full-length atg-16.2, atg-16.2(ΔWD40), or atg-16.2(Phe394A, Ile509Met) from the neuronal rgef-1 promoter. Schematic of atg-16.2 cDNA indicates the ok3224 deletion, the WD40 domain, the position of the Phe394Ala and Ile509Met point mutations, and the amplicons produced by the primers used in this experiment. Data are the mean and s.e.m. of three biological replicates, each with three technical replicates, and are normalized to the mean expression levels of three housekeeping genes. Amplicon 1: **P = 0.0013, ns P = 0.23, P = 0.36, P = 0.49, Amplicon 2: *P = 0.03, ns P = 0.16, *P = 0.03, ns P = 0.89 by unpaired two-sided t-test with control.
Supplementary Material
Acknowledgements
We thank members of the M.H. and C.K. laboratories for comments on the manuscript. We thank X. She, E. P. Tan, S. Lim and K. Schilling from the M.H. laboratory for creating some of the strains used in this study. We thank H. Zhang for sharing construct atg-16.2p::atg-16.2::gfp, R. Legouis for the GFP::LGG-1(Gly116Ala) strain and B. Grant for sharing the hygromycin resistance-containing plasmid. We thank O. Florey for help with designing the Phe394 mutation in the C. elegans ATG-16.2 WD40 domain. We thank N. Riehs and C. Kenyon for sharing unpublished information on neuronal-only RNAi of autophagy genes. Some of the nematode strains used in this work were provided by the Caenorhabditis Genetics Center (University of Minnesota), which is supported by the NIH Office of Research Infrastructure Program (P40OD010440). We acknowledge the C. elegans Gene Knockout Project at the Oklahoma Medical Research Foundation, which was part of the International C. elegans Gene Knockout Consortium, for generating atg-16.2(ok3224) mutation and WormBase WS283. This work was funded by NIA/NIH fellowship F31AG066405 to M.L.A., a Paul F. Glenn Center Fellowship and Larry L. Hillblom Fellowship 2023-A-019-FEL to H.E., a Taiwan Global Fellowship to L.-H.S., NIH/NIA grants R37AG056510 (to M.D.), R01AG038664 and R01AG039756 and Breakthrough in Gerontology Award BIG 20016 from American Association for Aging Research (to M.H.). The content of this article is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Footnotes
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Competing interests
The authors declare no competing interests. The funders had no role in the study design, data collection and analysis, decision to publish or preparation of the manuscript.
Additional information
Extended data is available for this paper at https://doi.org/10.1038/s43587-023-00548-1.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s43587-023-00548-1.
Data availability
The authors declare that all data supporting the findings of this study are available within this article, extended data and Supplementary Information. Source data are available with this paper. Additional information on data is available from the corresponding authors upon reasonable request. The unique identifiers for ATG-16.2 sequences are stated in the figure legends.
References
- 1.Hansen M, Rubinsztein DC & Walker DW Autophagy as a promoter of longevity: insights from model organisms. Nat. Rev. Mol. Cell Biol 19, 579–593 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Miller HA, Dean ES, Pletcher SD & Leiser SF Cell non-autonomous regulation of health and longevity. Elife 10.7554/eLife.62659 (2020). [DOI] [PMC free article] [PubMed]
- 3.Simonsen A. et al. Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184 (2008). [DOI] [PubMed] [Google Scholar]
- 4.Ulgherait M, Rana A, Rera M, Graniel J. & Walker DW AMPK modulates tissue and organismal aging in a non-cell-autonomous manner. Cell Rep. 8, 1767–1780 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hara T. et al. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441, 885–889 (2006). [DOI] [PubMed] [Google Scholar]
- 6.Komatsu M. et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441, 880–884 (2006). [DOI] [PubMed] [Google Scholar]
- 7.Jia K, Hart AC & Levine B. Autophagy genes protect against disease caused by polyglutamine expansion proteins in Caenorhabditis elegans. Autophagy 3, 21–25 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Kumsta C. et al. The autophagy receptor p62/SQST-1 promotes proteostasis and longevity in C. elegans by inducing autophagy. Nat. Commun 10, 5648 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kumsta C, Chang JT, Schmalz J. & Hansen M. Hormetic heat stress and HSF-1 induce autophagy to improve survival and proteostasis in C. elegans. Nat. Commun 8, 14337 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kamath RS & Ahringer J. Genome-wide RNAi screening in Caenorhabditis elegans. Methods 30, 313–321 (2003). [DOI] [PubMed] [Google Scholar]
- 11.Timmons L, Court DL & Fire A. Ingestion of bacterially expressed dsRNAs can produce specific and potent genetic interference in Caenorhabditis elegans. Gene 263, 103–112 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Winston WM, Molodowitch C. & Hunter CP Systemic RNAi in C. elegans requires the putative transmembrane protein SID-1. Science 295, 2456–2459 (2002). [DOI] [PubMed] [Google Scholar]
- 13.Feinberg EH & Hunter CP Transport of dsRNA into cells by the transmembrane protein SID-1. Science 301, 1545–1547 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Mizushima N, Yoshimori T. & Ohsumi Y. The role of Atg proteins in autophagosome formation. Annu. Rev. Cell Dev. Biol 27, 107–132 (2011). [DOI] [PubMed] [Google Scholar]
- 15.Noda T, Fujita N. & Yoshimori T. The late stages of autophagy: how does the end begin? Cell Death Differ. 16, 984–990 (2009). [DOI] [PubMed] [Google Scholar]
- 16.Noda NN & Inagaki F. Mechanisms of autophagy. Annu. Rev. Biophys 44, 101–122 (2015). [DOI] [PubMed] [Google Scholar]
- 17.Klionsky DJ et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang H. et al. Guidelines for monitoring autophagy in Caenorhabditis elegans. Autophagy 11, 9–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cadwell K. & Debnath J. Beyond self-eating: the control of nonautophagic functions and signaling pathways by autophagy-related proteins. J. Cell Biol 217, 813–822 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fletcher K. et al. The WD40 domain of ATG16L1 is required for its non-canonical role in lipidation of LC3 at single membranes. EMBO J. 10.15252/embj.201797840 (2018). [DOI] [PMC free article] [PubMed]
- 21.Martinez J. et al. Molecular characterization of LC3-associated phagocytosis reveals distinct roles for Rubicon, NOX2 and autophagy proteins. Nat. Cell Biol 17, 893–906 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 22.Heckmann BL et al. Noncanonical function of an autophagy protein prevents spontaneous Alzheimer’s disease. Sci. Adv 6, eabb9036 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nieto-Torres JL, Leidal AM, Debnath J. & Hansen M. Beyond autophagy: the expanding roles of ATG8 proteins. Trends Biochem. Sci 46, 673–686 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Melentijevic I. et al. C. elegans neurons jettison protein aggregates and mitochondria under neurotoxic stress. Nature 542, 367–371 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Turek M. et al. Muscle-derived exophers promote reproductive fitness. EMBO Rep. 22, e52071 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicolas-Avila JA et al. A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183, 94–109 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Siddique I. et al. Exophers are components of mammalian cell neurobiology in health and disease. Preprint at bioRxiv 10.1101/2021.12.06.471479 (2021). [DOI]
- 28.Calixto A, Chelur D, Topalidou I, Chen X. & Chalfie M. Enhanced neuronal RNAi in C. elegans using SID-1. Nat. Methods 7, 554–559 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roux AE et al. Individual cell types in C. elegans age differently and activate distinct cell-protective responses. Cell Rep. 42, 112902 (2023). [DOI] [PubMed] [Google Scholar]
- 30.WormBase WS283. http://www.wormbase.org/db/get?name=WBGene00006843;class=Gene (2023).
- 31.Wilhelm T. et al. Neuronal inhibition of the autophagy nucleation complex extends lifespan in post-reproductive C. elegans. Genes Dev. 31, 1561–1572 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Apfeld J. & Kenyon C. Regulation of lifespan by sensory perception in Caenorhabditis elegans. Nature 402, 804–809 (1999). [DOI] [PubMed] [Google Scholar]
- 33.Tank EM, Rodgers KE & Kenyon C. Spontaneous age-related neurite branching in Caenorhabditis elegans. J. Neurosci 31, 9279–9288 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toth ML et al. Neurite sprouting and synapse deterioration in the aging Caenorhabditis elegans nervous system. J. Neurosci 32, 8778–8790 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Melendez A. et al. Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301, 1387–1391 (2003). [DOI] [PubMed] [Google Scholar]
- 36.Gelino S. et al. Intestinal autophagy improves healthspan and longevity in C. elegans during dietary restriction. PLoS Genet. 12, e1006135 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Manil-Segalen M. et al. The C. elegans LC3 acts downstream of GABARAP to degrade autophagosomes by interacting with the HOPS subunit VPS39. Dev. Cell 28, 43–55 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Chang JT, Kumsta C, Hellman AB, Adams LM & Hansen M. Spatiotemporal regulation of autophagy during Caenorhabditis elegans aging. Elife 10.7554/eLife.18459 (2017). [DOI] [PMC free article] [PubMed]
- 39.Kuma A, Matsui M. & Mizushima N. LC3, an autophagosome marker, can be incorporated into protein aggregates independent of autophagy: caution in the interpretation of LC3 localization. Autophagy 3, 323–328 (2007). [DOI] [PubMed] [Google Scholar]
- 40.Chang JT, Hansen M. & Kumsta C. Assessing tissue-specific autophagy flux in adult Caenorhabditis elegans. Methods Mol. Biol 2144, 187–200 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yamamoto A. et al. Bafilomycin A1 prevents maturation of autophagic vacuoles by inhibiting fusion between autophagosomes and lysosomes in rat hepatoma cell line, H-4-II-E cells. Cell Struct. Funct 23, 33–42 (1998). [DOI] [PubMed] [Google Scholar]
- 42.Wilkinson DS et al. Phosphorylation of LC3 by the Hippo kinases STK3/STK4 is essential for autophagy. Mol. Cell 57, 55–68 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Arnold ML, Cooper J, Grant BD & Driscoll M. Quantitative approaches for scoring in vivo neuronal aggregate and organelle extrusion in large exopher vesicles in C. elegans. J. Vis. Exp 10.3791/61368 (2020). [DOI] [PMC free article] [PubMed]
- 44.Wu F, Li Y, Wang F, Noda NN & Zhang H. Differential function of the two Atg4 homologues in the aggrephagy pathway in Caenorhabditis elegans. J. Biol. Chem 287, 29457–29467 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H. et al. The two C. elegans ATG-16 homologs have partially redundant functions in the basal autophagy pathway. Autophagy 9, 1965–1974 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsushita M. et al. Structure of Atg5.Atg16, a complex essential for autophagy. J. Biol. Chem 282, 6763–6772 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Arnold ML et al. Intermediate filaments associate with aggresome-like structures in proteostressed C. elegans neurons and influence large vesicle extrusions as exophers. Nat. Commun 14, 4450 (2023). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Durgan J. et al. Non-canonical autophagy drives alternative ATG8 conjugation to phosphatidylserine. Mol. Cell 81, 2031–2040 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hooper KM et al. V-ATPase is a universal regulator of LC3-associated phagocytosis and non-canonical autophagy. J. Cell Biol 10.1083/jcb.202105112 (2022). [DOI] [PMC free article] [PubMed]
- 50.Ishibashi K, Uemura T, Waguri S. & Fukuda M. Atg16L1, an essential factor for canonical autophagy, participates in hormone secretion from PC12 cells independently of autophagic activity. Mol. Biol. Cell 23, 3193–3202 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Guo H. et al. Atg5 disassociates the V1V0-ATPase to promote exosome production and tumor metastasis independent of canonical macroautophagy. Dev. Cell 43, 716–730 (2017). [DOI] [PubMed] [Google Scholar]
- 52.Varga K. et al. Loss of Atg16 delays the alcohol-induced sedation response via regulation of Corazonin neuropeptide production in Drosophila. Sci. Rep 6, 34641 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Choi AM, Ryter SW & Levine B. Autophagy in human health and disease. N. Engl. J. Med 368, 651–662 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Wong E. & Cuervo AM Autophagy gone awry in neurodegenerative diseases. Nat. Neurosci 13, 805–811 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cortes CJ & La Spada AR Autophagy in polyglutamine disease: imposing order on disorder or contributing to the chaos? Mol. Cell Neurosci 66, 53–61 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kim DK et al. Cell-to-cell transmission of polyglutamine aggregates in C. elegans. Exp. Neurobiol 26, 321–328 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Pearce MMP, Spartz EJ, Hong W, Luo L. & Kopito RR Prion-like transmission of neuronal huntingtin aggregates to phagocytic glia in the Drosophila brain. Nat. Commun 6, 6768 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jeon I. et al. Human-to-mouse prion-like propagation of mutant huntingtin protein. Acta Neuropathol. 132, 577–592 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Takeuchi T. & Nagai Y. Emerging roles of extracellular vesicles in polyglutamine diseases: mutant protein transmission, therapeutic potential, and diagnostics. Neurochem. Int 157, 105357 (2022). [DOI] [PubMed] [Google Scholar]
- 60.Zhang X. et al. Potential transfer of polyglutamine and CAG-repeat RNA in extracellular vesicles in Huntington’s disease: background and evaluation in cell culture. Cell Mol. Neurobiol 36, 459–470 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Fussi N. et al. Exosomal secretion of alpha-synuclein as protective mechanism after upstream blockage of macroautophagy. Cell Death Dis. 9, 757 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Abdulrahman BA, Abdelaziz DH & Schatzl HM Autophagy regulates exosomal release of prions in neuronal cells. J. Biol. Chem 293, 8956–8968 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Miranda AM et al. Neuronal lysosomal dysfunction releases exosomes harboring APP C-terminal fragments and unique lipid signatures. Nat. Commun 9, 291 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura S. et al. Suppression of autophagic activity by Rubicon is a signature of aging. Nat. Commun 10, 847 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Brenner S. The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Shen K. & Bargmann CI The immunoglobulin superfamily protein SYG-1 determines the location of specific synapses in C. elegans. Cell 112, 619–630 (2003). [DOI] [PubMed] [Google Scholar]
- 67.Rual JF et al. Toward improving Caenorhabditis elegans phenome mapping with an ORFeome-based RNAi library. Genome Res. 14, 2162–2168 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hansen M, Hsu AL, Dillin A. & Kenyon C. New genes tied to endocrine, metabolic, and dietary regulation of lifespan from a Caenorhabditis elegans genomic RNAi screen. PLoS Genet. 1, 119–128 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y. et al. Large vesicle extrusions from C. elegans neurons are consumed and stimulated by glial-like phagocytosis activity of the neighboring cell. Elife 10.7554/eLife.82227 (2023). [DOI] [PMC free article] [PubMed]
- 70.Kauffman A. et al. C. elegans positive butanone learning, short-term, and long-term associative memory assays. J. Vis. Exp 10.3791/2490 (2011). [DOI] [PMC free article] [PubMed]
- 71.Kauffman AL, Ashraf JM, Corces-Zimmerman MR, Landis JN & Murphy CT Insulin signaling and dietary restriction differentially influence the decline of learning and memory with age. PLoS Biol. 8, e1000372 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Han SK et al. OASIS 2: online application for survival analysis 2 with features for the analysis of maximal lifespan and healthspan in aging research. Oncotarget 7, 56147–56152 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within this article, extended data and Supplementary Information. Source data are available with this paper. Additional information on data is available from the corresponding authors upon reasonable request. The unique identifiers for ATG-16.2 sequences are stated in the figure legends.