

Abstract
Absorption, distribution, metabolism, and excretion (ADME) are the key biologic processes for determination of a drug’s pharmacokinetic parameters, which have direct impacts on efficacy and adverse drug reactions (ADRs). The chemical structures, dosage forms, and sites and routes of administration are the principal determinants of ADME profiles and consequent impacts on their efficacy and ADRs. Newly developed large molecule biologic antisense oligonucleotide (ASO) drugs have completely unique ADME that is not fully defined. ASO-based drugs are single-stranded synthetic antisense nucleic acids with diverse modes of drug actions from induction of mRNA degradation, exon skipping and restoration, and interactions with proteins. ASO drugs have a great potential to treat certain human diseases that have remained untreatable with small molecule-based drugs. The ADME of ASO drugs contributes to their unique set of ADRs and toxicity. In this review, to better understand their ADME, the 10 US Food and Drug Administration (FDA)-approved ASO drugs were selected: fomivirsen, pegaptanib, mipomersen, nusinersen, inotersen, defibrotide, eteplirsen, golodirsen, viltolarsen, and casimersen. A meta-analysis was conducted on their formulation, dosage, sites of administration, local and systematic distribution, metabolism, degradation, and excretion. Membrane permeabilization through endocytosis and nucleolytic degradation by endonucleases and exonucleases are major ADME features of the ASO drugs that differ from small-molecule drugs. The information summarized here provides comprehensive ADME characteristics of FDA-approved ASO drugs, leading to a better understanding of their therapeutic efficacy and their potential ADRs and toxicity. Numerous knowledge gaps, particularly on cellular uptake and subcellular trafficking and distribution, are identified, and future perspectives and directions are discussed.
SIGNIFICANCE STATEMENT
Through a systematic analysis of the existing information of absorption, distribution, metabolism, and excretion (ADME) parameters for 10 US Food and Drug Administration (FDA)-approved antisense oligonucleotide (ASO) drugs, this review provides an overall view of the unique ADME characteristics of ASO drugs, which are distinct from small chemical drug ADME. This knowledge is useful for discovery and development of new ASO drugs as well as clinical use of current FDA-approved ASO drugs.
Introduction
Absorption, distribution, metabolism, and excretion (ADME) of a drug affect the biologic and physiologic processes that determine the drug concentrations reaching to its organ(s) and tissue(s) over time. ADME parameters influence whether desired therapeutic goals can be reached while undesired adverse drug reactions (ADRs) can be avoided. ADRs can often be reduced by choosing the correct administration dose and route of administration, including newly developed classes of biologic drugs, such as antisense oligonucleotide (ASO) drugs (Amantana and Iversen, 2005).
ASO drugs are single-stranded nucleic acid polymers intended for the treatment of common and rare human diseases (Smith and Zain, 2019). Their principles of drug action involve the modulation of the production or function of disease-causing proteins via alterations of pre-mRNA splicing, mRNA degradation, regulation of protein translation, or direct interactions with proteins (Di Fusco et al., 2019). Because of the ability to directly alter the production of disease-causing proteins, the ASO drugs have shown significant potential to bring transformative treatments to a variety of human diseases (Bennett et al., 2019), particularly some diseases, such as Duchenne muscular dystrophy (DMD), for which small chemical drugs are not available (Quemener et al., 2020). ADME processes of ASO drugs have direct impacts on therapeutic efficacy and ADRs; therefore, the understanding of general ADME features of ASO drugs and their unique characteristics is vital. Because the ADME characteristics of ASO drugs differ substantially from small chemical drugs, understanding of ADME is critical for the discovery and development of new ASO drugs with greater efficacy and improved safety profiles as well as for the clinical practice of approved ASO drugs. By analyzing all ASO drugs approved by the US Food and Drug Administration (FDA), this article aims to provide an overview of both class-wide and ASO-specific ADME features of the 10 FDA-approved ASO drugs and how these features differ from those of small molecule drugs.
Brief Historical Perspective
The discovery and development of ASO drugs have a history of nearly half a century (Oberemok et al., 2018), dating back to 1978 when a specific oligonucleotide was first discovered to be able to inhibit Rous sarcoma viral RNA translation (Stephenson and Zamecnik, 1978). After nearly 20 years of effort by the pharmaceutical industry, the FDA approved the first ASO drug, fomivirsen, for the treatment of cytomegalovirus (CMV) retinitis in 1998 (Fig. 1A) (Roehr, 1998). However, fomivirsen was withdrawn from the market by the European Union in 2002 and in the US in 2006 due to the success of other highly active antiretroviral therapy and the reduced incidence of CMV infection (Shahryari et al., 2019). After a gap of more than a decade with only one drug (pegaptanib) approved in 2004, the FDA has approved eight ASO drugs since 2013, including mipomersen, nusinersen, inotersen, defibrotide, eteplirsen, golodirsen, viltolarsen, and casimersen (Fig. 1A). However, when mipomersen was approved by the FDA for the treatment of familial hypercholesterolemia, a black box warning was placed in the drug label describing the risk of hepatotoxicity (Dixon et al., 2014). Because of this serious safety warning and unacceptable risk, the FDA withdrew mipomersen from the US market in 2019 only after 6 years of use. Recently, further refinement of the ASO drug discovery and development platform has led to a much faster pace in the emergence of new members of this class of drugs used in the treatments of muscular disorders, neurologic disorders, and various other diseases (Bennett et al., 2017; Dhuri et al., 2020) (Fig. 1). More than 100 ASO drugs have been enrolled in various phases of clinical trials, with some close to be approved within a few years (Crooke et al., 2021). The annual market sales of ASO drugs have been estimated to be approximately $1 billion in 2021 and are expected to increase to $1.62 billion by 2025 with a steady annual growth rate of approximately 13%.
Fig. 1.

Timeline of the 10 FDA-approved antisense oligonucleotide (ASO) drugs with FDA approval dates, companies that developed the drugs, and target diseases. FH, familial hypercholesterolemia; PNA, polyneuropathy of amyloidosis; SMA, spinal muscular atrophy; VOD, veno-occlusive disease.
Based on their main modes of drug action, the 10 FDA-approved ASO drugs can be classified into five groups (Fig. 2). 1) Group 1 drugs induce RNase H-mediated mRNA degradation, including fomivirsen targeting on CMV IE2 mRNA, mipomersen on apolipoprotein B (Apo B) mRNA, and inotersen on transthyretin (TTR) mRNA (Fig. 2A). 2) Group 2 drugs induce exon skipping, including eteplirsen for dystrophin exon 51 skipping, golodirsen for dystrophin exon 53 skipping, viltolarsen for dystrophin exon 53 skipping, and casimersen for dystrophin exon 45 skipping (Fig. 2B). 3) Nusinersen in Group 3 works as exon restoration. The target of nusinersen is the pre-mRNA of survival motor neuron 2 (SMN2) (Wurster and Ludolph, 2018). Upon ASO binding, SMN2 pre-mRNA then undergoes an ASO-induced splicing event, resulting in inclusion of exon 7 (Wurster and Ludolph, 2018). With exon 7 included, a fully functional SMN2 mRNA is transcribed and levels of SMN protein are increased (Fig. 2C). 4) Pegaptanib in Group 4 works via an aptamer-protein complex by interacting with vascular endothelial growth factor (VEGF) receptor (Fig. 2D). 5) Defibrotide in Group 5 targets on multiple targets. Defibrotide works on veno-occlusive disease by antagonistic mechanisms. Defibrotide is a polydisperse mixture of various single-stranded oligonucleotide particles. By binding to adenosine receptors A1 and A2, defibrotide reduces systemic levels of plasminogen activator inhibitor 1 and increases concentrations of endogenous prostaglandins (I2 and E2), which modulates thrombomodulin, platelets, and fibrinolysis and thus results in the anti-inflammatory, antiatherosclerotic, anti-ischemic, and antithrombotic property of defibrotide (Baker and Demaris, 2016).
Fig. 2.
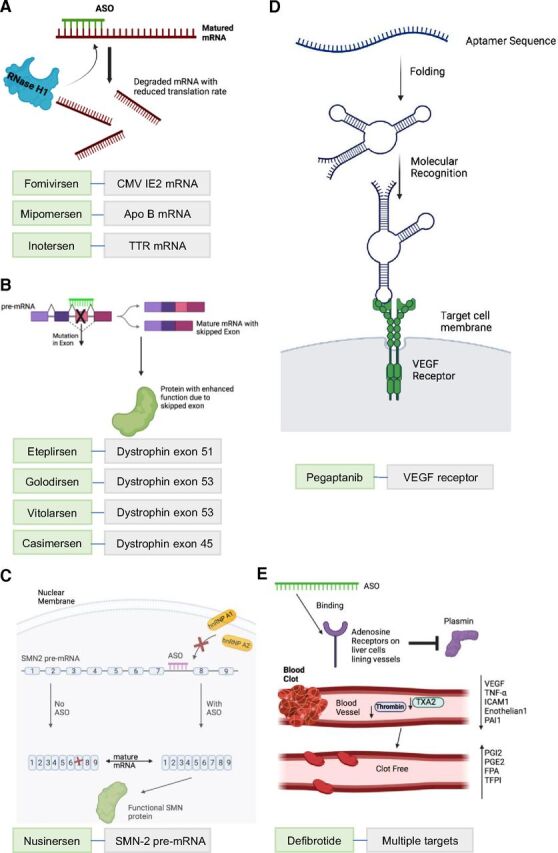
The mode of drug action of the 10 FDA-approved ASO drugs. (A) ASOs with RNase H-mediated mRNA degradation. By binding to their target mRNAs, ASO drugs cause the targeted mRNAs to be broken down into smaller pieces within the cell. ASO drugs in this modality are fomivirsen targeting on CMV IE2 mRNA, mipomersen targeting on Apo B mRNA, and inotersen targeting on transthyretin (TTR) mRNA. (B) ASO-mediated exon skipping. By sterically blocking a splicing site in the exon with mutations, ASO drugs result in skipping of the mutated exon and increased production of protein with enhanced function due to the skipped exon. ASO drugs in this mode include eteplirsen for exon 51 skipping, golodirsen and viltolarsen for exon 53 skipping, and casimersen for exon 45 skipping in Dystrophin mRNA. (C) ASO-mediated exon inclusion. By sterically blocking the splicing site in intron 7 of the SMN2 pre-mRNA, nusinersen blocks key intrinsic splicing factors heterogeneous nuclear ribonucleoprotein A1 (hnRNP A1) and heterogeneous nuclear ribonucleoprotein A2 (hnRNP A2) and results in translation of full-length, functional SMN2 protein. (D) Aptamer ASO. Pegaptanib is a polynucleotide aptamer, which undergoes folding to bind to targeted vascular endothelial growth factor (VEGF). This prevents a cascade of kinase-mediated cell signaling, which then results in angiogenesis. (E) ASO with multiple modes. Defibrotide aims to bind to adenosine receptors and trigger a cascade of events. Plasmin is inhibited and can no longer hydrolyze clots. Key proclotting factors such as VEGF, tumor necrosis factor (TNF-α), intracellular adhesion molecule 1 (ICAM1), endothelium, plasminogen activator inhibitor 1 (PAI1), thromboxane A2 (TXA2), and thrombin are decreased. Anticlotting factors such as prostaglandins I2 and E2 and tissue factor pathway inhibitor (TFPI) all are increased. Fibrinopeptide A levels are increased due to chronic coagulation from disease. Clot-free blood vessels lead to general healing of damaged endothelial cells and tissue (Pescador et al., 2013).
As with any other drug under discovery and development, the information available on the ADME features for these ASO drugs has been part of the vast battery of studies of an investigational drug with the anticipation that some ADME principles would vary for ASO drug molecules from traditional drugs (Weidolf et al., 2021). ADME properties are useful for the generation of preclinical pharmacokinetic predictions of drug efficacy at specific doses and frequency of treatment in clinical studies (Reichel and Lienau, 2016; Wang et al., 2019b).
Key Recent Advances
The ADME features of the 10 FDA-approved ASO drugs were investigated to find common class-wide characteristics and unique features of individual ASO drugs. This analysis was based on available information from the FDA ASO drug labeling, the outcomes of clinical trials from ASO investigational new drug (IND) applications, and the published literature.
Administration
The FDA-approved ASO drugs can be divided into four groups based on their administration routes: (A) intravitreal injection for fomivirsen and pegaptanib (Fig. 3A), (B) intrathecal injection for nusinersen (Fig. 3B), (C) subcutaneous injection (s.c.) for mipomersen and inotersen (Fig. 3C), and (D) intravenous infusion (i.v.) for defibrotide, eteplirsen, golodirsen, viltolarsen, and casimersen (Fig. 3D). It should be noted that defibrotide may be administered orally for long-term usage, though this formulation is not available yet. Each ASO drug uses a specific route of administration to treat its target disease or condition.
Fig. 3.

Administration paths, formulation, and dosing information of the FDA-approved ASO drugs. The ASO drugs are divided based on their administration paths with (A) intravitreal injection, (B) intrathecal injection, (C) subcutaneous injection, and (D) intravenous infusion. The information for formulation and dosing is extracted from the FDA drug labels at Drugs@FDA: FDA-Approved Drugs (https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm).
Fomivirsen is used to treat CMV retinitis in the eye and is administered by direct intravitreal injection with a 50 μl solution containing 0.33 mg of the drug (de Smet et al., 1999). Fomivirsen has two phases of administration: loading and maintenance phases. The loading dose consists of two intravitreal injections every two weeks, and the maintenance dose is given only once every 4 weeks. Pegaptanib is used to treat neovascular macular degeneration in the back of the eye; therefore, it is administered by direct intravitreal injection of a 90 μl solution containing 0.3 mg pegaptanib once every 4 weeks.
Nusinersen was designed for the treatment of spinal muscular atrophy (Goodkey et al., 2018). A direct intrathecal injection of a 5 ml solution containing 12 mg drug is used to deliver the drug to the spinal cord. Nusinersen also has two phases of administration: loading and maintenance phases. In the loading phase, the first 3 doses are given at 14-day intervals with the fourth dose given 30 days after the third dose. Maintenance doses are subsequently provided once every 4 months. Special instructions for sedating patients must be followed when performing the intrathecal injections of nusinersen and are guided by ultrasound or other imaging techniques. A removal of 5 ml cerebrospinal fluid over 1–3 minutes using a spinal anesthesia needle is recommended prior to the nusinersen administration.
Mipomersen is the ASO drug developed for the treatment of familial hypercholesterolemia. It works by targeting Apo B mRNA and stimulating its degradation in the liver. The ensuing reduction in Apo B protein is responsible for the lipid-lowering effect of mipomersen (Parham and Goldberg, 2019). An s.c. injection of 200 mg mipomersen in 1 ml solution is applied once weekly for up to 6 months to achieve the therapeutic goal. Mipomersen should not be administered by intramuscular or intravenous injection according to the FDA drug label (Wong and Goldberg, 2014). Inotersen was developed to treat polyneuropathy of amyloidosis caused by over-deposits of TTR protein in tissues, which is mainly produced in the liver (Benson et al., 2018; Keam, 2018). Inotersen is administered by an s.c. injection with a 1 ml solution containing 284 mg drug once weekly. Just like mipomersen, inotersen must be injected subcutaneously.
Defibrotide was approved to treat hepatic veno-occlusive disease, and the mechanism of action of defibrotide involves an enhancement of the enzymatic activity of plasmin to hydrolyze fibrin clots (Richardson et al., 2017). Defibrotide is given as a 2-hour i.v. infusion every 6 hours at a dose of 6.25 mg/kg for a total of 21 days (Richardson et al., 2021).
Eteplirsen, golodirsen, viltolarsen, and casimersen were all developed to treat DMD. They all target the dystrophin gene pre-mRNA, resulting in the skipping of specific mutation-containing exons. Their administration is via i.v. infusion for approximately 30–60 minutes once weekly at a dose of 30 mg/kg for eteplirsen, golodirsen, and casimersen and 80 mg/kg for viltolarsen. In addition, viltolarsen is infused via i.v. using a peripheral or central venous catheter and cannot be infused concomitantly with other medications through the same i.v. access line.
Small chemical drugs typically have a high oral availability compared to large molecule therapeutics despite being metabolized by phase I/II enzymes in the liver, intestine, and kidney. Due to oral dosing, the half-life is normally very short and requires daily dosing, sometimes multiple times daily. The longer half-life of ASO drugs allows less frequent dosing requirements, such as weekly, monthly, and even yearly.
Absorption
Absorption is a key parameter that affects pharmacokinetics and defines the movement of the unmetabolized drug from the site of administration to the local or systematic circulation (Alagga and Gupta, 2021). Different factors can affect the absorption of drugs into the body. These include drug administration routes and first-pass metabolism. Both factors have an impact on drug bioavailability, measured as percentage of the bioactive form of drug out of the total amount of the parent drug administered. Absorption will result in a peak time of plasma drug concentration. The peak plasma time is the time needed to achieve the highest level of drug present in plasma reflective of absorption of the drug into the body. The 10 FDA-approved ASO drugs investigated in this review have various absorption patterns related to the disease state intended to treat, how the drug is administered, and peak plasma concentration time (Table 1).
TABLE 1.
Local and systematic absorption of the FDA-approved ASO drugs
The data are extracted from the FDA drug labels of each ASO drug.
| Drug | AdministrationSite and Route | Local vs. Systematic Absorption | Ability to Cross Blood-Brain Barrier | Tissues with Highest Drug Concentrations | Peak Plasma Concentration Time (Hours) |
|---|---|---|---|---|---|
| Fomivirsen | Intravitreal injection | Local eye | No | Retina, iris; systemic exposure: minimal (below limits of quantitation) | Very low |
| Pegaptanib | Intravitreal injection | Local eye | No | Vitreous fluid, retina, aqueous fluid; systemic exposure: kidney | Slowly absorbed into systemic circulation |
| Nusinersen | Intrathecal injection | CNS and systematic | Yes | CNS, plasma, peripheral tissue (skeletal muscle, liver, kidney), fat, bone marrow, spleen | 1.7–6 |
| Mipomersen | S.C. injection | Systematic | No | Liver, kidney, bone marrow, adipose tissue, lymph nodes | 3–4 |
| Inotersen | S.C. injection | Systematic | No | Liver, kidney, broad tissues | 2–4 |
| Defibrotide | I.V. infusion | Systematic | No | Cells lining blood vessel in liver | 2 |
| Eteplirsen | I.V. infusion | Systematic | No | Kidney, skeletal muscles | 1–2 |
| Golodirsen | I.V. infusion | Systematic | No | Kidney, all tissue except CNS | 1 |
| Viltolarsen | I.V. infusion | Systematic | No | Kidney, skeletal muscle | 1 |
| Casimersen | I.V. infusion | Systematic | No | Kidney, skeletal muscle | 1 |
Both fomivirsen and pegaptanib are intended to treat ocular diseases, which require the drug target and optimization approach to be more local than systematic. Based on animal studies on rabbits, absorption of fomivirsen is highest in the retina and iris (Henry et al., 2001). Fomivirsen was detectable in the retina within hours of injection, and concentrations still increased over 3–5 days. Due to the low doses used for intravitreal administration and slow disposition of fomivirsen from the eye, there is limited systemic drug exposure. Similarly, animal studies indicated that the highest concentration of pegaptanib was found in the eyes of rabbits, especially in the vitreous fluids, retina, and aqueous fluid (Foy et al., 2007). Pegaptanib is slowly absorbed into the systemic circulation after intravitreal administration and, as for many ASO drugs, results in ADRs involving the kidney.
Nusinersen administration methods circumvent the blood-brain barrier through intrathecal injection, which then affects the central nervous system (CNS). Nusinersen is distributed and absorbed in the CNS with the help of cerebrospinal fluid after intrathecal administration. The plasma concentration is relatively low compared with cerebrospinal fluid concentration (Luu et al., 2017). The median terminal half-life of nusinersen in the cerebrospinal fluid was determined to be 163 days, which supported infrequent dosing of once every 4–6 months in patients with spinal muscular atrophy. After administration, the peak plasma nusinersen concentrations can be detected within a few hours (Chiriboga et al., 2016).
The remaining ASO drugs, namely mipomersen, inotersen, defibrotide, eteplirsen, golodirsen, viltolarsen, and casimersen, have primarily systemic absorption and affect a range of organs in the body. After subcutaneous administration, peak plasma concentrations of mipomersen and inotersen were observed at 2–4 hours (Li et al., 2014; Yu et al., 2020). With intravenous infusion, peak plasma concentrations of defibrotide, eteplirsen, golodirsen, viltolarsen, and casimersen can be reached within 1–2 hours. Defibrotide stands alone, reaching its peak plasma concentration at 2 hours. It is important to mention that the effects of the ASO drugs on the kidneys are prominent with most ASO drugs investigated in this review.
Distribution
Distribution refers to the movement of a drug between intravascular (blood/plasma) and extravascular compartments of the body. Protein bound and unbound forms of drugs affect their distribution in the body and absorption to tissues and organs (Mansoor and Mahabadi, 2021). Chemical properties of the ASO drugs affect their distribution. This includes factors such as acid-base characteristics, lipophilicity, size, and protein binding. Biodistribution of the 10 ASO drugs is quite different (Table 2). Of these ASO drugs, inotersen, nusinersen in plasma, defibrotide, and mipomersen have high protein-binding potential (>90%), whereas viltolarsen, golodirsen, nusinersen, eteplirsen, and fomivirsen exhibit relatively lowered protein-binding potential (<40%). The freer form pool of the drug in circulation, the more likely the drug will be absorbed into tissues for a drug response. Bioavailability of nusinersen, defibrotide, eteplirsen, golodirsen, viltolarsen, and casimersen after administration through intrathecal or intravenous routes is 100% because the drugs are directly administered into the bloodstream, thus bypassing first-pass metabolism in the liver. Bioavailability is also related to the range of distribution (Price and Patel, 2021). A drug with a wider distribution is more likely to leave the plasma and enter extravascular compartments of the body (the tissues). Comparing the availability data on the range of distribution at steady state, the rank ordering of the ASO drugs is golodirsen > eteplirsen > casimersen, viltolarsen, inotersen, and nusinersen. The extent of exposure of the drug in the body is demonstrated by the area under the curve (AUC) of a specific drug. Specific AUC data for golodirsen, viltolarsen, and casimersen were not found, but intersubject variability of AUC is 34%–44%, 16%–27%, and 16%–34%, respectively. AUC values for defibrotide showed a 50%–60% increase in patients with renal impairment and hemodialysis-dependent end-stage renal disease. Peak concentration was also 35%–37% higher in patients with renal impairment after both single and multidose regimen. This variability is important to be addressed in the monitoring of ADRs and toxicity in patients with increased AUC.
TABLE 2.
Biodistribution of the FDA-approved ASO drugs
The data are extracted from the FDA drug labels of each ASO drug.
| Biodistribution | ||||
|---|---|---|---|---|
| Drug | Protein Binding | Bioavailability | AUC | Volume of Distribution |
| Fomivirsen | 40% (from analysis of vitreous samples from treated rabbits and monkey) | N/A | N/A | N/A |
| Pegaptanib | N/A | N/A | 25 μg/h per ml (at 3 mg monocular dose) | N/A |
| Nusinersen | CSF: <25%; plasma: >94% | 100% (intrathecal) | N/A | CSF: 0.4 L; plasma: 29 L |
| Mipomersen | ≥90% at clinically relevant concentrations (1–8 μg/ml) | 54% to 78% | N/A | N/A |
| Inotersen | >94% (independent of dosage) | N/A | 90 mcg/h per ml | 293 L |
| Defibrotide | Average of 93% | 100% (i.v.) | 26.9–48.1 μg/ml × h | 8.1–9.1 L |
| Eteplirsen | 6% to 17% | 100% (i.v.) | N/A | 600 ml/kg |
| Golodirsen | 33% to 39% (independent of dosage) | 100% (i.v.) | 34% to 44%a | 668 ml/kg (at dose of 30 mg/kg) |
| Viltolarsen | ∼40% | 100% (i.v.) | 16% to 27%a | 300 ml/kg |
| Casimersen | 8% to 32% | 100% (i.v.) | 16% to 34%a | 367 ml/kg |
N/A, no specific information available.
aIntersubject variability
Metabolism
Metabolism is the process in which each drug becomes chemically modified and broken down by the body. All 10 FDA-approved ASO drugs have mechanisms of degradation in the bloodstream and target cells. These mechanisms are quite different from the conventional metabolic pathways that are used for small molecule drugs.
During clinical trials, fomivirsen did not undergo biotransformation through oxidative metabolic pathways; instead, the main pathway was found to be nuclease degradation, including both endonuclease and exonuclease activities in the bloodstream and target cells (Geary et al., 2002). Exonucleases are a class of enzymes that are responsible for cleaving the phosphodiester bonds between adjacent nucleotides at the ends of the nucleic acid chains. Endonucleases are similar in function, but nucleolytic cleavage occurs at an internal location in the nucleic acid chain. Nuclease degradation of fomivirsen begins once it has been distributed throughout the retina. The resulting metabolites have not been detected in the blood plasma, thus suggesting that there is no significant systemic absorption of the metabolites (Grillone and Lanz, 2001). Pegaptanib also does not undergo cytochrome P450 (P450)-mediated metabolism. Instead, its primary degradation pathway is endonuclease and exonuclease activity in blood (Kourlas and Schiller, 2006). Mipomersen has the same degradation pathway as well. After metabolic degradation, the metabolites of mipomersen are referred to as “chain shortened” because they range between 7 and 14 nucleotides and have no pharmacological function (Bell et al., 2011). Although the parent drug reaches plasma concentrations within hours of administration, the metabolites have not been found in the plasma, either free or bound to plasma proteins (Crooke and Geary, 2013). During the in vitro testing for eteplirsen, hepatic microsomes did not have a significant effect on the metabolism of the drug. It was determined that metabolism of the drug does not occur in the liver (Lim et al., 2017). There was also no accumulation of the drug or its metabolites, and minimal drug-drug interactions with plasma proteins or other coadministered drugs were detected (Syed, 2016). Nusinersen has an exonuclease-dependent metabolic pathway. The exonuclease activity specifically involves hydrolysis reactions as each individual nucleotide is cleaved off from the ends of the ASO molecule. There are no known interactions with the P450 enzymes, and thus the parent drug is not considered a substrate for hepatic oxidative metabolism (Neil and Bisaccia, 2019). Inotersen is metabolized by both endonucleases and exonucleases. The parent drug is first subject to endonuclease degradation, where the phosphodiester bonds toward the middle of the nucleic acid are cleaved. The metabolites of this reaction have been found to be inactive, thus suggesting that it does not have another pharmacological use as a metabolite. Then, these short metabolites are subject to even more nuclease degradation in the form of exonuclease activity. There is little or no interaction with coadministered drugs that are subject to oxidative metabolism because inotersen is not a substrate of the hepatic metabolic pathway (Gertz et al., 2019). Golodirsen, viltolarsen, and casimersen are all FDA-approved ASO drugs developed for the treatment of DMD. These drugs are not subject to hepatic metabolism and can be considered metabolically stable. The metabolites were not found in the plasma or urine. In vitro studies showed that these drugs had little to no interactions with P450 enzymes or drug transporters since they were not subject to typical drug metabolism pathways.
By analyzing the metabolic pathways, there are some commonalities that can be identified between all FDA-approved ASO drugs. The primary method of metabolism is nuclease degradation via endonuclease and exonuclease activities. For the majority of these drugs, endonuclease activity occurs first, thus cleaving the oligonucleotides into fragments. Then exonuclease activity can take place on the 3′ and 5′ ends via hydrolysis reactions. To increase stability of ASO drugs from nuclease degradation, they have been modified with chemical groups that allow them to remain stable enough to reach their target tissues. In general, ASO drugs can have phosphorothioate linkages in the phosphodiester backbone, 2'-O-methoxyethyl ribose gapmer modifications, and 2-4’ methylene-bridged nucleic acids, which are also referred to as locked nucleic acids (Swayze et al., 2007). These modifications allow the molecules to have better resistance to nuclease degradation. The FDA-approved ASO drugs summarized above also have such modifications that likely contribute to their stability and longer residency at their target tissues. Fomivirsen is considered a first-generation oligonucleotide, as it has phosphorothioate linkages in the backbone. This means that one of the oxygen atoms in the phosphate group at the phosphodiester bond is replaced by a sulfur atom (de Smet et al., 1999). Mipomersen and inotersen, on the other hand, are considered second-generation ASOs because of the 2'-O-methoxyethyl ribose modification (Crooke and Geary, 2013). Second-generation ASO drugs have been found to have better resistance against nuclease degradation; hence, these drugs have longer half-lives as well and increased stability within the target cell (Swayze et al., 2007). Another similarity between these drugs is that they are not subject to metabolism by the liver or hepatic microsomes. This is due to the drugs not being substrates for the major class of drug-metabolizing enzymes, P450s. There is also little to no potential for most of these drugs of having interactions with other proteins or drug transporters because of their distinct metabolic pathways. The metabolites for these drugs have not been found to be pharmacologically active or to impact cellular processes in a manner that could be of safety concern.
Although this information about the metabolism of ASO drugs provides insight into the primary pathways of drug degradation, there are some areas that deserve more research. There is no detailed information on the chemical and physical properties of ASO drug metabolites that are the products of nuclease degradation. This topic could be studied more to better understand the role of metabolites. The specific rates of the hydrolysis reactions for exonuclease activity could be useful in improving the stability of the oligonucleotides via chemical modifications. A possible future direction would be to investigate how the stability of ASO drugs can be increased and how it would affect resistance to nuclease degradation and therapeutic efficacy. The frequency and different combinations of chemical modifications can be studied to investigate whether the dosing intervals could be changed as well.
Excretion
Each individual ASO drug has its excretion features. While conducting preclinical studies of fomivirsen on rabbits, a small amount of administrated fomivirsen was shown to be eliminated through urine (16%) and feces (3%). Fomivirsen is administered intravitreally, and the half-life of fomivirsen is approximately 55 hours in humans. In monkeys, the half-life is estimated to be 78 hours (Perry and Balfour, 1999). This is different from the route of excretion for mipomersen, which is primarily through renal filtration (Visser et al., 2012) and urine by “nuclease digested metabolites” (Crooke and Geary, 2013). This route of excretion seems to be constant among species. Eteplirsen is also excreted via the renal system. The renal system accounts for two-thirds of the excretion of the administered dose within 24 hours post–intravenous administration. The total body clearance for eteplirsen is 339 ml/h per kg after 12 weeks of drug therapy with 30 mg/kg per week (Baker, 2017). The peak time is 1.1–1.2 hours, and the half-life is around 3–4 hours. Nusinersen also primarily utilizes the urinary route to excretion. It is excreted by the kidneys as pharmacologically inactive chain-shortened oligonucleotides (Li, 2020). The half-life is expected to be 135–177 days in cerebrospinal fluid (CSF) and 63–87 days in plasma (Shorrock et al., 2018). For CSF clearance, it is assumed that the transport from the CSF to the plasma compartment is the predominant mechanism (Luu et al., 2017). Inotersen is metabolized and excreted through the urine. The duration of the 284 mg/1.5 ml dose is 1 week. The total body clearance rate and half-life are 3.18 l/h and 32.3 days. Since one of the target organs of inotersen is the kidney, the urinary route to excretion is an efficient method of drug delivery to its target. During the first 24 hours, less than 1% of the drug is excreted unchanged. Golodirsen also uses the kidney for excretion. However, golodirsen is mainly excreted unchanged by the renal system. Viltolarsen is mainly excreted in the urine unchanged as well. The elimination half-life is approximately 2.5 hours (8%), and the plasma clearance is 217 ml/h per kg (22%). Patients taking 1.25, 5, or 20 mg/kg viltolarsen weekly for 12 weeks had a total clearance of between 149 ± 21 and 239 ± 97 ml/h per kg (Komaki et al., 2020). Lastly, casimersen also primarily uses the urinary route for excretion. Its excretion is completed by 336 hours postdose. The excretion method for casimersen is similar to that of eteplirsen and golodirsen. Collectively, the commonality in excretion for all ASO drugs is the predominant use of the urinary system. Even fomivirsen, which is administered intravitreally, has trace amounts detected in urine. This suggests a prominent preference for excretion via the urinary route for the ASO drugs.
Differences in ADME between Small Molecule Drugs and ASO Drugs
Based on the above analysis on the information from the FDA drug label, clinical studies, and published literature, ADME features of the FDA-approved ASO drugs are quite different from small chemical drugs with their unique characteristics for ADME. The ADME behavior of small chemical drugs is mediated by uptake and efflux transporters, phase I and II drug-metabolizing enzymes, transcription factors, and xenobiotic-activated nuclear receptors (Fig. 4A). The ADME of ASO drugs is determined by completely different pathways (Fig. 4B). Absorption of ASOs into target cells involves uptake via endocytic internalization into early endosomes (EEs). ASOs are then moved within the cell by subcellular trafficking. Release of ASOs from endosomes has significant impact on therapeutic efficacy and the occurrence of ADRs. Metabolism of ASO drugs is mainly catalyzed by exonucleases and endonucleases, resulting in various sizes of shortened oligonucleotides. Either intact or shortened ASOs are excreted by membrane leakage, vesicle release, or exosome release to the urinary tract. These features determine the half-life of ASO drugs in the circulatory system and frequency of administration. To optimize therapeutic efficacy, studies on the ADME features for ASO drugs should continue in real-world settings to monitor therapeutic efficacy and ADRs.
Fig. 4.
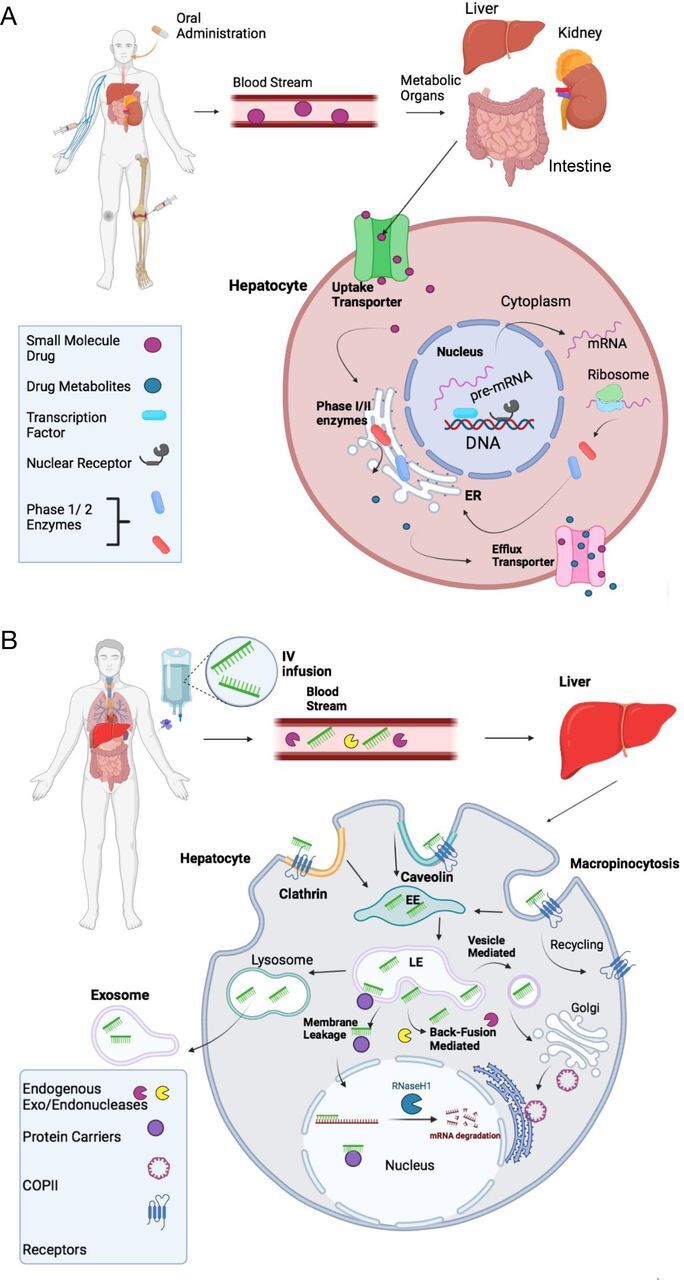
Comparison of absorption, distribution, metabolism, and excretion (ADME) of small chemical drugs and ASO drugs. (A) ADME of an orally administrated small chemical drug. After absorption, a small chemical drug is normally distributed to metabolic organs of intestine, liver, and kidney via the bloodstream. In the metabolic organs, such as the liver, the drug is taken up by hepatocytes through either passive or active transport against concentration gradients with the help of uptake transporters. Normally, the lipophilic drug is biotransformed by either Phase I or II metabolizing enzymes to more polar water-soluble metabolites in rough endoplasmic reticulum (ER) membrane. The parental drug and metabolites are excreted from hepatocyte cells via efflux transporters into the bloodstream and then become available as an active form to target organs. Therapeutic concentrations of the drug in its target organs are determined by uptake and efflux transporters and Phase I and II metabolizing enzymes, which are tightly regulated at transcriptional and translation levels by transcription factors and nuclear receptors. (B) ADME of an ASO drug. After an intravenous infusion, subcutaneous injection, or direct injection, ASOs travel through the bloodstream, where they can be broken down via endogenous exo- and endonucleases. Once ASOs reach their target organ/cell, such as the liver, they enter cells through various endocytic pathways, including clathrin-mediated, caveolin-mediated, and micropinocytosis endocytosis, into early endosomes (EEs). After being internalized into EEs, ASO drugs go through subcellular trafficking to the late endosomes. A proportion of ASO drugs (maybe very small proportion) is released from late endosomes to cytoplasm to target mRNAs or pre-mRNAs in either the cytoplasm or the nucleus, where they execute their therapeutic actions. Many small cellular proteins including COPII can assist subcellular trafficking to the nucleus. However, the mechanism of nuclear trafficking is not fully understood. Another proportion of ASO drugs undergo into lysosomes or further are released out of the cell via three proposed mechanisms: membrane leakage, back-fusion mediated release, and vesicle-mediated release. Back-fusion mediated release is believed to help endogenous exo- and endonucleases break the ASO down further. ASO drugs may also leave the cell completely via the exosome release from the cell.
Current Challenges, Knowledge Gaps, and Future Perspective
Several knowledge gaps for the ADME features of the FDA-approved ASO drugs were identified, including cellular uptake, subcellular trafficking, and cellular efflux. Almost no information is available on how these FDA-approved drugs are taken up into cells after local or systematic distribution. Existing data on uptake mechanisms of ASO molecules are mainly based on in vitro models of cultured cells. Commonly accepted theories are that ASOs undergo cellular uptake by two competing pathways: the productive pathway bringing the drugs into their targets and the nonproductive pathway directing the drugs into saturable sinks (Khorkova and Wahlestedt, 2017). Both pathways take up ASOs by clathrin-coated endocytic vesicles from plasma membrane. Few factors have been strongly identified as key players for these pathways, such as TCP1 complex, Hsp90, SSB, NPM1, and AP2M1 in the productive pathway and paraspeckle proteins, Ku70, and Ku80 in the nonproductive pathway (Li, 2020). However, very limited information is available for identifying key determinant factors in the intracellular endosomal packing of ASO drugs and what proportion of these drugs go into the productive versus nonproductive pathway. These are important considerations since these variables dictate in part the amount of an ASO drug that reaches its therapeutic targets. Furthermore, some surface receptors may be critical for this endocytosis process. For example, adenosine receptors in endothelial cells may have a potential role in the endocytic uptake of defibrotide (Palomo et al., 2016). Future studies should employ FDA-approved ASO drugs as experimental tools to understand cellular uptake mechanisms for this class of drugs, which is completely different from the transporter-mediated uptake of small chemical drugs. Significant variations on the cellular uptake of ASO drugs may exist in the patient population, resulting in differences on therapeutic efficacy and safety.
Another knowledge gap is the lack of information on subcellular trafficking and distribution of the FDA-approved ASO drugs and their impact on therapeutic efficacy. Unlike small chemical drugs, which can quickly reach different subcellular regions after uptake by transporters or non–transporter-dependent diffusion across the cell membrane, ASO drugs are normally first encapsulated in endosomes after uptake into cells by endocytosis. Different ASO drugs have their targets located at different subcellular locations. For RNase H-mediated mRNA degradation, the ASO drugs need to reach either the cytoplasm (ribosomes) or nucleus (Liang et al., 2017). For exon skipping, the ASO drugs should be present in spliceosomes in the nucleus (Echevarria et al., 2018). Endosomal release and intracellular trafficking are key determinants of the pharmacological actions of the ASO drugs (Juliano, 2018). Numerous factors have been identified as mediators of these events, which can enhance endosomal release and intracellular trafficking (Ono et al., 2020). For example, Hsc70 (Liang et al., 2021), M6PR (Liang et al., 2020), and lipid conjugates (Wang et al., 2019a) are proposed contributors of endosomal release and ASO trafficking. Future studies should pay particular attention to how ASO drugs are taken up and released from endosomes and what assists them in reaching their therapeutic targets. Equally important is the identification of mechanisms that can lead to the optimization of intracellular trafficking of ASO drugs toward pharmacological targets to enhance therapeutic outcomes. It is also imperative to know which intracellular locations may be associated with ASO-mediated cell damage and toxicity to avoid ASO drugs from reaching and accumulating in these intracellular locations, which are distinct from their therapeutic targets.
Regarding ASO drugs biotransformation, the mechanism by which the remaining parent and shortened ASO drugs are effluxed out from cells after breakdown by endonucleases and exonucleases is completely unknown. It is also unknown whether efflux is a critical component. Unlike many small chemical drugs that are eliminated from the cells by efflux transporters, ASOs use completely different cellular exit mechanisms, which may have considerable impact on plasma concentrations and duration of ASO drugs in their target cells. It can be hypothesized that lack of efflux activity could have substantial effects on accumulation of ASO drugs in liver and kidney. Hepatotoxicity and kidney toxicity are a major safety concern for the FDA-approved ASO drugs (Alhamadani et al., 2022). Determination of efflux mechanisms may provide better understanding of interindividual variability of ASO-mediated drug toxicity.
Conclusions
The discovery and development of ASO drugs have moved at a much faster pace in recent years. Numerous new ASO drugs are expected to be approved in the next decade, and the use of ASO drugs for a variety of indication will become more commonplace. However, before this relatively new class of drugs is more widely accepted, a better understanding of the ADME characteristic of these drugs is needed to optimize their therapeutic efficacy and safety profiles. The current review summarizes administration, absorption, distribution, metabolism (degradation), and excretion of the 10 FDA-approved ASO drugs. Specifically, this review explores the unique set of ADME features for ASO drugs that differs quite dramatically from the more traditional ADME profiles of small chemical drugs. Several knowledge gaps are identified on cellular uptake by endocytosis, intracellular endosomal release, intracellular/nuclear trafficking, and cellular efflux for these ASO drugs, and future directions are discussed.
Abbreviations
- ADME
absorption, distribution, metabolism, and excretion
- ADR
adverse drug reaction
- Apo B
apolipoprotein B
- ASO
antisense oligonucleotide
- AUC
area under the curve
- CMV
cytomegalovirus
- CNS
central nervous system
- CSF
cerebrospinal fluid
- DMD
Duchenne muscular dystrophy
- EE
early endosome
- FDA
US Food and Drug Administration
- P450
cytochrome P450
- TTR
transthyretin
- VEGF
vascular endothelial growth factor
Authorship Contributions
Participated in research design: Bahal, Rasmussen, Manautou, Zhong.
Performed data analysis: Migliorati, S. Liu, A. Liu, Gogate, Nair.
Wrote or contributed to the writing of the manuscript: Migliorati, S. Liu, A. Liu, Gogate, Nair, Bahal, Rasmussen, Manautou, Zhong.
Footnotes
This study was partly supported by National Institutes of Health National Institute of General Medical Sciences [Grant R35-GM140862] (to X.Z.) and National Cancer Institute [Grant R01-CA241194] (to R.B.).
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Alagga AA, Gupta V (2021) Drug absorption, in StatPearls [Internet], StatPearls Publishing, Treasure Island, FL. [PubMed] [Google Scholar]
- Alhamadani F, Zhang K, Parikh R, Wu H, Rasmussen TP, Bahal R, Zhong X-B, Manautou JE (2022) Adverse drug reactions and toxicity of the FDA-approved antisense oligonucleotide drugs. Drug Metab Dispos 50:880–888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amantana A, Iversen PL (2005) Pharmacokinetics and biodistribution of phosphorodiamidate morpholino antisense oligomers. Curr Opin Pharmacol 5:550–555. [DOI] [PubMed] [Google Scholar]
- Baker DE (2017) Eteplirsen. Hosp Pharm 52:302–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DE, Demaris K (2016) Defibrotide. Hosp Pharm 51:847–854 10.1310/hpj5110-847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bell DA, Hooper AJ, Burnett JR (2011) Mipomersen, an antisense apolipoprotein B synthesis inhibitor. Expert Opin Investig Drugs 20:265–272. [DOI] [PubMed] [Google Scholar]
- Bennett CF, Baker BF, Pham N, Swayze E, Geary RS (2017) Pharmacology of antisense drugs. Annu Rev Pharmacol Toxicol 57:81–105. [DOI] [PubMed] [Google Scholar]
- Bennett CF, Krainer AR, Cleveland DW (2019) Antisense oligonucleotide therapies for neurodegenerative diseases. Annu Rev Neurosci 42:385–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benson MDWaddington-Cruz MBerk JLPolydefkis MDyck PJWang AKPlanté-Bordeneuve VBarroso FAMerlini GObici L, et al. (2018) Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med 379:22–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiriboga CA, Swoboda KJ, Darras BT, Iannaccone ST, Montes J, De Vivo DC, Norris DA, Bennett CF, Bishop KM (2016) Results from a phase 1 study of nusinersen (ISIS-SMN(Rx)) in children with spinal muscular atrophy. Neurology 86:890–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST, Geary RS (2013) Clinical pharmacological properties of mipomersen (Kynamro), a second generation antisense inhibitor of apolipoprotein B. Br J Clin Pharmacol 76:269–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooke ST, Liang XH, Crooke RM, Baker BF, Geary RS (2021) Antisense drug discovery and development technology considered in a pharmacological context. Biochem Pharmacol 189:114196. [DOI] [PubMed] [Google Scholar]
- de Smet MD, Meenken CJ, van den Horn GJ (1999) Fomivirsen - a phosphorothioate oligonucleotide for the treatment of CMV retinitis. Ocul Immunol Inflamm 7:189–198. [DOI] [PubMed] [Google Scholar]
- Dhuri K, Bechtold C, Quijano E, Pham H, Gupta A, Vikram A, Bahal R (2020) Antisense oligonucleotides: an emerging area in drug discovery and development. J Clin Med 9:2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Fusco D, Dinallo V, Marafini I, Figliuzzi MM, Romano B, Monteleone G (2019) Antisense oligonucleotide: basic concepts and therapeutic application in inflammatory bowel disease. Front Pharmacol 10:305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon DL, Sisson EM, Butler M, Higbea A, Muoio B, Turner B (2014) Lomitapide and mipomersen: novel lipid-lowering agents for the management of familial hypercholesterolemia. J Cardiovasc Nurs 29:E7–E12. [DOI] [PubMed] [Google Scholar]
- Echevarría L, Aupy P, Goyenvalle A (2018) Exon-skipping advances for Duchenne muscular dystrophy. Hum Mol Genet 27:R163–R172. [DOI] [PubMed] [Google Scholar]
- Foy JW, Rittenhouse K, Modi M, Patel M (2007) Local tolerance and systemic safety of pegaptanib sodium in the dog and rabbit. J Ocul Pharmacol Ther 23:452–466. [DOI] [PubMed] [Google Scholar]
- Geary RS, Henry SP, Grillone LR (2002) Fomivirsen: clinical pharmacology and potential drug interactions. Clin Pharmacokinet 41:255–260. [DOI] [PubMed] [Google Scholar]
- Gertz MA, Scheinberg M, Waddington-Cruz M, Heitner SB, Karam C, Drachman B, Khella S, Whelan C, Obici L (2019) Inotersen for the treatment of adults with polyneuropathy caused by hereditary transthyretin-mediated amyloidosis. Expert Rev Clin Pharmacol 12:701–711. [DOI] [PubMed] [Google Scholar]
- Goodkey K, Aslesh T, Maruyama R, Yokota T (2018) Nusinersen in the treatment of spinal muscular atrophy. Methods Mol Biol 1828:69–76. [DOI] [PubMed] [Google Scholar]
- Grillone LR, Lanz R (2001) Fomivirsen. Drugs Today (Barc) 37:245–255. [DOI] [PubMed] [Google Scholar]
- Henry SP, Miner RC, Drew WL, Fitchett J, York-Defalco C, Rapp LM, Levin AA (2001) Antiviral activity and ocular kinetics of antisense oligonucleotides designed to inhibit CMV replication. Invest Ophthalmol Vis Sci 42:2646–2651. [PubMed] [Google Scholar]
- Juliano RL (2018) Intracellular trafficking and endosomal release of oligonucleotides: what we know and what we don’t. Nucleic Acid Ther 28:166–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keam SJ (2018) Inotersen: first global approval. Drugs 78:1371–1376. [DOI] [PubMed] [Google Scholar]
- Khorkova O, Wahlestedt C (2017) Oligonucleotide therapies for disorders of the nervous system. Nat Biotechnol 35:249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komaki HTakeshima YMatsumura TOzasa SFunato MTakeshita EIwata YYajima HEgawa YToramoto T, et al. (2020) Viltolarsen in Japanese Duchenne muscular dystrophy patients: a phase 1/2 study. Ann Clin Transl Neurol 7:2393–2408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kourlas H, Schiller DS (2006) Pegaptanib sodium for the treatment of neovascular age-related macular degeneration: a review. Clin Ther 28:36–44. [DOI] [PubMed] [Google Scholar]
- Li Q (2020) Nusinersen as a therapeutic agent for spinal muscular atrophy. Yonsei Med J 61:273–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Z, Hard ML, Andersen G, Pabst G, Wagener G, Singh T, Chin W, Culm-Merdek K, Boltje I, von Moltke LL (2014) Pharmacokinetics, safety and tolerability of mipomersen in healthy Japanese volunteers and comparison with Western subjects. Int J Clin Pharmacol Ther 52:314–320. [DOI] [PubMed] [Google Scholar]
- Liang XH, Nichols JG, Hsu CW, Crooke ST (2021) Hsc70 facilitates mannose-6-phosphate receptor-mediated intracellular trafficking and enhances endosomal release of phosphorothioate-modified antisense oligonucleotides. Nucleic Acid Ther 31:284–297. [DOI] [PubMed] [Google Scholar]
- Liang XH, Sun H, Hsu CW, Nichols JG, Vickers TA, De Hoyos CL, Crooke ST (2020) Golgi-endosome transport mediated by M6PR facilitates release of antisense oligonucleotides from endosomes. Nucleic Acids Res 48:1372–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang XH, Sun H, Nichols JG, Crooke ST (2017) RNase H1-dependent antisense oligonucleotides are robustly active in directing RNA cleavage in both the cytoplasm and the nucleus. Mol Ther 25:2075–2092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim KR, Maruyama R, Yokota T (2017) Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des Devel Ther 11:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luu KT, Norris DA, Gunawan R, Henry S, Geary R, Wang Y (2017) Population pharmacokinetics of nusinersen in the cerebral spinal fluid and plasma of pediatric patients with spinal muscular atrophy following intrathecal administrations. J Clin Pharmacol 57:1031–1041. [DOI] [PubMed] [Google Scholar]
- Mansoor A, Mahabadi N (2021) Volume of distribution, in StatPearls [Internet], StatPearls Publishing, Treasure Island, FL. [PubMed] [Google Scholar]
- Neil EE, Bisaccia EK (2019) Nusinersen: a novel antisense oligonucleotide for the treatment of spinal muscular atrophy. J Pediatr Pharmacol Ther 24:194–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberemok VVLaikova KVRepetskaya AIKenyo IMGorlov MVKasich INKrasnodubets AMGal’chinsky NVFomochkina IIZaitsev AS, et al. (2018) A half-century history of applications of antisense oligonucleotides in medicine, agriculture and forestry: we should continue the journey. Molecules 23:1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ono D, Asada K, Yui D, Sakaue F, Yoshioka K, Nagata T, Yokota T (2020) Separation-related rapid nuclear transport of DNA/RNA heteroduplex oligonucleotide: unveiling distinctive intracellular trafficking. Mol Ther Nucleic Acids 23:1360–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palomo M, Mir E, Rovira M, Escolar G, Carreras E, Diaz-Ricart M (2016) What is going on between defibrotide and endothelial cells? Snapshots reveal the hot spots of their romance. Blood 127:1719–1727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parham JS, Goldberg AC (2019) Mipomersen and its use in familial hypercholesterolemia. Expert Opin Pharmacother 20:127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry CM, Balfour JA (1999) Fomivirsen. Drugs 57:375–380, discussion 381). [DOI] [PubMed] [Google Scholar]
- Pescador N, Pérez-Barba M, Ibarra JM, Corbatón A, Martínez-Larrad MT, Serrano-Ríos M(2013) Serum circulating microRNA profiling for identification of potential type 2 diabetes and obesity biomarkers. PLoS One 8:e77251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price G, Patel DA (2021) Drug bioavailability, in StatPearls [Internet], StatPearls Publishing, Treasure Island, FL. [PubMed] [Google Scholar]
- Quemener AM, Bachelot L, Forestier A, Donnou-Fournet E, Gilot D, Galibert MD (2020) The powerful world of antisense oligonucleotides: From bench to bedside. Wiley Interdiscip Rev RNA 11:e1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichel A, Lienau P (2016) Pharmacokinetics in drug discovery: an exposure-centred approach to optimising and predicting drug efficacy and safety. Handb Exp Pharmacol 232:235–260. [DOI] [PubMed] [Google Scholar]
- Richardson PG, Grupp SA, Pagliuca A, Krishnan A, Ho VT, Corbacioglu S (2017) Defibrotide for the treatment of hepatic veno-occlusive disease/sinusoidal obstruction syndrome with multiorgan failure. Int J Hematol Oncol 6:75–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PG, Smith AR, Kernan NA, Lehmann L, Ryan RJ, Grupp SA (2021) Analysis of time to complete response after defibrotide initiation in patients with hepatic veno-occlusive disease/sinusoidal obstruction syndrome after hematopoietic cell transplantation. Transplant Cell Ther 27:88.e1–88.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehr B (1998) Fomivirsen approved for CMV retinitis. J Int Assoc Physicians AIDS Care 4:14–16. [PubMed] [Google Scholar]
- Shahryari A, Saghaeian Jazi M, Mohammadi S, Razavi Nikoo H, Nazari Z, Hosseini ES, Burtscher I, Mowla SJ, Lickert H (2019) Development and clinical translation of approved gene therapy products for genetic disorders. Front Genet 10:868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shorrock HK, Gillingwater TH, Groen EJN (2018) Overview of current drugs and molecules in development for spinal muscular atrophy therapy. Drugs 78:293–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CIE, Zain R (2019) Therapeutic oligonucleotides: state of the art. Annu Rev Pharmacol Toxicol 59:605–630. [DOI] [PubMed] [Google Scholar]
- Stephenson ML, Zamecnik PC (1978) Inhibition of Rous sarcoma viral RNA translation by a specific oligodeoxyribonucleotide. Proc Natl Acad Sci USA 75:285–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swayze EE, Siwkowski AM, Wancewicz EV, Migawa MT, Wyrzykiewicz TK, Hung G, Monia BP, Bennett CF (2007) Antisense oligonucleotides containing locked nucleic acid improve potency but cause significant hepatotoxicity in animals. Nucleic Acids Res 35:687–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed YY (2016) Eteplirsen: first global approval. Drugs 76:1699–1704. [DOI] [PubMed] [Google Scholar]
- Visser ME, Witztum JL, Stroes ES, Kastelein JJ (2012) Antisense oligonucleotides for the treatment of dyslipidaemia. Eur Heart J 33:1451–1458. [DOI] [PubMed] [Google Scholar]
- Wang S, Allen N, Prakash TP, Liang XH, Crooke ST (2019a) Lipid conjugates enhance endosomal release of antisense oligonucleotides into cells. Nucleic Acid Ther 29:245–255. [DOI] [PubMed] [Google Scholar]
- Wang Y, Yu RZ, Henry S, Geary RS (2019b) Pharmacokinetics and clinical pharmacology considerations of GalNAc3-conjugated antisense oligonucleotides. Expert Opin Drug Metab Toxicol 15:475–485. [DOI] [PubMed] [Google Scholar]
- Weidolf L, Björkbom A, Dahlén A, Elebring M, Gennemark P, Hölttä M, Janzén D, Li X, Andersson S (2021) Distribution and biotransformation of therapeutic antisense oligonucleotides and conjugates. Drug Discov Today 26:2244–2258. [DOI] [PubMed] [Google Scholar]
- Wong E, Goldberg T (2014) Mipomersen (kynamro): a novel antisense oligonucleotide inhibitor for the management of homozygous familial hypercholesterolemia. P&T 39:119–122. [PMC free article] [PubMed] [Google Scholar]
- Wurster CD, Ludolph AC (2018) Nusinersen for spinal muscular atrophy. Ther Adv Neurol Disord 11:1756285618754459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RZ, Wang Y, Norris DA, Kim TW, Narayanan P, Geary RS, Monia BP, Henry SP (2020) Immunogenicity assessment of inotersen, a 2′-O-(2-methoxyethyl) antisense oligonucleotide in animals and humans: effect on pharmacokinetics, pharmacodynamics, and safety. Nucleic Acid Ther 30:265–275. [DOI] [PubMed] [Google Scholar]