

Abstract
Our laboratory has shown that activation of transforming growth factor-β (TGF-β)/activin receptor-like kinase 1 (ALK1) signaling can increase protein expression and transport activity of organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier (BBB). These results are relevant to treatment of ischemic stroke because Oatp transport substrates such as 3-hydroxy-3-methylglutaryl coenzyme A reductase inhibitors (i.e., statins) improve functional neurologic outcomes in patients. Advancement of our work requires determination if TGF-β/ALK1 signaling alters Oatp1a4 functional expression differently across brain regions and if such disparities affect central nervous system (CNS) statin disposition. Therefore, we studied regulation of Oatp1a4 by the TGF-β/ALK1 pathway, in vivo, in rat brain microvessels isolated from cerebral cortex, hippocampus, and cerebellum using the ALK1 agonist bone morphogenetic protein-9 (BMP-9) and the ALK1 inhibitor 4-[6-[4-(1-piperazinyl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]quinoline dihydrochloride 193189. We showed that Oatp1a4 protein expression and brain distribution of three currently marketed statin drugs (i.e., atorvastatin, pravastatin, and rosuvastatin) were increased in cortex relative to hippocampus and cerebellum. Additionally, BMP-9 treatment enhanced Oatp-mediated statin transport in cortical tissue but not in hippocampus or cerebellum. Although brain drug delivery is also dependent upon efflux transporters, such as P-glycoprotein and/or Breast Cancer Resistance Protein, our data showed that administration of BMP-9 did not alter the relative contribution of these transporters to CNS disposition of statins. Overall, this study provides evidence for differential regulation of Oatp1a4 by TGF-β/ALK1 signaling across brain regions, knowledge that is critical for development of therapeutic strategies to target Oatps at the BBB for CNS drug delivery.
SIGNIFICANCE STATEMENT
Organic anion transporting polypeptides (Oatps) represent transporter targets for brain drug delivery. We have shown that Oatp1a4 statin uptake is higher in cortex versus hippocampus and cerebellum. Additionally, we report that the transforming growth factor-β/activin receptor-like kinase 1 agonist bone morphogenetic protein-9 increases Oatp1a4 functional expression, but not efflux transporters P-glycoprotein and Breast Cancer Resistance Protein, in cortical brain microvessels. Overall, this study provides critical data that will advance treatment for neurological diseases where drug development has been challenging.
Introduction
Blood-brain barrier (BBB) transporters are critical determinants of brain drug disposition. This principle emphasizes the need to rigorously study regulation, expression, and activity of these transport proteins so that optimized treatments for neurologic diseases can be developed. Our laboratory has shown that organic anion transporting polypeptides (OATPs in humans; Oatps in rodents) are expressed at the brain microvascular endothelium and can be targeted to facilitate uptake of currently marketed drugs (Ronaldson et al., 2011; Thompson et al., 2014; Abdullahi et al., 2018; Brzica et al., 2018a). In these studies, we have focused our attention on Oatp1a4, the primary drug transporting Oatp isoform expressed at the rat BBB (Ronaldson et al., 2011; Ronaldson and Davis, 2013). Specifically, our experimental work has described the Oatp-mediated transport of 3-hydroxyl-3-methylglutaryl coenzyme A (HMG-CoA) reductase inhibitors (i.e., statins) at the BBB (Thompson et al., 2014; Abdullahi et al., 2018; Brzica et al., 2018a). We conducted detailed transport studies in human umbilical vein endothelial cells (HUVECs) and reported that molecular regulation and statin transport properties of OATP1A2 are comparable with those of its rodent ortholog Oatp1a4 (Ronaldson et al., 2021). Indeed, others have corroborated our observations by demonstrating Oatp-mediated drug disposition in brain microvascular endothelial cells (Ose et al., 2010; Albekairi et al., 2019). Blood-to-brain uptake of statins is relevant to pharmacotherapy of neurologic diseases, such as ischemic stroke. Indeed, clinical studies have demonstrated that statins are unique in their ability to reduce physical disability in stroke patients (Ishikawa et al., 2016; Malhotra et al., 2019) as well as to decrease risk of recurrent stroke (Lee et al., 2017). This proven efficacy of statins is in contrast to many other compounds developed for ischemic stroke treatment where clinical success in phase III clinical trials has not yet been achieved (Shi et al., 2018).
P-glycoprotein (P-gp) and Breast Cancer Resistance Protein (BCRP in humans; Bcrp in rodents) also are critical determinants of brain statin disposition. P-gp is believed to be the most critical BBB transporter due to its role in restricting brain permeability of various structurally diverse drugs. The substrate profile for BCRP/Bcrp greatly overlaps with that of P-gp, thereby enabling these two transporters to function synergistically to limit central nervous system (CNS) drug uptake (Polli et al., 2009; Williams et al., 2020). Many currently marketed statins are transport substrates for these two critical efflux transporters. For example, atorvastatin and rosuvastatin are known P-gp substrates (Hochman et al., 2004; Li et al., 2011; Ronaldson et al., 2021). BCRP/Bcrp is known to efflux both pravastatin and rosuvastatin (Hirano et al., 2005; Safar et al., 2019; Ronaldson et al., 2021). Therefore, a detailed assessment of Oatp-mediated statin uptake at the BBB must consider the concept of the multitransporter environment, particularly the contributions of those transporters that restrict CNS drug delivery, such as P-gp and BCRP/Bcrp.
Another critical consideration in understanding the role of Oatp1a4 in blood-to-brain transport of statins is the potential for brain regional differences in transporter expression. This possibility is exemplified by two proteomics studies conducted in brain microvasculature. Al-Majdoub and colleagues reported that OATP1A2 was the most abundant OATP isoform present in frontal cortex microvessels (Al-Majdoub et al., 2019). In contrast, Billington and colleagues reported that OATP1A2 protein expression at the BBB in Brodmann Areas 17 and 39 (Billington et al., 2019) was below the limit of detection in their targeted proteomic analysis. In rodent tissue, brain regional differences in Oatp1a4 regulation or functional expression have not been evaluated in detail. Using a preplanned study design, we addressed this critical issue in female Sprague-Dawley rats by testing the hypothesis that Oatp1a4 regulation and functional expression is different between cerebral cortex, hippocampus, and cerebellum. These brain regions were selected due to their susceptibility to injury after acute ischemic stroke or cerebellar stroke (Ramos-Cabrer et al., 2011; Sarikaya and Steinlin, 2018; Higashi et al., 2021). Furthermore, we postulate that such variability will affect CNS disposition of atorvastatin, pravastatin, and rosuvastatin. Our data showed that brain uptake of all three statins is highest in cerebral cortex. We also report, for the first time, that activation of transforming growth factor-β (TGF-β)/activin receptor-like kinase 1 (ALK1) signaling using a specific agonist [bone morphogenetic protein-9 (BMP-9)] increased Oatp1a4 functional expression and CNS statin delivery in cortex but not in hippocampus or cerebellum. This effect likely results from regional differences in BBB expression of endoglin (ENG), a TGF-β/ALK1 coreceptor that binds ALK1 ligands (i.e., BMP-9) to form a protein complex that is then recruited to ALK1 receptors (Lee et al., 2008; van Meeteren et al., 2012; Lawera et al., 2019).
Materials and Methods
Animals and Drug Treatments
Animal experiments were approved by the University of Arizona Institutional Animal Care and Use Committee and were designed to comply with the Animal Research: Reporting In Vivo Experiments guidelines. Female Sprague-Dawley rats (200–250g; 3 months old; Envigo, Denver, CO) were intentionally selected for these studies to enable comparison with our past studies on Oatp1a4 regulation and functional expression at the BBB (Ronaldson et al., 2011; Thompson et al., 2014; Abdullahi et al., 2017, 2018). Additionally, our previous work on transporter sex differences in rat brain microvessels showed, for the first time, that Oatp1a4 protein expression and transport activity were significantly higher in female Sprague-Dawley rats compared with age-matched males (Brzica et al., 2018a). Observations from this study suggested that testosterone signaling may be involved in repression of Oatp1a4 functional expression at the BBB in 3-month-old male Sprague-Dawley rats (Brzica et al., 2018a). A detailed understanding of testosterone signaling and its regulation of BBB transporters is required before conducting analyses of Oatp1a4 expression/activity in male Sprague-Dawley rats. This is particularly relevant for studies involving other intracellular pathways, such as TGF-β/ALK1 signaling. Age-matched rats were randomized into treatment groups and injected with either BMP-9 (1.0 μg/kg (1.0 mL/kg) in 0.9% saline, i.p.; R&D Systems, Minneapolis, MN) or vehicle (0.9% saline, i.p.) according to our previously defined dosing paradigm (Abdullahi et al., 2018). Inhibition experiments were performed using the established ALK1 receptor antagonist 4-[6-[4-(1-piperazinyl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]quinoline dihydrochloride (LDN193189; 10 mg/kg (1.0 mL/kg) in 0.9% saline i.p.; Sigma-Aldrich, St. Louis, MO), which was administered 1 hour prior to injection with BMP-9. After a 6-hour BMP-9 treatment or 1-hour LDN193189/6-hour BMP-9, animals were anesthetized (100 mg/kg Ketamine, 20 mg/kg Xylazine, i.p.) and prepared for brain microvessel isolation. These time points were selected to enable detailed comparison with our previous study (Abdullahi et al., 2018).
Brain Microvessel Isolation
Brain microvessels were isolated from rat brain tissue using our previously published protocol (Brzica et al., 2018b). All steps were performed on ice at 4°C to minimize degradation of proteins during tissue processing. After euthanasia by decapitation, brains were isolated from the skull, and meninges and choroid plexus were removed. Cerebral hemispheres were homogenized at 3,700 x g in 5 ml brain microvessel buffer (300 mM mannitol, 5 mM EGTA, 12 mM Tris HCl, pH 7.4) containing 0.1% protease inhibitor cocktail (Sigma-Aldrich). For brain regional studies, brain tissue was separated into cerebral cortices, hippocampus, and cerebellum prior to homogenization. After homogenization, 8 ml 26% (w/v) dextran (MW 75,000; Spectrum Chemical Manufacturing Corporation, Gardena, CA) solution was added to each sample. Samples were then vortexed and centrifuged at 5,000 x g for 15 minutes at 4°C. At this time, the supernatant was aspirated, and capillary pellets were resuspended in 5 ml of brain microvessel buffer. Dextran homogenization and centrifugation steps were repeated an additional three times to ensure appropriate microvessel quality. After completion of dextran homogenization and centrifugation, the supernatant was aspirated and the microvessel pellet was resuspended in 5 ml of brain microvessel buffer. Enriched whole microvessel samples were homogenized at 3,700 x g, placed into ultracentrifuge tubes, and centrifuged at 150,000 x g for 60 minutes at 4°C for isolation of cellular membranes. Crude membrane pellets were resuspended in 500 μl of storage buffer (50% brain microvessel isolation buffer; 50% diH2O, v/v) containing 0.1% protease inhibitor cocktail. Samples were stored at −80°C until further use. We have previously confirmed purity of our microvessel preparations by demonstrating enrichment in platelet endothelial cell adhesion molecule-1 compared with expression of astrocyte marker proteins (i.e., glial fibrillary acidic protein) or neuronal marker proteins (i.e., synaptophysin) (Abdullahi et al., 2017; Brzica et al., 2018a,b).
Western Blot Analysis
Western blotting was performed as previously described (Abdullahi et al., 2018) with a few modifications. Crude membrane samples from isolated microvessels were quantified for total protein using the Pierce BCA Protein Assay (ThermoFisher Scientific, Waltham, MA), normalized across samples, and heated at 95°C for 5 minutes or 70°C for 10 minutes under reducing conditions [i.e., 2.5% (v/v) 2-mercaptoethanol (Sigma-Aldrich) in 1X Laemmli sample buffer (Bio-Rad, Hercules, CA)]. After SDS-PAGE and transfer, polyvinylidene difluoride (PVDF) membranes were incubated overnight at 4°C with primary antibodies against Oatp1a4 (anti-Oatp1, Invitrogen #PA5-42445; 0.5 mg/ml at 1:500 dilution), P-gp (anti-P-gp C219, Invitrogen #MA1-26528; 0.72 mg/ml at 1:500 dilution), Bcrp (anti-BCRP, Abcam #ab191812; 50 μg/mL at 1:500 dilution; Abcam #ab207732 50 μg/mL at 1:1000 dilution), claudin-5 (anti-claudin-5 4C3C2, ThermoFisher Scientific #35-2500; 0.5 mg/mL at 1:2,000 dilution), occludin (anti-occludin, ThermoFisher Scientific #40-6100; 0.25 mg/mL at 1:500), ALK1 (anti-ACVRL1, Invitrogen # MA5-38212, 1.4 mg/mL at 1:1000 dilution), ENG (anti-endoglin/CD105, Proteintech #1082-1-AP; 50 μg/mL at 1:1000 dilution), sodium-potassium ATPase (anti-sodium-potassium ATPase EP1845Y, Abcam #ab76020; 0.563 mg/ml at 1:20,000 dilution), and tubulin (anti-α tubulin, Abcam #ab7291, 1 mg/mL at 1:20,000 dilution). Membranes were washed using Tris-buffered saline containing 0.1% (v/v) Tween 20 detergent and incubated with horseradish peroxidase-conjugated anti-rabbit IgG (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA; 1:40,000 dilution) or anti-mouse IgG (Jackson ImmunoResearch, 1:50,000 dilution) for 60 minute at room temperature. Control western blot experiments to confirm antibody specificity were performed in the absence of primary antibody (i.e., used secondary antibody only). Protein bands were visualized using enhanced chemiluminesence (Super Signal West Pico, ThermoFisher Scientific, Waltham, MA). Bands were quantitated using ImageJ software (Wayne Rasband, Research Services Branch, National Institute of Mental Health, Bethesda, MD), normalized to tubulin, and reported as relative values.
Measurement of Vascular Density in Brain Tissue
Rats were anesthetized with ketamine/xylazine, decapitated, and the brains were immediately snap-frozen in isopentane on dry ice, and stored at −80°C. Cryosections (20 μm) were mounted onto glass slides, fixed in ice-cold methanol, and immunostained for the vascular marker rat endothelial cell antigen-1 (RECA-1; abcam #ab9774). Briefly, sections were blocked for 1 hour in PBS w/0.3% Tx-100 + 5% goat serum and then incubated overnight with RECA-1 (1:500). Sections were washed in PBS then incubated in Alexa 488 goat anti-mouse (Thermo Fisher # A11029; 1:500) for 1 hour. After washing in PBS and incubating in 4′,6-diamidino-2-phenylindole, the sections were mounted on coverslips in ProLong Diamond and imaged on a Leica SP8 scanning confocal microscope. Ten randomly chosen regions of interest in the hippocampus, cortex, and cerebellum from four different rats were imaged and the number of capillaries (vessels <10 μm diameter) per region were quantified.
In Situ Perfusion
In situ brain perfusion was performed using the approach described by our laboratory (Abdullahi et al., 2018; Brzica et al., 2018a). After treatment with BMP-9 (1.0 μg/kg) or LDN193189 (10 mg/kg)/BMP-9 (1.0 μg/kg), animals were anesthetized and heparinized (10,000 U/kg i.p.). Common carotid arteries were cannulated with silicone tubing connected to a perfusion circuit. Perfusion pressure and flow rate were tightly maintained between 95 to 105 mmHg and 3.1 ml/min, respectively. Both jugular veins were severed to allow for perfusate drainage. The perfusate used in these experiments was an erythrocyte-free modified mammalian Ringer's solution consisting of 117 mM sodium chloride, 4.7 mM potassium chloride, 0.8 mM magnesium sulfate, 1.2 mM potassium dihydrogen phosphate, 2.5 mM calcium chloride, 10 mM d-glucose, 3.9% (w/v) dextran (molecular weight 60,000), and 1.0 g/liter bovine serum albumin (type IV), pH 7.4, warmed to 37°C and continuously oxygenated with 95% oxygen/5% carbon dioxide. Evan’s blue dye (55 mg/liter) was added to the perfusate as a marker of BBB integrity. Using a slow-drive dual syringe pump (Harvard Apparatus Inc., Holliston, MA), [3H]atorvastatin (0.5 μCi/ml; 0.013 μM total concentration; American Radiolabeled Chemicals, Inc., St. Louis, MO), [3H]pravastatin (0.2 μCi/ml; 0.013 μM total concentration; American Radiolabeled Chemicals, Inc.), [3H]rosuvastatin (0.5 μCi/ml; 0.013 μM total concentration; PerkinElmer) or [carbon-14 (14C)]sucrose (0.5 μCi/ml; 0.92 μM total concentration; PerkinElmer) was added to the inflowing perfusion solution at a rate of 0.5 ml/min per cerebral hemisphere, which resulted in a total flow rate of 3.6 ml/min. For inhibition studies, animals were perfused with Ringer’s solution containing transport inhibitor [i.e., 100 μM fexofenadine (FEX), 5 μM PSC 833 (valspodar), 10 μM fumitremorgin C (FTC), or 10 μM GF 120918 (elacridar)] for 10 minutes prior to perfusion with [3H]atorvastatin, [3H]pravastatin, or [3H]rosuvastatin. We have previously confirmed the stability of Oatp transport substrates and sucrose in both perfusion medium and in jugular vein venous outflow and have shown that they remain intact in our in situ perfusion experiments (Ronaldson et al., 2009; Ronaldson et al., 2011; Thompson et al., 2014). Additionally, we have monitored EKG and respiratory waveforms in animals subjected to in situ brain perfusion for up to 30 minutes. In all animals, these physiologic parameters remained within normal limits, which implies that our in situ brain perfusion method allows for the evaluation of BBB transport mechanisms in a stable, well-controlled environment that maintains viability of experimental animals for the entire duration of the perfusion.
Immediately after perfusion for the desired time (i.e., 2.5, 5, 10, or 20 minutes), brain tissue was removed from the skull of an experimental animal. Meninges and choroid plexus were excised, and cerebral hemispheres were sectioned. TS2 tissue solubilizer (1.0 ml; Research Products International, Mount Prospect, IL) was added to each sample, and these were permitted to solubilize for 2 days at room temperature. To eliminate chemiluminescence, 30% glacial acetic acid (100 μl) was added, along with 2.0 ml of Optiphase SuperMix liquid scintillation cocktail (PerkinElmer Life and Analytical Sciences, Boston, MA). Radioactivity was measured using a model 1450 Liquid Scintillation and Luminescence Counter (PerkinElmer Life and Analytical Sciences). Results were reported as picomoles of radiolabeled transport substrate (i.e., atorvastatin, pravastatin, rosuvastatin, or sucrose) per milligram of brain tissue (C; pmol/mg tissue), which is equal to the total amount of radioisotope in the brain [CBrain; dpm/mg tissue] divided by the amount of radioisotope in the perfusate [CPerfusate; dpm/pmol]:
The brain vascular volume in rats has been previously shown to range between 6 and 9 μl/g brain tissue in perfusion studies utilizing a saline-based bicarbonate buffer (Takasato et al., 1984). Since brain tissue was processed immediately after perfusion with radiolabeled substrate, all whole-brain uptake values for radiolabeled statins or sucrose require correction for brain vascular volume (i.e., 8.0 μl/g brain tissue as calculated from data reported by Takasato and colleagues).
Multiple time uptake data were best fit to a nonlinear least-squares regression model, using the following equation, as described previously (Ronaldson et al., 2011):
where C is the concentration of drug per gram of brain tissue, t is time in min, KIN is the calculated uptake transfer constant, and kout is the estimated brain efflux rate coefficient. KIN was determined from the slope of the linear portion of the uptake curve according to:
The estimated brain volume of distribution (VBr) was calculated using the following equation as described previously (Ronaldson et al., 2011):
Using Eqs. 2–4, we can fit a nonlinear least-squares regression curve to our multiple-time uptake data. This results in the ability to determine both KIN and VBr. From these values, we can rearrange Eq. 4 to solve for kout:
Area under the curve (AUC) analysis was conducted on multiple time uptake data over the 0- to 20-minute perfusion interval (i.e., AUC0–20) using the trapezoidal method as an indicator of CNS drug exposure. All kinetic parameters and AUC0–20 values were calculated using Prism 9.0.1 software (GraphPad Software, La Jolla, CA).
Statistical Analysis
Western blot data are presented as mean ± S.D. of three independent experiments where each treatment group consisted of 3–4 individual animals (n = 4–6). In situ brain perfusion data are reported as mean ± S.D. of six individual animals per treatment group (n = 6). These sample sizes were based on the ability to detect a 35% difference between treatment with 20% variability. Statistical significance was determined using one-way ANOVA followed by post hoc Dunnett’s Multiple Comparison test. A value of p < 0.05 was accepted as statistically significant.
Results
BMP Treatment Modulates Brain Uptake and Exposure to Statins via Modulation of Oatp-Mediated Transport
Previous work in our laboratory has demonstrated that commonly prescribed statins, such as atorvastatin and pravastatin, are Oatp transport substrates and that their blood-to-brain uptake is increased in response to BMP-9 treatment (Abdullahi et al., 2017, 2018; Brzica et al., 2018a). Since these observations were obtained at a single time point, we conducted a time course (i.e., multiple-time uptake) study on whole brain tissue to assess the relationship between BMP-9 administration and changes in Oatp-mediated statin transport. In these multiple-time uptake experiments, female Sprague-Dawley rats were perfused with a radiolabeled statin drug (i.e., [tritiated (3H)]atorvastatin, [3H]pravastatin, or [3H]rosuvastatin) at various time points (i.e., 2.5, 5, 10, or 20 minutes). The time course of uptake for [3H]atorvastatin (0.013 μM), [3H]pravastatin (0.013 μM), or [3H]rosuvastatin (0.013 μM) in whole brain tissue isolated from control animals showed increasing statin accumulation over the entire 20-minute duration of the in situ brain perfusion experiment (Fig. 1, A–C). In animals administered BMP-9 (i.e., 1.0 μg/kg, i.p.; 6-hour treatment prior to perfusion), an increase (p < 0.01) in uptake was observed for all three radiolabeled statin drugs. These increases were up to 61% greater than control for [3H]atorvastatin, 60% greater than control for [3H]pravastatin, and 67% greater than control for [3H]rosuvastatin. In contrast, treatment with the ALK1 inhibitor LDN193189 (10 mg/kg, i.p.) 1 hour before dosing with BMP-9 completely attenuated increases in whole brain [3H]atorvastatin, [3H]pravastatin, or [3H]rosuvastatin uptake observed in animals treated with BMP-9 only (Fig. 1, A–C). The linear phase of uptake for [3H]atorvastatin, [3H]pravastatin, or [3H]rosuvastatin is shown in Fig. 1, D–F. These data clearly show that BMP-9 increased the rate of uptake for all three statin drugs, whereas LDN193189 attenuated BMP-9 effects on drug uptake transport.
Fig. 1.

Brain uptake of currently marketed statins after treatment with BMP-9. Effect of BMP-9 treatment on Oatp-mediated uptake of [3H]atorvastatin (A), [3H]pravastatin (B), and [3H]rosuvastatin (C) was measured via a multiple-time uptake in situ brain perfusion study. Animals were treated with BMP-9 (1 μg/kg, i.p.; 6-hour treatment) in the presence and absence of LDN193189 (10 mg/kg, i.p.; 1-hour pretreatment) and perfused with equal concentrations of radiolabeled statins (0.013 μM total concentration) for 2.5, 5, 10, and 20 minutes. Linear phases of uptake for [3H]atorvastatin (D), [3H]pravastatin (E), and [3H]rosuvastatin (F) are also shown. Results are expressed as mean ± S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; **p < 0.01).
Changes in whole brain exposure to statin drugs that resulted from altered Oatp-mediated transport at the BBB were measured by AUC0–20 analysis of our multiple-time uptake data (Fig. 2, A–C). After BMP-9 treatment, calculated AUC0–20 values were increased by 60% for [3H]atorvastatin [987.9 ± 53.41 pmol x min/mg brain tissue for control (95% CI: 931.8, 1044.0), 1581.0 ± 52.26 pmol x min/mg brain tissue for BMP-9 (95% CI: 1526.0, 1636.0; p < 0.01)] compared with saline controls, which suggests that activation of the ALK1 receptor leads to a significant increase in brain atorvastatin exposure. Similarly, calculated AUC0–20 values were increased by 69% for [3H]pravastatin [800.0 ± 47.41 pmol x min/mg brain tissue for control (95% CI: 750.2, 849.8), 1349.0 ± 48.00 pmol x min/mg brain tissue for BMP-9 (95% CI: 1299.0, 1399.0; p < 0.01)] and by 74% for [3H]rosuvastatin [836.8 ± 50.53 pmol x min/mg brain tissue for control (95% CI: 783.8, 889.8), 1459.0 ± 53.51 pmol x min/mg brain tissue for BMP-9 (95% CI: 1403.0, 1515.0; p < 0.01)]. Pharmacological inhibition of the ALK1 receptor with LDN193189 resulted in attenuation of the increased brain exposure observed in the presence of BMP-9 only for all three statin drugs. Kinetic analysis of multiple-time uptake data showed increases in both KIN and VBr for [3H]atorvastatin, [3H]pravastatin, and [3H]rosuvastatin after BMP-9 treatment (Table 1), results that reflect enhanced distribution of statin drugs to the brain after BMP-9 administration.
TABLE 1.
Kinetic analysis of multiple-times uptake data of Oatp-mediated statin transport in rat brain
| VBr (pmol/mg) | KIN (pmol/mg·min) |
kout (min−1) |
VBr (pmol/mg) |
KIN (pmol/mg·min) |
kout (min−1) |
VBr (pmol/mg) |
KIN (pmol/mg·min) |
kout (min−1) |
|
|---|---|---|---|---|---|---|---|---|---|
| Control | 82.62 ± 5.55 (95% CI: 71.52, 93.72) |
9.91 ± 0.22 (95% CI: 9.47, 10.35) |
0.12 ± 0.04 (95% CI: 0.04, 0.20) |
69.41 ± 4.50 (95% CI: 60.41, 78.41) |
7.64 ± 0.18 (95% CI: 7.28, 8.00) |
0.11 ± 0.04 (95% CI: 0.03, 0.19) |
75.37 ± 5.12 (95% CI: 65.13, 85.61) |
7.54 ± 0.10 (95% CI: 7.34, 7.74) |
0.10 ± 0.02 (95% CI: 0.06, 0.14) |
| BMP-9 (1.0 μg/kg) | 115.00 ± 6.52** (95% CI: 101.96, 128.04) |
18.40 ± 0.26** (95% CI: 17.88, 18.92) |
0.16 ± 0.04 (95% CI: 0.08, 0.24) |
92.43 ± 7.12** (95% CI: 78.19, 106.67) |
17.56 ± 0.57** (95% CI: 16.42, 18.70) |
0.19 ± 0.08 (95% CI: 0.03, 0.35) |
101.40 ± 2.87** (95% CI: 95.66, 107.14) |
18.25 ± 0.03** (95% CI: 18.19, 18.31) |
0.18 ± 0.03 (95% CI: 0.12, 0.24) |
| BMP-9 (1.0 μg/kg) + LDN193189 (10 mg/kg) | 77.07 ± 9.10 (95% CI: 58.87, 95.27) |
10.79 ± 0.36 (95% CI: 10.07, 11.51) |
0.14 ± 0.04 (95% CI: 0.06, 0.22) |
73.08 ± 4.81 (95% CI: 63.46, 92.32) |
7.31 ± 0.14 (95% CI: 7.03, 7.59) |
0.10 ± 0.03 (95% CI: 0.04, 0.16) |
71.74 ± 5.20 (95% CI: 61.34, 82.14) |
7.89 ± 0.10 (95% CI: 7.69, 8.09) |
0.11 ± 0.02 (95% CI: 0.07, 0.15) |
**p < 0.01 relative to control. VBr, brain volume of distribution; KIN, calculated uptake transfer constant; kout, brain efflux rate coefficient.
Fig. 2.

AUC analysis of multiple-time uptake data for currently marketed statin drugs. AUC analysis was conducted on multiple-time uptake data for atorvastatin (A), pravastatin (B), and rosuvastatin (C) using the trapezoidal method to obtain an indicator of CNS total drug exposure. Results are expressed as mean ± S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; **p < 0.01).
To confirm specificity of Oatp-mediated transport, we performed in situ brain perfusion studies in control rats, BMP-9–treated animals, and animals administered LDN193189 and BMP-9 in the presence and absence of the established Oatp transport inhibitor FEX. Statistical analysis of this experiment was conducted using three separate one-way ANOVA tests where control, BMP-9, and LDN193189/BMP-9 treatments were compared independently. As shown in Fig. 3A, whole brain uptake of [3H]atorvastatin in control animals (63.72 ± 9.78 pmol/mg brain tissue; 95% CI: 44.16, 83.28) was reduced by 39% (p < 0.01) in the presence of 100 μM FEX (24.89 ± 7.55 pmol/mg brain tissue; 95% CI: 9.79, 39.99). Comparable responses were observed in BMP-9–treated animals and in animals administered LDN193189/BMP-9. Similarly, whole brain uptake of [3H]pravastatin in control animals [54.98 ± 6.37 pmol/mg brain tissue for control (95% CI: 42.24, 67.72), 12.39 ± 4.81 pmol/mg brain tissue for control animals receiving FEX (95% CI: 2.77, 22.01; p < 0.01), Fig. 3B] and whole brain uptake of [3H]rosuvastatin in control animals [55.83 ± 7.84 pmol/mg brain tissue for control (95% CI: 40.15, 71.51) 10.54 ± 3.65 pmol/mg brain tissue for control animals receiving FEX (95% CI: 3.24, 17.84; p < 0.01), Fig. 3C] were reduced in the presence of FEX, suggesting an Oatp-dependent transport mechanism. As with [3H]atorvastatin, comparable responses for [3H]pravastatin and [3H]rosuvastatin were observed in BMP- 9–treated animals and in animals administered LDN193189/BMP-9.
Fig. 3.

Accumulation of currently marketed statins in whole brain tissue is determined by OATP-mediated transport at the BBB. Effect of an established Oatp transport inhibitor (i.e., FEX) on the uptake of [3H]atorvastatin (A), [3H]pravastatin (B), and [3H]rosuvastatin (C) was assessed by in situ brain perfusion studies. Accumulation of statin drugs was measured in animals injected with BMP-9 (1 μg/kg, i.p.; 6-hour treatment) in the presence and absence of LDN193189 (10 mg/kg, i.p.; 1-hour pretreatment) where animals were perfused with FEX (100 μM) prior to perfusion with radiolabeled atorvastatin, pravastatin, or rosuvastatin. Results are expressed as mean ± S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; **p < 0.01).
Vascular Density Is Highest in Cerebellum, Followed by Cerebral Cortex and Hippocampus
We performed immunofluorescence analysis on brain sections stained with an antibody to the vascular marker RECA-1. Ten randomly chosen regions of the cortex, hippocampus, and cerebellum were imaged from four different rats and the capillary density was quantified. We observed statistically significant differences in capillary density in the cortex (129.1 ± 30.5 capillaries/mm2), hippocampus (84.6 ± 15.5 capillaries/mm2), and the cerebellum (167.6 ± 36.3 capillaries/mm2) (Fig. 4; p < 0.0001). These numbers are in general agreement with previous observations of vascular density in different brain regions (Cavaglia et al., 2001).
Fig. 4.
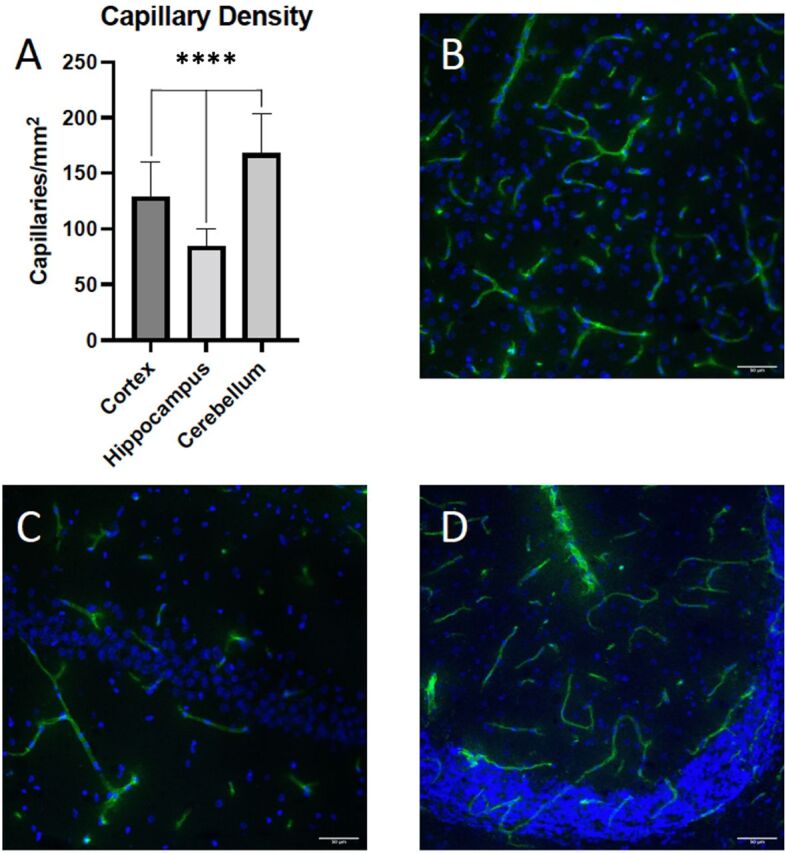
Brain vascular density in cerebral cortex, hippocampus, and cerebellum. Ten randomly chosen regions in the cortex, hippocampus, and cerebellum from four different rats (n = 40) were imaged and the number of RECA-1 positive capillaries were quantified (A). Representative images of the cortex (B), hippocampus (C), and cerebellum (D) showing RECA-1 immunofluorescence in green and 4′,6-diamidino-2-phenylindole (DAPI) in blue. Results are expressed as mean ± S.D. of 10 vessels per brain region. Asterisks indicate data points that were significantly different from control animals (****P < 0.0001).
BMP-9 Treatment Alters Oatp1a4 Protein Expression in Cerebral Microvessels Isolated from Cortex but Not in Hippocampus or Cerebellum
Previously, we demonstrated that BMP-9 treatment increases Oatp1a4 protein expression in cerebral microvessels prepared from whole brain tissue collected from female Sprague-Dawley rats (Abdullahi et al., 2017, 2019). Therefore, we sought to determine if BMP-9-induced changes in BBB Oatp1a4 expression varied across brain regions. To assess such regional differences, we isolated brain microvessels from cortical, hippocampal, and cerebellar tissue and used western blot analysis to evaluate Oatp1a4 expression in rats administered BMP-9 or LDN193189/BMP-9 (Fig. 5A). Densitometric analysis of our western blot data showed increased Oatp1a4 protein expression (normalized to tubulin) in cortical microvessels from female Sprague-Dawley rats administered a single dose of BMP-9 (1.0 μg/kg, i.p.; 6-hour time point) (Fig. 5B). This increase was attenuated in animals administered both LDN193189 and BMP-9, further supporting involvement of TGF-β/ALK1 signaling in mediating this response. Interestingly, neither hippocampal microvessels nor cerebellar microvessels showed an increase in Oatp1a4 protein expression in response to treatment with BMP-9 (Fig. 5B). The relative magnitude of Oatp1a4 protein changes induced by BMP-9 are presented in Supplemental Table 1. Since we have previously demonstrated that BMP-9 can directly induce mRNA and functional protein expression via activation of the TGF-β/ALK1 canonical Suppressor of Mothers against Decapentaplegic (Smad) 1/5/8 signaling pathway with minimal off-target effects (Abdullahi et al., 2017, 2018), we also evaluated protein expression of the ALK1 receptor in cortical, hippocampal, and cerebellar microvessels. Interestingly, ALK1 expression was comparable in microvessels derived from these three brain regions and did not change in response to either BMP-9 or LDN193189/BMP-9 treatment (Fig. 6, A–B). The magnitude of ALK1 protein changes induced by BMP-9 relative to control are presented in Supplemental Table 2. With growing evidence elucidating the importance of ENG in downstream ALK1 signaling (Lee et al., 2008; Lawera et al., 2019), we determined ENG expression in rat brain microvessels isolated from cerebral cortex, hippocampus, and cerebellum. We observed elevated ENG protein expression in cortical microvessels that was further increased after BMP-9 treatment (Fig. 6, C–D). In contrast, ENG expression was lower in microvessels from hippocampus and cerebellum compared with microvessels isolated from cerebral cortex. Furthermore, BMP-9 treatment did not alter ENG expression in either hippocampal or cerebellar microvessels (Fig. 6D).
Fig. 5.

BMP-9 treatment increases Oatp1a4 protein expression in cerebral cortex but not in hippocampus or cerebellum. (A) Animals were administered a single dose of BMP-9 (1.0 μg/ml) or LDN193189 (10 mg/kg) at 1 hour prior to receiving a single dose of BMP-9 (1.0 μg/ml). After 6 hours, animals were euthanized and brain microvessels isolated and prepared for western blot analysis. Isolated microvessels (10 μg) were resolved on a 10% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of Oatp1a4 or tubulin (i.e., the loading control). Each lane pair on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. (B) Relative levels of Oatp1a4 protein expression were determined by densitometric analysis and normalized to tubulin. Results are expressed as mean ± SD from at least two independent experiments, where each treatment group consisted of normalized data from eight individual animals (n = 8). Asterisks represent data points that were significantly different from control (**p < 0.01).
Fig. 6.

Expression of ALK1 receptors and ENG in cerebral cortex, hippocampus, and cerebellum after treatment with BMP-9. (A) Animals were administered a single dose of BMP-9 (1.0 μg/ml) or LDN193189 (10 mg/kg) at 1 hour prior to receiving a single dose of BMP-9 (1.0 μg/ml). After 6 hours, animals were euthanized and brain microvessels isolated and prepared for western blot analysis. Isolated microvessels (10 μg) were resolved on a 10% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of ALK1 or tubulin (i.e., the loading control). Each lane pair on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. (B) Relative levels of ALK1 protein expression were determined by densitometric analysis and normalized to tubulin. Results are expressed as mean ± S.D. from at least two independent blots, where each treatment group consisted of normalized data from eight individual animals (n = 8). (C) Isolated microvessels (10 μg) were resolved on a 7.5% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of ENG or tubulin (i.e., the loading control). Each lane pair on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. (D): Relative levels of ENG protein expression were determined by densitometric analysis and normalized to tubulin. Results are expressed as mean ± S.D. where each treatment group consisted of normalized data from four individual animals (n = 4). Asterisks represent data points that were significantly different from control (**p < 0.01).
Increased Cortical Microvascular Oatp1a4 Protein Expression after BMP-9 Treatment Correlates with Enhanced Drug Uptake of Currently Marketed Statins
To determine whether increased cortical microvascular Oatp1a4 expression after BMP-9 treatment correlates with enhanced blood-to-brain transport of statins, we measured brain uptake of equimolar concentrations of [3H]atorvastatin, [3H]pravastatin, and [3H]rosuvastatin by in situ brain perfusion. Brain accumulation of these three statin drugs was studied in control rats, BMP-9–treated animals, and animals administered LDN193189 and BMP-9 in the presence and absence of the established Oatp inhibitor FEX. Cortical brain uptake of [3H]atorvastatin in control animals was 87.06 ± 8.66 pmol/mg brain tissue (95% CI: 69.74, 104.38) after a 10-minute perfusion (Fig. 7A). Cortical accumulation of [3H]pravastatin in control animals was 69.21 ± 7.23 pmol/mg brain tissue (95% CI: 54.75, 83.67; Fig. 7B), whereas cortical uptake of [3H]rosuvastatin was 75.79 ± 8.14 pmol/mg brain tissue (95% CI: 59.51, 92.07; Fig. 7C), levels that were comparable with [3H]atorvastatin. BMP-9 treatment increased cortical brain uptake of [3H]atorvastatin (132.56 ± 8.88 pmol/mg brain tissue; 95% CI: 114.80, 150.32; p < 0.01), [3H]pravastatin (99.05 ± 8.52 pmol/mg brain tissue; 95% CI: 82.01, 116.09; p < 0.01), and [3H]rosuvastatin (100.79 ± 7.23 pmol/mg brain tissue; 95% CI: 86.33, 115.25; p < 0.01). Pretreatment with LDN193189 in BMP-9–treated animals attenuated cortical brain uptake for [3H]atorvastatin (84.43 ± 9.40 pmol/mg brain tissue; 95% CI: 65.63, 103.23), [3H]pravastatin (66.44 ± 8.22 pmol/mg brain tissue; 95% CI: 50.00, 82.88), and [3H]rosuvastatin (70.94 ± 8.62 pmol/mg brain tissue; 95% CI: 53.70, 88.18). In the presence of FEX, cortical brain uptake of [3H]atorvastatin was reduced in control rats (22.78 ± 8.05 pmol/mg brain tissue; 95% CI: 6.68, 38.88; p < 0.01), BMP-9–treated animals (26.66 ± 8.78 pmol/mg brain tissue; 95% CI: 9.10, 44.22; p < 0.01), and in rats pretreated with LDN193189 prior to BMP-9 administration (23.67 ± 9.01 pmol/mg brain tissue; 95% CI: 5.65, 41.69; p < 0.01). FEX inhibited cortical accumulation of [3H]pravastatin in control animals (17.04 ± 5.00 pmol/mg brain tissue; 95% CI: 7.04, 27.04; p < 0.01), rats administered BMP-9 (17.43 ± 5.79 pmol/mg brain tissue; 95% CI: 5.85, 29.01; p < 0.01), and in LDN193189/BMP-9–treated animals (16.42 ± 4.70 pmol/mg brain tissue; 95% CI: 7.02, 25.82; p < 0.01). Similar results were also obtained for [3H]rosuvastatin where cortical drug accumulation was decreased by FEX in control rats (9.77 ± 3.21 pmol/mg brain tissue; 95% CI: 3.35, 16.19; p < 0.01), BMP-9–treated animals (12.68 ± 3.97 pmol/mg brain tissue; 95% CI: 4.74, 20.62; p < 0.01), and in rats administered LDN193189 and BMP-9 (10.45 ± 3.00 pmol/mg brain tissue; 95% CI: 4.45, 16.45; p < 0.01). Interestingly, BMP-9 treatment did not change brain uptake of either [3H]atorvastatin, [3H]pravastatin, or [3H]rosuvastatin in hippocampus or in cerebellum (Fig. 7, B–C). Statin accumulation was significantly reduced (p < 0.01) by FEX in hippocampal and cerebellar tissue, suggesting a role for Oatp1a4 in determining drug distribution to these important brain regions.
Fig. 7.

Oatp-mediated statin accumulation is increased in cerebral cortex, but not in hippocampus or cerebellum, after BMP-9 treatment. Blood-to-brain transport of [3H]atorvastatin (A), [3H]pravastatin (B), and [3H]rosuvastatin (C) were measured by the in situ brain perfusion approach. Animals were treated with BMP-9 (1 μg/kg, i.p.; 6-hour treatment) in the presence and absence of LDN193189 (10 mg/kg, i.p.; 1-hour pretreatment) and perfused with equal concentrations of radiolabeled statins (0.013 μM total concentration). Each experiment was conducted in the presence and absence of FEX (100 μM) to confirm specificity of Oatp-mediated transport. At the conclusion of the experiment, concentrations of each currently marketed statin were measured in cerebral cortex, hippocampus, and cerebellum. Results are expressed as mean ± S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; **p < 0.01).
CNS Disposition of Statins Is also Dependent on Functional Expression of P-gp and Bcrp at the BBB
Interpretation of blood-to-brain transport data for statins requires the appreciation that multiple transporters play a critical role in determining their brain disposition. Indeed, our previous work in HUVECs has shown that atorvastatin, pravastatin, and rosuvastatin are transport substrates for P-gp and/or BCRP (Ronaldson et al., 2021). Therefore, we wanted to determine if these critical efflux transporters played a role in the CNS disposition of statins in our rodent model. To demonstrate specificity of transport for P-gp and/or Bcrp, we used an established P-gp inhibitor (i.e., PSC 833), a known Bcrp inhibitor (i.e., FTC), and a dual P-gp/Bcrp inhibitory drug (i.e., GF120918). Whole brain uptake of [3H]atorvastatin in control animals (60.04 ± 8.55 pmol/mg brain tissue; 95% CI: 42.94, 77.14) was significantly increased (p < 0.01) by 44% in the presence of PSC833 (86.35 ± 7.56 pmol/mg brain tissue; 95% CI: 71.23, 101.47), by 33% in the presence of FTC (79.56 ± 7.21 pmol/mg brain tissue; 95% CI: 65.14, 93.98), and by 79% in the presence of GF120918 (107.45 ± 8.40 pmol/mg brain tissue; 95% CI: 90.65, 124.25; Fig. 8A). In animals treated with BMP-9, the magnitude of increase in CNS atorvastatin drug delivery caused by P-gp and/or Bcrp transport inhibitors was less than that measured in control animals. Specifically, [3H]atorvastatin in rats administered BMP-9 was enhanced (p < 0.01) by 23% in the presence of PSC833 (116.39 ± 11.57 pmol/mg brain tissue; 95% CI: 93.25, 139.53), by 21% in the presence of FTC (114.28 ± 5.85 pmol/mg brain tissue; 95% CI: 102.58, 125.98), and by 40% in the presence of GF120918 (132.78 ± 8.92 pmol/mg brain tissue; 95% CI: 114.94, 150.62) compared with [3H]atorvastatin alone (94.54 ± 10.57 pmol/mg brain tissue; 95% CI: 73.40, 115.68). These data suggest that efflux transporters play a reduced role in the CNS disposition of [3H]atorvastatin in the setting of BMP-9 treatment. This was supported by our observation that the magnitude of inhibition caused by PSC833, FTC, and GF120918 was restored to 38% (85.00 ± 8.78 pmol/mg brain tissue; 95% CI: 67.44, 102.56), 33% (81.39 ± 7.83 pmol/mg brain tissue; 95% CI: 65.73, 97.05), and 70% (103.10 ± 6.52 pmol/mg brain tissue; 95% CI: 90.06, 116.14), respectively, in animals pretreated with LDN193189 prior to BMP-9 administration (Fig. 8A). These values were determined based on the uptake of [3H]atorvastatin in the absence of P-gp/Bcrp inhibitors in rats administered LDN193189 and BMP-9 (64.38 ± 9.67 pmol/mg brain tissue; 95% CI: 45.04, 83.72).
Fig. 8.

P-gp and Bcrp are critical determinants of BBB permeability to currently marketed statin drugs. Brain uptake of [3H]atorvastatin (A), [3H]pravastatin (B), and [3H]rosuvastatin (C) were measured by the in situ brain perfusion approach. Animals were treated with BMP-9 (1 μg/kg, i.p.; 6-hour treatment) in the presence and absence of LDN193189 (10 mg/kg, i.p.; 1-hour pretreatment) and perfused with equal concentrations of radiolabeled statins (0.013 μM total concentration). Each experiment was conducted in the presence and absence of PSC833 (5 μM), FTC (10 μM), or GF120918 (10 μM) to limit the involvement of P-gp and/or Bcrp in the brain microvascular transport of statin drugs. Results are expressed as mean ±S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; ** p< 0.01).
Our ATP-binding cassette (ABC) transporter inhibitor data with [3H]pravastatin suggests involvement of Bcrp, but not P-gp, in the CNS disposition of this statin drug (Fig. 8B). Specifically, blood-to-brain uptake of [3H]pravastatin was 49.56 ± 9.80 pmol/mg brain tissue (95% CI: 29.96, 69.16), which was increased to 69.28 ± 8.56 pmol/mg brain tissue (95% CI: 52.16, 86.40; p < 0.05) in the presence of FTC and 75.39 ± 7.88 pmol/mg brain tissue (95% CI: 59.63, 91.15; p < 0.01) in response to GF120918 treatment. The P-gp inhibitor PSC833 had no effect on brain uptake of [3H]pravastatin. In animals treated with BMP-9, a significant increase (p < 0.01) in [3H]pravastatin accumulation (87.65 ± 9.42 pmol/mg brain tissue; 95% CI: 68.81, 106.49) was achieved with both FTC (108.56 ± 6.29 pmol/mg brain tissue; 95% CI: 95.98, 121.14) and GF120918 (116.28 ± 10.56 pmol/mg brain tissue; 95% CI: 95.16, 137.40) but not with PSC833 (94.06 ± 13.57 pmol/mg brain tissue; 95% CI: 66.92, 121.20). In animals treated with LDN193189 and BMP-9, [3H]pravastatin uptake values were comparable to those obtained in control rats (48.67 ± 8.17 pmol/mg brain tissue; 95% CI: 32.33, 65.01). In presence of FTC, brain accumulation was increased (p < 0.01) by 45% to 70.56 ± 7.83 pmol/mg brain tissue (95% CI: 54.90, 86.22) in the presence of FTC and by 47% to 71.29 ± 10.01 pmol/mg brain tissue (95% CI: 51.27, 91.31) in the presence of GF120918. Similar to control rats and animals administered BMP-9, PSC833 had no effect on [3H]pravastatin uptake (59.26 ± 11.56 pmol/mg brain tissue; 95% CI: 36.14, 82.38).
In contrast to [3H]pravastatin, brain disposition of [3H]rosuvastatin is dependent on transport processes mediated by both P-gp and Bcrp (Fig. 8C). In control rats, [3H]rosuvastatin uptake was 58.29 ± 9.01 pmol/mg brain tissue (95% CI: 40.27, 76.31); this value was increased by 73% in the presence of PSC833 (100.70 ± 10.43 pmol/mg brain tissue; 95% CI: 79.84, 121.56; p < 0.01), by 59% in the presence of FTC (92.67 ± 11.56 pmol/mg brain tissue; 95% CI: 69.55, 115.79; p < 0.01), and by 114% in the presence of GF120918 (124.54 ± 11.14 pmol/mg brain tissue; 95% CI: 102.26, 146.82; p < 0.01). Pharmacological inhibition of P-gp and/or Bcrp caused a substantial enhancement in [3H]rosuvastatin uptake in animals subjected to BMP-9 treatment. Although [3H]rosuvastatin accumulation was 96.43 ± 9.04 pmol/mg brain tissue (95% CI: 78.35, 114.51) in the absence of inhibitors, uptake values were increased by 100% in the presence of PSC833 (193.08 ± 11.00 pmol/mg brain tissue; 95% CI: 171.08, 215.08; p < 0.01), by 92% in the presence of FTC (185.29 ± 15.97 pmol/mg brain tissue; 95% CI: 153.35, 217.23; p < 0.01), and by 116% in the presence of GF120918 (207.85 ± 14.56 pmol/mg brain tissue; 95% CI: 178.73, 236.97; p < 0.01). The magnitude of enhancement of [3H]rosuvastatin accumulation in rats treated with LDN193189 and BMP-9 was comparable to those obtained in control animals. Specifically, blood-to-brain uptake of [3H]rosuvastatin by itself was 59.22 ± 7.52 pmol/mg brain tissue (95% CI: 44.18, 74.26). ABC transporter inhibitors increased this uptake as evidenced by a 77% enhancement in the presence of PSC833 (104.78 ± 13.78 pmol/mg brain tissue; 95% CI: 77.22, 132.34; p < 0.01), by a 67% increase in the presence of FTC (98.78 ± 9.80 pmol/mg brain tissue; 95% CI: 79.18, 11.38; p < 0.01), and by a 102% enhancement in the presence of GF120918 (119.68 ± 14.79 pmol/mg brain tissue; 95% CI: 90.12, 149.24; p < 0.01).
BMP-9 Administration Does Not Alter Protein Expression of P-gp or Bcrp in Rat Brain Microvessels
A critical consideration in the interpretation of our transport data is whether BMP-9 treatment could modulate protein expression of ABC transporters at the BBB. Therefore, we conducted a time course study to assess the temporal relationship between BMP-9 administration and changes in microvascular expression of P-gp and/or Bcrp. The time points evaluated in these experiments were 0, 2, 4, and 6 hours, which were the same time points that we used to study the effect of BMP-9 treatment on Oatp1a4 in our previous publication (Abdullahi et al., 2018). Densitometric analysis of our western blot data showed no change in either P-gp or Bcrp protein expression at any of the time points that were studied (Fig. 9, A–B). We confirmed this response at the 6-hour time point using the pharmacological ALK1 inhibitor LDN193189 (Fig. 9, C–D). In these experiments, we observed via densitometric analysis that protein expression of P-gp and Bcrp were the same in brain microvessels isolated from control animals, BMP-9–treated rats, and from animals administered both LDN193189 and BMP-9. Since these data were derived from microvessels isolated from whole brain tissue, we also sought to determine if BMP-9 could alter P-gp and/or Bcrp protein expression in different brain regions. In these experiments, we evaluated protein expression of these critical ABC transporters in cerebral cortex, hippocampus, and cerebellum. In animals treated with BMP-9, we did not observe any change in P-gp or Bcrp expression in microvessels isolated from cortex, hippocampus, or cerebellum compared with untreated controls (Fig. 10, A–B). Additionally, LDN193189 administration prior to BMP-9 treatment did not affect protein expression of either efflux transporter. All western blot expression data for P-gp and Bcrp were normalized to tubulin (i.e., the loading control). Taken together, these data suggest that activation of TGF-β/ALK1 signaling via a selective agonist, such as BMP-9, does not affect BBB protein expression of ABC transporters, such as P-gp and Bcrp. The relative magnitude of P-gp and Bcrp protein changes induced by BMP-9 across a 0- to 6- hour time course or in cortex, hippocampus, and cerebellum are presented in Supplemental Tables 3–6.
Fig. 9.

P-gp and Bcrp expression in rat brain microvessels is not altered in response to BMP-9 treatment. (A) Animals were administered a single dose of BMP-9 4(1.0 μg/ml). After 2 hours, 4 hours, or 6 hours, animals were euthanized and brain microvessels isolated and prepared for western blot analysis. Isolated microvessels (10 μg) were resolved on a 10% or 4%–12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane, and analyzed for expression of P-gp, Bcrp, or tubulin (i.e., the loading control). Each lane pair on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. Relative levels of P-gp and Bcrp protein expression were determined by densitometric analysis and normalized to tubulin. Results are expressed as mean ± S.D. from at least two independent experiments, where each treatment group consisted of normalized data from six individual animals (n = 6). Asterisks represent data points that were significantly different from control (**p < 0.01). (B) Animals were administered a single dose of BMP-9 (1.0 μg/ml) or LDN193189 (10 mg/kg) at 1 hour prior to receiving a single dose of BMP-9 (1.0 μg/ml). After 6 hours, animals were euthanized and brain microvessels isolated and prepared for western blot analysis. Isolated microvessels (10 μg) were resolved on a 10% or 4%–12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane and analyzed for expression of P-gp, Bcrp, or tubulin (i.e., the loading control). Each lane pair on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. Relative levels of P-gp and Bcrp protein expression were determined by densitometric analysis and normalized to tubulin. Results are expressed as mean ± S.D. from at least two independent experiments, where each treatment group consisted of normalized data from six individual animals (n = 6). Asterisks represent data points that were significantly different from control (**p < 0.01).
Fig. 10.

P-gp and Bcrp expression in cerebral cortex, hippocampus, and cerebellum after BMP-9 treatment. Animals were administered a single dose of BMP-9 (1.0 μg/ml) or LDN193189 (10 mg/kg) at 1 hour prior to receiving a single dose of BMP-9 (1.0 μg/ml). After 6 hours, animals were euthanized and brain microvessels isolated and prepared for western blot analysis. Isolated microvessels (10 μg) were resolved on a 10% or 4%–12% SDS-polyacrylamide gel, transferred to a polyvinylidene difluoride membrane and analyzed for protein expression of P-gp (A) or Bcrp (B) in cerebral cortex, hippocampus, or cerebellum (C). Each lane pair on the depicted western blot corresponds to a microvessel sample obtained from a single experimental animal. Relative levels of P-gp or Bcrp protein expression were determined by densitometric analysis and normalized to tubulin (i.e., the loading control). Results are expressed as mean ± S.D. from at least two independent experiments, where each treatment group consisted of normalized data from eight individual animals (n = 8). Asterisks represent data points that were significantly different from control (**p < 0.01).
Brain Regional Uptake of Statin Drugs Is Increased by GF120918 at the Same Magnitude in Control, BMP-9–Treated, and LDN193189/BMP-9–Treated Animals
To complement our protein expression results, we conducted in situ perfusion experiments using the dual P-gp/Bcrp transport inhibitor GF120918. This strategy enabled us to evaluate the effect of blocking P-gp/Bcrp transport activity on CNS statin uptake in control, BMP-9–treated, and LDN193189/BMP-9–treated rats. For [3H]atorvastatin (Fig. 11A), brain accumulation in cortical tissue from control rats (87.06 ± 8.68 pmol/mg brain tissue; 95% CI: 69.70, 104.42) was increased by 37% (119.46 ± 7.96 pmol/mg brain tissue; 95% CI: 103.54, 135.38; p < 0.01). Similarly, brain [3H]atorvastatin uptake was increased by 50% (p < 0.01) in hippocampus (54.28 ± 9.00 pmol/mg brain tissue (95% CI: 36.28, 72.28) for control versus 81.34 ± 8.05 pmol/mg protein (95% CI: 65.24, 97.44) for animals perfused with GF120918) and by 41% (p < 0.01) in cerebellum (30.93 ± 6.54 pmol/mg brain tissue (95% CI: 17.85, 44.01) for control versus 43.60 ± 6.85 pmol/mg brain tissue (95% CI: 29.90, 57.30) for animals perfused with GF120918) (Fig. 11A). Although there was an increase in cortical [3H]atorvastatin uptake in response to BMP-9 treatment relative to control (132.56 ± 8.88 pmol/mg brain tissue; 95% CI: 114.80, 150.32; p < 0.01), the magnitude of effect of GF120918 was 32% (175.29 ± 9.01 pmol/mg brain tissue; 95% CI: 157.27, 193.31; p < 0.01), which is comparable to that measured in control animals. In hippocampal and cerebellar brain tissue, BMP-9 administration caused neither a change in [3H]atorvastatin uptake nor an alteration in the magnitude of GF120918 effect. In animals administered both LDN193189 and BMP-9, the magnitude of effect of GF120918 on [3H]atorvastatin accumulation was restored to levels observed in control (i.e., untreated) rats.
Fig. 11.

P-gp and Bcrp have a similar effect on statin disposition in cerebral cortex, hippocampus, and cerebellum. Brain uptake of [3H]atorvastatin (A), [3H]pravastatin (B), and [3H]rosuvastatin (C) were measured by the in situ brain perfusion approach. Animals were treated with BMP-9 (1 μg/kg, i.p.; 6-hour treatment) in the presence and absence of LDN193189 (10 mg/kg, i.p.; 1-hour pretreatment) and perfused with a radiolabeled statin (0.013 μM total concentration). Each experiment was conducted in the presence and absence of GF120918 (10 μM) to determine involvement of the critical BBB efflux transporters P-gp and Bcrp in the brain microvascular transport of statin drugs. At the conclusion of this experiment, drug concentrations were measured in cerebral cortex, hippocampus, and cerebellum to evaluate brain regional differences in statin transport. Results are expressed as mean ± S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; **p < 0.01).
With respect to [3H]pravastatin (Fig. 11B) and [3H]rosuvastatin (Fig. 11C), similar effects of GF120918 on statin uptake in cerebral cortex, hippocampus, and cerebellum were detected. In the absence of GF120918, cortical brain uptake of [3H]pravastatin was 69.21 ± 7.23 pmol/mg brain tissue (95% CI: 54.75, 83.67) and cortical accumulation of [3H]rosuvastatin was 75.79 ± 8.14 pmol/mg brain tissue (95% CI: 59.51, 92.07). In animals treated with BMP-9 but not perfused with GF120918, cortical uptake of both [3H]pravastatin and [3H]rosuvastatin was significantly increased (p < 0.01) to 99.05 ± 8.52 pmol/mg brain tissue (95% CI: 82.01, 116.09) and 100.79 ± 7.23 pmol/mg brain tissue (95% CI: 86.33, 115.25), respectively. Inclusion of GF120918 in our perfusion experiments increased [3H]pravastatin uptake in cerebral cortex by 48% in control rats (102.78 ± 8.00 pmol/mg brain tissue; 95% CI: 86.78, 118.78; p < 0.01) and by 31% in BMP-treated animals (129.67 ± 6.79 pmol/mg brain tissue; 95% CI: 116.09, 143.25; p < 0.01) (Fig. 11B). Co-administration of LDN193189 and BMP-9 did not lead to an increase in [3H]pravastatin uptake in the absence of GF120918 (66.44 ± 8.22 pmol/mg brain tissue; 95% CI: 50.00, 82.88). Although GF120918 increased [3H]pravastatin uptake by 50% (99.87 ± 6.28 pmol/mg brain tissue; 95% CI: 87.31, 112.43) in animals treated with BMP-9 and LDN193189, this value was not different from [3H]pravastatin brain accumulation in animals that were not treated with BMP-9. In the case of [3H]rosuvastatin, addition of GF120918 to the perfusate in our in situ perfusion system resulted in enhanced uptake in cerebral cortex by 93% in control rats (146.58 ± 11.28 pmol/mg brain tissue; 95% CI: 124.02, 169.14; p < 0.01) and by 98% in BMP-9–treated animals (199.58 ± 12.57 pmol/mg brain tissue; 95% CI: 174.44, 224.72; p < 0.01) (Fig. 11B). Coadministration of LDN193189 and BMP-9 did not lead to an increase in [3H]rosuvastatin uptake in the absence of GF120918 (70.94 ± 8.62 pmol/mg brain tissue; 95% CI: 53.70, 88.18). GF120918 did increase [3H]rosuvastatin uptake by 112% (150.63 ± 14.56 pmol/mg brain tissue; 95% CI: 121.51, 179.75) in animals treated with BMP-9 and LDN193189, but this value was not different from [3H]rosuvastatin brain accumulation in animals that were not treated with BMP-9. In both the hippocampus and cerebellum, the magnitude of GF120918 effect on statin drug uptake was not different between control animals, rats administered BMP-9, and animals subjected to LDN193189/BMP-9 treatment (Fig. 11, B–C).
BMP-9 Treatment Does Not Alter Expression of Transmembrane Tight Junction Proteins or Paracellular Permeability in Rat Brain Microvessels
A thorough understanding of the effects of BMP-9 treatment on CNS disposition of statins requires assessment of BBB integrity. Indeed, pathophysiological and pharmacological stressors can change tight junction protein expression, an effect that can lead to paracellular “leak” between adjacent endothelial cells and increase the ability of drugs, metabolites, and toxicants to access the brain. With respect to TGF-β/ALK1 signaling, the effect of this pathway activation on BBB integrity is unknown. To address this critical issue, we measured protein expression of the transmembrane tight junction proteins claudin-5 and occludin, which perform a “sealing function” at the BBB. We observed that BMP-9 (1.0 μg/kg, i.p.) did not alter protein expression of claudin-5 or monomeric occludin at time points up to 6 hours postadministration (Fig. 12A). Furthermore, brain uptake of [14C]sucrose, a vascular marker that does not cross the intact BBB, was not changed at 2 hours, 4 hours, or 6 hours after intraperitoneal injection of BMP-9 (Fig. 12B). Overall, these data indicate that activation of TGF-β/ALK1 signaling by a specific agonist (i.e., BMP-9) does not modulate tight junction protein expression or paracellular permeability (i.e., leak) at the cerebral microvasculature. Taken together with all data presented in this study, we have demonstrated that the effect of BMP-9 on BBB transport mechanisms is highly selective for Oatp1a4 mediated uptake of small molecules including drugs.
Fig. 12.

BMP-9 Treatment does not alter BBB expression of critical tight junction proteins or paracellular permeability in rat brain microvessels. (A) Animals were administered a single dose of BMP-9 (1 μg/kg, i.p.). After the time course of 2 to 6 hours, animals were euthanized and brain microvessels were isolated for western blot analysis. Isolated microvessels (10 μg) were resolved on a 4%–12% SDS-polyacrylamide gel, transferred to a PVDF membrane, and analyzed for expression of claudin-5 and occludin monomers. Relative levels of claudin-5 and occludin monomers were determined by densitometric analysis and normalized to sodium-potassium ATPase. Western blot data are reported as mean ±S.D. of three independent experiments where each treatment group consisted of three individual animals (n = 3). Asterisks represent data points that were significantly different from control (* p < 0.05; ** p < 0.01). (B) Animals were administered a single dose of BMP-9 (1 μg/kg, i.p.). After 2 to 6 hours BMP-9 exposure, paracellular permeability to [14C]sucrose, a vascular marker that does not typically cross the BBB, was measured by in situ brain perfusion. Results are expressed as mean ± S.D. of six animals per time point. Asterisks represent data points that were significantly different from control animals (*p < 0.05; **p< 0.01).
Discussion
Brain delivery of therapeutics requires attainment of clinically relevant concentrations at specific molecular targets. Since this objective continues to be a challenge in treatment of neurologic disorders, our laboratory’s research is focused on evaluation of BBB transport mechanisms that can be exploited for this purpose. Much of our work has focused on Oatps, in part because their substrate profile includes statins, drugs that confer neuroprotection independent of their well-established effects on circulating cholesterol (Prinz et al., 2008; Zhang et al., 2009; Fang et al., 2015; Christophe et al., 2020). This is apparent in ischemic stroke treatment where these drugs are routinely administered during the poststroke recovery period to promote neurocognitive improvement (Ni Chroinin et al., 2013; Montaner et al., 2016; Malhotra et al., 2019). Such a therapeutic goal requires the ability of statins to cross the BBB. In human endothelial cells, we have shown that luminal-to-abluminal transport of statins is facilitated by OATP1A2 (Ronaldson et al., 2021). Localization and expression of OATP1A2 at the human BBB is supported by immunohistochemical studies (Bronger et al., 2005; Lee et al., 2005) and proteomic analysis (Al-Majdoub et al., 2019).
One of the most critical considerations in the design of preclinical transport studies is the potential for differences in transporter substrate profiles between rodents and humans. An excellent example of this issue comes from a previous paper by Liu and colleagues who reported distinct differences in transport properties of antimigraine triptan drugs (Liu et al., 2015). Specifically, this work showed no difference in uptake transport for almotriptan, naratriptan, sumatriptan, rizatriptan, and zolmitriptan between human embryonic kidney cells transfected with Oatp1a4 and/or wild-type mice and Oatp1a4(−/−) mice, suggesting that this therapeutic class does not include Oatp1a4 substrates (Liu et al., 2015). In contrast, zolmitriptan was demonstrated to be a transport substrate for OATP1A2, thereby showing a clear difference in triptan transport properties between rodents and humans (Liu et al., 2015). For statins (i.e., atorvastatin, pravastatin, rosuvastatin), both our current study and our previous work (Thompson et al., 2014; Abdullahi et al., 2018; Brzica et al., 2018a; Ronaldson et al., 2021) have demonstrated that these drugs are transport substrates for both OATP1A2 and Oatp1a4. An additional strength of our study is that we have compared Oatp transport properties of statins with different physicochemical properties. We purposely selected one lipophilic statin (i.e., atorvastatin) and two hydrophilic statins (i.e., pravastatin, rosuvastatin) for evaluation. Although our data showed that Oatp-mediated transport is required for brain distribution of all three statins, whole brain uptake and brain exposure was highest for atorvastatin, followed by rosuvastatin and pravastatin. These results likely reflect the fact that passive transcellular diffusion makes a greater contribution to CNS distribution of atorvastatin compared with more hydrophilic molecules, such as pravastatin and rosuvastatin. These data are also remarkably congruent with our in vitro transport experiments in HUVECs that showed that endothelial cell exposure was greatest for atorvastatin compared with pravastatin or rosuvastatin (Ronaldson et al., 2021). Furthermore, the consistency between OATP1A2 transport data in human endothelial cells and Oatp1a4 uptake results in female Sprague-Dawley rats emphasizes that preclinical utility of utilizing in vivo rodent models to discover and characterize transport properties of statins at the BBB.
We also evaluated the effect of in vivo efflux transporter liability for atorvastatin, pravastatin, and rosuvastatin. ABC transporters, such as P-gp and Bcrp, are critical determinants of CNS drug disposition (Kikuchi et al., 2013; Dallas et al., 2016; Hernandez-Lozano et al., 2021). Atorvastatin is an established transport substrate for both P-gp and Bcrp (Hochman et al., 2004; Wang et al., 2019; Yang et al., 2020). Similarly, rosuvastatin is known to be transported by P-gp and Bcrp (Li et al., 2011; Elsby et al., 2016; Safar et al., 2019). In contrast, efflux transporter liability for pravastatin includes Bcrp but not P-gp (Hirano et al., 2005; Shirasaka et al., 2011; Afrouzian et al., 2018). To evaluate the role of P-gp and Bcrp on transport and, by extension, CNS disposition of statins, we used specific pharmacological inhibitors. Accumulation of both atorvastatin and rosuvastatin were increased in the presence of a P-gp inhibitor (i.e., PSC833), a Bcrp inhibitor (i.e., FTC), and a dual P-gp/Bcrp inhibitor (i.e., GF120918). In contrast, blood-to-brain transport of pravastatin was enhanced by FTC and GF120918 only, further confirming that this drug is not a P-gp transport substrate. In a manner consistent with our recent in vitro data in HUVECs, P-gp/Bcrp inhibition had the largest effect on rosuvastatin brain uptake compared with atorvastatin and pravastatin. Furthermore, we also observed that Bcrp is involved in limiting CNS uptake of pravastatin while both P-gp and Bcrp can determine CNS disposition of atorvastatin. Indeed, atorvastatin is known to have comparable efflux transporter liability as rosuvastatin (i.e., cellular uptake restricted by both P-gp and Bcrp) (Hochman et al., 2004; Wang et al., 2019; Yang et al., 2020).
Our laboratory has demonstrated, for the first time, that TGF-β/ALK1 signaling is involved in regulation of Oatp1a4 in rat brain microvessels (Abdullahi et al., 2017, 2018) and in human endothelial cells (Ronaldson et al., 2021). This mechanism was uncovered using the established ALK1 agonist BMP-9, which has an EC50 of 50 pg/ml (David et al., 2008). In our present study, we used a pharmacological dose of BMP-9 (1.0 μg/kg) to induce Oatp1a4 protein expression in rat brain microvessels at 6 hours postinjection so we could study effects of TGF-β/ALK1 pathway activation on CNS delivery of statins. We conducted these experiments in cortex, hippocampus, and cerebellum to determine if Oatp1a4 regulation via TGF-β/ALK1 signaling varied across brain regions. Interestingly, activation of the TGF-β/ALK1 pathway using BMP-9 resulted in enhanced Oatp1a4 protein expression and Oatp-mediated statin delivery in cerebral cortex but not in hippocampus or cerebellum. Specificity for the TGF-β/ALK1 pathway was established using the competitive ALK1 receptor inhibitor LDN193189, which attenuated changes in Oatp1a4 protein expression in cortical microvessels and blood-to-brain uptake of atorvastatin, pravastatin, and rosuvastatin. These findings are relevant to treatment of neurological diseases, such as ischemic stroke, where injury to the frontal cortex results in both cognitive and motor impairment. Indeed, targeting TGF-β/ALK1 signaling for control of Oatp-mediated transport activity could prove to be a viable strategy for optimizing cortical distribution of neuroprotective drugs, such as statins. Our results indicate that the molecular pharmacology of TGF-β/ALK1 signaling in brain microvessels is complex and provides impetus to study this pathway in intricate detail. This concept requires appreciation that the BMP-9 interaction with ALK1 is dependent upon expression and activity of ENG (Luo et al., 2010; van Meeteren et al., 2012). ENG binds to ligands, such as BMP-9, which form a protein complex that is then recruited to the ALK1 receptor (Lee et al., 2008; van Meeteren et al., 2012). This mechanism facilitates phosphorylation of ALK1-associated second messengers (i.e., Smad1/5/8) and subsequent activation of TGF-β/ALK1 signaling in endothelial cells (Lee et al., 2008; Luo et al., 2010; van Meeteren et al., 2012). Our study provided the first evidence for elevated ENG expression in cortical microvessels compared with hippocampal or cerebellar microvessels. This observation provides a mechanistic explanation for the BMP-9-mediated increase in Oatp1a4 functional expression in cortex but not the other brain regions that were evaluated. Previous immunodetection of ENG has shown both single and double bands (Lee et al., 2012). In our microvessel samples, the alterations in ENG molecular weight between brain regions may be explained by post-translational modifications. Changes in ENG glycosylation can lead to altered BMP-9 binding and could play a critical role in the observed differences in regional regulation of Oatp1a4 expression (Alt et al., 2012). Indeed, a more thorough understanding of TGF-β/ALK1 signaling at the BBB can be achieved by evaluation of ENG expression/activity. Ongoing work in our laboratory is aimed at uncovering how ENG and the ALK1 receptor interact with each other to regulate drug transport mechanisms (i.e., Oatp1a4) in cerebral microvessels. With respect to ABC transporters, BMP-9 treatment had no effect on functional expression of either P-gp or Bcrp in cortex, hippocampus, or cerebellum. Additionally, activation of the ALK1 receptor did not alter expression of critical tight junction proteins (i.e., claudin-5, monomeric occludin) or paracellular “leak” at the BBB. These are critical data because they suggest that Oatp1a4 is a unique transporter target for the TGF-β/ALK1 pathway.
Overall, this study describes increased functional expression of Oatp1a4 in cortical microvessels after dosing with the selective ALK1 agonist BMP-9. These data also demonstrate, for the first time, that direct activation of the TGF-β/ALK1 signaling pathway does not affect protein expression or transport activity of efflux transporters (i.e., P-gp, Bcrp) or BBB tight junction integrity. These observations are critical because they provide support for our hypothesis that TGF-β/ALK1 signaling can be exploited to control drug delivery to brain regions relevant to neurologic diseases, such as the cerebral cortex in ischemic stroke. Furthermore, data presented in this paper strengthen evidence primarily generated by our laboratory that Oatp1a4 can be targeted for delivery of therapeutic drugs used to treat several known neurological disease states.
Abbreviations
- 14C
carbon-14
- 3H
tritium
- ABC
ATP-binding cassette
- ALK1
activin receptor-like kinase 1
- AUC
area under the curve
- BBB
blood-brain barrier
- BCRP
Breast Cancer Resistance Protein
- BMP-9
bone morphogenetic protein-9
- CI
confidence interval
- CNS
central nervous system
- ENG
endoglin
- FEX
fexofenadine
- FTC
fumitrimorgin C
- HUVEC
human umbilical vein endothelial cell
- LDN193189
4-[6-[4-(1-piperazinyl)phenyl]pyrazolo[1,5-a]pyrimidin-3-yl]quinoline dihydrochloride
- OATP
organic anion transporting polypeptide
- P-gp
P-glycoprotein
- RECA-1
rat endothelial cell antigen-1
- Smad
Suppressor of Mothers Against Decapentaplegic
- TGF-β
transforming growth factor-β
Authorship Contributions
Participated in research design: Betterton, Abdullahi, Lochhead, Brzica, Davis, Ronaldson.
Conducted experiments: Betterton, Abdullahi, Williams, Lochhead, Brzica, Stanton, Reddell, Ogbonnaya, Ronaldson.
Performed data analysis: Betterton, Abdullahi, Lochhead, Brzica, Ronaldson.
Wrote or contributed to the writing of the manuscript: Betterton, Davis, Ronaldson.
Footnotes
This work was supported by National Institutes of Health National Institute of Neurological Disorders and Stroke [Grant R01-NS084941] (to P.T.R.), the American Heart Association [Grant 19TPA34910113] (to P.T.R.), and National Institutes of Health National Institute on Drug Abuse [Grant R01-DA051812] (to T.P.D. and P.T.R.).
No author has an actual or perceived conflict of interest with the contents of this article.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
Bibliography
- Abdullahi W, Brzica H, Hirsch NA, Reilly BG, Ronaldson PT (2018) Functional expression of organic anion transporting polypeptide 1a4 is regulated by transforming growth factor-β/activin receptor-like kinase 1 signaling at the blood-brain barrier. Mol Pharmacol 94:1321–1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdullahi W, Brzica H, Ibbotson K, Davis TP, Ronaldson PT (2017) Bone morphogenetic protein-9 increases the functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier via the activin receptor-like kinase-1 receptor. J Cereb Blood Flow Metab 37:2340–2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afrouzian M, Al-Lahham R, Patrikeeva S, Xu M, Fokina V, Fischer WG, Abdel-Rahman SZ, Costantine M, Ahmed MS, Nanovskaya T (2018) Role of the efflux transporters BCRP and MRP1 in human placental bio-disposition of pravastatin. Biochem Pharmacol 156:467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albekairi TH, Vaidya B, Patel R, Nozohouri S, Villalba H, Zhang Y, Lee YS, Al-Ahmad A, Abbruscato TJ (2019) Brain delivery of a potent opioid receptor agonist, biphalin during ischemic stroke: role of organic anion transporting polypeptide (OATP). Pharmaceutics 11:467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Majdoub ZM, Al Feteisi H, Achour B, Warwood S, Neuhoff S, Rostami-Hodjegan A, Barber J (2019) Proteomic quantification of human blood-brain barrier SLC and ABC transporters in healthy individuals and dementia patients. Mol Pharm 16:1220–1233. [DOI] [PubMed] [Google Scholar]
- Alt A, Miguel-Romero L, Donderis J, Aristorena M, Blanco FJ, Round A, Rubio V, Bernabeu C, Marina A (2012) Structural and functional insights into endoglin ligand recognition and binding. PLoS One 7:e29948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington SSalphati LHop CECAChu XEvers RBurdette DRowbottom CLai YXiao GHumphreys WG, et al. (2019) Interindividual and regional variability in drug transporter abundance at the human blood-brain barrier measured by quantitative targeted proteomics. Clin Pharmacol Ther 106:228–237. [DOI] [PubMed] [Google Scholar]
- Bronger H, König J, Kopplow K, Steiner HH, Ahmadi R, Herold-Mende C, Keppler D, Nies AT (2005) ABCC drug efflux pumps and organic anion uptake transporters in human gliomas and the blood-tumor barrier. Cancer Res 65:11419–11428. [DOI] [PubMed] [Google Scholar]
- Brzica H, Abdullahi W, Reilly BG, Ronaldson PT (2018a) Sex-specific differences in organic anion transporting polypeptide 1a4 (Oatp1a4) functional expression at the blood-brain barrier in Sprague-Dawley rats. Fluids Barriers CNS 15:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brzica H, Abdullahi W, Reilly BG, Ronaldson PT (2018b) A simple and reproducible method to prepare membrane samples from freshly isolated rat brain microvessels. J Vis Exp (135):57698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavaglia M, Dombrowski SM, Drazba J, Vasanji A, Bokesch PM, Janigro D (2001) Regional variation in brain capillary density and vascular response to ischemia. Brain Res 910:81–93. [DOI] [PubMed] [Google Scholar]
- Christophe B, Karatela M, Sanchez J, Pucci J, Connolly ES (2020) Statin therapy in ischemic stroke models: a meta-analysis. Transl Stroke Res 11:590–600. [DOI] [PubMed] [Google Scholar]
- Dallas SSalphati LGomez-Zepeda DWanek TChen LChu XKunta JMezler MMenet MCChasseigneaux S, et al. (2016) Generation and characterization of a breast cancer resistance protein humanized mouse model. Mol Pharmacol 89:492–504. [DOI] [PubMed] [Google Scholar]
- David L, Mallet C, Keramidas M, Lamandé N, Gasc JM, Dupuis-Girod S, Plauchu H, Feige JJ, Bailly S (2008) Bone morphogenetic protein-9 is a circulating vascular quiescence factor. Circ Res 102:914–922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsby R, Martin P, Surry D, Sharma P, Fenner K (2016) Solitary inhibition of the breast cancer resistance protein efflux transporter results in a clinically significant drug-drug interaction with rosuvastatin by causing up to a 2-fold increase in statin exposure. Drug Metab Dispos 44:398–408. [DOI] [PubMed] [Google Scholar]
- Fang X, Tao D, Shen J, Wang Y, Dong X, Ji X (2015) Neuroprotective effects and dynamic expressions of MMP9 and TIMP1 associated with atorvastatin pretreatment in ischemia-reperfusion rats. Neurosci Lett 603:60–65. [DOI] [PubMed] [Google Scholar]
- Hernández-Lozano I, Mairinger S, Traxl A, Sauberer M, Filip T, Stanek J, Kuntner C, Wanek T, Langer O (2021) Assessing the functional redundancy between P-gp and BCRP in controlling the brain distribution and biliary excretion of dual substrates with PET imaging in mice. Pharmaceutics 13:1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higashi Y, Aratake T, Shimizu T, Shimizu S, Saito M (2021) Protective role of glutathione in the hippocampus after brain ischemia. Int J Mol Sci 22:7765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano M, Maeda K, Matsushima S, Nozaki Y, Kusuhara H, Sugiyama Y (2005) Involvement of BCRP (ABCG2) in the biliary excretion of pitavastatin. Mol Pharmacol 68:800–807. [DOI] [PubMed] [Google Scholar]
- Hochman JH, Pudvah N, Qiu J, Yamazaki M, Tang C, Lin JH, Prueksaritanont T (2004) Interactions of human P-glycoprotein with simvastatin, simvastatin acid, and atorvastatin. Pharm Res 21:1686–1691. [DOI] [PubMed] [Google Scholar]
- Ishikawa HWakisaka YMatsuo RMakihara NHata JKuroda JAgo TKitayama JNakane HKamouchi M, et al. ; Fukuoka Stroke Registry Investigators (2016) Influence of statin pretreatment on initial neurological severity and short-term functional outcome in acute ischemic stroke patients: The Fukuoka Stroke Registry. Cerebrovasc Dis 42:395–403. [DOI] [PubMed] [Google Scholar]
- Kikuchi R, de Morais SM, Kalvass JC (2013) In vitro P-glycoprotein efflux ratio can predict the in vivo brain penetration regardless of biopharmaceutics drug disposition classification system class. Drug Metab Dispos 41:2012–2017. [DOI] [PubMed] [Google Scholar]
- Lawera ATong ZThorikay MRedgrave RECai Jvan Dinther MMorrell NWAfink GBCharnock-Jones DSArthur HM, et al. (2019) Role of soluble endoglin in BMP9 signaling. Proc Natl Acad Sci USA 116:17800–17808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee MSaver JLWu YLTang SCLee JDRao NMWang HHJeng JSLee THChen PC, et al. (2017) Utilization of statins beyond the initial period after stroke and 1-year risk of recurrent stroke. J Am Heart Assoc 6:e005658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NY, Golzio C, Gatza CE, Sharma A, Katsanis N, Blobe GC (2012) Endoglin regulates PI3-kinase/Akt trafficking and signaling to alter endothelial capillary stability during angiogenesis. Mol Biol Cell 23:2412–2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee NY, Ray B, How T, Blobe GC (2008) Endoglin promotes transforming growth factor beta-mediated Smad 1/5/8 signaling and inhibits endothelial cell migration through its association with GIPC. J Biol Chem 283:32527–32533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee W, Glaeser H, Smith LH, Roberts RL, Moeckel GW, Gervasini G, Leake BF, Kim RB (2005) Polymorphisms in human organic anion-transporting polypeptide 1A2 (OATP1A2): implications for altered drug disposition and central nervous system drug entry. J Biol Chem 280:9610–9617. [DOI] [PubMed] [Google Scholar]
- Li J, Volpe DA, Wang Y, Zhang W, Bode C, Owen A, Hidalgo IJ (2011) Use of transporter knockdown Caco-2 cells to investigate the in vitro efflux of statin drugs. Drug Metab Dispos 39:1196–1202. [DOI] [PubMed] [Google Scholar]
- Liu HYu NLu SIto SZhang XPrasad BHe ELu XLi YWang F, et al. (2015) Solute carrier family of the organic anion-transporting polypeptides 1A2- Madin-Darby canine kidney II: a promising in vitro system to understand the role of organic anion-transporting polypeptide 1A2 in blood-brain barrier drug penetration. Drug Metab Dispos 43:1008–1018. [DOI] [PubMed] [Google Scholar]
- Luo JTang MHuang JHe BCGao JLChen LZuo GWZhang WLuo QShi Q, et al. (2010) TGFbeta/BMP type I receptors ALK1 and ALK2 are essential for BMP9-induced osteogenic signaling in mesenchymal stem cells. J Biol Chem 285:29588–29598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra KSafouris AGoyal NArthur ALiebeskind DSKatsanos AHSargento-Freitas JRibo MMolina CChung JW, et al. (2019) Association of statin pretreatment with collateral circulation and final infarct volume in acute ischemic stroke patients: a meta-analysis. Atherosclerosis 282:75–79. [DOI] [PubMed] [Google Scholar]
- Montaner JBustamante AGarcía-Matas SMartínez-Zabaleta MJiménez Cde la Torre JRubio FRSegura TMasjuán JCánovas D, et al. ; STARS Investigators (2016) Combination of thrombolysis and statins in acute stroke is safe: results of the STARS randomized trial (stroke treatment with acute reperfusion and simvastatin). Stroke 47:2870–2873. [DOI] [PubMed] [Google Scholar]
- Ní Chróinín DAsplund KÅsberg SCallaly ECuadrado-Godia EDíez-Tejedor EDi Napoli MEngelter STFurie KLGiannopoulos S, et al. (2013) Statin therapy and outcome after ischemic stroke: systematic review and meta-analysis of observational studies and randomized trials. Stroke 44:448–456. [DOI] [PubMed] [Google Scholar]
- Ose A, Kusuhara H, Endo C, Tohyama K, Miyajima M, Kitamura S, Sugiyama Y (2010) Functional characterization of mouse organic anion transporting peptide 1a4 in the uptake and efflux of drugs across the blood-brain barrier. Drug Metab Dispos 38:168–176. [DOI] [PubMed] [Google Scholar]
- Polli JW, Olson KL, Chism JP, John-Williams LS, Yeager RL, Woodard SM, Otto V, Castellino S, Demby VE (2009) An unexpected synergist role of P-glycoprotein and breast cancer resistance protein on the central nervous system penetration of the tyrosine kinase inhibitor lapatinib (N-{3-chloro-4-[(3-fluorobenzyl)oxy]phenyl}-6-[5-({[2-(methylsulfonyl)ethyl]amino} methyl)-2-furyl]-4-quinazolinamine; GW572016). Drug Metab Dispos 37:439–442. [DOI] [PubMed] [Google Scholar]
- Prinz V, Laufs U, Gertz K, Kronenberg G, Balkaya M, Leithner C, Lindauer U, Endres M (2008) Intravenous rosuvastatin for acute stroke treatment: an animal study. Stroke 39:433–438. [DOI] [PubMed] [Google Scholar]
- Ramos-Cabrer P, Campos F, Sobrino T, Castillo J (2011) Targeting the ischemic penumbra. Stroke 42(Suppl 1):S7–S11. [DOI] [PubMed] [Google Scholar]
- Ronaldson PT, Brzica H, Abdullahi W, Reilly BG, Davis TP (2021) Transport properties of statins by organic anion transporting polypeptide 1A2 and regulation by transforming growth factor-β signaling in human endothelial cells. J Pharmacol Exp Ther 376:148–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Davis TP (2013) Targeted drug delivery to treat pain and cerebral hypoxia. Pharmacol Rev 65:291–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Demarco KM, Sanchez-Covarrubias L, Solinsky CM, Davis TP (2009) Transforming growth factor-beta signaling alters substrate permeability and tight junction protein expression at the blood-brain barrier during inflammatory pain. J Cereb Blood Flow Metab 29:1084–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronaldson PT, Finch JD, Demarco KM, Quigley CE, Davis TP (2011) Inflammatory pain signals an increase in functional expression of organic anion transporting polypeptide 1a4 at the blood-brain barrier. J Pharmacol Exp Ther 336:827–839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safar Z, Kis E, Erdo F, Zolnerciks JK, Krajcsi P (2019) ABCG2/BCRP: variants, transporter interaction profile of substrates and inhibitors. Expert Opin Drug Metab Toxicol 15:313–328. [DOI] [PubMed] [Google Scholar]
- Sarikaya H, Steinlin M (2018) Cerebellar stroke in adults and children. Handb Clin Neurol 155:301–312. [DOI] [PubMed] [Google Scholar]
- Shi L, Rocha M, Leak RK, Zhao J, Bhatia TN, Mu H, Wei Z, Yu F, Weiner SL, Ma F, Jovin TG, Chen J (2018) A new era for stroke therapy: integrating neurovascular protection with optimal reperfusion. J Cereb Blood Flow Metab 38:2073–2091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shirasaka Y, Suzuki K, Nakanishi T, Tamai I (2011) Differential effect of grapefruit juice on intestinal absorption of statins due to inhibition of organic anion transporting polypeptide and/or P-glycoprotein. J Pharm Sci 100:3843–3853. [DOI] [PubMed] [Google Scholar]
- Takasato Y, Rapoport SI, Smith QR (1984) An in situ brain perfusion technique to study cerebrovascular transport in the rat. Am J Physiol 247:H484–H493. [DOI] [PubMed] [Google Scholar]
- Thompson BJ, Sanchez-Covarrubias L, Slosky LM, Zhang Y, Laracuente ML, Ronaldson PT (2014) Hypoxia/reoxygenation stress signals an increase in organic anion transporting polypeptide 1a4 (Oatp1a4) at the blood-brain barrier: relevance to CNS drug delivery. J Cereb Blood Flow Metab 34:699–707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meeteren LA, Thorikay M, Bergqvist S, Pardali E, Stampino CG, Hu-Lowe D, Goumans MJ, ten Dijke P (2012) Anti-human activin receptor-like kinase 1 (ALK1) antibody attenuates bone morphogenetic protein 9 (BMP9)-induced ALK1 signaling and interferes with endothelial cell sprouting. J Biol Chem 287:18551–18561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZYang HXu JZhao KChen YLiang LLi PChen NGeng DZhang X, et al. (2019) Prediction of atorvastatin pharmacokinetics in high-fat diet and low-dose streptozotocin-induced diabetic rats using a semiphysiologically based pharmacokinetic model involving both enzymes and transporters. Drug Metab Dispos 47:1066–1079. [DOI] [PubMed] [Google Scholar]
- Williams EI, Betterton RD, Davis TP, Ronaldson PT (2020) Transporter-mediated delivery of small molecule drugs to the brain: a critical mechanism that can advance therapeutic development for ischemic stroke. Pharmaceutics 12:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Li P, Zhang Z, Wang Z, Liu L, Liu X (2020) Prediction of cyclosporin-mediated drug interaction using physiologically based pharmacokinetic model characterizing interplay of drug transporters and enzymes. Int J Mol Sci 21:7023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Chopp M, Jia L, Cui Y, Lu M, Zhang ZG (2009) Atorvastatin extends the therapeutic window for tPA to 6 h after the onset of embolic stroke in rats. J Cereb Blood Flow Metab 29:1816–1824. [DOI] [PMC free article] [PubMed] [Google Scholar]