

Abstract
(S)-Efavirenz (EFV) is a reverse transcriptase inhibitor and an antiviral drug. In addition, (S)-EFV can interact off target with CYP46A1, the major cholesterol hydroxylating enzyme in the mammalian brain, and allosterically activate CYP46A1 at a small dose in mice and humans. Studies with purified CYP46A1 identified two allosteric sites on the enzyme surface, one for (S)-EFV and the second site for L-glutamate (Glu), a neurotransmitter that also activates CYP46A1 either alone or in the presence of (S)-EFV. Previously, we found that racemic (rac)-7-hydroxyefavirenz, (rac)-8-hydroxyefavirenz, (S)-8-hydroxyefavirenz, and (rac)-8,14-dihydroxyefavirenz, compounds with the hydroxylation positions corresponding to the metabolism of (S)-EFV in the liver, activated CYP46A1 in vitro. Yet, these compounds differed from (S)-EFV in how they allosterically interacted with CYP46A1. Herein, we further characterized (rac)-7-hydroxyefavirenz, (rac)-8-hydroxyefavirenz, (S)-8-hydroxyefavirenz, and (rac)-8,14-dihydroxyefavirenz, and, in addition, (R)-EFV, (S)-7-hydroxyefavirenz, (rac)-7,8-dihydroxyefavirenz, (S)-7,8-dihydroxyefavirenz, and (S)-8,14-dihydroxyefavirenz for activation and binding to CYP46A1 in vitro. We found that the spatial configuration of all tested compounds neither affected the CYP46A1 activation nor the sites of binding to CYP46A1. Yet, the hydroxylation position determined whether the hydroxylated metabolite interacted with the allosteric site for (S)-EFV [(R)-EFV, (rac)-7,8-dihydroxyefavirenz, and (S)-7,8-dihydroxyefavirenz], L-Glu [(rac)- and (S)-8,14-dihydroxyefavirenz], or both [(rac)-7-hydroxyefavirenz, (S)-7-hydroxyefavirenz, (rac)-8-hydroxyefavirenz, and (S)-8-hydroxyefavirenz]. This difference in binding to the allosteric sites determined, in turn, how CYP46A1 activity was changed in the coincubations with (S)-EFV and either its metabolite or L-Glu. The results suggest EFV metabolites that could be more potent for CYP46A1 activation in vivo than (S)-EFV.
SIGNIFICANCE STATEMENT
This study found that not only efavirenz but also all its hydroxylated metabolites allosterically activate CYP46A1 in vitro. The enzyme activation depended on the hydroxylation position but not the metabolite spatial configuration and involved either one or two allosteric sites—for efavirenz, L-glutamate, or both. The results suggest that the hydroxylated efavirenz metabolites may differ from efavirenz in how they interact with the CYP46A1 allosteric and active sites.

Introduction
(S)-Efavirenz (EFV) is a non-nucleoside inhibitor of the reverse transcriptase enzyme encoded by human immunodeficiency virus. Accordingly, (S)-EFV is approved by the U.S. Food and Drug Administration for reduction of a viral load in human immunodeficiency virus-infected subjects. Previously, we found that small-dose (S)-EFV has the off-target interaction with CYP46A1 in mice (Mast et al., 2014, 2017b; Petrov et al., 2019a) and humans (ClinicalTrials.gov NCT03706885) and allosterically increases cholesterol 24-hydroxylation by this P450 (Mast et al., 2014; Anderson et al., 2016). Under normal conditions, CYP46A1 is a central nervous system (CNS)-specific enzyme residing in the endoplasmic reticulum of specific neurons (Lund et al., 1999; Ramirez et al., 2008). Yet, under pathologic conditions, CYP46A1 could be additionally expressed in other cell types as exemplified by the enzyme ectopic expression in astrocytes in Alzheimer’s disease (AD) (Bogdanovic et al., 2001; Brown et al., 2004).
CYP46A1 expression or activity is decreased in the human brain in AD, Huntington’s and prion diseases as well as spinocerebellar ataxia type 3 and glioblastoma (Boussicault et al., 2016; Testa et al., 2016; Han et al., 2020; Nobrega et al., 2019; Ali et al., 2021; Varma et al., 2021). Hence, increasing CYP46A1 expression or activity by gene therapy or treatment with (S)-EFV, respectively, is an emerging strategy for treatment of not only AD but other brain disorders as suggested by studies in mouse models (Hudry et al., 2010; Boussicault et al., 2016; Mast et al., 2017b; Han et al., 2020; Nobrega et al., 2019; Petrov et al., 2019a,b; Ali et al., 2021). We used 5XFAD mice, a model of AD, and found that CYP46A1 activation by (S)-EFV improved animal performance in memory tasks and had treatment-specific effects on the brain amyloid β load, astrocyte and microglia activation, and expression of essential synaptic proteins (Mast et al., 2017b; Petrov et al., 2019a,b).
Remarkably, besides the exogenous compound (S)-EFV, CYP46A1 could be activated, at least in vitro, by some endogenous compounds, including the neurotransmitter L-glutamate (Glu) (Mast et al., 2017a). The allosteric sites for (S)-EFV and L-Glu were identified and appeared to be in a spatial proximity to each other, separated only by the loop (Fig. 1). Hence, when simultaneously coincubated, (S)-EFV and L-Glu synergistically activate purified CYP46A1 in vitro (Anderson et al., 2016; Mast et al., 2017a, 2020).
Fig. 1.

Computational models of (S)-EFV (A) and L-Glu (B) binding to the allosteric sites on CYP46A1, showing some of the amino acid residues involved in the interaction with these compounds. The loop (from G417 to T426) separating the two allosteric sites is also shown and colored in magenta. The nitrogen, oxygen, chlorine, and fluorine atoms are in blue, red, light green, and cyan, respectively. The allosteric site mapping and compound docking were carried out in our previous work (Anderson et al., 2016; Mast et al., 2017a, 2020).
In mammals, (S)-EFV is mostly cleared by the liver with the first steps being the hydroxylation reactions catalyzed by several P450 enzymes (Fig. 2). Both monohydroxy, 7-hydroxyefavirenz (7OH) and 8-hydroxyefavirenz (8OH EFV), and dihydroxy metabolites, 7,8-dihydroxyefavirenz (7,8diOH) and 8,14-dihydroxyefavirenz, (8,14diOH EFV) are generated, and of them, 8-hydroxylation represents the predominant route for the phase 1 drug biotransformation (Ward et al., 2003; Bumpus et al., 2006; Ogburn et al., 2010; Avery et al., 2013a,b). Then, once hydroxylated, EFV metabolites become glucuronidated and/or sulfated and represent the major circulating efavirenz species in humans (Mutlib et al., 1999; Aouri et al., 2016). Previously, we tested either the (S) enantiomer or racemic (rac) mixtures of some of the phase I EFV metabolites [(rac)-7OH EFV, (rac)-8OH EFV, (S)-8OH EFV, and (rac)-8,14-diOH EFV] and established that they also activate purified CYP46A1 in vitro by binding either to the allosteric site for L-Glu [(rac)-8,14-diOH EFV] or both (S)-EFV and L-Glu [(rac)-7OH EFV, (rac)-8OH EFV, and (S)-8OH EFV]. In the present work, we continued our studies of the hydroxylated EFV metabolites (either as (S) enantiomers or racemates) and finished acquisition of the CYP46A1 activation and binding data for all known hydroxylated EFV metabolites. We enhanced our understanding of how the hydroxylated EFV metabolites interact with CYP46A1 in vitro and conceptualized subsequent investigations of the hydroxylated EFV metabolites in vivo to develop better CYP46A1 activators than (S)-EFV.
Fig. 2.

Phase 1 metabolic products of (S)-EFV generated by cytochrome P450 enzymes as suggested by previous studies (Avery et al., 2013b). The major P450s metabolizing (S)-EFV are shown in bold.
Materials and Methods
Materials
(S)-EFV and (R)-EFV were purchased from Toronto Research Chemicals (E425000, Toronto, ON, Canada) and Carbosynth (154801-74-8, Compton, United Kingdom), respectively. All EFV metabolites were obtained from Toronto Research Chemicals (H805345, H941820, H941830, H941825, D452979, D46250, D468240, D452800). Methanol was used to prepare 5–20 mM stock solutions of EFV and its metabolites. L-Glu was from MilliporeSigma (1446600, St. Louis, MO) and was dissolved in water to prepare a 2 mM stock solution. Cholesterol was obtained from Steraloids (C6760, Newport, RI), and 24-hydroxy-[25,26,26,26,27,27,27-2H7]-cholesterol was from Medical Isotopes (D17258, Pelham, NH). Cholesterol was added from a 1 mM stock in 4.5%, w/v, aqueous 2-hydroxypropyl-β-cyclodextrin (332607, MilliporeSigma); deuterated 24-hydroxycholesterol was added from a 0.1 mM stock in methanol. Human truncated Δ(2–50) CYP46A1 with a four-histidine tag on the C terminus wild type (WT) and rat cytochrome P450 oxidoreductase were expressed in Escherichia coli and purified as described (Hanna et al., 1998; White et al., 2008). All CYP46A1 mutants, except Q440M, were used in our previous studies (Mast et al., 2017a, 2020); the Q440M CYP46A1 mutant was generated in the present work. An in vitro QuikChange site-directed mutagenesis kit (#200522, Stratagene, San Diego, CA) was used according to the manufacturer’s instructions. The correct generation of the desired mutation and absence of undesired mutations were confirmed by nucleotide sequencing of the entire CYP46A1 coding region as well as by the restriction analysis.
Enzyme Assays
Incubations for the concentration dependence curves were carried out as described (Mast et al., 2020) in 1 mL of 50 mM potassium phosphate buffer (pH 7.2) containing 100 mM sodium chloride, 40 μg/mL L-α-1,2-dilauroyl-sn-glycero-3-phosphocholine, 0.5 µM CYP46A1, 1.0 µM cytochrome P450 oxidoreductase, 40 µM cholesterol, varying concentrations of a test compound (0–100 µM, the 2% final methanol concentration), 2 units of catalase, and an NADPH-regenerating system (1 mM NADPH, 10 mM glucose-6-phosphate, and 2 units of glucose-6-phosphate dehydrogenase). The reaction time was 30 minutes at 37°C, and sterol extraction was with 5 mL of dichloromethane containing 3 nmol of 24-hydroxy-[25,26,26,26,27,27,27-2H7]-cholesterol, which served as an internal standard. Sterol extracts were then processed and analyzed by gas chromatography-mass spectrometry as described (Mast et al., 2011). The apparent binding constant (Kd) of cholesterol for CYP46A1 is 2.6 μM (Mast et al., 2014). Hence, the 40 μM cholesterol concentration used in the enzyme assays or the 20 μM sterol concentration used in the spectral assay (see below) were saturating for CYP46A1.
The measurements of CYP46A1 activity in coincubations with a test compound and either (S)-EFV (20 µM) or L-Glu (100 µM) were carried out similarly, except a fixed concentration of a test compound was used (20 µM). For studies of the CYP46A1 mutants, the same concentrations of (S)-EFV (20 µM), L-Glu (100 µM), and a test compound (20 µM) were used as at these concentrations, (S)-EFV and L-Glu elicit the maximal CYP46A1 activation in the presence of 20 µM or 40 µM cholesterol (Mast et al., 2014, 2017a).
Spectral Assays
Spectral titrations of substrate-free CYP46A1 were carried out at 18°C as described (Mast et al., 2020) in 1 mL of 50 mM potassium phosphate buffer (pH 7.2) containing 100 mM sodium chloride and 0.4–0.5 µM enzyme. To obtain cholesterol-bound CYP46A1, 20 μM cholesterol was added to the titration buffer. Spectral changes were fit by the GraphPad Prism software to either of the following equations:
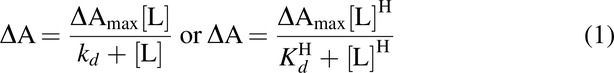 |
in which ΔA is the spectral response at different ligand concentrations [L], ΔAmax is the maximal amplitude of the spectral response, Kd is the ligand dissociation constant, and H is the Hill coefficient.
Statistics
The results represent the mean ±S.D. of the measurements in three independent experiments. Statistical significance of mean differences was determined either by a two-tailed unpaired Student’s t test, one-way ANOVA with Tuckey multiple comparisons, or two-way ANOVA with Bonferroni multiple comparisons. The significance was defined as follows: *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.001.
Results
The Concentration Dependence Curves
To compare the effect of different spatial configurations of EFV and its hydroxylated metabolites on activity of the same CYP46A1 preparation, we repeated the enzyme assays with (S)-EFV, (rac)-7OH EFV, (S)-8OH EFV, (rac)-8OH EFV, and (rac)-8,14diOH EFV, which were carried out in our previous investigation (Mast et al., 2020). In addition, we tested (R)-EFV, (S)-7OH EFV, (S)-7,8diOH, (rac)-7,8diOH EFV, and (S)-8,14diOH EFV that became commercially available. The concentration dependence curves for the five compounds that we evaluated previously (Mast et al., 2020) and in the present work were similar, thus confirming our previous results and enabling comparisons with the curves of the newly evaluated compounds. Specifically, the curve shape of (R)- versus (S)-EFV was similar, and the major difference was that the maximal CYP46A1 activation by (R)-EFV was at a lower concentration (10 μM) than that by (S)-EFV (20 μM) (Fig. 3A). CYP46A1 activation by the (S)- versus (rac)-compound was similar for each EFV metabolite as well, and no significant differences were found between the two in the extent of CYP46A1 activation (Fig. 3, B and C). Yet, unlike EFV, none of the drug metabolites inhibited CYP46A1 at a high 100 μM concentration, even though the extent of CYP46A1 activation started to decrease from the 60 μM metabolite concentration. The latter was reflected in the shape of the CYP46A1 activation curves that seemed to be different among EFV metabolites. In the case of 7OH and 8OH EFV, the drug monohydroxylated metabolites, the shape was more similar to that of EFV, i.e., was bell-shaped. In the case of 7,8diOH and 8,14diOH EFV, the drug dihydroxylated metabolites, the CYP46A1 activation curves were more of a hyperbolic type. A combination of different factors, namely how EFV and its metabolites interact with the CYP46A1 allosteric sites at low concentrations and in addition, with the P450 active site at high concentrations, could determine in part the shape of the concentration dependence curves as suggested by our previous investigation (Mast et al., 2020). Therefore, we next used six CYP46A1 mutants to map the allosteric site(s) for binding of EFV and its metabolites.
Fig. 3.

Dependence of in vitro activation of CYP46A1 on the concentration of EFV (A), its mono- (B) and dihydroxylated (C) metabolites. The Y-axis is identical for all three graphs and represents CYP46A1 activity as nanomoles of 24-hydoxycholsterol (24HC) formed per nmole of CYP46A1 per minute. The results are the mean ±S.D. of the measurements from the three independent experiments. Statistically significant differences between (S)-EFV versus (R)-EFV were assessed by two-way ANOVA with Bonferroni multiple comparisons. No significant differences were found between the (S) enantiomer versus racemate of the same EFV metabolite * P ≤ 0.05.
Activation of the CYP46A1 Mutants
The CYP46A1 mutants with replacements within the allosteric sites for (S)-EFV (F405A and R415A) and L-Glu (K358A, F416A, Y427A, and Q440M) were used (Fig. 4). The (S)-EFV site mutants showed enzyme activation only by L-Glu, (rac)-8,14diOH EFV, and (S)-8,14diOH EFV and were either not activated by (S)-EFV, (R)-EFV, racemates, and (S) enantiomers of 7OH EFV, 8OH EFV, and 7,8diOH EFV or were activated by these compounds to a lesser extent (up to 1.9-fold) than WT (up to 5.9-fold). This result suggested that L-Glu, (rac)-8,14diOH EFV, and (S)-8,14diOH EFV do not bind to the allosteric site for (S)-EFV, whereas the remaining tested compounds do bind to this allosteric site.
Fig. 4.

Effect of the allosteric site mutations on basal CYP46A1 activity and enzyme activation by L-Glu, EFV, and EFV metabolites. CYP46A1 activity is presented as nanomoles of 24-hydoxycholsterol (24HC) formed per nmole of CYP46A1 per minute. The results are the mean ±S.D. of the measurements from the three independent experiments. Statistically significant differences were assessed by one-way ANOVA with Tuckey multiple comparisons versus basal activity of WT or the CYP46A1 mutant. ** P ≤ 0.01; *** P ≤ 0.001.
The activation pattern of the L-Glu site CYP46A1 mutants (K358A, F416A, Y427A, and Q440M) was different. Either they were not activated by L-Glu (Y427A and Q440M), (rac)-8,14diOH EFV (K358A) and (S)-8,14diOH EFV (K358A) or were activated by these compounds to a much lesser extent (up to 2.0-fold) than WT (K358A and F416A by L-Glu; F416A, Y427A, and Q440M by (rac)-8,14diOH EFV and (S)-8,14diOH EFV) (up to 6.3-fold). In addition, the K358A, F416A, and Y427A mutants, but not the Q440M mutant, were activated to a lesser extent than WT by (rac)- or (S)-7OH EFV and (rac)- or (S)-8OH EFV. Yet, all of the L-Glu site mutants were activated by (S)- and (R)-EFV as well as (rac)- or (S)-7,8-diOH EFV. Collectively, these data suggested that EFV and 7,8-diOH EFV mainly bind to the allosteric site for (S)-EFV; 8,14diOH EFV mainly binds to the allosteric site for L-Glu, whereas 7OH EFV and 8OH EFV bind to both allosteric sites (Table 1). Furthermore, our results indicated that different amino acid residues seemed to be important for compound binding within each allosteric site: F405 and R415 are likely involved in the interaction with EFV, 7OH EFV, 8OH EFV, and 7,8diOH EFV in the (S)-EFV site, whereas K358 and F416 are likely necessary for the interaction with the 8,14-dihydroxy EFV metabolites in the L-Glu site, which also includes Y427 and Q440 involved in the interaction with L-Glu.
TABLE 1.
A summary of compound activation and binding to purified CYP46A1
| EFV Metabolite | Interacts via the Allosteric Site for | Maximal Individual Activation (-fold) |
Activation in the Presence of (S)-EFV (-fold) | Activation in the Presence of l-Glu (-fold) |
Binding to Substrate-Free CYP46A1 (Response Type; Kd, μM) |
Binding to Cholesterol -Bound CYP46A1 (Response Type; Curve Fit) |
|
|---|---|---|---|---|---|---|---|
| (S)-EFV | L-Glu | ||||||
| (S)-EFV | + | − | 3.9 | 3.2 | 5.0 | Type 2; 0.8a | r.t. 1; S |
| (R)-EFV | + | − | 3.9 | 3.0 | 4.9 | Type 1; 9.2 | r.t. 1; S |
| (rac)-7OH | + | + | 5.5 | 3.9 | 6.9 | Type 2; 3.3a | r.t. 1; Sa |
| (S)-7OH | + | + | 5.8 | 3.5 | 7.2 | Type 2; 4.8 | r.t. 1; S |
| (rac)-8OH | + | + | 5.8 | 3.1 | 7.0 | Type 2; 2.2a | r.t. 1; Sa |
| (S)-8OH | + | + | 5.9 | 3.3 | 7.4 | Type 2; 4.1a | r.t. 1; Sa |
| (rac)-7,8diOH | + | − | 5.6 | 1.2 | 8.5 | Type 1; 30.5 | type 1; H |
| (S)-7,8diOH | + | − | 5.6 | 2.3 | 10.3 | Type 1; 15.7 | type 1; H |
| (rac)-8,14diOH | − | + | 5.9 | 7.6 | 4.6 | Type 2; 1.6a | r.t. 1; Ha |
| (S)-8,14diOH | − | + | 6.4 | 7.4 | 5.1 | Type 2; 2.2 | r.t. 1; H |
| L-Glu | − | + | 2.8 | 5.0 | ND | ND | ND |
H, hyperbolic; ND, not detectable; r.t., reverse type 1; S, sigmoidal.
aTaken from reference (Mast et al., 2020).
Activation of CYP46A1 in Compound Coincubations with (S)-EFV or L-Glu
This approach was complimentary to studies with the CYP46A1 mutants to map the allosteric sites for compound binding to CYP46A1. We used the activator concentrations eliciting maximal CYP46A1 activation in the individual incubations: 20 μM compound in question, 20 μM (S)-EFV, and 100 μM L-Glu. The major assumptions were that if a tested compound binds to the same allosteric site as (S)-EFV (or L-Glu), then the CYP46A1 activation by this compound in coincubations with (S)-EFV (or L-Glu) will be lower because of the compound competition with (S)-EFV (or L-Glu) for the same allosteric site. Conversely, if a tested compound and (S)-EFV (or L-Glu) bind to different allosteric sites, then the CYP46A1 activation by this compound in coincubations with (S)-EFV (or L-Glu) will be higher. Yet, if a tested compound binds to both (S)-EFV and L-Glu sites, then the CYP46A1 activation by this compound in coincubations with (S)-EFV and L-Glu may reveal whether one of the allosteric sites represent the preferential site of compound binding. Consistent with our assumptions, all compounds [(R)-EFV, (rac)-7OH EFV, (S)-7OH EFV, (rac)-8OH EFV, (S)-8OH EFV, (rac)-7,8diOH EFV, and (S)-7,8diOH EFV)] that were suggested by the site-directed mutagenesis data to bind to the allosteric site for (S)-EFV activated CYP46A1 to a smaller extent in the presence of (S)-EFV (Fig. 5, Table 1). Yet CYP46A1 activation by all these compounds in the presence of L-Glu was higher. Similarly, (rac)-8,14diOH EFV and (S)-8,14diOH EFV that were suggested to bind to the L-Glu site, activated CYP46A1 to a higher extent in the presence of (S)-EFV and to a lower extent in the presence of L-Glu. Thus, the coincubation studies supported the site-directed mutagenesis data and also suggested that (rac)-7OH EFV, (S)-7OH EFV, (rac)-8OH EFV, and (S)-8OH EFV may have a higher affinity for the (S)-EFV site than the L-Glu site and perhaps bind to the (S)-EFV site stronger than (S)-EFV.
Fig. 5.

Effect of L-Glu or (S)-EFV on CYP46A1 activation by EFV metabolites. CYP46A1 activity is presented as nanomoles of 24-hydoxycholsterol (24HC) formed per nmole of CYP46A1 per minute. The results are the mean ±S.D. of the measurements from the three independent experiments. Statistically significant differences were assessed by one-way ANOVA with Tuckey multiple comparisons versus CYP46A1 activity when only compound in question was present. *** P ≤ 0.001.
Compound Binding to CYP46A1 as Assessed by Spectral Assays
Compound binding to the CYP46A1 active site frequently induces a spectral shift, which provides insight into the coordination of the heme group, located in the P450 active site, at the sixth axial position of the heme iron. Type I spectral response is typically induced by P450 substrates, which displace a water molecule that coordinates the heme iron in substrate-free P450s. Reverse type 1 spectral response is believed to reflect the formation of an iron-oxygen (Fe-O) bond (Mailman et al., 1974; Kumaki et al., 1978). Type 2 spectral response is usually produced by P450 inhibitors, which bind to the enzyme active site and coordinate the heme iron with their nitrogen atom (Remmer et al., 1966; Schenkman et al., 1967; Mailman et al., 1974; Mast et al., 2008, 2010). Previously (Table 1), substrate-free and cholesterol-bound forms of CYP46A1 were titrated with (S)-EFV, (rac)-7OH EFV, (S)-8OH EFV, (rac)-8OH EFV, and (rac)-8,14diOH EFV (Mast et al., 2014, 2020). Herein, substrate-free and cholesterol-bound forms of CYP46A1 were titrated with (R)-EFV, (S) enantiomers of 7OH EFV and 8,14diOH EFV plus (S)-7,8diOH EFV and (rac)-7,8diOH EFV. Unlike (S)-EFV, (R)-EFV elicited type 1 spectral response (a peak at 387 nm and a trough at 421 nm in the P450 difference spectrum) in substrate-free CYP46A1 and had a 9.2 μM Kd, which was >10-fold higher than that of (S)-EFV (Fig. 6A). (Rac)-7,8diOH and (S)-7,8diOH EFV also elicited type I spectral response but had the medium micromolar apparent Kds (30.5 μM and 15.7 μM, respectively). Yet, (S)-7OH and (S)-8,14diOH EFV elicited type 2 spectral response (a trough at 412–415 nm and a peak at 430–438 nm in the P450 difference spectrum) and had the low micromolar apparent Kd values (4.8 μM and 2.2 μM, respectively). This spectral response and apparent Kds were similar to those of (rac)-7OH EFV, (rac)-8,14diOH EFV, (S)-8OH EFV, and (rac)-8OH EFV (Table 1), which also induced type 2 spectral response and had the apparent Kd values in a low micromolar range of 1.6–4.1 μM (Mast et al., 2020). Thus, both racemates and (S) enantiomers of all hydroxylated EFV metabolites can bind to the CYP46A1 active site in the absence of cholesterol, yet the binding modes of (R)-EFV, (rac)- and (S)-7,8diOH EFV are different from that of other EFV metabolites and likely lead to a lower compound affinity to CYP46A1.
Fig. 6.
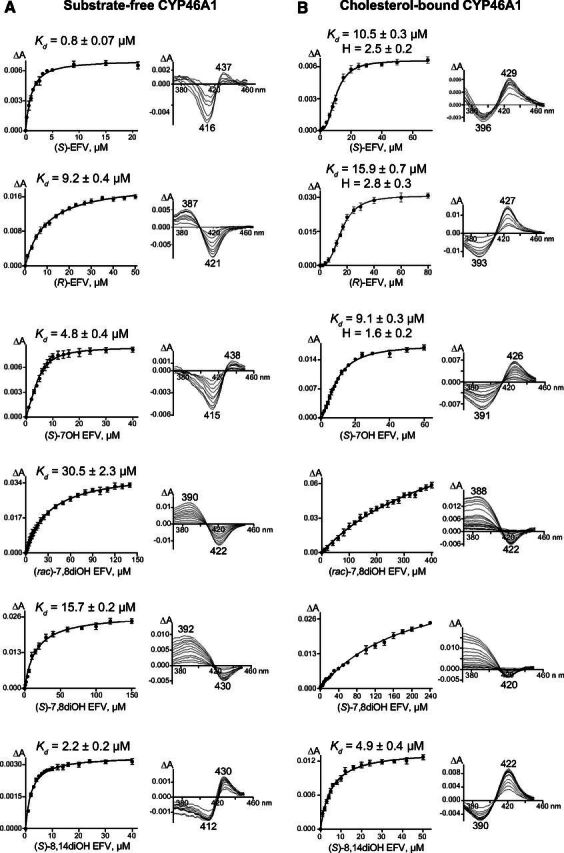
Spectral titrations of substrate-free CYP46A1 (A) and cholesterol-bound CYP46A1 (B) by EFV and some of its metabolites. Fit of spectral changes (ΔA, the amplitude of spectral response in the CYP46A1 difference spectrum) either to a hyperbolic equation or to the Hill equation, when cooperative binding was observed, is shown on the left, and the spectral response type in the CYP46A1 difference spectrum is shown on the right. The results are the mean ±S.D. of the measurements from the three independent titrations. Data for (S)-EFV are taken from (Mast et al., 2020).
Titrations of substrate-bound CYP46A1, conducted at the saturating 20 μM cholesterol concentration, were carried out to ascertain whether (R)-EFV and the tested EFV hydroxylation products can displace cholesterol from the CYP46A1 active site and have, like (S)-EFV, sigmoidal binding curves suggestive of allosteric binding (Fig. 6B). Indeed, in the case of (R)-EFV and (S)-7OH, the curve fit was sigmoidal, and the spectral parameters of binding were similar to that of (S)-EFV. The spectral response type was of reverse type 1 (a trough at 391–393 nm and a peak at 426–427 nm in the P450 difference spectrum), the spectral Kds were in the 9–16 micromolar range, and the Hill coefficient was ≥2. However, in the case of (S)- and (rac)-7,8diOH as well as (S)-8,14diOH EFV, the curve fit was hyperbolic. Both (rac)- and (S)-7,8diOH EFV elicited type 1 spectral response, whereas (S)-8,14diOH EFV elicited reverse type 1 spectral response. Notably, like in titrations of substrate-free CYP46A1, binding of (rac)- and (S)-7,8diOH EFV to the active site of cholesterol-bound CYP46A1 was weak, and compound binding curves did not even reach the plateau. Thus, spectral titrations of substrate-free and cholesterol-bound CYP46A1 identified stereoisomers of 7,8diOH EFV as weak binders to the CYP46A1 active site, a potential advantage for CYP46A1 activation in vivo, as it reduces competition with cholesterol for the enzyme active site.
Discussion
Herein, we completed our in vitro characterizations of different hydroxylated EFV metabolites for activation and binding to CYP46A1, an emerging pharmacologic target for different brain disorders (Pikuleva, 2021; Pikuleva and Cartier, 2021). Although both inhibition and activation of CYP46A1 could be of therapeutic value (Halford et al., 2021; Pikuleva and Cartier, 2021), we are focused on CYP46A1 activation by low-dose (S)-EFV, which at high doses inhibits the P450 in mice (Mast et al., 2014). The latter as well as the ability of (S)-EFV to accumulate in the brain (Dirson et al., 2006) are potential limitations of (S)-EFV for clinical studies requiring CYP46A1 activation. Hence, we are looking for EFV-related compounds that will not inhibit CYP46A1 at higher doses and may even be stronger CYP46A1 activators than (S)-EFV. Collectively, this and our previous investigation provided several important insights.
First, by assessing enzymatic activity of purified CYP46A1, we obtained evidence that the maximal CYP46A1 activation by all known phase 1 (hydroxylated) (S)-EFV metabolites is at least 1.4-fold higher than that of the parent compound (Fig. 3). In humans, hydroxylated metabolites of (S)-EFV are detected in the cerebrospinal fluid (Avery et al., 2011, 2013b; Best et al., 2011; Winston et al., 2015; Aouri et al., 2016; Nightingale et al., 2016), a marker for compound presence in the brain. Hence, it is feasible that the hydroxymetabolites of (S)-EFV activate CYP46A1 in vivo as well, thus explaining why only a very small dose of the drug is required for CYP46A1 activation. However, the phase I (S)-EFV metabolites are more polar than (S)-EFV, and their direct measurements in the brain have not yet been carried out (to the best of our knowledge). Therefore, it is not clear whether these metabolites reach the brain as efficiently as the parent compound, and CYP46A1 will have a similar exposure to them as to (S)-EFV. This is a potential limitation of the phase I (S)-EFV metabolites as alternative therapeutics to (S)-EFV. Apparently, the measurements of the (S)-EFV hydroxylation products in the brain are required to address this limitation experimentally.
Second, the spatial configuration of EFV and its hydroxy metabolites did not seem to affect CYP46A1 activation and metabolite binding in vitro as indicated by experiments with (R)-EFV and (S)-EFV or racemates and (S) enantiomers of EFV metabolites (Figs. 3, 5, and 6). This finding could have an important practical implication, suggesting that replacing the (S) enantiomer with a racemate or the (R) enantiomer and using either EFV or EFV hydroxy metabolite could minimize the known side effects of large-dose (S)-EFV in the CNS and reported toxicity of (S)-7OH EFV and (S)-8OH EFV in cell culture. The latter, however, was not supported by the subsequent investigation of the correlation between the plasma or cerebrospinal fluid (CSF) levels of these metabolites and CNS toxicity (Bumpus, 2011; Tovar-y-Romo et al., 2012; Brandmann et al., 2013; Aouri et al., 2016; Apostolova et al., 2017). Of pertinence is that the rate of (R)-EFV hydroxylation by CYP2B6 in vitro is approximately one-tenth that of (S)-EFV, and that (R)-8OH EFV is the only metabolite formed (Wang et al., 2019). Accordingly, the half-life of (R)-EFV in the plasma could be longer, thus requiring either a lower drug dose or a less frequent administration.
Third and perhaps the most fascinating finding was that both dihydroxy EFV metabolites (7,8diOH and 8,14diOH EFV) had very similar concentration dependence curves with only a small decrease in CYP46A1 activation at high compound concentrations relative to the maximal enzyme activation (Fig. 3). This finding suggests that in vivo, the two EFV metabolites may not have such a narrow therapeutic window for CYP46A1 activation as (S)-EFV, and of all phase 1 EFV metabolites, these compounds should be tested in vivo (on mice) first. Although either (S)-enantiomers or racemates could be used, ideally both forms should be compared. Mechanistically, lack of CYP46A1 inhibition at high metabolite concentrations could be because of the low metabolite affinity (as compared with (S)-EFV and the monohydroxylated EFV metabolites) to the CYP46A1 active site as well as inability (likely due to the two hydroxy groups) to form the iron-nitrogen (Fe-N) bond, which inhibits the P450-mediated catalysis.
Fourth, is it possible that simultaneously with binding to the CYP46A1 allosteric sites, the two cytosolic regions away from the membrane-bound enzyme active site (Anderson et al., 2016; Mast et al., 2017a), (S)-EFV and its metabolites also bind to the CYP46A1 active site? Such a possibility was suggested previously for the heteroactivator binding to the drug-metabolizing P450s 3A4 and 2C9 (Shou et al., 1994; Hutzler et al., 2001; Korzekwa, 2021; Seibert and Tracy, 2021). The active sites of CYP3A4 and CYP2C9 are large and can accommodate several molecules (Williams et al., 2003; Yano et al., 2004; Ekroos and Sjogren, 2006), i.e., both the heteroactivator and substrate, thus reducing the substrate motion and thereby uncoupling, leading to enhanced metabolite formation (Locuson et al., 2006). Yet the active site of substrate-bound CYP46A1 represents a banana-shaped channel and does not have enough space for binding of a second molecule (Mast et al., 2008). Therefore, it is unlikely, in our opinion, that both an activator and cholesterol can simultaneously bind to the CYP46A1 active site. However, we cannot completely rule out this possibility as the CYP46A1 active site is plastic and can bind molecules of different shapes, sizes, and polarity (Mast et al., 2010, 2012, 2013a,b). Hence, efforts are underway in this laboratory to cocrystallize CYP46A1 in complex with cholesterol and a coactivator either (S)-EFV or one of its hydroxymetabolites.
Finally, our in vitro data revealed that depending on the hydroxylation positions, dihydroxy EFV metabolites bind either to the allosteric site for (S)-EFV (7,8diOH EFV) or L-Glu (8,14diOH EFV) and therefore increase or decrease, respectively, CYP46A1 activation in vitro in the presence of L-Glu (Figs. 4 and 5), an excitatory neurotransmitter involved in memory and learning. Accordingly, we hypothesize that 7,8diOH EFV and 8,14diOH EFV may differ in their effects on animal performance in memory tasks. We have already begun testing this hypothesis on mice.
In summary, we established that all phase 1 EFV metabolites allosterically activate purified CYP46A1 in vitro both as (S) enantiomers and racemates. The dihydroxy metabolites preferentially bind to one allosteric site, either for (S)-EFV (7,8diOH EFV) or L-Glu (8,14diOH EFV), whereas 7- and 8-monohydroxy metabolites bind to both allosteric sites. This difference in the allosteric site binding likely determines in part how EFV metabolites activate CYP46A1 in vitro in the coincubations with (S)-EFV and L-Glu. We also established that of all hydroxylated EFV metabolites, (S)- and (rac)-7,8diOH EFV are the weakest binders to the CYP46A1 active site. This property along with binding to the allosteric site for (S)-EFV make 7,8diOH EFV the first candidate for testing in vivo as potentially a better CYP46A1 activator than (S)-EFV. 8,14diOH EFV should be tested in vivo as well to determine whether binding to a different allosteric site is translated into different metabolite effects on animal performance in behavioral tests. The present work determines the direction of our future studies of the hydroxylated EFV metabolites in vivo, which we have already started.
Acknowledgments
The authors thank John Denker from the Molecular Biology and Genotyping Core of the Visual Sciences Research Center Core Facilities (supported by National Institutes of Health Grant P30- EY11373) for the generation of the Q440M CYP46A1 mutant.
Abbreviations
- AD
Alzheimer’s disease
- CNS
central nervous system
- EFV
efavirenz
- Glu
glutamate
- 24HC
24-hydroxycholesterol
- rac
racemic
- WT
wild type
Authorship Contributions
Participated in research design: Mast, Pikuleva.
Conducted experiments: Mast, Fotinich.
Performed data analysis: Mast, Pikuleva.
Wrote or contributed to the writing of the manuscript: Mast, Pikuleva.
Footnotes
This work was supported in part by National Institutes of Health National Institute on Aging [Grant AG-067552] (to I.A.P) and National Institutes of Health National Eye Institute [Grant EY-011373] (to I.A.P). I.A.P. is a Carl F. Asseff Professor of Ophthalmology.
No author has an actual or perceived conflict of interest with the contents of this article.
References
- Ali T, Hannaoui S, Nemani S, Tahir W, Zemlyankina I, Cherry P, Shim SY, Sim V, Schaetzl HM, Gilch S (2021) Oral administration of repurposed drug targeting Cyp46A1 increases survival times of prion infected mice. Acta Neuropathol Commun 9:58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson KW, Mast N, Hudgens JW, Lin JB, Turko IV, Pikuleva IA (2016) Mapping of the allosteric site in cholesterol hydroxylase CYP46A1 for efavirenz, a drug that stimulates enzyme activity. J Biol Chem 291:11876–11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aouri MBarcelo CTernon BCavassini MAnagnostopoulos AYerly SHugues HVernazza PGünthard HFBuclin T, et al. ; Swiss HIV Cohort Study (2016) In vivo profiling and distribution of known and novel phase I and phase II metabolites of efavirenz in plasma, urine, and cerebrospinal fluid. Drug Metab Dispos 44:151–161. [DOI] [PubMed] [Google Scholar]
- Apostolova N, Blas-Garcia A, Galindo MJ, Esplugues JV (2017) Efavirenz: what is known about the cellular mechanisms responsible for its adverse effects. Eur J Pharmacol 812:163–173. [DOI] [PubMed] [Google Scholar]
- Avery LB, Bakshi RP, Cao YJ, Hendrix CW (2011) The male genital tract is not a pharmacological sanctuary from efavirenz. Clin Pharmacol Ther 90:151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery LB, Sacktor N, McArthur JC, Hendrix CW (2013a) Protein-free efavirenz concentrations in cerebrospinal fluid and blood plasma are equivalent: applying the law of mass action to predict protein-free drug concentration. Antimicrob Agents Chemother 57:1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avery LB, VanAusdall JL, Hendrix CW, Bumpus NN (2013b) Compartmentalization and antiviral effect of efavirenz metabolites in blood plasma, seminal plasma, and cerebrospinal fluid. Drug Metab Dispos 41:422–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Best BMKoopmans PPLetendre SLCapparelli EVRossi SSClifford DBCollier ACGelman BBMbeo GMcCutchan JA, et al. ; CHARTER Group (2011) Efavirenz concentrations in CSF exceed IC50 for wild-type HIV. J Antimicrob Chemother 66:354–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogdanovic N, Bretillon L, Lund EG, Diczfalusy U, Lannfelt L, Winblad B, Russell DW, Björkhem I (2001) On the turnover of brain cholesterol in patients with Alzheimer’s disease. Abnormal induction of the cholesterol-catabolic enzyme CYP46 in glial cells. Neurosci Lett 314:45–48. [DOI] [PubMed] [Google Scholar]
- Boussicault LAlves SLamazière APlanques AHeck NMoumné LDespres GBolte SHu APagès C, et al. (2016) CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 139:953–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandmann M, Nehls U, Dringen R (2013) 8-Hydroxy-efavirenz, the primary metabolite of the antiretroviral drug Efavirenz, stimulates the glycolytic flux in cultured rat astrocytes. Neurochem Res 38:2524–2534. [DOI] [PubMed] [Google Scholar]
- Brown J IIITheisler CSilberman SMagnuson DGottardi-Littell NLee JMYager DCrowley JSambamurti KRahman MM, et al. (2004) Differential expression of cholesterol hydroxylases in Alzheimer’s disease. J Biol Chem 279:34674–34681. [DOI] [PubMed] [Google Scholar]
- Bumpus NN (2011) Efavirenz and 8-hydroxyefavirenz induce cell death via a JNK- and BimEL-dependent mechanism in primary human hepatocytes. Toxicol Appl Pharmacol 257:227–234. [DOI] [PubMed] [Google Scholar]
- Bumpus NN, Kent UM, Hollenberg PF (2006) Metabolism of efavirenz and 8-hydroxyefavirenz by P450 2B6 leads to inactivation by two distinct mechanisms. J Pharmacol Exp Ther 318:345–351. [DOI] [PubMed] [Google Scholar]
- Dirson G, Fernandez C, Hindlet P, Roux F, German-Fattal M, Gimenez F, Farinotti R (2006) Efavirenz does not interact with the ABCB1 transporter at the blood-brain barrier. Pharm Res 23:1525–1532. [DOI] [PubMed] [Google Scholar]
- Ekroos M, Sjögren T (2006) Structural basis for ligand promiscuity in cytochrome P450 3A4. Proc Natl Acad Sci USA 103:13682–13687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halford JJ, Sperling MR, Arkilo D, Asgharnejad M, Zinger C, Xu R, During M, French JA (2021) A phase 1b/2a study of soticlestat as adjunctive therapy in participants with developmental and/or epileptic encephalopathies. Epilepsy Res 174:106646. [DOI] [PubMed] [Google Scholar]
- Han MWang SYang NWang XZhao WSaed HSDaubon THuang BChen ALi G, et al. (2020) Therapeutic implications of altered cholesterol homeostasis mediated by loss of CYP46A1 in human glioblastoma. EMBO Mol Med 12:10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna IH, Teiber JF, Kokones KL, Hollenberg PF (1998) Role of the alanine at position 363 of cytochrome P450 2B2 in influencing the NADPH- and hydroperoxide-supported activities. Arch Biochem Biophys 350:324–332. [DOI] [PubMed] [Google Scholar]
- Hudry EVan Dam DKulik WDe Deyn PPStet FSAhouansou OBenraiss ADelacourte ABougnères PAubourg P, et al. (2010) Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther 18:44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutzler JM, Hauer MJ, Tracy TS (2001) Dapsone activation of CYP2C9-mediated metabolism: evidence for activation of multiple substrates and a two-site model. Drug Metab Dispos 29:1029–1034. [PubMed] [Google Scholar]
- Korzekwa K (2021) Enzyme kinetics of oxidative metabolism-cytochromes P450. Methods Mol Biol 2342:237–256. [DOI] [PubMed] [Google Scholar]
- Kumaki K, Sato M, Kon H, Nebert DW (1978) Correlation of type I, type II, and reverse type I difference spectra with absolute changes in spin state of hepatic microsomal cytochrome P-450 iron from five mammalian species. J Biol Chem 253:1048–1058. [PubMed] [Google Scholar]
- Locuson CW, Gannett PM, Tracy TS (2006) Heteroactivator effects on the coupling and spin state equilibrium of CYP2C9. Arch Biochem Biophys 449:115–129. [DOI] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW (1999) cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA 96:7238–7243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman RB, Kulkarni AP, Baker RC, Hodgson E (1974) Cytochrome P-450 difference spectra: effect of chemical structure on type II spectra in mouse hepatic microsomes. Drug Metab Dispos 2:301–308. [PubMed] [Google Scholar]
- Mast N, Anderson KW, Johnson KM, Phan TTN, Guengerich FP, Pikuleva IA (2017a) In vitro cytochrome P450 46A1 (CYP46A1) activation by neuroactive compounds. J Biol Chem 292:12934–12946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Charvet C, Pikuleva IA, Stout CD (2010) Structural basis of drug binding to CYP46A1, an enzyme that controls cholesterol turnover in the brain. J Biol Chem 285:31783–31795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Li Y, Linger M, Clark M, Wiseman J, Pikuleva IA (2014) Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J Biol Chem 289:3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Linger M, Clark M, Wiseman J, Stout CD, Pikuleva IA (2012) In silico and intuitive predictions of CYP46A1 inhibition by marketed drugs with subsequent enzyme crystallization in complex with fluvoxamine. Mol Pharmacol 82:824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Reem R, Bederman I, Huang S, DiPatre PL, Björkhem I, Pikuleva IA (2011) Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest Ophthalmol Vis Sci 52:594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Saadane A, Valencia-Olvera A, Constans J, Maxfield E, Arakawa H, Li Y, Landreth G, Pikuleva IA (2017b) Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology 123:465–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Verwilst P, Wilkey CJ, Guengerich FP, Pikuleva IA (2020) In vitro activation of cytochrome P450 46A1 (CYP46A1) by efavirenz-related compounds. J Med Chem 63:6477–6488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, White MA, Björkhem I, Johnson EF, Stout CD, Pikuleva IA (2008) Crystal structures of substrate-bound and substrate-free cytochrome P450 46A1, the principal cholesterol hydroxylase in the brain. Proc Natl Acad Sci USA 105:9546–9551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Zheng W, Stout CD, Pikuleva IA (2013a) Antifungal azoles: structural insights into undesired tight binding to cholesterol-metabolizing CYP46A1. Mol Pharmacol 84:86–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Zheng W, Stout CD, Pikuleva IA (2013b) Binding of a cyano- and fluoro-containing drug bicalutamide to cytochrome P450 46A1: unusual features and spectral response. J Biol Chem 288:4613–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutlib AE, Chen H, Nemeth GA, Markwalder JA, Seitz SP, Gan LS, Christ DD (1999) Identification and characterization of efavirenz metabolites by liquid chromatography/mass spectrometry and high field NMR: species differences in the metabolism of efavirenz. Drug Metab Dispos 27:1319–1333. [PubMed] [Google Scholar]
- Nightingale SChau TTFisher MNelson MWinston AElse LCarr DFTaylor SUstianowski ABack D, et al. (2016) Efavirenz and metabolites in cerebrospinal fluid: relationship with CYP2B6 c.516G→T genotype and perturbed blood-brain barrier due to tuberculous meningitis. Antimicrob Agents Chemother 60:4511–4518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nóbrega CMendonça LMarcelo ALamazière ATomé SDespres GMatos CAMechmet FLangui Dden Dunnen W, et al. (2019) Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol 138:837–858. [DOI] [PubMed] [Google Scholar]
- Ogburn ET, Jones DR, Masters AR, Xu C, Guo Y, Desta Z (2010) Efavirenz primary and secondary metabolism in vitro and in vivo: identification of novel metabolic pathways and cytochrome P450 2A6 as the principal catalyst of efavirenz 7-hydroxylation. Drug Metab Dispos 38:1218–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Lam M, Mast N, Moon J, Li Y, Maxfield E, Pikuleva IA (2019a) CYP46A1 activation by efavirenz leads to behavioral improvement without significant changes in amyloid plaque load in the brain of 5XFAD mice. Neurotherapeutics 16:710–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Mast N, Li Y, Pikuleva IA (2019b) The key genes, phosphoproteins, processes, and pathways affected by efavirenz-activated CYP46A1 in the amyloid-decreasing paradigm of efavirenz treatment. FASEB J 33:8782–8798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikuleva IA (2021) Targeting cytochrome P450 46A1 and brain cholesterol 24-hydroxylation to treat neurodegenerative diseases. Explor Neuroprotective Ther 1:159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pikuleva IA, Cartier N (2021) Cholesterol hydroxylating cytochrome P450 46A1: from mechanisms of action to clinical applications. Front Aging Neurosci 13:696778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez DM, Andersson S, Russell DW (2008) Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol 507:1676–1693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remmer H, Schenkman J, Estabrook RW, Sasame H, Gillette J, Narasimhulu S, Cooper DY, Rosenthal O (1966) Drug interaction with hepatic microsomal cytochrome. Mol Pharmacol 2:187–190. [PubMed] [Google Scholar]
- Schenkman JB, Remmer H, Estabrook RW (1967) Spectral studies of drug interaction with hepatic microsomal cytochrome. Mol Pharmacol 3:113–123. [PubMed] [Google Scholar]
- Seibert E, Tracy TS (2021) Fundamentals of enzyme kinetics: Michaelis-Menten and non-Michaelis–type (atypical) enzyme kinetics, in Enzyme Kinetics in Drug Metabolism: Fundamentals and Applications (Nagar S, Argikar UA, Tweedie D, eds) pp 3–27, Springer US, New York. [DOI] [PubMed] [Google Scholar]
- Shou M, Grogan J, Mancewicz JA, Krausz KW, Gonzalez FJ, Gelboin HV, Korzekwa KR (1994) Activation of CYP3A4: evidence for the simultaneous binding of two substrates in a cytochrome P450 active site. Biochemistry 33:6450–6455. [DOI] [PubMed] [Google Scholar]
- Testa G, Staurenghi E, Zerbinati C, Gargiulo S, Iuliano L, Giaccone G, Fantò F, Poli G, Leonarduzzi G, Gamba P (2016) Changes in brain oxysterols at different stages of Alzheimer’s disease: their involvement in neuroinflammation. Redox Biol 10:24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar-y-Romo LB, Bumpus NN, Pomerantz D, Avery LB, Sacktor N, McArthur JC, Haughey NJ (2012) Dendritic spine injury induced by the 8-hydroxy metabolite of efavirenz. J Pharmacol Exp Ther 343:696–703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma VRBüşra Lüleci HOommen AMVarma SBlackshear CTGriswold MEAn YRoberts JAO’Brien RPletnikova O, et al. (2021) Abnormal brain cholesterol homeostasis in Alzheimer’s disease—a targeted metabolomic and transcriptomic study. NPJ Aging Mech Dis 7:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang PF, Neiner A, Kharasch ED (2019) Efavirenz metabolism: influence of polymorphic CYP2B6 variants and stereochemistry. Drug Metab Dispos 47:1195–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward BA, Gorski JC, Jones DR, Hall SD, Flockhart DA, Desta Z (2003) The cytochrome P450 2B6 (CYP2B6) is the main catalyst of efavirenz primary and secondary metabolism: implication for HIV/AIDS therapy and utility of efavirenz as a substrate marker of CYP2B6 catalytic activity. J Pharmacol Exp Ther 306:287–300. [DOI] [PubMed] [Google Scholar]
- White MA, Mast N, Björkhem I, Johnson EF, Stout CD, Pikuleva IA (2008) Use of complementary cation and anion heavy-atom salt derivatives to solve the structure of cytochrome P450 46A1. Acta Crystallogr D Biol Crystallogr 64:487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams PA, Cosme J, Ward A, Angove HC, Matak Vinković D, Jhoti H (2003) Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature 424:464–468. [DOI] [PubMed] [Google Scholar]
- Winston A, Amin J, Clarke A, Else L, Amara A, Owen A, Barber T, Jessen H, Avihingsanon A, Chetchotisakd P,, et al. ; ENCORE Cerebrospinal Fluid (CSF) Substudy Team; ENCORE Cerebrospinal Fluid CSF Substudy Team (2015) Cerebrospinal fluid exposure of efavirenz and its major metabolites when dosed at 400 mg and 600 mg once daily: a randomized controlled trial [published correction appears in Clin Infect Dis (2015) 61:143]. Clin Infect Dis 60:1026–1032. [DOI] [PubMed] [Google Scholar]
- Yano JK, Wester MR, Schoch GA, Griffin KJ, Stout CD, Johnson EF (2004) The structure of human microsomal cytochrome P450 3A4 determined by X-ray crystallography to 2.05-A resolution. J Biol Chem 279:38091–38094. [DOI] [PubMed] [Google Scholar]