

Abstract
It was recently disclosed that CYP3A is responsible for the tertiary stereoselective oxidations of deoxycholic acid (DCA), which becomes a continuum mechanism of the host-gut microbial cometabolism of bile acids (BAs) in humans. This work aims to investigate the species differences of BA redox metabolism and clarify whether the tertiary metabolism of DCA is a conserved pathway in preclinical animals. With quantitative determination of the total unconjugated BAs in urine and fecal samples of humans, dogs, rats, and mice, it was confirmed that the tertiary oxidized metabolites of DCA were found in all tested animals, whereas DCA and its oxidized metabolites disappeared in germ-free mice. The in vitro metabolism data of DCA and the other unconjugated BAs in liver microsomes of humans, monkeys, dogs, rats, and mice showed consistencies with the BA-profiling data, confirming that the tertiary oxidation of DCA is a conserved pathway. In liver microsomes of all tested animals, however, the oxidation activities toward DCA were far below the murine-specific 6β-oxidation activities toward chenodeoxycholic acid (CDCA), ursodeoxycholic acid, and lithocholic acid (LCA), and 7-oxidation activities toward murideoxycholic acid and hyodeoxycholic acid came from the 6-hydroxylation of LCA. These findings provided further explanations for why murine animals have significantly enhanced downstream metabolism of CDCA compared with humans. In conclusion, the species differences of BA redox metabolism disclosed in this work will be useful for the interspecies extrapolation of BA biology and toxicology in translational researches.
SIGNIFICANCE STATEMENT
It is important to understand the species differences of bile acid metabolism when deciphering biological and hepatotoxicology findings from preclinical studies. However, the species differences of tertiary bile acids are poorly understood compared with primary and secondary bile acids. This work confirms that the tertiary oxidation of deoxycholic acid is conserved among preclinical animals and provides deeper understanding of how and why the downstream metabolism of chenodeoxycholic acid dominates that of cholic acid in murine animals compared with humans.
Introduction
Bile acids (BAs) are amphipathic steroids produced almost exclusively by the liver from cholesterol. Most BAs are trapped in the bile acid pool by the process of enterohepatic circulation driven by a series of host-gut microbial metabolism and transport mechanisms (Russell, 2003; Halilbasic et al., 2013; Dawson and Karpen, 2015). As shown in Fig. 1, this cometabolism involves metabolic pathways from primary to secondary BAs and from secondary to tertiary BAs, and with this, the nomenclature of BAs in our work follows Hofmann’s proposal (Hofmann et al., 1992). During the enterohepatic circulation of BAs, BA-mediated host-gut microbial crosstalk is important for maintaining a healthy gut microbiota (Theriot et al., 2014; Buffie et al., 2015), balanced lipid and carbohydrate metabolism (de Aguiar Vallim et al., 2013; Wahlstrom et al., 2016), insulin sensitivity (Maruyama et al., 2002; Kawamata et al., 2003), and innate immunity (Inagaki et al., 2006; Vavassori et al., 2009). Drug-induced disturbance of BA homeostasis may result in cholestasis, and it induces hepatic and extrahepatic toxicities known as drug-induced liver injuries. Because there are significant species differences along the host-gut microbiota cometabolism axis of BAs, drug-induced liver injury is often a poorly predicted adverse event during preclinical studies, and sometimes it is unnoticed until later clinical trials (Chen et al., 2014). As such, it is of great importance to seriously take into account the species differences when deciphering biologic and toxicological findings from preclinical animal species.
Fig. 1.

Metabolic scheme of the primary BAs to secondary BAs to tertiary BAs pathways. The abbreviated nomenclature of unconjugated bile acids follows Hofmann’s proposal (Hofmann et al., 1992). The structures of conjugated forms and oxo- or keto- intermediates were not shown.
The species differences of primary BA come firstly from oxidative on the steroid nucleus and secondly from conjugation with glycine, taurine, sulfate, and/or glucuronic acid. Cholic acid (CA) and chenodeoxycholic acid (CDCA) are the primary BAs for most mammals (Hofmann et al., 2010). CDCA may be partly 6α-hydroxylated into α-hyocholic acid (HCA) by CYP3A (Deo and Bandiera, 2008). Because of the presence of the hepatic 6β-hydroxylase, murine animals have additional primary BAs known as muricholic acids (MCAs), αMCA and βMCA (Hsla et al., 1957; Mahowald et al., 1957a). The murine 6β-hydroxylase was recently identified as CYP2C22 in rats and Cyp2c70 in mice, whose homologous enzyme in humans, CYP2C9, is a drug-metabolizing enzyme without BA oxidation activities (Takahashi et al., 2016). Ursodeoxycholic acid (UDCA) is also a primary BA of mice (Sayin et al., 2013) since mice Cyp2c70 is also able to transform CDCA into UDCA (Takahashi et al., 2016). The BAs in humans, minipigs, and hamsters conjugate mainly with glycine, whereas taurine amidation is predominant in mice, rats, and dogs. It has been also discovered that less sulfation occurs in rats and mice compared with humans and chimpanzees (Thakare et al., 2018a,b).
The reactions of secondary BA metabolism greatly enlarge the structural diversity. The major pathway for secondary BA metabolism is the deconjugation of the amidated primary BAs by bacteria with bile salt hydrolase activity (Huijghebaert and Hofmann, 1986; Gopal-Srivastava and Hylemon, 1988) and the subsequent 7-dehydroxylation of the unconjugated forms by bacteria that carry bile acid–inducible genes (Stellwag and Hylemon, 1979; Hirano et al., 1981; Batta et al., 1990). CA and CDCA are 7-dehydroxylated into deoxycholic acid (DCA) and lithocholic acid (LCA), respectively. UDCA is dehydroxylated into LCA at a similar fashion. The murine-specific BAs, α-MCA and β-MCA, are 7-dehydroxylated into murideoxycholic acid (MDCA) (Zhang et al., 2012). Another type of reaction of secondary BA metabolism is epimerization of hydroxyl groups catalyzed by hydroxysteroid dehydrogenases (HSDHs) (Macdonald et al., 1976; MacDonald et al., 1977). CDCA and CA are epimerized into UDCA and ursocholic acid (UCA) and, subsequently, into isoUDCA and isoUCA, respectively (Zhu et al., 2018). CA and DCA are epimerized into 12-epiCA and 12-epiDCA, respectively (Zhu et al., 2018). To the best of our knowledge, evidence is not yet available for 6-HSDH that may catalyze epimerization between β-MCA and β-HCA (also termed ω-MCA in literature) in murine animals (Ridlon et al., 2006, 2016). Some of the secondary BAs can be reabsorbed into the liver, where they undergo N-acylamidation metabolism.
Tertiary BAs are redox metabolites of the reabsorbed secondary BAs catalyzed by host enzymes. The term of “tertiary BA” was proposed for the first time based on the identified 7β-oxidation pathway from hyodeoxycholic acid (HDCA) to β-HCA after oral administration of HDCA to germ-free rats (Madsen et al., 1975). As a matter of fact, earlier studies have provided evidence for the tertiary BA metabolism by administrating the radiolabeled secondary BAs to bile fistula rats. CDCA, MDCA, α-MCA, and β-MCA were identified as metabolites of LCA (Thomas et al., 1964); MDCA and β-HCA were identified as metabolites of HDCA (Matschiner et al., 1957, 1958); β-MCA was identified as a major metabolite of MDCA (Thomas et al., 1965); and CA and DCA-6β-ol were identified as metabolites of DCA (Mahowald et al., 1957b; Ratliff et al., 1961). The murine-specific 7α-hydroxylase, which catalyzes the conversion of DCA and LCA to CA and CDCA in taurine-conjugated forms, respectively, was recently identified as Cyp2a12 in mice (Honda et al., 2020). However, it is much more challenging to study tertiary BA metabolism in larger animals, particularly in humans, because of lack of germ-free models and ethnic problems of radiolabeled interventions. Although several BAs with unusual oxidation sites have been identified in humans in the last century (Sjövall et al., 2010), only two CYP3A-catalyzed pathways, 6α-hydroxylation of LCA to HDCA (Trulzsch et al., 1974; Araya and Wikvall, 1999; Deo and Bandiera, 2009) and 1β-hydroxylation of DCA to DCA-1β-ol (Gustafsson et al., 1985; Bodin et al., 2005; Hayes et al., 2016), have been confirmed. The species differences of tertiary BAs are still poorly understood compared with primary and secondary BAs.
We have recently extended the scope of tertiary BA metabolism in humans based on BA metabolome analysis in combination with in vitro metabolism assays. As shown in Fig. 1, a series of tertiary oxidation reactions of DCA, glycodeoxycholic acid (GDCA), and taurodeoxycholic acid (TDCA) regioselectively at C-3β, C-1β, C-5β, C-6α, C19, C-4β, and C-6β were characterized to be exclusively catalyzed by CYP3A4 and CYP3A7 (Zhang et al., 2019). The 19-hydroxylation of DCA, GDCA, and TDCA was identified as a novel in vitro marker of CYP3A7 activity (Chen et al., 2019). After this observation, we confirmed in this work that the tertiary metabolism of DCA is a conserved pathway in commonly used preclinical animal models. A series of murine-specific metabolic pathways that may be responsible for the predominance of downstream metabolism of CDCA in murine animals compared with humans were identified and illustrated in Fig. 1.
Materials and Methods
Materials and Reagents.
Authentic standards of 25 unconjugated BAs and four stable isotope-labeled internal standards were obtained from Steraloids (Newport, RI), TRC (Toronto, Canada), Santa Cruz (Dallas, TX), or Sigma-Aldrich (St. Louis, MO) as previously described (Zhu et al., 2018). Six DCA metabolites, DCA-1β-ol, DCA-4β-ol, DCA-5β-ol, DCA-6β-ol, DCA-6α-ol, and DCA-19-ol, were synthesized as described in our recent report (Zhang et al., 2019). Sulfatase from Helix pomatia Type H-1, β-glucuronidase from Helix pomatia Type H-1, and choloylglycine hydrolase from clostridium perfringens were purchased from Sigma-Aldrich. Human liver microsomes from 150 (76 female and 74 male) mixed-gender pooled donors, dog liver microsomes from seven (three female and four male) mixed-gender pooled donors, rat liver microsomes from 181 (21 female and 160 male) mixed-gender pooled donors, mice liver microsomes from 170 (150 female and 20 male) mixed-gender pooled donors, NADPH-regenerating system solution A (containing 26 mM NADP+, 66 mM glucose-6-phosphate, and 66 mM MgCl2 in water), NADPH-regenerating system solution B (containing 40 U/ml glucose-6-phosphate dehydrogenase in 5 mM sodium citrate), and 0.5 M pH 7.4 PBS were purchased from Corning (Tewksbury, MA). Cynomolgus monkey liver microsomes from 10 (seven female and three male) mixed-gender pooled donors were purchased from Research Institute for Liver Diseases (Shanghai, China). Sodium acetate, glacial acetic acid, ammonium hydroxide, liquid chromatography and mass spectrometry–grade methanol, acetonitrile, and formic acid were purchased from Sigma-Aldrich. DMSO was purchased from Thermo Fisher Scientific (Waltham, MA). Ultra-pure water was obtained by using a Milli-Q system (Bedford, MA).
Fecal and Urine Samples.
Human fecal and urine samples were collected from healthy volunteers (n = 6, five female and one male) in our laboratory who had signed informed consents. Fecal and urine samples from Beagle dogs (n = 6, three male and three female), Sprague-Dawley rats (n = 6, three male and three female), and C57BL/6 mice (n = 8, four male and four female) were gifted from West China-Frontier PharmaTech (Chengdu, China). Fecal and urine samples of germ-free C57BL/6 mice (n = 3, male) were gifted from Shanghai SLAC Laboratory Animal Co., Ltd (Shanghai, China). The collected samples were stored at −80°C until analysis.
Sample Preparation for BA Profile Analysis.
Quantitative analysis of BA profile was performed using the enzyme digestion techniques (Zhu et al., 2018). In brief, urine and fecal samples were thawed on ice bath. All urine samples were centrifuged at 4°C at 12,000g for 5 minutes, and 100 µl supernatants from the same species were mixed into a pooled urine sample. Each 50-mg fecal sample was homogenized with 500 µl 0.1% (v/v) ammonium hydroxide solution. The homogenized samples were frozen at −40°C for 10 minutes for protein precipitation. The processed samples were centrifuged at 4°C at 12,000g for 20 minutes, and 100 µl supernatants from the same species were mixed into a pooled fecal sample. An aliquot (50 µl) of the pooled urine or fecal samples was transferred to 700-µl round well 96-well plate in triplicate. A volume of 150 µl sodium acetate buffer (pH 5.0) containing 50 U of sulfatase, 500 U of β-glucuronidase, and 100 U of choloylglycine hydrolase was added to digest the conjugated BAs. The plate was incubated at 37°C for 6 hours and subsequently lyophilized. Then, 200 µl of acetonitrile containing 1% formic acid and 100 nM internal standards (LCA-2,2,4,4-D4, DCA-2,2,4,4-D4, UDCA-2,2,4,4-D4, and CA-2,2,4,4-D4) were added in each well. The plate was mixed by vortexing at 1500 rpm for 30 minutes at 10°C and then centrifuged at 3000g for 20 minutes at 4°C. The 200 μl supernatant was transferred to another plate and vacuum evaporated at 30°C. The residue was reconstituted with 50 μl of acetonitrile and 50 μl of water and then mixed by vortexing at 900 rpm for 20 minutes at 10°C. After centrifugation, the plate was placed into an autosampler for analysis.
In Vitro Oxidation Metabolism Assay.
In vitro metabolism was performed using the protocol described in our recent report (Chen et al., 2019; Zhang et al., 2019). In brief, stock solutions of unconjugated BAs were prepared in DMSO. Incubations with an initial substrate concentration of 1.0, 5.0, 25, 50, 100, 200, 300, 500, 750, 1000, 3000, and 5000 μM for DCA were performed in 96-well plates in a shaking incubator at 37°C. Incubations for the other unconjugated BAs were performed with an initial substrate concentration of 50 μM. The 100-μl incubation solution contained 0.1 M PBS (pH 7.4), 5.0 μl NADPH-regenerating system solution A, 1.0 μl NADPH-regenerating system solution B, 0.5 µl DCA working solution, 0.5 µl blank solvent, and 2.5 μl liver microsomes (protein concentration of 20 mg/ml). The final protein concentration in the incubation media was 0.5 mg/ml. All incubations were performed in triplicate, and the DMSO concentration in all incubations was 1% (v/v). The reactions were initiated after 5 minutes preincubation at 37°C by adding liver microsomes and stopped at a preset time point by adding 300 µl of ice-cold acetonitrile containing 0.1% formic acid and 50 µM CA-2,2,4,4-D4 as an internal standard. The reaction mixture was then centrifuged at 4°C at 4000g for 20 minutes. The supernatant (50 µl) was diluted with 50 µl water and subjected to liquid chromatography with tandem mass spectrometry (LC-MS/MS) analysis.
Quantitative LC-MS/MS Analysis.
Quantitative analysis of unconjugated BAs was performed on ACQUITY ultra-performance liquid chromatography coupled to a Xevo TQ-S mass spectrometer (Waters, Milford, MA) (Yin et al., 2017; Zhu et al., 2018). The mobile phases consisted of 0.01% formic acid in water (mobile phase A) and acetonitrile (mobile phase B). The 5 μl of each sample was injected onto an ACQUITY BEH C18 column (1.7 μm, 100 × 2.1 mm) (Waters). The flow rate was 0.45 ml/min with the following mobile phase gradient: 0.0–0.5 (95% A), 0.5–1.0 (95%–64% A), 1.0–2.0 (64%–74% A), 2.0–4.0 (74%–70% A), 4.0–6.0 (70% A), 6.0–7.0 (70%–62% A), 7.0–9.0 (62%–55% A), 9.0–12.5 (55%–30% A), 12.5–13.0 (30%–0% A), 13.0–14.0 (0% A), 14.0–14.1 (0%–95% A), and 14.1–15.0 minutes (95% A). The mass spectrometer was operated in the negative mode with a 3.0-kV capillary voltage. Selected ion recording for quantification and identification of unconjugated BAs was described in our previous reports (Yin et al., 2017; Zhu et al., 2018; Zhang et al., 2019). The source and desolvation temperatures were set at 150 and 550°C, respectively. The collision energy was set at 27 V. Nitrogen and argon were used as cone and collision gases, respectively. The cone gas flow and desolvation gas flow were set at 150 and 950 l/h, respectively.
Data Processing.
The LC-MS/MS raw data were processed by UNIFI (V1.8; Waters). The metabolite formations as a function of substrate concentrations were fit to Hill (sigmoidal) equation and hyperbolic (Michaelis-Menten) equation using GraphPad Prism software (version 7.0; GraphPad Software, La Jolla, CA).
Results
Tertiary Oxidations of Deoxycholate Are a Conserved Pathway among the Tested Animals.
In this work, we used a pooled-sample strategy to attenuate the intraspecies differences and highlight the interspecies differences of the BA profile. Serum or plasma samples were not included in this study because of the known limited exposure of BAs in circulation. A total of 31 unconjugated BAs were quantitated in the pooled urine and fecal samples collected from humans, dogs, rats, specific-pathogen-free (SPF) mice, and germ-free mice. These BA metabolites were classified into secondary or tertiary metabolites for the downstream of CA or CDCA (Fig. 2) based on the known host-gut microbial cometabolism network of BAs. It was clear that BAs are excreted mainly via feces and minorly via urine. The phenomenon was more significantly observed in murine animals than in humans and dogs. The primary BAs, CA and CDCA, and the corresponding secondary BAs, DCA and LCA, respectively, were prevalent in mice, rats, dogs, and humans. α-MCA and β-MCA, the 6β-hydroxylated metabolites of CDCA and UDCA, were almost exclusively detected in murine animals, except that α-MCA was detected to a much lesser extent in dogs. β-MCA, the 6-hydroxyl epimer of β-MCA, was exclusively detected in rats and mice. αHCA, a CYP3A-catalyzed 6α-oxidized metabolite of CDCA (Deo and Bandiera, 2008), was detected in humans and mice. Comparison between SPF and germ-free mice confirmed that CA, CDCA, UDCA, α-MCA, and β-MCA are the primary BAs of mice, whereas the other BAs detected in mice are more or less associated with the modification of primary BAs by the host, the secondary metabolism of primary BAs by the gut microbes, and/or the tertiary metabolism of reabsorbed secondary BAs by the host.
Fig. 2.
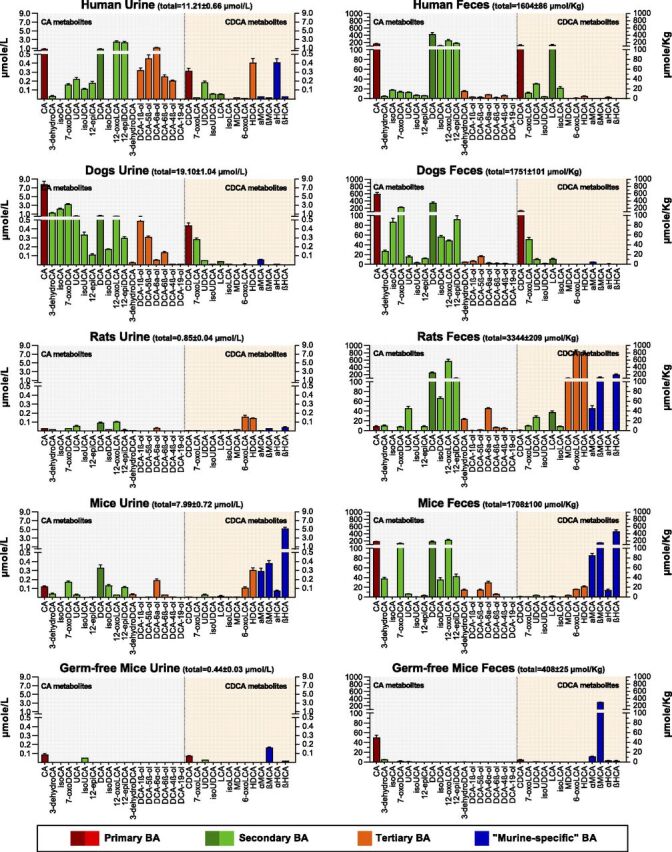
Quantitative profiles of the total unconjugated BAs in the pooled urine and fecal samples of humans (n = 6), dogs (n = 6), rats (n = 6), SPF mice (n = 8), and germ-free mice (n = 3). The unconjugated BAs were quantitatively determined by treating samples with choloylglycine hydrolase, sulfatase, and β-glucuronidase as the method described previously. Data are shown as mean ± S.D. for triplicate analysis of the pooled samples.
We have recently characterized both in vitro and in vivo the tertiary oxidations of DCA, GDCA, and TDCA regioselectively at C-1β, C-3β, C-4β, C-5β, C-6α, C-6β, and C-19, in which the 3β-hydroxylated metabolite rapidly dehydrates into 3-dehydroDCA (Zhang et al., 2019). The finding that 3-dehydroDCA, DCA-5β-ol, DCA-6α-ol, DCA-4β-ol, and DCA-6β-ol were also detected in the feces of dogs, rats, and mice was also confirmed in this study (Fig. 2). DCA-1β-ol was detected in dogs but not in rats and mice. Similar to our recent BA-profiling data of human urine, DCA-19-ol, the human CYP3A7-specific tertiary BA metabolite characterized in vitro, was not detected in human feces and absent in dogs, rats, and mice. Comparison between SPF and germ-free mice showed that both DCA and its downstream tertiary metabolites were absent in germ-free mice. The same pattern was also observed for both LCA and its downstream tertiary metabolites, HDCA and MDCA. Our data have provided preliminary in vivo evidence that the tertiary metabolism of LCA and DCA can be catalyzed by host enzymes.
In vitro metabolite screening of DCA and the other unconjugated monohydroxy or dihydroxy BAs was performed in liver microsomes of dogs, rats, mice, monkeys, and humans to confirm whether these tertiary BAs found in animals were metabolites uniquely from DCA. Similar to our recent observations in human liver microsomes (Zhang et al., 2019), these tertiary BAs were identified to be neither the di-oxidized metabolites of LCA nor the mono-oxidized metabolites of the other unconjugated dihydroxy BAs (unpublished data). As shown in Fig. 3, a total of eight oxidized metabolites were continuously produced within 0–720 minutes after an incubation of 50 μM DCA in liver microsomes of the test species. The DCA oxidations at C-3β, C-1β, C-5β, C-6α, C-4β, and C-6β were detected in all the test species, whereas the human CYP3A7-specific metabolite, DCA-19-ol, was not found at a substrate level of 50 μM in the test species. Moreover, rats preferred 7β-oxidation from DCA to UCA, which was commonly considered to be the epimerized metabolite of CA mediated by gut microbes. In brief, these results have made it clear that the tertiary oxidation of DCA is a conserved pathway in humans, monkeys, dogs, rats, and mice.
Fig. 3.

Time-dependent metabolite formation after incubation of DCA (50 μM) in the liver microsomes (protein level 0.5 mg/ml) of humans, monkeys, dogs, rats, and mice. Data are shown as mean ± S.D. (n = 3).
Species Differences of the Regioselective Oxidation Activities of DCA in Liver Microsomes.
Figure 4 shows the apparent kinetic plots of DCA oxidation activities in the liver microsomes of humans, monkeys, dogs, rats, and mice. Consistent with previous results observed from the recombinant human CYP3A enzymes, oxidation activities gradually reduced to zero for all the species when the concentration of substrates were above 300 μM. The activity loss was probably caused by the protein denaturation of enzymes and/or the formation of multimers and micelles of DCA molecules (Chen et al., 2019). The Hill equation (Supplemental Table 1) provides a better fit than the hyperbolic equation (Supplemental Table 2) for most kinetic plots within the concentration range from 1 to 300 μM for DCA. The R2 coefficient for most fittings was >0.99 but lower than that for those with no reaction or less productive reactions.
Fig. 4.

Kinetic plots of DCA oxidations in the liver microsomes (protein level 0.5 mg/ml) of humans, monkeys, dogs, rats, and mice. The dotted extrapolations of Hill curves demonstrate a violation of enzyme kinetics at higher substrate levels (>300 μM). Data are shown as mean ± S.D. (n = 3). CLint, intrinsic clearance; CMC, critical micelle concentration; ND, not detected; S50, substrate concentration occupying half of the binding sites.
Figure 5 illustrates the Vmax, substrate concentration occupying half of the binding sites, and intrinsic clearance data obtained from the best curve fitting. Data were calculated predominately using the Hill equation, except that the results for 6β-oxidation in dogs, 3β-oxidation in mice, and 4β-oxidation in mice were calculated using the hyperbolic equation because the Hill-fitting was not applicable. The fitting threshold (300 μM) and critical micelle concentration (about 1500 μM) of DCA in the incubation media are marked on the substrate concentration occupying half of the binding sites plot, indicating that all reactions may not achieve the maximum productivity because of the activity loss at substrate levels higher than 300 μM. Anyway, the in vitro intrinsic clearance data represent the intrinsic hepatic metabolic clearance toward DCA under physiologic levels. As shown in Fig. 5, the total clearance of DCA decreased in turn in the liver microsomes of humans, monkeys, rats, mice, and dogs. In the liver microsomes of the test animals, C-3β, C-1β, and C-7β are the major oxidation sites of DCA, whereas oxidations at the other sites have minor or minimum contribution to the total clearance. A trace activity toward the 19-oxidation of DCA, a human CYP3A7-specific reaction, was detected only in monkeys. The 7β-oxidation of DCA was a rat-specific reaction, which explained well why UCA was found with the highest level in the rat fecal sample. Murine animals showed no activity or only a trace activity toward the 1β-oxidation of DCA compared with humans, monkeys, and dogs, which was consistent with DCA-1β-ol not being detected in rats and mice.
Fig. 5.
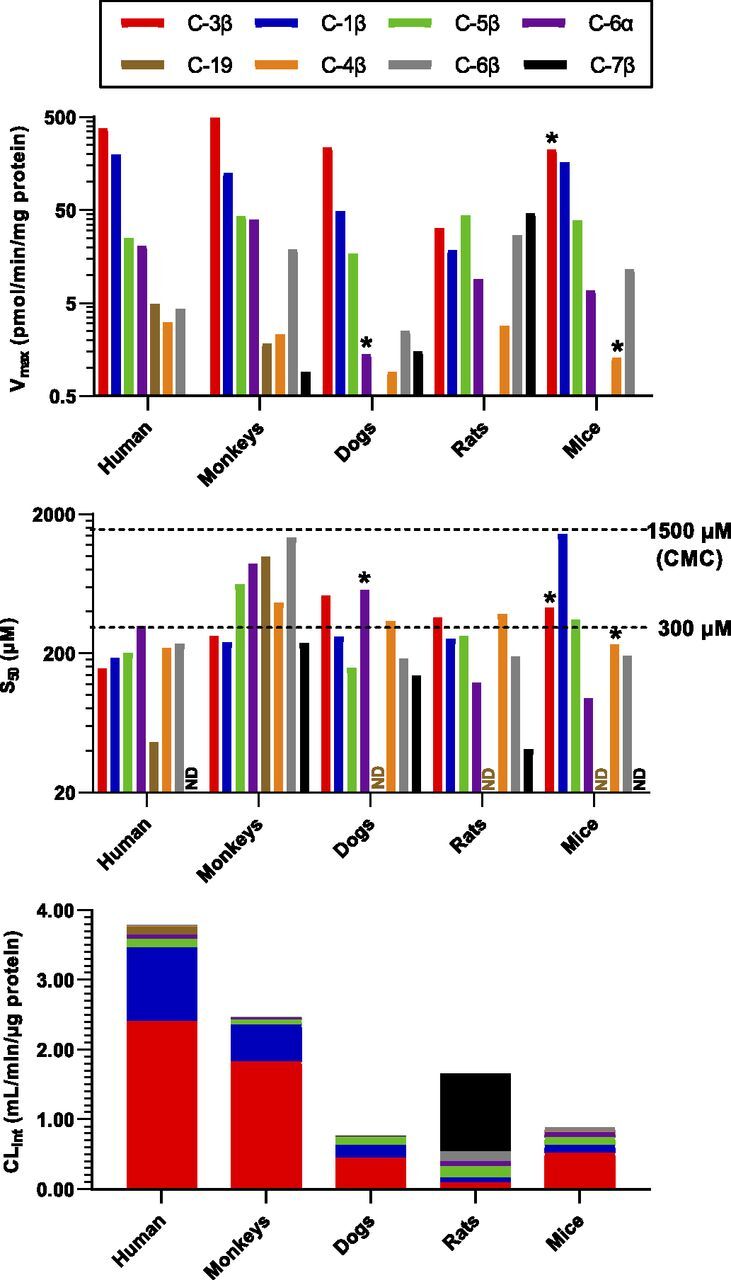
Apparent Hill parameters of DCA oxidation in the liver microsomes (protein level 0.5 mg/ml) of humans, monkeys, dogs, rats, and mice fitted with Hill Eq. (1–300 μM).
In Vitro Reactions Responsible for the Murine-Specific Downstream Metabolism of CDCA.
It was recently discovered that rat CYP2C22 and mouse Cyp2c70 are responsible for the murine-specific 6β-oxidation from CDCA to α-MCA and UDCA to β-MCA (Takahashi et al., 2016). Cyp2a12 was recently reported to be responsible for the 7α-oxidation from TDCA to TCA and TLCA to TCDCA (Honda et al., 2020). Thus, the species differences of BA metabolism between murine and the others observed in our study may also indicate that CDCA metabolism appears to be superior to CA metabolism in murine animals. We therefore investigated the oxidation metabolism of the downstream metabolites of CDCA, including CDCA, UDCA, LCA, HDCA, and MDCA (50 μM), in liver microsomes of murine animals in contrast to the other species. In this way, a series of murine-specific metabolic reactions were identified. As shown in Fig. 6, these reactions revealed not only the known 6β-oxidation from CDCA to α-MCA and UDCA to β-MCA but also the 6β-oxidation from LCA to MDCA, the 7α-oxidation from MDCA to α-MCA, and the rat-specific 7β-oxidation from MDCA to β-MCA and HDCA to β-HCA. Theses murine-specific oxidation activities were much higher than their activities toward DCA oxidations (Fig. 2). The 7α-oxidation from DCA to CA and LCA to CDCA was not detected, which is consistent with conjugated BAs being much better substrates for Cyp2a12 than unconjugated BAs in mice (Honda et al., 2020).
Fig. 6.

Metabolite formation after incubation of LCA, CDCA, UDCA, HDCA, and MDCA (50 μM) in the liver microsomes (protein level 0.5 mg/ml) of humans, monkeys, dogs, rats, and mice for 60 minutes. Data are shown as mean ± S.D. (n = 3) in pmol/min/mg protein.
Discussion
By using BA-profiling technique in combination with the in vitro metabolism assays, this work has confirmed that tertiary oxidation of DCA is a conserved pathway in humans, monkeys, dogs, rats, and mice. The BA-profiling data between SPF mice and germ-free mice have provided preliminary in vivo evidence that the host liver is responsible for the tertiary metabolism of DCA. Future works addressing dispositions of oral DCA in germ-free mice may provide direct evidence to identify how the host eliminates DCA without colonization of gut microbiota. Species differences in the oxidation regioselectivity of DCA were shown by the apparent oxidation kinetics of DCA in the pooled liver microsomes of animals. Most of the in vitro kinetic data were consistent with the BA-profiling data, in which all conjugated forms (N-amidated forms, sulfated forms, and glucuronidated forms) were detected as unconjugated forms by using the enzyme digestion techniques in the present work. Some in vitro–in vivo data inconsistency may be associated with the N-acylamidation metabolism of BAs, which has been shown to have an impact on the activity and regioselectivity of DCA oxidation in humans (Zhang et al., 2019). Deeper understanding of the species differences relies on development of the next-generation BA-profiling techniques, including not only N-amidated forms but also sulfated forms and glucuronidated forms of BAs.
Our previous work has demonstrated that renal clearance of the tertiary metabolites of DCA is significantly higher than that of DCA and CA, indicating that tertiary BA metabolism facilitates, at least in part, the renal excretion of DCA (Zhang et al., 2019). Similarly, 12-epiDCA, an epimerized microbial metabolite of DCA, was also inclined to be excreted in urine, in which it was detected mainly as amidated forms (Zhu et al., 2018). In this work, a total of nine downstream metabolites of DCA were detected in human urine and feces, including three microbial metabolites (isoDCA, 12-epiDCA, and 12-oxoLCA) and six tertiary metabolites (3-dehydroDCA, DCA-6β-ol, DCA-5β-ol, DCA-6α-ol, DCA-1β-ol, and DCA-4β-ol). Two microbial metabolites of DCA, 12-epiDCA and 12-oxoLCA, were detected in both urine and feces, whereas isoDCA was only found in feces. Consistent with our previous data, all these tertiary metabolites of DCA were detected in urine except for 3-dehydroDCA, which was detected with much higher level in feces than the other tertiary metabolites. Nevertheless, the microbial metabolites of DCA had higher levels than the tertiary metabolites in urine and particularly feces. Therefore, DCA appeared to be disposed mainly by microbial metabolism and fecal excretion and, complementally, by hepatic tertiary metabolism and renal excretion in humans. As illustrated in Fig. 2, the disposition pathways of DCA are relatively conserved in the tested animals, which might have explained why a slight decrease of TDCA was observed without significance in the liver and gallbladder of Cyp3a knockout (KO) mice compared with the wild-type littermate controls (Wahlstrom et al., 2017).
This work also has provided deeper understanding of how and why the species differences of BA metabolism between murine and the other animals occurs more in the downstream metabolism of CDCA than that of CA. As shown in Fig. 2, CA metabolism prevails over CDCA metabolism in humans and dogs, whereas CDCA metabolism is significantly enhanced in murine animals, particularly in rats. The underlying mechanism is clearly shown by the murine animals having strong 6β-hydroxylation activities toward the downstream metabolites of CDCA. In liver microsomes of all tested animals, the oxidation activities toward DCA (Fig. 3) are far below the murine-specific oxidation activities toward CDCA, UDCA, and LCA (Fig. 6). Therefore, the 6β-oxidation of CDCA and UDCA constitute the major pathway to synthesis of α-MCA and β-MCA. Moreover, murine animals also have a specific tertiary pathway for the 6β-oxidation from LCA to MDCA and the 7α- and 7β-oxidation from MDCA to α-MCA and β-MCA, which furnish an alternative pathway to synthesize α-MCA and β-MCA. In these ways, CDCA, UDCA, and LCA in murine animals are almost completely transformed into the more polar 6β-hydroxylated BAs, α-MCA, β-MCA, and MDCA. In contrast, because of the lack of 6β-oxidation activities, humans and dogs have evolved the BA metabolism patterns dominated by the downstream metabolism of CA and DCA.
However, the host-responsible in vitro reactions carried out in this work did not well explain the murine-specific synthesis of β-HCA. Although rat liver microsomes were able to oxidize HDCA at C-7β into β-HCA, both rats and mice had limited activities toward the 6α-oxidation from LCA to HDCA. A recent study also found no significant differences of β-HCA and TβHCA in Cyp3a KO mice compared with the wild-type controls (Wahlstrom et al., 2017). Therefore, we deduced that tertiary metabolism of LCA contributes less to the production of HDCA and β-HCA compared with the microbial modifications of α-MCA and β-MCA (Eyssen et al., 1999). Our deduction was consistent with that from a recent study of BA metabolism in Cyp2a12 KO, Cyp2c70 KO, and Cyp2a12/Cyp2c70 double-KO mice (Honda et al., 2020). Future studies are required to characterize the microbial 6-HSDHs that are potentially responsible for the 6-epimerization reactions between β-MCA and β-HCA in murine animals.
In conclusion, this work demonstrates that the tertiary oxidation of DCA is a conserved BA metabolism pathway in preclinical animals. The orthologous genes of human CYP3A are believed to be responsible for the tertiary oxidation of DCA in animals. In the tested animals, the majority of DCA synthesized by microbes in lower gut is disposed by microbial metabolism and undergoes fecal excretion, whereas the tertiary metabolism of DCA might have physiologically constituted a complementary disposition mechanism of DCA. The conclusion was consistent with Cyp3a KO not significantly altering the major BA composition in mice (Wahlstrom et al., 2017). Compared with humans and dogs, murine animals favored the downstream metabolism of CDCA, mainly because of the presence of the strong hepatic 6β-oxidation activities toward CDCA, UDCA, LCA, and 7-oxidation activities toward MDCA and HDCA, the 6-hydroxylated metabolites of LCA. The murine-specific 6β-hydroxylase was identified as CYP2C22 in rats and Cyp2c70 in mice (Takahashi et al., 2016). The mouse-specific 7α-hydroxylase responsible for hydroxylating TDCA into TCA and TLCA into TCDCA was identified as Cyp2a12 (Honda et al., 2020). These P450s are believed to produce a murine-specific hydrophilic BA composition, which partly explains why murine animals are relatively resistant to hepatobiliary toxicities caused by accumulation of hydrophobic BAs. Cyp2c70 KO, Cyp2a12 KO, and Cyp2c70/Cyp2a12 double-KO mice have been characterized to produce more human-like hydrophobic BA composition (de Boer et al., 2020; Honda et al., 2020). Although the tertiary BAs were not studied in these works, the expression and activity of Cyp3a11 were found to be complementarily induced in Cyp2c70/Cyp2a12 double-KO mice via activation of pregnane X receptor and/or constitutive androstane receptor. Collectively based on this evidence, the genetic modified murine animal models that knock out murine-specific BA metabolizing P450 genes are believed to be promising interspecies scaling tools for future studies of BA associated signaling, metabolism, drug metabolism, and toxicology.
Acknowledgments
We are grateful to Bing Liu and Lian Yang (West China-Frontier PharmaTech) for the gift of animal urine and fecal samples.
Abbreviations
- BA
bile acid
- CA
cholic acid
- CDCA
chenodeoxycholic acid
- DCA
deoxycholic acid
- GDCA
glycodeoxycholic acid
- HCA
hyocholic acid
- HDCA
hyodeoxycholic acid
- HSDH
hydroxysteroid dehydrogenase
- KO
knockout
- LCA
lithocholic acid
- LC-MS/MS
liquid chromatography with tandem mass spectrometry
- MCA
muricholic acid
- MDCA
murideoxycholic acid
- SPF
specific-pathogen-free
- TDCA
taurodeoxycholic acid
- UCA
ursocholic acid
- UDCA
ursodeoxycholic acid
Authorship Contributions
Participated in research design: Lan.
Conducted experiments: Lin, Tan, Wang, Zeng, Gui, Su, Lan.
Contributed metabolite synthesis: Xu.
Performed data analysis: Lin, Tan, Lan.
Wrote or contributed to the writing of the manuscript: Lin, Tan, Su, Liu, Jia, Lan.
Footnotes
This work was supported partly by the Fundamental Research Funds for the Central Universities and the 111 Project of the National Ministry of Education (B18035).
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Araya Z, Wikvall K (1999) 6α-hydroxylation of taurochenodeoxycholic acid and lithocholic acid by CYP3A4 in human liver microsomes. Biochim Biophys Acta 1438:47–54. [DOI] [PubMed] [Google Scholar]
- Batta AK, Salen G, Arora R, Shefer S, Batta M, Person A (1990) Side chain conjugation prevents bacterial 7-dehydroxylation of bile acids. J Biol Chem 265:10925–10928. [PubMed] [Google Scholar]
- Bergström S, Danielsson H, Göransson A (1959) On the bile acid metabolism in the pig. Bile acids and steroids 81. Acta Chem Scand 13:776–783. [Google Scholar]
- Bodin K, Lindbom U, Diczfalusy U (2005) Novel pathways of bile acid metabolism involving CYP3A4. Biochim Biophys Acta 1687:84–93. [DOI] [PubMed] [Google Scholar]
- Buffie CG, Bucci V, Stein RR, McKenney PT, Ling L, Gobourne A, No D, Liu H, Kinnebrew M, Viale A, et al. (2015) Precision microbiome reconstitution restores bile acid mediated resistance to Clostridium difficile. Nature 517:205–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen M, Borlak J, Tong W (2014) Predicting idiosyncratic drug-induced liver injury: some recent advances. Expert Rev Gastroenterol Hepatol 8:721–723. [DOI] [PubMed] [Google Scholar]
- Chen YJ, Zhang J, Zhu PP, Tan XW, Lin QH, Wang WX, Yin SS, Gao LZ, Su MM, Liu CX, et al. (2019) Stereoselective oxidation kinetics of deoxycholate in recombinant and microsomal CYP3A enzymes: deoxycholate 19-hydroxylation is an in vitro marker of CYP3A7 activity. Drug Metab Dispos 47:574–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson PA, Karpen SJ (2015) Intestinal transport and metabolism of bile acids. J Lipid Res 56:1085–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Aguiar Vallim TQ, Tarling EJ, Edwards PA (2013) Pleiotropic roles of bile acids in metabolism. Cell Metab 17:657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Boer JF, Verkade E, Mulder NL, de Vries HD, Huijkman NCA, Koehorst M, Boer T, Wolters JC, Bloks VW, van de Sluis B, et al. (2020) A human-like bile acid pool induced by deletion of hepatic Cyp2c70 modulates effects of FXR activation in mice. J Lipid Res 61:291–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deo AK, Bandiera SM (2008) Identification of human hepatic cytochrome p450 enzymes involved in the biotransformation of cholic and chenodeoxycholic acid. Drug Metab Dispos 36:1983–1991. [DOI] [PubMed] [Google Scholar]
- Deo AK, Bandiera SM (2009) 3-ketocholanoic acid is the major in vitro human hepatic microsomal metabolite of lithocholic acid. Drug Metab Dispos 37:1938–1947. [DOI] [PubMed] [Google Scholar]
- Eyssen HJ, De Pauw G, Van Eldere J (1999) Formation of hyodeoxycholic acid from muricholic acid and hyocholic acid by an unidentified gram-positive rod termed HDCA-1 isolated from rat intestinal microflora. Appl Environ Microbiol 65:3158–3163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gopal-Srivastava R, Hylemon PB (1988) Purification and characterization of bile salt hydrolase from Clostridium perfringens. J Lipid Res 29:1079–1085. [PubMed] [Google Scholar]
- Gustafsson J, Andersson S, Sjövall J (1985) Bile acid metabolism during development: metabolism of taurodeoxycholic acid in human fetal liver. Biol Neonate 47:26–31. [DOI] [PubMed] [Google Scholar]
- Halilbasic E, Claudel T, Trauner M (2013) Bile acid transporters and regulatory nuclear receptors in the liver and beyond. J Hepatol 58:155–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslewood GA (1954) “Hyocholic acid”, a trihydroxy bile acid from pig bile. Biochem J 56 (325th Meeting):xxxviii. [PubMed] [Google Scholar]
- Hayes MA, Li XQ, Grönberg G, Diczfalusy U, Andersson TB (2016) CYP3A specifically catalyzes 1β-hydroxylation of deoxycholic acid: characterization and enzymatic synthesis of a potential novel urinary biomarker for CYP3A activity. Drug Metab Dispos 44:1480–1489. [DOI] [PubMed] [Google Scholar]
- Hirano S, Nakama R, Tamaki M, Masuda N, Oda H (1981) Isolation and characterization of thirteen intestinal microorganisms capable of 7 alpha-dehydroxylating bile acids. Appl Environ Microbiol 41:737–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF, Hagey LR, Krasowski MD (2010) Bile salts of vertebrates: structural variation and possible evolutionary significance. J Lipid Res 51:226–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann AF, Sjövall J, Kurz G, Radominska A, Schteingart CD, Tint GS, Vlahcevic ZR, Setchell KD (1992) A proposed nomenclature for bile acids. J Lipid Res 33:599–604. [PubMed] [Google Scholar]
- Honda A, Miyazaki T, Iwamoto J, Hirayama T, Morishita Y, Monma T, Ueda H, Mizuno S, Sugiyama F, Takahashi S, et al. (2020) Regulation of bile acid metabolism in mouse models with hydrophobic bile acid composition. J Lipid Res 61:54–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsla SL, Matschiner JT, Mahowald TA, Elliott WH, Doisy EA Jr, Thayer SA, Doisy EA (1957) Bile acids. VI. The structure and synthesis of acid II. J Biol Chem 226:667–671. [PubMed] [Google Scholar]
- Huijghebaert SM, Hofmann AF (1986) Influence of the amino acid moiety on deconjugation of bile acid amidates by cholylglycine hydrolase or human fecal cultures. J Lipid Res 27:742–752. [PubMed] [Google Scholar]
- Inagaki T, Moschetta A, Lee YK, Peng L, Zhao G, Downes M, Yu RT, Shelton JM, Richardson JA, Repa JJ, et al. (2006) Regulation of antibacterial defense in the small intestine by the nuclear bile acid receptor. Proc Natl Acad Sci USA 103:3920–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, Fukusumi S, Habata Y, Itoh T, Shintani Y, et al. (2003) A G protein-coupled receptor responsive to bile acids. J Biol Chem 278:9435–9440. [DOI] [PubMed] [Google Scholar]
- Lundell K (2004) The porcine taurochenodeoxycholic acid 6alpha-hydroxylase (CYP4A21) gene: evolution by gene duplication and gene conversion. Biochem J 378:1053–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lundell K, Hansson R, Wikvall K (2001) Cloning and expression of a pig liver taurochenodeoxycholic acid 6alpha-hydroxylase (CYP4A21): a novel member of the CYP4A subfamily. J Biol Chem 276:9606–9612. [DOI] [PubMed] [Google Scholar]
- MacDonald IA, Mahony DE, Jellet JF, Meier CE (1977) NAD-dependent 3alpha- and 12alpha-hydroxysteroid dehydrogenase activities from Eubacterium lentum ATCC no. 25559. Biochim Biophys Acta 489:466–476. [DOI] [PubMed] [Google Scholar]
- Macdonald IA, Meier EC, Mahony DE, Costain GA (1976) 3alpha-, 7alpha- and 12alpha-hydroxysteroid dehydrogenase activities from Clostridium perfringens. Biochim Biophys Acta 450:142–153. [DOI] [PubMed] [Google Scholar]
- Madsen DC, Chang L, Wostmann B (1975) Omega-muricholate: a tertiary bile acid of the Wistar rat. Proc Indiana Acad Sci 84:416–420. [Google Scholar]
- Mahowald TA, Matschiner JT, Hsia SL, Doisy EA Jr, Elliott WH, Doisy EA (1957a) Bile acids. III. Acid I; the principal bile acid in urine of surgically jaundiced rats. J Biol Chem 225:795–802. [PubMed] [Google Scholar]
- Mahowald TA, Matschiner JT, Hsia SL, Richter R, Doisy EA Jr, Elliott WH, Doisy EA (1957b) Bile acids. II. Metabolism of deoxycholic acid-24-C14 and chenodeoxycholic acid-24-C14 in the rat. J Biol Chem 225:781–793. [PubMed] [Google Scholar]
- Maruyama T, Miyamoto Y, Nakamura T, Tamai Y, Okada H, Sugiyama E, Nakamura T, Itadani H, Tanaka K (2002) Identification of membrane-type receptor for bile acids (M-BAR). Biochem Biophys Res Commun 298:714–719. [DOI] [PubMed] [Google Scholar]
- Matschiner JT, Mahowald TA, Hsia SL, Doisy EA Jr, Elliott WH, Doisy EA (1957) Bile acids. IV. The metabolism of hyodeoxycholic acid-24-C14. J Biol Chem 225:803–810. [PubMed] [Google Scholar]
- Matschiner JT, Ratliff RL, Mahowald TA, Doisy EA Jr, Elliott WH, Hsia SL, Doisy EA (1958) Bile acids. IX. Metabolites of hyodeoxycholic acid-24-C14 in surgically jaundiced rats. J Biol Chem 230:589–596. [PubMed] [Google Scholar]
- Ratliff RL, Matschiner JT, Doisy EA Jr, Hsia SL, Thayer SA, Elliott WH, Doisy EA (1961) Bile acids. XV. Partial synthesis of the new metabolite of deoxycholic acid, 3 alpha,6 beta,12 alpha-trihydroxycholanic acid. J Biol Chem 236:685–687. [PubMed] [Google Scholar]
- Ridlon JM, Harris SC, Bhowmik S, Kang DJ, Hylemon PB (2016) Consequences of bile salt biotransformations by intestinal bacteria. Gut Microbes 7:22–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridlon JM, Kang DJ, Hylemon PB (2006) Bile salt biotransformations by human intestinal bacteria. J Lipid Res 47:241–259. [DOI] [PubMed] [Google Scholar]
- Russell DW (2003) The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 72:137–174. [DOI] [PubMed] [Google Scholar]
- Sayin SI, Wahlström A, Felin J, Jäntti S, Marschall HU, Bamberg K, Angelin B, Hyötyläinen T, Orešič M, Bäckhed F (2013) Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell Metab 17:225–235. [DOI] [PubMed] [Google Scholar]
- Sjövall J, Griffiths WJ, Setchell KD, Mano N, Goto J (2010) Analysis of bile acids, in Steroid Analysis (Makin HL and Gower D eds) pp 837–966, Springer, Dordrecht, the Netherlands. [Google Scholar]
- Stellwag EJ, Hylemon PB (1979) 7alpha-Dehydroxylation of cholic acid and chenodeoxycholic acid by Clostridium leptum. J Lipid Res 20:325–333. [PubMed] [Google Scholar]
- Takahashi S, Fukami T, Masuo Y, Brocker CN, Xie C, Krausz KW, Wolf CR, Henderson CJ, Gonzalez FJ (2016) Cyp2c70 is responsible for the species difference in bile acid metabolism between mice and humans. J Lipid Res 57:2130–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thakare R, Alamoudi JA, Gautam N, Rodrigues AD, Alnouti Y (2018a) Species differences in bile acids I. Plasma and urine bile acid composition. J Appl Toxicol 38:1323–1335. [DOI] [PubMed] [Google Scholar]
- Thakare R, Alamoudi JA, Gautam N, Rodrigues AD, Alnouti Y (2018b) Species differences in bile acids II. Bile acid metabolism. J Appl Toxicol 38:1336–1352. [DOI] [PubMed] [Google Scholar]
- Theriot CM, Koenigsknecht MJ, Carlson PE Jr, Hatton GE, Nelson AM, Li B, Huffnagle GB, Z Li J, Young VB (2014) Antibiotic-induced shifts in the mouse gut microbiome and metabolome increase susceptibility to Clostridium difficile infection. Nat Commun 5:3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas PJ, Hsia SL, Matschiner JT, Doisy EA Jr, Elliott WH, Thayer SA, Doisy EA (1964) Bile acids. Xix. Metabolism of lithocholic acid-24-14c in the rat. J Biol Chem 239:102–105. [PubMed] [Google Scholar]
- Thomas PJ, Hsia SL, Matschiner JT, Thayer SA, Elliott WH, Doisy EA Jr, Doisy EA (1965) Bile acids. Xxi. Metabolism of 3-alpha,6-beta-dihydroxy-5-beta-cholanoic acid-24-C 14-6-alpha-H3 in the rat. J Biol Chem 240:1059–1063. [PubMed] [Google Scholar]
- Trülzsch D, Roboz J, Greim H, Czygan P, Rudick J, Hutterer F, Schaffner F, Popper H (1974) Hydroxylation of taurolithocholate by isolated human liver microsomes. I. Identification of metabolic product. Biochem Med 9:158–166. [DOI] [PubMed] [Google Scholar]
- Vavassori P, Mencarelli A, Renga B, Distrutti E, Fiorucci S (2009) The bile acid receptor FXR is a modulator of intestinal innate immunity. J Immunol 183:6251–6261. [DOI] [PubMed] [Google Scholar]
- Wahlström A, Al-Dury S, Ståhlman M, Bäckhed F, Marschall HU (2017) Cyp3a11 is not essential for the formation of murine bile acids. Biochem Biophys Rep 10:70–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wahlström A, Sayin SI, Marschall HU, Bäckhed F (2016) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24:41–50. [DOI] [PubMed] [Google Scholar]
- Yin S, Su M, Xie G, Li X, Wei R, Liu C, Lan K, Jia W (2017) Factors affecting separation and detection of bile acids by liquid chromatography coupled with mass spectrometry in negative mode. Anal Bioanal Chem 409:5533–5545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Gao LZ, Chen YJ, Zhu PP, Yin SS, Su MM, Ni Y, Miao J, Wu WL, Chen H, et al. (2019) Continuum of host-gut microbial Co-metabolism: host CYP3A4/3A7 are responsible for tertiary oxidations of deoxycholate species. Drug Metab Dispos 47:283–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Limaye PB, Lehman-McKeeman LD, Klaassen CD (2012) Dysfunction of organic anion transporting polypeptide 1a1 alters intestinal bacteria and bile acid metabolism in mice. PLoS One 7:e34522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu P, Zhang J, Chen Y, Yin S, Su M, Xie G, Brouwer KLR, Liu C, Lan K, Jia W (2018) Analysis of human C24 bile acids metabolome in serum and urine based on enzyme digestion of conjugated bile acids and LC-MS determination of unconjugated bile acids. Anal Bioanal Chem 410:5287–5300. [DOI] [PMC free article] [PubMed] [Google Scholar]