

Abstract
Fibroblast growth factors 15 (FGF15) and 19 (FGF19) are endocrine growth factors that play an important role in maintaining bile acid homeostasis. FGF15/19-based therapies are currently being tested in clinical trials for the treatment of nonalcoholic steatohepatitis and cholestatic liver diseases. To determine the physiologic impact of long-term elevations of FGF15/19, a transgenic mouse model with overexpression of Fgf15 (Fgf15 Tg) was used in the current study. The RNA sequencing (RNA-seq) analysis revealed elevations of the expression of several genes encoding phase I drug metabolizing enzymes (DMEs), including Cyp2b10 and Cyp3a11, in Fgf15 Tg mice. We found that the induction of several Cyp2b isoforms resulted in increased function of CYP2B in microsomal metabolism and pharmacokinetics studies. Because the CYP2B family is known to be induced by constitutive androstane receptor (CAR), to determine the role of CAR in the observed inductions, we crossed Fgf15 Tg mice with CAR knockout mice and found that CAR played a minor role in the observed alterations in DME expression. Interestingly, we found that the overexpression of Fgf15 in male mice resulted in a phenotypical switch from the male hepatic expression pattern of DMEs to that of female mice. Differences in secretion of growth hormone (GH) between male and female mice are known to drive sexually dimorphic, STAT5b-dependent expression patterns of hepatic genes. We found that male Fgf15 Tg mice presented with many features similar to GH deficiency, including lowered body length and weight, Igf-1 and Igfals expression, and STAT5 signaling.
SIGNIFICANCE STATEMENT
The overexpression of Fgf15 in mice causes an alteration in DMEs at the mRNA, protein, and functional levels, which is not entirely due to CAR activation but associated with lower GH signaling.
Introduction
Drug metabolism is the process by which exogenous compounds undergo biotransformation to facilitate their removal from the body. The process of drug metabolism is described in three phases: functional conversion, conjugation, and transport/excretion. The transcriptional regulation of genes encoding hepatic drug metabolizing enzymes and transporters (DMETs) is a critical mechanism capable of responding to various challenges during development, exposure to xenobiotics, and alterations in physiology and pathology.
The liver is the primary site of drug metabolism. The efficiency or rate at which the liver is able to process xenobiotic biotransformation is dependent upon the relative abundance of DMETs. To regulate the expression of DMETs, the liver is capable of responding to xenobiotic exposure through the activation of xenobiotic-sensing nuclear receptors (NRs), such as the constitutive androstane receptor (CAR, NR1I3), pregnane X receptor, glucocorticoid receptor, and vitamin D receptor.
CAR is a well-studied xenobiotic sensing NR in the regulation of DMET gene expression. Under normal conditions, CAR is bound to the cytosol by a complex of heat-shock protein 90 and CAR cytoplasmic retention protein. Xenobiotics can activate CAR by both direct ligand binding and indirect protein modification mechanisms, causing dissociation from its chaperone proteins, followed by translocation to the nucleus (Mackowiak and Wang, 2016). Additionally, CAR is known to be inactivated by the androstane metabolites, androstenol and androstanol, which act as inverse agonists (Kobayashi et al., 2015). Following translocation to the nucleus, CAR forms a heterodimer with the retinoid X receptor, binds to its response elements, and recruits coactivators SRC1 and GRIP1 to regulate target gene transcription (Mackowiak and Wang, 2016). CAR activation results in the differential regulation of over 2000 genes in mice and has overlap with pregnane X receptor and farnesoid X receptor (FXR, NR1H4) signaling (Cui and Klaassen, 2016). CAR activation is well known for mediating robust inductions of CYP2B6 in humans and Cyp2b10 in mice.
FXR is an NR that serves as a master regulator in bile acid (BA) homeostasis. BAs are amphipathic molecules that aid in the digestion and absorption of lipids and lipid-soluble vitamins in the small intestine. BAs are reabsorbed in the distal small bowel where they activate FXR to initiate a negative feedback mechanism, controlling their own synthesis (Makishima et al., 1999). Activation of intestinal FXR by BAs results in a strong induction of fibroblast growth factor 15 (FGF15) in mice and FGF19 in humans. FGF15/19 act as endocrine molecules, traveling to the liver where they interact with fibroblast growth factor receptor 4 (FGFR4) on hepatocytes. Activation of FGFR4 leads to the activation of the mitogen-activated protein kinases signaling pathway, including extracellular signal regulated kinase (ERK) and c-Jun N-terminal kinase, to reduce BA production by suppressing the expression of genes involved in BA synthesis, including CYP7A1/Cyp7a1 and CYP8B1/Cyp8b1 (Kong et al., 2012).
Many liver diseases are initiated and/or worsened by BA dysregulation, including cholestasis, nonalcoholic fatty liver diseases, and liver tumors (hepatocellular carcinoma and cholangiocarcinoma). Nonalcoholic steatohepatitis (NASH) is within the more severe spectrum of nonalcoholic fatty liver diseases, which includes liver steatosis and inflammation. Dysregulations of many of the metabolic pathways governed by FGF15/19 have been found to be sequelae of NASH. NASH is rapidly becoming a major health issue affecting approximately 3–12% of the US population (Spengler and Loomba, 2015) and is projected to overtake hepatitis C virus as the leading indication of liver transplant in the United States (Noureddin et al., 2018). Currently there are no approved drug therapies for the treatment of NASH but synthetic FXR ligands and modified FGF19 proteins are in clinical trials for the treatment of NASH and cholestatic liver diseases.
As pharmaceutical companies continue to investigate the potential of FXR agonism and FGF19 therapies for disease intervention, it is important to understand the long-term effects of these therapies on liver function. The impact of sustained elevations of plasma FGF15/19 protein on xenobiotic metabolism is unknown. Alterations to DMETs can lead to drug-drug interactions, in which a perpetrator drug alters the disposition and/or action of a victim drug when taken in combination. Drug-drug interactions can lead to toxicity or the loss of efficacy in patients; therefore it is important to properly evaluate the potential of new therapies to alter DMETs. In the current study, using the Fgf15 transgenic mice (Fgf15 Tg) and intestine-specific Fgf15 knockout mice (Fgf15int−/−), as well as in vitro–synthesized FGF19 protein, we have determined the effect of modulating FGF15/19 levels on the hepatic expression and function of DMET genes. Additionally, using in vitro primary human hepatocytes (PHH) and in vivo CAR knockout mice (CAR−/−), we have examined the role of CAR in mediating the alterations of gene expression following FGF15/FGF19 overexpression.
Materials and Methods
Animals and Treatment
Male 8–12-week-old wild type (WT), Fgf15 Tg, and Fgf15int−/− mice, all on C57BL/6J genetic background, were used (n = 3–5). The generation of Fgf15int−/− mice is detailed in Supplemental Fig. 4 and Supplemental Doc. 1. Car−/− mice on a mixed genetic background were generously gifted by Dr. Wen Xie from the University of Pittsburgh (Saini et al., 2004). F1 heterozygotes mice were obtained by crossing the Car−/− mice with Fgf15 Tg mice, then intercross breeding of F1 heterozygotes was adapted to obtain genetic background matched WT, Car−/−, Fgf15 Tg, and Car−/−/Fgf15 Tg mice. Male WT, Car−/−, Fgf15 Tg, and Car−/−/Fgf15 were 12–16 weeks of age at time of necropsy. All animal experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee (IACUC) at Rutgers University. For positive controls of Cyp2b10 and Cyp3a11 induction in the liver, WT mice were treated with an intraperitoneal injection of phenobarbital (PB, 50 mg/kg) for 3 days, and the control group was treated with PBS. All mice were euthanized for tissue collection between 10:00–11:00 AM without fasting. Methods for serum biochemical assays and liver histopathological examination have been described previously (Schumacher et al., 2020). Additional animal information can be found in Supplemental Fig. 1.
In Vitro Treatment with Recombinant FGF19 Protein
HepaRG cells were cultured as previously described (Hart et al., 2010; Pande et al., 2020). PHHs were from a cryopreserved pool (5 donors) of human hepatocytes (PHH8007A, IVAL, Columbia, MD). PHHs were plated into 6- or 24-well collagen-coated plates at a density of 0.7 × 106 cells/ml and allowed to attach for 4 hours. Following attachment, plating medium was removed and the cells were changed to hepatocyte induction medium (HIM, IVAL, Columbia, MD). The plate was cultured in an incubator maintained with a humidified atmosphere of 95% air and 5% CO2. HepaRG and PHH were treated with recombinant FGF19 (Kong and Guo, 2014) at concentrations of 5 or 50 ng/ml for 24 and 48 hours. Recombinant FGF19 was diluted in PBS and PBS was used as the vehicle control.
RNA-seq Analysis
Total liver RNA from WT, Fgf15 Tg, or Fgf15int−/− male mice (n = 3 per group) was extracted from frozen tissue by TRIzol method (Thermo Fisher Scientific, Waltham, MA). Whole-transcriptome cDNA libraries were prepared by NuGEN Mondrian system. Paired-end sequencing was performed using the Illumina HiSEq 2000 platform (Illumina, Inc., San Diego, CA). Alignment and read quantification were performed for all samples and FPKM values for each liver sample were calculated based on the method for RNA-seq data analysis described previously (Peng et al., 2012).
Gene Expression
Relative gene expression was determined as previously described (Rizzolo et al., 2019). Briefly, total RNA was extracted from frozen liver or ileum tissue using TRIzol reagent. Reverse transcription was performed to acquire cDNA. Real-time quantitative polymerase chain reaction was performed on the ViiA7 Real-Time PCR System (Life Technologies, Grand Island, NY) using SYBR green to determine relative gene expression. Ct values were converted to delta delta Ct values and normalized to β-actin. Primer sequences used in this study can be found in Supplementary Fig. 1.
Western Blot Analysis
Livers were homogenized and lysed in 1X radioimmunoprecipitation assay buffer with protease and phosphatase inhibitors (Thermo Fisher Scientific, Waltham, MA). Proteins (20 µg per well) were separated on a 10% sodium dodecyl sulfate-polyacrylamide gel and transferred to a polyvinylidene difluoride membrane. The blots were blocked for 2-hours at room temperature with 5% nonfat milk, incubated with primary antibody overnight at 4°C, incubated with species-specific secondary antibody for 1 hour at room temperature, and visualized using enhanced chemiluminescence substrates (Thermo Fisher Scientific, Waltham, MA). A list of antibodies used in this study can be found in Supplemental Fig. 1. Protein loading was normalized to levels of GAPDH or β-actin.
CYP2B and CYP3A Enzyme Activity
Liver microsomes were isolated from liver tissue, as previously described (Tien et al., 2015). Pentoxyresorufin and midazolam were used as probe substrates for the reactions pentoxyresorufin O-dealkylation and midazolam 1’-hydroxylation, which were used to detect the enzymatic activities of CYP2B and CYP3A, respectively. In brief, the incubation of 50 μg of mouse liver microsomes was carried out in 1x phosphate-buffered saline (pH 7.4) with 30 μM of substrate concentration in a total volume of 95 μL. The reactions were initiated with 5 μL of 20 mM NADPH. Reactions containing pentoxyresorufin were carried out for 30 minutes and reactions containing midazolam were carried out for 10 minutes. All reactions were terminated by the addition of 100 μL of ice-cold acetonitrile. The samples were vortexed for 30 seconds, centrifuged at 15,000 rpm for 10 minutes, and 1.0 μL aliquots of the supernatant were injected into Waters Synapt G2-S QTOFMS system (Waters, Milford, MA) for metabolite analysis. Chromatographic separation of metabolites was performed on an Acquity UPLC BEH C18 column (2.1 × 100 mm, 1.7 µm, Waters). Details on the analytical methodology can be found in Supplemental Doc. 1.
LC-MS/MS-Based Protein Quantification
Mouse liver microsome samples were digested for LC-MS/MS-based proteomic analysis as previously described (Shi et al., 2018). 80 μg of protein from liver microsomes were mixed with 0.2 μg of bovine serum albumin (BSA) internal standard. A detailed description of sample preparation and digestion can be found in Supplemental Doc. 1. The digested samples were analyzed on a TripleTOF 5600+ mass spectrometer (AB Sciex, Farmingham, MA) coupled with an Eksigent 2D plus LC system (Ekseigent Technologies, Dublin, CA). LC separation was performed via a trap-elute configuration including a trapping column (ChromXP C18-CL, 120 Å, 5μm, 10 × 0.3 mm, Eksigent Technologies, Dublin, CA) and an analytical column (ChromXP C18-CL, 120 Å, 150 × 0.3 mm, 5μm, Eksigent Technologies, Dublin, CA). A detailed description of the analytical methodology can be found in Supplemental Doc. 1. The surrogate peptides used for quantification of CYP2B9, CYP2B10, CYP2B19, CYP3A11, CYP3A13, CYP3A16, and CYP3A25 are listed in Supplemental Table 1. These peptides were selected based on their uniqueness and chromatographic performance. The peak areas of the top 3 to 5 fragment ions were summed up and normalized to the internal standard BSA. The BSA-normalized peak area of each peptide was further divided by the average of the 18 samples to calculate the relative abundance of the peptide. The average of relative abundance of all surrogate peptides of a protein was used to determine the relative abundance across different microsome samples.
CAR Nuclear Translocation by FGF19
Adenovirus expressing enhanced yellow fluorescent protein-tagged hCAR (Ad/EYFP-hCAR) was used in PHHs (Bioreclamation In Vitro Technologies, Baltimore, MD). PHHs with over 90% viability were seeded at 0.25 × 106 cells/well in 24-well biocoated plates in INVITROGROCP Medium (Bioreclamation In Vitro Technologies, Baltimore, MD) and infected with Ad/EYFP-hCAR (6 µL/mL) as described previously (Li et al., 2009). Twenty-four hours after infection, PHHs were treated with vehicle control (0.1% DMSO), PB (1 mM), 6-(4-Chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde-O-(3,4-dichlorobenzyl)oxime (CITCO) (1 µM), or FGF19 (40 and 200 ng/mL) for 8 hours. EYFP-hCAR localization in hepatocytes was visualized on a Nikon Eclipse TI fluorescent microscope (Nikon, Melville, NY).
Body Composition CT Imaging
Male 5-month-old WT and Fgf15 Tg mice (n = 5/group) were imaged using an Albira PET/CT (Bruker, Billerica, MA) as previously described (Metzinger et al., 2014; Murray et al., 2020). In brief, anesthetized mice were scanned using the following settings: Good Low High scan setting (400 slices), field of view (120mm2) with tube current (200 μA) and voltage (45kV). After the scans were completed, they were reconstructed to a “Best” setting, which increased the total slices to 600. The images were then analyzed using VivoQuant 2.0 imaging software. Hounsfield unit (HU) histograms were produced using 1000 bins describing a range of -1000 to 4000 HU. HU distribution was divided into three regions: fat (-350 to -75 HU), muscle (-75 to 200 HU), and bone (200 to 1000 HU). HU for each region was summed, divided by 512 (image matrix), corrected for bin total (1000), and then multiplied by tissue density factors of 0.9, 1.0, and 1.9 (g/cm3) for fat, muscle, and bone, respectively. To determine percent represented by each tissue, the previously described determination of mass was divided by the animal’s body weight.
Pharmacokinetic Study
Male 4-month-old WT and Fgf15 Tg mice were injected intravenously with 1 mg/kg of bupropion (free base). Twenty μL of whole blood were collected with K3EDTA at 3, 30, 60, 120, 180, and 240 minutes. Samples were stored on ice and spun for 10 minutes at 10,000 g for plasma collection. 10 µL of mouse K3EDTA plasma was spiked with 10 µL of a mixture of internal standards (250 ng/mL HBUP-D6 and 1000 ng/mL BUP-D9 in MeOH) and 10 µL of 10% trichloroacetic acid. Samples were vortexed for 5 minutes and centrifuged for 5 minutes at 10°C with 15,700 g. The resulted supernatant was filtered through 0.2 µm PVDF filters at 10°C. The filtrate was then transferred to the HPLC vials with high-recovery inserts, and 10 µL were injected into the column. Bupropion and hydroxybupropion concentrations were determined by LC-MS/MS. A detailed description of the analytical methodology can be found in Supplemental Doc. 1. Pharmacokinetic parameters were calculated by noncompartmental analysis with intravenous bolus dosing using PK Solver 2.0.
Serum GH and IGF-1 ELISA Measurements
Blood samples were collected every 30 minutes from 9:30 AM to 2:30 PM from WT and Fgf15 Tg mice (male n = 5/genotype, female n = 3/genotype). In each collection, approximately 20 μL of blood was collected from the lateral tail vein at each designated time point. After collection, blood samples were allowed to coagulate at room temperature for 10 minutes, followed by centrifugation at 8000 g for 10 minutes to prepare for serum. The serum samples were transferred to a fresh tube and stored at -80°C until assay. Commercially available ELISA kits for growth hormone (GH) (EZRMGH-45K, Millipore-Sigma) and insulin-like growth factor-1 (IGF-1) (DY791, R&D Systems) were used to determine their levels in the serum. To reduce intersample dilution effects, serum samples were diluted identically 2 and 500 times before performing GH and IGF-1 ELISA assays, respectively. The sensitivities for the ELISA assays are 70 pg/ml and 3.5 pg/ml for GH and IGF-1, respectively.
Statistical Analysis
The data are presented as mean ± 1 S.D. Groups were compared using a one-way ANOVA followed by Tukey post-hoc unless otherwise noted. Data that failed assumptions of parametric statistics were run as Kruskal-Wallis. Statistical analysis was run using SAS Studio. Data were considered significant at P-values < 0.05.
Results
Transcriptomes in FGF15-Modified Mouse Livers
We have previously reported on the development of Fgf15 Tg mice (Kong et al., 2018). The enterocyte-specific Fgf15-deficient mice (Fgf15int−/−) were generated in our laboratory by the recombineering technology (Supplemental Fig. 4 and Supplemental Doc. 1). In Fig. 1A, quantitative polymerase chain reaction confirmed the overexpression of Fgf15 in both the liver and ileum. The protein levels of FGF15 in serum of Fgf15 Tg mice were markedly increased, while serum FGF15 levels in WT mice were undetectable as measured by a commercially available ELISA kit (data not shown). As expected, the overexpression of Fgf15 resulted in a significant reduction of Cyp7a1 mRNA levels. This reduction is a result of functional FGF15 binding to FGFR4 and not a result of hepatic FXR activation as these mice have significantly reduced levels of BAs (Kong et al., 2018) and there was no change in the hepatic expression of FXR or a classic FXR target gene, Shp (Nr0b2). To further understand transcriptional regulation affected by FGF15, we profiled the liver transcriptomes of WT, Fgf15 Tg and Fgf15int−/− mice by RNA-seq analysis. Consistent with previous findings, the RNA-seq analysis (Fig. 1B) showed Fgf15 Tg mice had reductions of the expression of hepatic genes involved in BA synthesis, such as Cyp7a1 and Cyp8b1. Fgf15int−/− mice displayed slight inductions of Cyp7a1 and Cyp8b1, likely due to reduced FGF15-FGFR4 binding. Among genes that were induced in Fgf15 Tg mice were critical genes involved in phase I drug metabolism, such as Cyp2b10, Cyp2b9, and Cyp3a11.
Fig. 1.
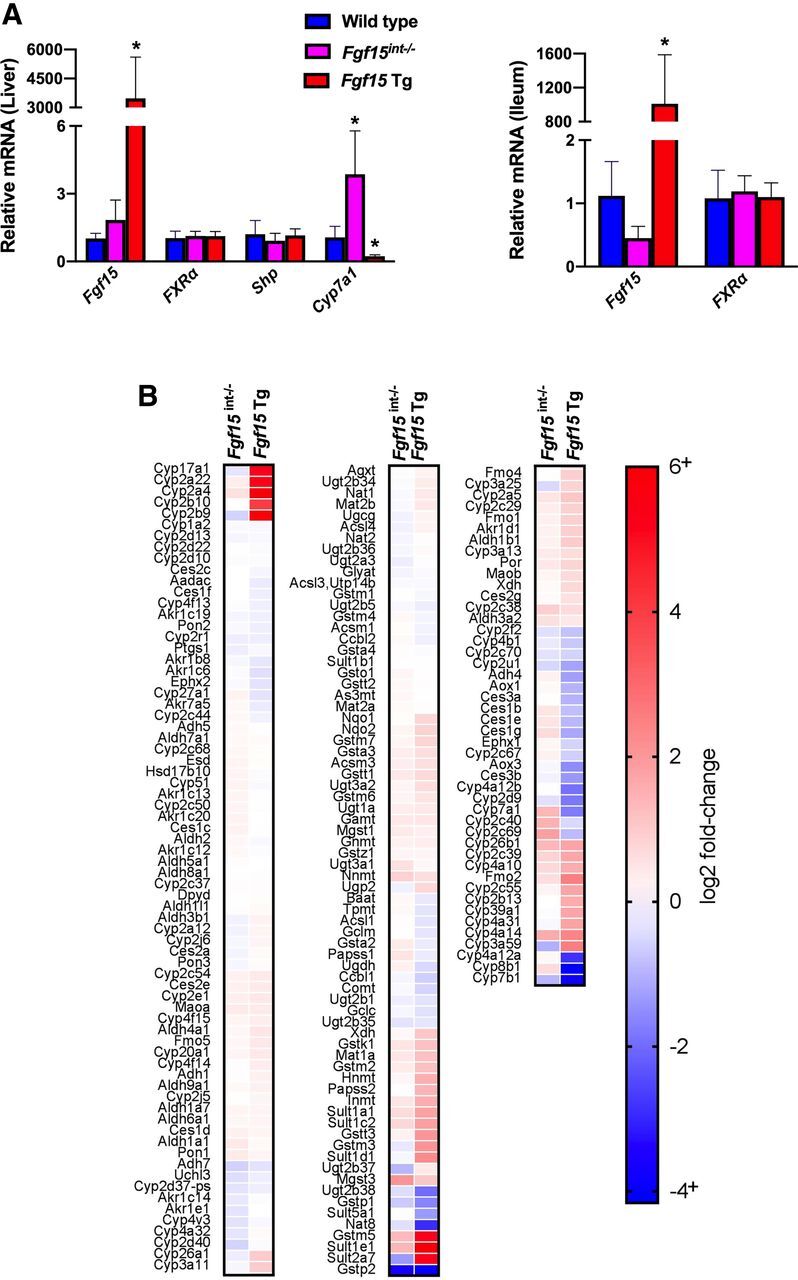
Relative hepatic and ileal mRNA values of Fgf15-related genes and RNA-seq analysis. (A) Hepatic and ileal gene expression was normalized to β-actin mRNA expression and graphs depict relative mRNA ± 1 S.D. (n: WT = 9, Fgf15int−/− = 6, Fgf15 Tg = 5; one-way ANOVA, Tukey post hoc). An asterisk denotes a significant difference from WT (P < 0.05). (B) Heat map illustrates logtwofold-change in RNAseq analysis as compared with WT (n = 3/group).
Gene and Protein Expression of Hepatic Drug Metabolizing Enzymes
RNA-seq analysis showed that the overexpression of Fgf15 led to differential expression patterns for several cytochrome P450 genes involved in drug metabolism. Fig. 2A shows the validation of the observed induction of Cyp2b9, Cyp2b10, Cyp3a11, and Cyp3a25 in Fgf15 Tg mice at mRNA levels by quantitative polymerase chain reaction. Compared with WT mice, the mRNA levels of Cyp2b9, Cyp2b10, Cyp3a11, and Cyp3a25 were all significantly increased in the livers of Fgf15 Tg mice, with no significant changes to those in the livers of Fgf15int−/− mice. To determine if the observed alterations at the mRNA level were manifested at the protein level, we determined the protein expression of CYP2B and CYP3A in these mice by western blots. Using antibodies targeting CYP2B and CYP3A, the results showed no change in the relative protein levels of CYP3A, an increase in CYP2B in Fgf15 Tg mice but not in Fgf15int−/− mice, compared with WT mice (Fig. 2B).
Fig. 2.

Relative hepatic mRNA, protein, and function of cytochrome P450s. (A) Relative hepatic mRNA expression normalized to β-actin mRNA expression (n: WT = 5, Fgf15int−/− = 6, Fgf15 Tg = 5; one-way ANOVA, Tukey post hoc). Graphs depict relative mRNA ± 1 S.D. An asterisk denotes a significant difference from WT (P < 0.05). Cyp2b9 and Cyp2b10 failed Levene’s test, therefore Kruskal-Wallis was used for analysis. (B) Western blot of CYP3A and CYP2B using 20μg of liver homogenate (n: WT = 4, Fgf15int−/− = 5, Fgf15 Tg = 5; one-way ANOVA, Tukey post hoc). (C) Liquid chromatograhy-mass spectrometry analysis of relative protein expression of CYP2B and CYP3A isoforms (n: WT = 5, Fgf15int−/− = 5, Fgf15 Tg = 5, WT-PB = 3; one-way ANOVA, Tukey post hoc). (D) Measurements of pentoxyresorufin and midazolam metabolite formation to assess CYP2B and CYP3A activity in liver microsomes of WT, Fgf15int−/−, Fgf15 Tg, and phenobarbital-treated WT mice (n: WT = 5, Fgf15int−/− = 5, Fgf15 Tg = 5, WT-PB = 3; one-way ANOVA, Tukey post hoc).
Determine the Protein Isoform Expression by LC-MS/MS-Based Proteomics
Because western blot analysis can be limited in its ability to differentiate between cytochrome P450 isoforms, we further determined the protein levels of individual CYP2B and CYP3A family members via LC-MS/MS. Fig. 2C shows the effects of FGF15 modulation on individual CYP2B and CYP3A isoforms in the liver. In male WT mice, the predominantly expressed CYP2B family members are CYP2B10 and CYP2B19, while CYP2B9 is undetectable as CYP2B9 is predominantly expressed in female mice. However, Fgf15 Tg male mice showed marked inductions of CYP2B9, as compared with WT. Fgf15int−/− mice generally had less CYP2B protein, regardless of isoform. Male WT mice had relatively equal amounts of CYP3A11, CYP3A13, and CYP3A25 while displaying no CYP3A16, which is generally found in neonates and female mice. Fgf15int−/− mice had minor reduction in CYP3A protein as compared with WT and also had no detectable CYP3A16. Fgf15 Tg mice had about a 50% reduction in CYP3A11 protein despite showing an induction in Cyp3a11 mRNA, as compared with WT mice. Fgf15 Tg mice also had a slight reduction in CYP3A13 protein as compared with WT and a relatively similar amount of CYP3A25 protein. Interestingly, the Fgf15 Tg mice did express CYP3A16 protein, which was not detected in the male WT mice.
Functional Activity of CYP2B and CYP3A Enzymes
The functional activities of CYP2B and CYP3A were measured by treating isolated microsomes with the CYP2B and CYP3A probe substrates pentoxyresorufin and midazolam, respectively. Specifically, we measured the reactions of pentoxyresorufin O-dealkylation and midazolam 1’-hydroxylation by LC/MS, which was used to assess the enzymatic activities of CYP2B and CYP3A, respectively. Microsomes isolated from Fgf15 Tg mice displayed a 1.70-fold increase in resorufin metabolite formation as compared with WT mice, suggesting the observed induction in CYP2B protein and mRNA resulted in an increase of CYP2B metabolic rate (P = 0.076). Microsomes from Fgf15int−/− mice showed no significant change from WT with a fold change of 0.93, as compared with WT (Fig. 2D). PB-treated mice were used as a positive control and displayed a 6.41-fold increase in pentoxyresorufin O-dealkylation. There was no observed difference in the rate of midazolam hydroxylation among WT, Fgf15int−/−, and Fgf15 Tg mice, suggesting there is no change to CYP3A hepatic microsomal activity.
FGF19 Did Not Induce CYP2B and CYP3A in HepaRG or PHH Cells
We have previously described the successful production of soluble recombinant FGF19 protein (Kong and Guo, 2014). We used the recombinant FGF19 protein to treat HepaRG and PHH cells at concentrations of 5 ng/mL and 50 ng/mL for 24 and 48 hours. Although FGF19 treatment led to activation of FGFR4 pathway (data not shown), it did not induce the mRNA expression of CYP2B6 or CYP3A4 regardless of the cell type, concentration, and time point (Supplemental Fig. 2).
Effects of FGF19 on CAR Nuclear Translocation In Vitro
CAR is a classic xenobiotic-sensing NR known to regulate the expression of many DMET genes. To determine whether FGF19 was capable of activating CAR in PHHs, an adenovirus expressing enhanced yellow fluorescent protein tagged hCAR (Ad/EYFP-hCAR) was used. Through the use of fluorescent microscopy, we were able to visualize the localization of the Ad/EYFP-hCAR following treatment with known activators of CAR (PB and CITCO) or recombinant FGF19 protein. Following treatment with PB (1mM) or CITCO (1μM), the Ad/EYFP-hCAR translocated from the cytosol (diffuse YFP seen in the vehicle control group) to the nucleus. The FGF19 (40 ng/mL or 200 ng/mL)-treated PHHs showed almost no nucleus translocation of the Ad/EYFP-hCAR, as compared with the PB- and CITCO-treated groups (Fig. 3A). These data suggested that recombinant FGF19 protein could not activate CAR in vitro.
Fig. 3.

Role of CAR in alterations brought about by the overexpression of Fgf15/19. (A) Fluorescent imaging of Ad/EYFP-hCAR PHHs treated with PB, CITCO, or FGF19 recombinant protein. (B-E) Hepatic gene expression was normalized to β-actin mRNA expression, and graphs depict relative mRNA ± 1 S.D. An asterisk denotes a significant difference from WT (P < 0.05). Cyp2b10, Cyp2b9, Cyp2b13, and Cyp17a1 failed Levene’s test, therefore Kruskal-Wallis was used for analysis. (B) Relative hepatic mRNA expression of genes regulated by CAR. (C) Relative hepatic mRNA expression of genes involved in drug metabolism. (D) Relative hepatic mRNA expression of genes predominantly expressed in male mice. (E) Relative hepatic mRNA expression of genes predominantly expressed in female mice (n: WT = 6, Fgf15 Tg = 7, CAR−/− = 6, Fgf15 Tg/CAR−/− = 7; one-way ANOVA, Tukey post hoc).
Effects of CAR on FGF15-Alteration of DMET Gene Expression In Vivo
Although FGF19 did not directly activate CAR in vitro, indirect activation of CAR in vivo remains a valid hypothesis. To determine the in vivo role of CAR on the observed inductions of drug metabolism genes in Fgf15 Tg mice, we crossed CAR−/− mice with the Fgf15 Tg mice to create Fgf15 Tg/CAR−/− mice. These mice develop and breed normally. Serum assays in WT, Fgf15 Tg, CAR−/−, and Fgf15 Tg/CAR−/− mice showed no significant differences between groups for total cholesterol, triglycerides, or ALT, AST, or ALP activities (Supplemental Fig. 3). Liver histology was examined by a board-certified pathologist. All WT and Fgf15 Tg mice had no noteworthy findings. Most Fgf15 Tg/CAR−/− mice displayed mild biliary hyperplasia. A detailed table of pathologic findings can be found in Supplemental Fig. 3.
In Fig. 3B we measured the gene expression of 4 genes that are positively regulated by CAR activation (Gsta1, Gstm3, Akr1b7, Cyp2b10) and 3 genes that are negatively regulated by CAR activation (Cyp2c55, Hsd3b5, Slco1a1). Of genes positively regulated by CAR, Fgf15 Tg mice displayed a 2.07 ± 1.17- and 1.93 ± 0.82-fold induction of Gstm3 and Akr1b7, respectively, as compared with WT mice. Fgf15 Tg/CAR−/− mice showed a slightly greater induction in the expression of Gstm3 and Akr1b7, with a significant fold change of 2.60 ± 1.45 and 2.58 ± 1.68, respectively. Cyp2b10, a prototypical CAR target gene is significantly induced 12.46 ± 6.11-fold in Fgf15 Tg mice, as compared with WT mice. In CAR−/− mice, the expression of Cyp2b10 was significantly reduced to 0.16 ± 0.16. In Fgf15 Tg/CAR−/− mice, Cyp2b10 expression was significantly induced 3.70 ± 2.22-fold as compared with WT, which is significantly less than that of the Fgf15 Tg mice (P = 0.0004). For genes negatively regulated by CAR activation, there was no significant difference in the expression of Hsd3b5 or Slco1a1 between WT and CAR−/− mice. However, Fgf15 Tg mice displayed a significant reduction in Hsd3b5 and Slco1a1 mRNA expression of 0.18 ± 0.36 and 0.24 ± 0.27, respectively. This reduction was further exacerbated in Fgf15 Tg/CAR−/− mice to 0.02 ± 0.02 and 0.09 ± 0.07, respectively, as compared with WT mice. In Fig. 3C, we measured the mRNA expression of 6 genes involved in phase I or phase II drug metabolism (Cyp1a, Cyp3a11, Cyp3a16, Cyp3a44, Ugt1a1, Sult1a1). There was no significant difference for any of the genes measured among WT, CAR−/−, Fgf15 Tg, and Fgf15 Tg/CAR−/− mice.
Among the differentially expressed cytochrome P450s described in Figs. 1 and 2, were Cyp2b9 and Cyp3a16, which are known to have sexually dimorphic expression patterns, with both being expressed higher in female than in male mice. Using male WT, Fgf15 Tg, CAR−/−, and Fgf15 Tg/CAR−/− mice, we measured the relative expression of 6 genes that are predominantly expressed in male mice (Fig. 3D; Hsd3b5, Ugt2b1, Srd5a1, Slco1a1, Mup1, Cyp2d9) and the relative gene expression of 5 genes that are predominantly expressed in female mice (Fig. 3E; Cyp2b9, Cyp2b13, Cyp17a1, Sult1e1, Hsd3b1). Fgf15 Tg mice displayed significant reductions in the expression of male dominant genes (Fig. 3D) Hsd3b5, Ugt2b1, Slco1a1, Mup1, and Cyp2d9, with a trend toward a significant reduction of Srd5a1 (P=0.0686). There was no significant difference between WT and CAR−/− mice for any of the genes measured; however, in Fgf15 Tg/CAR−/− mice the significant reductions of male dominant genes in Fgf15 Tg mice was further exacerbated in the absence of CAR. Male Fgf15 Tg and Fgf15 Tg/CAR−/− mice both displayed significant increases in the expression of Cyp2b9, Cyp2b13, and Cyp17a1, with trends for increases in Sult1e1 and Hsd3b1 (Fig. 3E), genes that are predominantly expressed in female mice.
The Impact of the Overexpression of Fgf15 on the Pharmacokinetics of Bupropion
To assess the functional implications of the induction of CYP2B mRNA and protein in the Fgf15 Tg mice, we performed a pharmacokinetic study using bupropion as a probe substrate to measure CYP2B activity in vivo. WT and Fgf15 Tg mice were dosed with 1 mg/kg of bupropion intravenously and plasma concentrations of bupropion and the metabolite hydroxybupropion were taken at 3, 30, 60, 120, 180, and 240 minutes. The mean plasma (nM) semilogarithmic concentration time plots are shown in Fig. 4A. A summary table (Fig. 4B) shows that Fgf15 Tg mice have a reduction (P = 0.163) in terminal half-life from 55.8 minutes to 46.8 minutes, as compared with WT. Additionally, Fgf15 Tg mice displayed a higher Cmax of hydroxybupropion (18.1 ng/mL) than WT mice (13.4 ng/mL) and Fgf15 Tg mice reached that maximum metabolite concentration 27.4 minutes faster (P = 0.077) than WT mice (a reduction of 28.5%). Taken together, these data suggest that Fgf15 Tg mice convert bupropion, a CYP2B probe substrate, to the metabolite hydroxybupropion at a faster rate than WT mice.
Fig. 4.

Impact of Fgf15 overexpression on the pharmacokinetics of bupropion in mouse plasma. (A) Average log concentrations of bupropion and hydroxybupropion (+S.D.) in WT and Fgf15 Tg mouse plasma. (B) Table summarizing pharmacokinetic parameters (±S.D.) of bupropion and hydroxybupropion in wild type and Fgf15 Tg mouse plasma (n: WT = 10, Fgf15 Tg = 8; Student’s t test).
Body Size and Composition
Gross observations suggested differences in the size of WT and Fgf15 Tg mice (Fig. 5A). Fgf15 Tg mice had a significantly shorter body length measured by nose-to-anus length (7.83 ± 0.41cm) than WT mice (9.15 ± 0.42cm). Fgf15 Tg mice also weighed significantly less (22.45 ± 2.18g) than WT mice (27.98 ± 0.94g) (Fig. 5B). CT imaging was used to determine if the overexpression of Fgf15 altered the disposition of fat, muscle, or bone (Fig. 5C). There were no noteworthy alterations in the distribution of fat, muscle, or bone between WT mice (6.9%, 76.2%, and 16.0% respectively) and Fgf15 Tg mice (7.4%, 74.0%, and 18.0% respectively).
Fig. 5.
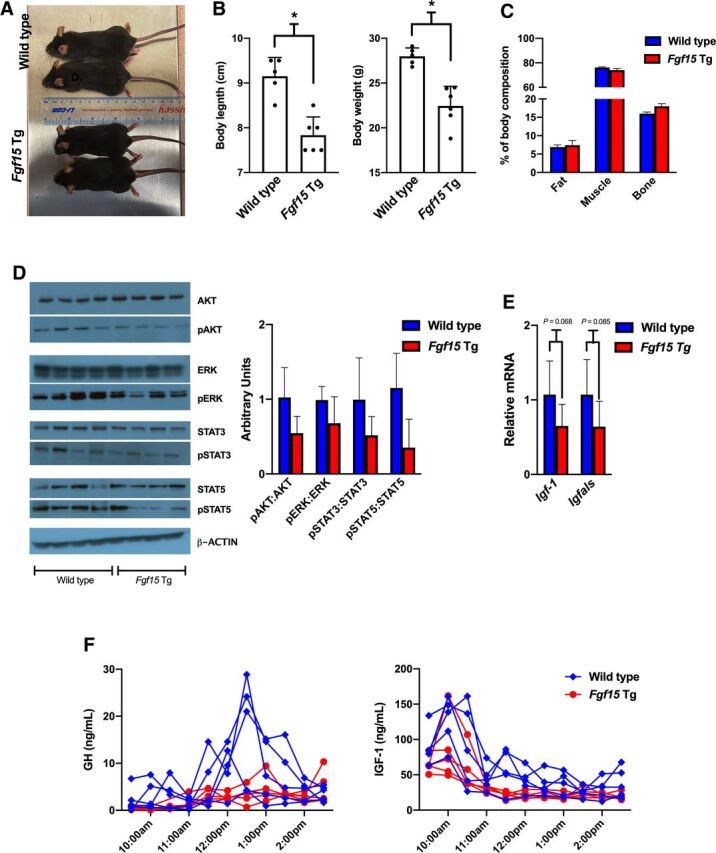
Body size and composition of WT and Fgf15 Tg mice. (A) Representative dorsal view of WT (top) and Fgf15 Tg (bottom) mice. (B) Quantification of nose to anus length (cm) and body weight (g) of WT and Fgf15 Tg mice (n: WT = 5, Fgf15 Tg = 6; Student’s t test). (C) Percent of total body weight distributed as fat, muscle, and bone (n: WT = 5, Fgf15 Tg = 5; Student’s t test). (D) Western blots and semiquantifications of cell signaling proteins in liver homogenates from WT and Fgf15 Tg mice (n: WT = 4, Fgf15 Tg = 4). (E) Relative hepatic gene expression of GH response genes Igf-1 and Igfals (n: WT = 6, Fgf15 Tg = 7; Student’s t test). (F) Serum GH (left) and IGF-1 (right) levels in WT and Fgf15 Tg mice measured by ELISA every 30 minutes from 9:30 AM to 2:30 PM.
Cell Signaling and Growth Hormone Signaling Pathways
FGFR4 activation by FGF15/19 is known to activate several intracellular signaling pathways, including ERK1/2 mitogen-activated protein kinases, PI3K-AKT, and JAK/STAT pathways (Liu et al., 2020). In Fig. 5D, we measured the modification of these pathways and semiquantified the western blot analysis. We observed an overall reduction in the phosphorylation of AKT, ERK, STAT3, and STAT5 in Fgf15 Tg mice, as compared with WT. The reduction may be a result of desensitization to FGFR4 activation as a result of the continuous overexpression of Fgf15. Of note is the drastic reduction of phosphorylated STAT5 in 3 out of 4 Fgf15 Tg samples measured. STAT5 signaling is well known to be activated by growth hormones and influence sexually dimorphic hepatic gene expression patterns and growth development (Zhang et al., 2012; Martinez et al., 2013). Measurements of the mRNA levels of GH response genes, Igf-1 and Igfals, showed reductions of gene expression in the livers of Fgf15 Tg mice (P = 0.068 and 0.085, respectively), as compared with WT mice (Fig. 5E).
Serum Levels of GH and IGF-1
To assess potential differences in GH release between WT and Fgf15 Tg mice, we collected blood from mice every 30 minutes from 9:30 AM to 2:30 PM. In Fig. 5F, we measured the serum levels of GH and the GH response gene, IGF-1, via ELISA assays. WT mice generally displayed a typical male GH release pattern with a robust GH pulse beginning around 12:00 PM and reaching a maximum concentration around 12:30 PM. Fgf15 Tg mice showed a GH release pattern more similar to female mice with low, steady levels detected. The serum GH levels for female mice were similar between WT and Fgf15 Tg mice, with levels generally being maintained between 2 and 4 ng/mL (Supplemental Fig. 5A). Serum IGF-1 concentrations were more varied, but WT mice tended to have higher levels of serum IGF-1 than Fgf15 Tg mice.
Discussion
FGF15 and its human ortholog FGF19 are endocrine FGFs that function to suppress BA production, reduce steatosis, regulate oxidative stress, promote liver growth and protein production, and improve insulin resistance in murine models of NASH (Henriksson and Andersen, 2020; Stofan and Guo, 2020). As such, FXR agonists, which are capable of producing robust inductions of FGF15/19 through ileael FXR activation, as well as FGF19 analogs/mimetics, have been in clinical trials for the treatments of NASH and cholestatic liver diseases. In the present study, we determine the effects of FGF15/19 overexpression on DMETs in vivo, through the use of an Fgf15 Tg mouse model, and in vitro, using recombinant FGF19 protein.
The overexpression of Fgf15 caused a broad alteration of the expression of DMETs revealed by RNA-seq transcriptomic analysis. Alterations of DMETs can result in unintended clinical consequences, such as loss of efficacy or toxicity. For this reason, we wanted to explore the functional consequences of these alterations, as well as investigate the potential mechanisms underlying this regulation. Among the genes with their expression altered, we confirmed inductions of Cyp2b9, Cyp2b10, Cyp3a11, and Cyp3a25 at the mRNA level and inductions of CYP2B9, CYP2B10, CYP2B19, and CYP3A16 at the protein level as a result of the overexpression of Fgf15. Through the use of isolated hepatic microsomes from Fgf15 Tg mice, we found an increase in the metabolism of pentoxyresorufin (P = 0.076), a probe substrate for CYP2B activity. Our pharmacokinetic study also found an increase (P = 0.163) in the rate of metabolism of bupropion, a CYP2B specific substrate, to the metabolite hydroxybupropion by Fgf15 Tg mice, as compared with WT mice.
CAR is a well-studied xenobiotic sensing NR known to induce human CYP2B6 and CYP3A4 (CYP2B10 and CYP3A11 in mice). Recent research has shown a connection between FGF15 and FXR or CAR activation in a tissue-specific manner (Weber et al., 2021). To determine if the observed alterations in Fgf15 Tg mice were a result of elevated CAR activation, we crossbred Fgf15 Tg mice with CAR−/− mice to create Fgf15 Tg/CAR−/− mice. We found that in some cases (Gstm3 and Akr1b7), the loss of CAR exacerbated inductions brought about by the overexpression of Fgf15. In other cases (Cyp2b10 and Cyp17a1), the loss of CAR-attenuated inductions caused by the overexpression of Fgf15. Taken together, these data suggest CAR may play a role in altering the expression pattern of DMETs in mice overexpressing Fgf15; however, there are clearly additional mechanisms driving the observed alterations.
Among the phase I metabolizing enzymes that were induced in male Fgf15 Tg mice was Cyp2b9. Cyp2b9 is a sexually dimorphic cytochrome P450 that is predominantly expressed in female mice (Wiwi et al., 2004). Looking at other hepatic genes known to be sexually dimorphic, we found that male Fgf15 Tg mice had large inductions of genes primarily expressed in the livers of female mice, e.g., Cyp2b9, Cyp2b13, Cyp17a1, Sult1e1, Hsd3b1. Conversely, we looked at the hepatic mRNA expression of male dominant genes and found that male Fgf15 Tg mice had a significantly lower expression pattern of many of these genes, including Hsd3b5, Ugt2b1, Slco1a1, Mup1, and Cyp2d9.
STAT5 is a transcription factor activated predominately by growth hormone and plays a key role in the sexually dimorphic expression of cytochrome P450s in the liver (Davey et al., 1999). Neuroendocrine factors regulate a pulsatile (in male) or continual (in female) GH release, which, among other things, regulate the tyrosine phosphorylation of STAT5b in the liver. In general, male mice go through cyclical periods of robust STAT5b phosphorylation/activation with periods of little or no activated STAT5b during the GH interpulse interval. Female mice have continual, low (but measurable) plasma GH and STAT5b activity (Waxman and O’Connor, 2006). Additionally, GH is known to influence body size and weight in mice (Kopchick et al., 2014). In line with this known mechanism of sexual dimorphism, our male Fgf15 Tg mice exhibited a low consistent concentration of serum GH, while our male WT mice displayed the prototypical male GH surge. As a likely result of altered GH signaling, our Fgf15 Tg mice had greatly reduced STAT5 phosphorylation as compared with WT mice. Downstream measurements of GH response genes Igf-1 and Igfals showed reduced mRNA expression in the livers of Fgf15 Tg mice (P = 0.068 and 0.085, respectively). Additionally, Fgf15 Tg mice have a shorter nose to anus length and reduced body weight compared with WT mice. Factors known to impact GH secretion patterns include stress, exercise, nutritional state, and metabolic signals (Kato et al., 2002). The observed alterations could be a result of reduced BAs impacting lipid and lipid-soluble vitamin absorption, causing changes to the nutritional state or metabolic signaling in the Fgf15 Tg mice. The impact of FGF15/19 on GH regulation and STAT5b activity is the subject of an ongoing investigation in our laboratory.
The influence of FXR and BAs in liver disease etiology is well established. As pharmaceutical companies continue to probe the FXR-BA-FGF15/19 pathway for the therapeutic intervention in treating diseases associated with BA dysregulation, it is important to understand the potential that manipulating these pathways has on the regulation of drug metabolizing enzymes. Using an Fgf15 Tg mouse model, we have shown that the overexpression of Fgf15 induces the expression of several phase I metabolizing enzymes and leads to a phenotypical switch from a male to female expression pattern in the livers of male Fgf15 Tg mice. The mechanism underlying this gender switch is unclear. However, the Fgf15 Tg mice have much lower BA contents than WT mice, which negatively affects lipid absorption. Lower lipid absorption is a form of nutrient deprivation known to be associated with growth retardation. In our study, we found shorter body length, reduced STAT5 activation, altered serum GH levels, and reduced GH target gene expression (Igf-1 and Igfals) as a result of Fgf15 overexpression. GH reduction may lead to CAR activation by releasing factors known to inhibit CAR activation, including EGFR signaling and production of CAR endogenous inhibitors. We will further determine the underling mechanism in future studies to determine the extent to which reduced GH and STAT5 signaling is responsible for the observed gender specific switch of DMET expression.
In summary, we have shown that overexpression of FGF15 led to a gender-specific switch of the expression of genes encoding DMETs in the liver. The mechanism responsible for this switch is unclear; however, initial data from the studies suggest that reduced GH signaling and increased CAR activation are associated with this switch.
Acknowledgments
The authors are grateful to Dr. Wen Xie for graciously providing CAR knockout mice. The authors appreciate the technical support by Katherine Otersen.
ABBREVIATIONS
- Ad/EYFP-hCAR
adenovirus expressing enhanced yellow fluorescent protein-tagged hCAR
- BA
bile acid
- CAR
constitutive androstane receptor
- CAR−/−
CAR knockout mice
- DMET
drug metabolizing enzymes and transporters
- ERK
extracellular signal regulated kinase
- FGF15
fibroblast growth factor 15
- Fgf15 Tg
FGF15 transgenic mice
- Fgf15int−/−
Intestine-specific FGF15 knockout mice
- FGF19
fibroblast growth factor 19
- FGFR4
fibroblast growth factor receptor 4
- FXR
farnesoid X receptor
- GH
growth hormone
- HU
Hounsfield unit
- NASH
nonalcoholic steatohepatitis
- NR
nuclear receptor
- PB
phenobarbital
- PHH
primary human hepatocytes
- WT
wild type
Authorship Contributions
Participated in research design: Rizzolo, Kong, Guo.
Conducted experiments: Rizzolo, Kong, Piekos, Chen, Lu, Siemiątkowska, Yang, A. Li, L. Li, Park, Shi.
Contributed new reagents or analytical tools: Kagan, Guo.
Performed data analysis: Rizzolo, Kong, Piekos, Siemiątkowska, Zhu, Zhong, Wang, Guo.
Wrote or contributed to the writing of the manuscript: Rizzolo, Guo.
Footnotes
This work was supported by the National Institutes of Health [GM135258, ES029258, DK122725], the VA [BX002741], and the Rutgers Center for Lipid Research graduate student small grant award.
The authors declare that they have no conflicts of interest with the contents of this article.
Part of this work was presented in the doctoral dissertation of D. Rizzolo (2021) Regulation of hepatic drug metabolizing enzymes by the bile acid-FXR- FGF15/19 pathway.
 This article has supplemental material available at dmd.aspetjournals.org.
This article has supplemental material available at dmd.aspetjournals.org.
References
- Cui JY, Klaassen CD (2016) RNA-Seq reveals common and unique PXR- and CAR-target gene signatures in the mouse liver transcriptome. Biochim Biophys Acta 1859:1198–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davey HW, Wilkins RJ, Waxman DJ (1999) STAT5 signaling in sexually dimorphic gene expression and growth patterns. Am J Hum Genet 65:959–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart SN, Li Y, Nakamoto K, Subileau EA, Steen D, Zhong XB (2010) A comparison of whole genome gene expression profiles of HepaRG cells and HepG2 cells to primary human hepatocytes and human liver tissues. Drug Metab Dispos 38:988–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriksson E, Andersen B (2020) FGF19 and FGF21 for the Treatment of NASH-Two Sides of the Same Coin? Differential and Overlapping Effects of FGF19 and FGF21 From Mice to Human. Front Endocrinol (Lausanne) 11:601349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato Y, Murakami Y, Sohmiya M, Nishiki M (2002) Regulation of human growth hormone secretion and its disorders. Intern Med 41:7–13. [DOI] [PubMed] [Google Scholar]
- Kobayashi K, Hashimoto M, Honkakoski P, Negishi M (2015) Regulation of gene expression by CAR: an update. Arch Toxicol 89:1045–1055. [DOI] [PubMed] [Google Scholar]
- Kong B, Guo GL (2014) Soluble expression of disulfide bond containing proteins FGF15 and FGF19 in the cytoplasm of Escherichia coli. PLoS One 9:e85890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Sun R, Huang M, Chow MD, Zhong XB, Xie W, Lee YH, Guo GL (2018) Fibroblast Growth Factor 15-Dependent and Bile Acid-Independent Promotion of Liver Regeneration in Mice. Hepatology 68:1961–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong B, Wang L, Chiang JY, Zhang Y, Klaassen CD, Guo GL (2012) Mechanism of tissue-specific farnesoid X receptor in suppressing the expression of genes in bile-acid synthesis in mice. Hepatology 56:1034–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopchick JJ, List EO, Kelder B, Gosney ES, Berryman DE (2014) Evaluation of growth hormone (GH) action in mice: discovery of GH receptor antagonists and clinical indications. Mol Cell Endocrinol 386:34–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Chen T, Cottrell J, Wang H (2009) Nuclear translocation of adenoviral-enhanced yellow fluorescent protein-tagged-human constitutive androstane receptor (hCAR): a novel tool for screening hCAR activators in human primary hepatocytes. Drug Metab Dispos 37: 1098–1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Cao M, Cai Y, Li X, Zhao C, Cui R (2020) Dissecting the Role of the FGF19-FGFR4 Signaling Pathway in Cancer Development and Progression. Front Cell Dev Biol 8:95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackowiak B, Wang H (2016) Mechanisms of xenobiotic receptor activation: Direct vs. indirect. Biochim Biophys Acta 1859:1130–1140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makishima M, Okamoto AY, Repa JJ, Tu H, Learned RM, Luk A, Hull MV, Lustig KD, Mangelsdorf DJ, Shan B (1999) Identification of a nuclear receptor for bile acids. Science 284:1362–1365. [DOI] [PubMed] [Google Scholar]
- Martinez CS, Piazza VG, Ratner LD, Matos MN, González L, Rulli SB, Miquet JG, Sotelo AI (2013) Growth hormone STAT5-mediated signaling and its modulation in mice liver during the growth period. Growth Horm IGF Res 23:19–28. [DOI] [PubMed] [Google Scholar]
- Metzinger MN, Miramontes B, Zhou P, Liu Y, Chapman S, Sun L, Sasser TA, Duffield GE, Stack MS, Leevy WM (2014) Correlation of X-ray computed tomography with quantitative nuclear magnetic resonance methods for pre-clinical measurement of adipose and lean tissues in living mice. Sensors (Basel) 14:18526–18542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray A, Gow AJ, Venosa A, Andres J, Malaviya R, Adler D, Yurkow E, Laskin JD, Laskin DL (2020) Assessment of mustard vesicant lung injury and anti-TNF-α efficacy in rodents using live-animal imaging. Ann N Y Acad Sci 1480:246–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noureddin MVipani ABresee CTodo TKim IKAlkhouri NSetiawan VWTran TAyoub WSLu SC, et al. (2018) NASH Leading Cause of Liver Transplant in Women: Updated Analysis of Indications For Liver Transplant and Ethnic and Gender Variances. Am J Gastroenterol 113:1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pande P, Zhong XB, Ku WW (2020) Histone Methyltransferase G9a Regulates Expression of Nuclear Receptors and Cytochrome P450 Enzymes in HepaRG Cells at Basal Level and in Fatty Acid Induced Steatosis. Drug Metab Dispos 48:1321–1329. [DOI] [PubMed] [Google Scholar]
- Peng L, Yoo B, Gunewardena SS, Lu H, Klaassen CD, Zhong X-B (2012) RNA sequencing reveals dynamic changes of mRNA abundance of cytochromes P450 and their alternative transcripts during mouse liver development. Drug Metab Dispos 40:1198–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzolo D, Buckley K, Kong B, Zhan L, Shen J, Stofan M, Brinker A, Goedken M, Buckley B, Guo GL (2019) Bile Acid Homeostasis in a Cholesterol 7α-Hydroxylase and Sterol 27-Hydroxylase Double Knockout Mouse Model. Hepatology 70:389–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini SPS, Sonoda J, Xu L, Toma D, Uppal H, Mu Y, Ren S, Moore DD, Evans RM, Xie W (2004) A novel constitutive androstane receptor-mediated and CYP3A-independent pathway of bile acid detoxification. Mol Pharmacol 65:292–300. [DOI] [PubMed] [Google Scholar]
- Schumacher JD, Kong B, Wu J, Rizzolo D, Armstrong LE, Chow MD, Goedken M, Lee Y-H, Guo GL (2020) Direct and Indirect Effects of Fibroblast Growth Factor (FGF) 15 and FGF19 on Liver Fibrosis Development. Hepatology 71:670–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Wang X, Lyu L, Jiang H, Zhu HJ (2018) Comparison of protein expression between human livers and the hepatic cell lines HepG2, Hep3B, and Huh7 using SWATH and MRM-HR proteomics: Focusing on drug-metabolizing enzymes. Drug Metab Pharmacokinet 33:133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spengler EK, Loomba R (2015) Recommendations for Diagnosis, Referral for Liver Biopsy, and Treatment of Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Mayo Clin Proc 90:1233–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stofan M, Guo GL (2020) Bile Acids and FXR: Novel Targets for Liver Diseases. Front Med (Lausanne) 7:544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tien Y-C, Liu K, Pope C, Wang P, Ma X, Zhong XB (2015) Dose of Phenobarbital and Age of Treatment at Early Life are Two Key Factors for the Persistent Induction of Cytochrome P450 Enzymes in Adult Mouse Liver. Drug Metab Dispos 43:1938–1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman DJ, O’Connor C (2006) Growth hormone regulation of sex-dependent liver gene expression. Mol Endocrinol 20:2613–2629. [DOI] [PubMed] [Google Scholar]
- Weber AA, Mennillo E, Yang X, van der Schoor LWE, Jonker JW, Chen S, Tukey RH (2021) Regulation of Intestinal UDP-Glucuronosyltransferase 1A1 by the Farnesoid X Receptor Agonist Obeticholic Acid Is Controlled by Constitutive Androstane Receptor through Intestinal Maturation. Drug Metab Dispos 49:12–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiwi CA, Gupte M, Waxman DJ (2004) Sexually dimorphic P450 gene expression in liver-specific hepatocyte nuclear factor 4alpha-deficient mice. Mol Endocrinol 18:1975–1987. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Laz EV, Waxman DJ (2012) Dynamic, sex-differential STAT5 and BCL6 binding to sex-biased, growth hormone-regulated genes in adult mouse liver. Mol Cell Biol 32:880–896. [DOI] [PMC free article] [PubMed] [Google Scholar]