

Abstract
Cytochrome P450 enzymes are responsible for the metabolism of >75% of marketed drugs, making it essential to identify the contributions of individual cytochromes P450 to the total clearance of a new candidate drug. Overreliance on one cytochrome P450 for clearance levies a high risk of drug-drug interactions; and considering that several human cytochrome P450 enzymes are polymorphic, it can also lead to highly variable pharmacokinetics in the clinic. Thus, it would be advantageous to understand the likelihood of new chemical entities to interact with the major cytochrome P450 enzymes at an early stage in the drug discovery process. Typical screening assays using human liver microsomes do not provide sufficient information to distinguish the specific cytochromes P450 responsible for clearance. In this regard, we experimentally assessed the metabolic stability of ∼5000 compounds for the three most prominent xenobiotic metabolizing human cytochromes P450, i.e., CYP2C9, CYP2D6, and CYP3A4, and used the data sets to develop quantitative structure-activity relationship models for the prediction of high-clearance substrates for these enzymes. Screening library included the NCATS Pharmaceutical Collection, comprising clinically approved low-molecular-weight compounds, and an annotated library consisting of drug-like compounds. To identify inhibitors, the library was screened against a luminescence-based cytochrome P450 inhibition assay; and through crossreferencing hits from the two assays, we were able to distinguish substrates and inhibitors of these enzymes. The best substrate and inhibitor models (balanced accuracies ∼0.7), as well as the data used to develop these models, have been made publicly available (https://opendata.ncats.nih.gov/adme) to advance drug discovery across all research groups.
SIGNIFICANCE STATEMENT
In drug discovery and development, drug candidates with indiscriminate cytochrome P450 metabolic profiles are considered advantageous, since they provide less risk of potential issues with cytochrome P450 polymorphisms and drug-drug interactions. This study developed robust substrate and inhibitor quantitative structure-activity relationship models for the three major xenobiotic metabolizing cytochromes P450, i.e., CYP2C9, CYP2D6, and CYP3A4. The use of these models early in drug discovery will enable project teams to strategize or pivot when necessary, thereby accelerating drug discovery research.
Introduction
Hepatic biotransformation of small-molecule therapeutics by cytochrome P450 enzymes continues to be the predominant route of metabolic clearance, highly impacting their bioavailability and systemic exposure. An in-depth analysis of clearance mechanisms is important because it will help predict human pharmacokinetics, indicate the probability of drug-drug interactions (DDI), and identify the potential for pharmacokinetic variability due to race, sex, age, and genetic polymorphisms (Roden and George, 2002; Sansone-Parsons et al., 2007). In this regard, it is necessary to determine the contributions of individual cytochromes P450 to the total clearance of a compound. When compounds have a high fraction metabolized by one enzyme, e.g., CYP2C9 for S-warfarin (Kaminsky and Zhang, 1997), variability in enzyme activity or expression can result in unanticipated low clearance or, conversely, undergo ultrarapid metabolism, a known issue for CYP2D6 gene variants (Ingelman-Sundberg, 2005). Inhibitors of an enzyme can lead to elevated circulatory concentrations—and toxicity, depending on the therapeutic index of the compound—with ensuing black box warnings on the drug label or withdrawal from the market (Layton et al., 2003; Di, 2017). Conversely, enzyme induction can diminish the efficacy of a compound when the increased expression and activity leads to rapid clearance. To address these concerns, compounds are sought that possess a well distributed metabolism profile across multiple enzymes and clearance mechanisms (Zientek and Youdim, 2015).
The current standard for preliminary estimation of the human metabolic stability of a compound at the discovery stage is the in vitro clearance assay using human liver microsomes (HLMs), which are enriched with various xenobiotic metabolizing cytochromes P450, including 1A2, 2C9, 2C19, 2D6, and 3A4, among others. However, a simple HLM clearance assay, which monitors the depletion of a compound over time, does not identify the cytochromes P450 responsible for the metabolism. Alternatively, assessing the clearance with individual enzymes for each new chemical entity would be an inefficient and costly approach, as medicinal chemists generate a multitude of compounds in their exploration of chemical space to develop novel therapies.
One focus at the National Center for Advancing Translational Sciences (NCATS) is to create and disseminate in silico tools that facilitate and accelerate translational research. To this end, NCATS, with support from the International Consortium for Innovation and Quality in Pharmaceutical Development, has endeavored to develop quantitative structure-activity relationship (QSAR) models capable of predicting the specific cytochrome P450 enzyme(s) responsible for clearance of new, unexplored compounds. Although predictive QSAR models for individual enzymes are commercially available, those highly regarded can be costly, thereby limiting this resource to the broader scientific community, which includes small companies, academic research institutes, and nonprofit patient-focused organizations. Furthermore, commercial models are often developed using small training data sets, and data are typically sourced from literature, which can introduce error through variability in methods from different laboratories with inconsistent expertise and foci. Alternatively, the robust quantitative high-throughput screening (qHTS) technologies at NCATS have enabled production of sizable databases from standardized protocols (Veith et al., 2009; Shah et al., 2016), which is the foundation for developing predictive QSAR models with improved accuracy.
Here, we report the in vitro activities of ∼5000 low-molecular-weight compounds with three major cytochrome P450 enzymes, i.e., CYP2C9, CYP2D6, and CYP3A4, which can be attributed with ∼75% of total cytochrome P450–mediated metabolism of clinical drugs (Guengerich, 2015). We focused on two primary cytochrome P450 endpoints used in the discovery stage, i.e., clearance and inhibition. The clearance assay typically used to assess the metabolic stability of a compound is dependent on complete enzymatic turnover, a process consisting of nine steps in the canonical cytochrome P450 oxidation reaction (Guengerich, 2018). Although informative, the knowledge garnered from this assay is limited to substrates, therefore obliging further studies to identify inhibitors. Notably, competitive inhibition assays that rely on probe conversion, such as P450-Glo, are alone unable to distinguish between substrates and inhibitors, given that both types of ligands can generate similar readouts through various cytochrome P450 enzyme binding mechanisms (Fig. 1B). By crossreferencing the compounds that exhibit probe inhibition with those that were metabolized, we identified the most probable inhibitors from our data sets.
Fig. 1.

Reaction schemes for most common scenarios in metabolic clearance and P450-Glo assays. (A) The clearance assay will identify the substrates that proceed through typical Michaelis-Menten kinetics (1) or multiligand processes (2) but is unable to identify either competitive (3) or mechanism-based (4) inhibitors. (B). Rationale for categorizing P450-Glo assay hits as either substrate or inhibitor based on crossreferencing the observations from the two assays. A test article is to be able to occlude the proluciferin probe by competing for the substrate pocket (i and ii, left) but may also generate an inhibitory multiligand complex (ii, middle and right). Alternatively, the test article may simply be a poor ligand (iii, left), form a noninhibitory multiligand complex (iii, right), or exhibit efficient clearance that does not impede probe metabolism (iv). E, enzyme; I, inhibitor; P, product; S, substrate.
The successful application of machine-learning approaches to develop predictive QSAR models for absorption, distribution, metabolism, elimination, and toxicity (ADMET) properties is well recognized (Kearnes et al., preprint, DOI: https://arxiv.org/abs/1606.08793; Wenzel et al., 2019) and is the impetus for the work reported herein. Using in-house-generated data sets, we developed conventional QSAR models, as well as multitask models, to predict cytochrome P450 substrates and inhibitors. Most importantly, the training data sets, and models with the greatest balanced accuracy, have been published (https://opendata.ncats.nih.gov/adme) to benefit and accelerate drug discovery across all research groups.
Material and Methods
P450-Glo assay kits were purchased from Promega Corporation (Madison, WI) for CYP3A4 (V9910), CYP2C9 (V9790), and CYP2D6 (V9890). NADPH Regenerating Solution A (catalog number 451220) and B (catalog number 451200), human CYP3A4 (456202), CYP2C9 (456258), and CYP2D6 (456217) Supersomes were purchased from Corning Life Sciences (Corning, NY). Ketoconazole, sulfaphenazole, quinidine, and albendazole were purchased from Sigma-Aldrich (St. Louis, MO).
Compound Library.
The ∼5000 compound library used for this publication encompasses the NCATS Pharmaceutical Collection (NPC) (Huang et al., 2011) and an annotated NCATS library. The NPC library contains ∼2800 compounds that have been approved for clinical use by United States, Canadian, Japanese, and European drug regulatory authorities. The NCATS annotated library comprises of ∼2200 diverse drug-like molecules. This annotated library consists of mostly investigational compounds that represent diverse target classes and disease areas. This combined library (∼5000 compounds) will henceforth be referred to as the NCATS-ADME library.
High-Throughput Metabolic Stability (Clearance) Assays.
The substrate depletion assay was employed to determine metabolic stability using an established mid-density (384-well format) protocol (Shah et al., 2016). The workflow included a robotic system for incubation and sample cleanup coupled with an automated ultra-high-performance liquid chromatography–high-resolution mass spectrometry method for sample analysis. Briefly, each 110 µl reaction mixture consisted of 1 µM test article, supersomes, and an NADPH regenerating system in 100 mM phosphate buffer at pH 7.4. The specific protein and enzyme concentrations, as well as the control compounds used, are listed in Table 1. Incubations were conducted at 37°C, with mixing, and reaction aliquots were quenched at 0, 5, 10, 15, 30, and 60 minutes by addition of cold acetonitrile with internal standard (IS), i.e., albendazole. Centrifugation at 3000g, 4°C, for 20 minutes was used to clear samples of precipitated protein and debris. Sample analysis in an ultra-high-performance liquid chromatography–high-resolution mass spectrometry instrument, data extraction, and half-life (t1/2) determinations were performed as previously described (Shah et al., 2016).
TABLE 1.
Summary of enzyme concentrations, cofactor activities, and controls used in the metabolic stability assays
| Matrix | Final Protein Concentration | Total Cytochrome P450 Content | Cytochrome c Reductase Activity | Cytochrome b5 Content | High-Clearance Controls | Moderate-Clearance Controls | Low-Clearance Controls |
|---|---|---|---|---|---|---|---|
| mg/ml | nM | nmol/(min × mg protein) | pmol/mg protein | ||||
| CYP3A4 | ∼0.2 | 30 | 2900 | 1090 | Buspirone, loperamide | Ketoconazole | Antipyrine, carbamazepine |
| CYP2C9 | ∼0.12 | 45 | 985 | 710 | Glyburide, glimepiride | Tamoxifen | Antipyrine, meloxicam |
| CYP2D6 | ∼0.38 | 60 | 3000 | — | Bufuralol, desipramine, amitriptyline | Mexiletine | Codeine |
The compounds were binned into clearance categories based on the observed t1/2 criteria outlined in Table 2. Data with below limit of quantitation (BLQ), inconclusive (INC), and not found (N/F) designations were excluded from further analysis. The complete data set, annotated with substrate class, is provided in the Supplemental Material.
TABLE 2.
Categorization of clearance data
| Observed t1/2 | Category | Substrate Class |
|---|---|---|
| t1/2 ≤ 30 min | Unstable | 1 |
| t1/2 > 30 min | Stable | 0 |
| Low signala | BLQ | Blank |
| Unable to reasonably fit line to datab | INC | |
| Analyte not detected in mass spectrometer | N/F |
y-intercept of line fitting the Ln(analyte/IS) versus time plot is ≤ −9.0.
Albendazole also assigned as INC due to its use as IS.
P450-Glo qHTS.
The P450-Glo inhibition assay is a luminescent technique used to detect cytochrome P450 activity through the liberation of luciferin from cytochrome P450 probe substrates. P450-Glo assays were performed using a previously described method with minor modifications (Veith et al., 2009). All assays were optimized by incubating positive control compounds at both room temperature (RT) and 37°C conditions. Since no difference in compound activities were found at RT and 37°C for CYP2D6 and CYP3A4, assays for these two enzymes were run at RT. Briefly, 2 µl of cytochrome P450 substrate mix was dispensed into medium-binding white/solid 1536-well plates using a Flying Reagent Dispenser (FRD; Aurora Discovery, Carlsbad, CA), with the exception of adding bovine serum albumin (BSA) to the mixture for CYP2C9. The initial optimization assays for CYP2C9 yielded lower signal-to-background ratios and higher well-to-well variation. To improve signal and prevent adhesion of protein to tubes of the plate dispenser, 0.4% BSA was added to the CYP2C9 enzyme assays. In total, 23 nl of each positive control (columns 1–4) and test compounds (columns 5–48) dissolved in DMSO were transferred to the assay plates using a Wako Pintool station (Wako Automation, San Diego, CA). The positive controls used in these experiments are listed in Table 3. After the control/test compounds were transferred, the assay plates were incubated at RT for 10 minutes before the addition of 2 µl NADPH regeneration solution using an FRD. The reaction incubation continued at either RT or 37°C for 60 minutes and was then quenched by FRD addition of 4 µl of the detection reagent. After a 20-minute incubation at room temperature, the luminescence intensity was measured and quantified using a ViewLux plate reader (PerkinElmer, Shelton, CT). Data were expressed as relative luminescence units.
TABLE 3.
Summary of incubation conditions and positive controls used in the P450-Glo assays
| Enzyme | Inhibitor | Dilution Format | Inhibitor Concentration | Incubation Conditions |
|---|---|---|---|---|
| CYP3A4 | Ketoconazole | 16 concentrations/2-fold dilution in duplicates | 57 µM to 1.8 nM | 1 h/RT |
| CYP2C9 | Sulfaphenazole | 57 µM to 1.8 nM | 1 h/ 37°C/ 0.4% BSA | |
| CYP2D6 | Quinidine | 1.4 µM to 0.04 nM | 1 h/RT |
The concentration-response activity data for each compound, relative to control, was fit with the four-parameter Hill equation to obtain percent activity and potency values. The complete P450-Glo qHTS data sets have been deposited to PubChem, with the following assay IDs: 1645841 (CYP3A4), 1645840 (CYP2D6), and 1645842 (CYP2C9). Compounds were classified as a hit in the P450-Glo screens when the inhibition efficacy was >65% and the potency was <10 µM.
Training Set and Test Set Preparation for QSAR.
The NCATS-ADME library was preprocessed to eliminate entries containing duplicates, inorganic compounds, noncovalent complexes, and mixtures. Furthermore, salts and compounds containing organometals were removed. The chemical structures were then standardized using Francis Atkinson Standardizer tool. To estimate the statistical performance in a robust way, we used a 5-fold crossvalidation routine. The final data set (Table 4) was then split 5-fold while retaining the initial ratio of active/inactive (stratified sampling). For each fold, four-fifths of the data set was used as the training set, and the remaining one-fifth was used as the test set, sliding over folds.
TABLE 4.
Summary of substrate and inhibitor data sets used in this study
| Type | Data Set Name | Total Number of Compounds | Number of Actives | Number of Inactives | Imbalance Ratio (Inactives/Actives) |
|---|---|---|---|---|---|
| Substrate data | CYP2C9 | 3966 | 1126 | 2840 | 3:1 |
| CYP2D6 | 3946 | 1318 | 2628 | 2:1 | |
| CYP3A4 | 3974 | 1883 | 2091 | 1:1 | |
| Inhibitor data | CYP2C9 | 3288 | 570 | 2718 | 5:1 |
| CYP2D6 | 3187 | 367 | 2820 | 8:1 | |
| CYP3A4 | 2794 | 340 | 2454 | 7:1 |
Parsing of Substrates and Inhibitors by Process of Elimination.
The hits in the P450-Glo data sets were crossreferenced with the substrate classifications from the clearance assay. The compounds were binned into four different categories using the classification criteria outlined in Table 5.
TABLE 5.
Parsing rationale for substrate and inhibitors
| Category | Clearance/P450-Glo | Classification | Parsing Rationale |
|---|---|---|---|
| 1 | +/+ | Substrate | Exhibiting activity in both assays, the compound is a clear ligand for the enzyme(s). It is unclear whether the parent, product, or both are responsible for the inhibition. |
| 2 | −/+ | Inhibitor | The compound is able to inhibit the enzyme metabolism of a probe substrate but is not itself cleared, indicating that the parent molecule is an effective inhibitor. |
| 3 | −/− | Noncompetitor | The lack of activity in both assays signifies either that binding does not occur or that the interaction does not generate a catalytically competent or inhibitory complex. |
| 4 | +/− | Substrate | Although a clear substrate, the binding kinetics of the parent compound and its metabolites do not preclude the concomitant metabolism of the P450-Glo probe. |
Molecular Descriptor Calculation.
The following sets of descriptors were calculated for each of the data sets:
The combination of fingerprints with five physicochemical properties, i.e., molecular weight, atom-based calculated partition coefficient (Slog P) (Wildman and Crippen, 1999), topological polar surface area (TPSA), number of H-bond donor, and number of H-bond acceptor, was reported to provide superior performance for prediction of cytochrome P450–mediated properties (Zakharov et al., 2019a). Hence, we used Avalon fingerprints (1024 bits) and Morgan fingerprints [calculated using RDKit (Landrum; http://www.rdkit.org)] in combination with the abovementioned five physicochemical properties.
Dragon descriptors: The Dragon package provided us with 3840 descriptors (https://chm.kode-solutions.net/products_dragon.php). Constant value descriptors (0 throughout) and descriptors with low variance (<0.4) were removed. For the final modeling exercise, we used 1164 descriptors.
Machine Learning Methods—Stratified Bagging with Random Forest and Multi-Task Deep Neural Networks.
Random forest (with default parameters) was used as a base classifier (Breiman, 2001). The number of trees was arbitrarily set to 100 (default), since it has been shown that the optimal number of trees usually falls between 64 and 128 and increasing the number of trees does not necessarily improve model performance (Oshiro et al., 2012). The problem of data imbalance was overcome using undersampling stratified bagging (SB) (He and Garcia, 2009; Tetko et al., 2013), which has been proven to be one of the best-performing methods for dealing with imbalanced data sets (Tetko et al., 2013; Jain et al., 2018). SB is a machine-learning technique that is based on an ensemble of models developed using multiple training data sets sampled from the original training set. This technique uses minority class samples to create the training set of positive samples using the traditional bagging approach (resampling with replacement) and then randomly selects the same number of samples from majority class. Thus, the total bagging training set size was double the minority class. Several models are then calculated and averaged to produce a final ensemble model (Tetko et al., 2013). Because of random sampling, about 37% of the compounds are left out in each run, creating “out-of-the-bag” sets that are used for testing the performance of the final model (Tetko et al., 2013). Although a small set of samples are selected each time, a majority of compounds contribute to the overall bagging procedure, given that data sets were generated randomly. Further, an earlier study by Tetko et al., (2013) showed that larger numbers of models per ensemble (e.g., 128, 256, 512, and 1024) did not significantly increase the balanced accuracy of models. Thus, in this study, we built a total of 64 models per ensemble. All models using Random Forest in combination with stratified bagging were developed and deployed by using the data analytics platform KNIME (Berthold et al., 2008).
The performance of the multitask deep neural network (DNN) method on our data sets was also evaluated. DNN has gained prestige and has been widely applied across different domains of science and technology (Korotcov et al., 2017; Zakharov et al., 2019b). DNN is a variation of an artificial neural network that consists of several sequential hidden layers. Each layer is represented by a linear vector transformation, Wx + b (where W is a matrix of tunable weights, and b is a bias vector), followed by a nonlinear transformation function, i.e., sigmoid. In this study, multitask DNN models (MT-DNN v1) were developed using the multilayer feedforward neural networks implemented in Keras using the Tensorflow back end. The loss function was minimized using the Adam algorithm. All models developed in this study were evaluated by 5-fold crossvalidation (Tropsha, 2010).
Model Performance Assessment.
The performance of each classification model was assessed based on sensitivity (eq. 1), specificity (eq. 2), accuracy (eq. 3), balanced accuracy (BACC; eq. 4), and the Matthews correlation coefficient (MCC; eq. 5). Accuracy may be misleading for a highly imbalanced data set, which makes BACC and MCC more appropriate performance measures to compare different classifiers, given their ability to handle skewed data sets.
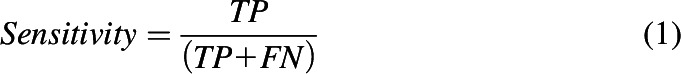 |
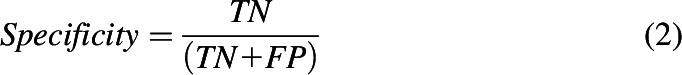 |
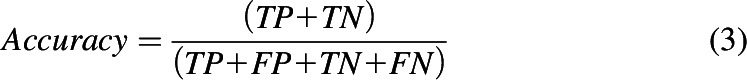 |
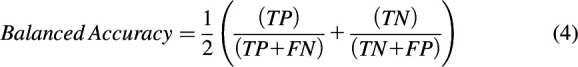 |
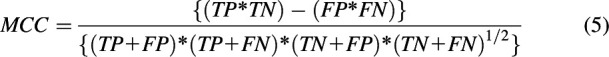 |
In the above equations, TP refers to true positives, TN refers to true negatives, FP refers to false positives, and FN refers to false negatives.
Results
Data Curation for the Clearance Assays.
The compound library was annotated with structural information using the simplified molecular-input line-entry system and LyChI (NCATS; https://github.com/ncats/lychi/) notation formats. During compound batch review, a small percentage (∼3%) were found to have inconsistent structural annotations, which created missing data among the three cytochrome P450 enzyme data sets when pivoted by either simplified molecular-input line-entry system or LyChI. The majority of misannotations were introduced through the vendor-supplied information, which is difficult to detect at purchase, or receipt, considering that the bulk of compounds included in the study was procured within large commercial compound libraries. Structural information was verified with the vendor, and the library annotations were updated accordingly.
Of the ∼5000 compounds screened in the clearance assay, 80% were assigned a usable t1/2 within each cytochrome P450 data set, following exclusion of the compounds with BLQ, INC, and N/F designations. The unreliable data (20% unusable data) generated in this study are typical of high-throughput mass spectrometry-based assays, which are afflicted by erratic pipetting errors common to liquid handlers performing incubations in 384-well format and weak mass spectrometry signal due to inefficient ionization or unmonitored adduct formation.
Cytochrome P450 Substrates Identified in High-Throughput Clearance Assays.
As expected, the highest number of substrates (t1/2 < 30 minutes) were identified in the CYP3A4 screen (45%), followed by CYP2D6 (33%) and CYP2C9 (27%) (Table 6). Overall, only an 11% substrate overlap was observed between all three enzymes.
TABLE 6.
Percentage of high-clearance compounds across three cytochrome P450 enzymes
| CYP3A4 | CYP2C9 | CYP2D6 | |
|---|---|---|---|
| % | |||
| CYP3A4 | 47 | 21 | 19 |
| CYP2C9 | 28 | 13 | |
| CYP2D6 | 33 | ||
Physicochemical Distribution of High- and Low-Clearance Compounds.
Molecular properties of compounds, such as Slog P, TPSA, and molecular weight, were calculated using an in-house compound data set annotation tool known as NCATS Find (NCATS). For all three enzymes, a large proportion of substrates (t1/2 < 30 minutes) fell within the 250–550 mol. wt. range and had Slog P values between 2 and 6, TPSA values less than 100, 0–2 hydrogen bond donors (data not shown), and 1–8 hydrogen bond acceptors (data not shown). No major difference was observed in the physicochemical property distributions between substrates of the enzymes except for CYP2D6 substrates, which displayed lower molecular weight and lower TPSA compared with CYP2C9 and CYP3A4 (Fig. 2). Furthermore, a direct correlation between the calculated t1/2 values and the abovementioned molecular descriptors was not apparent. Additionally, we did not find the established charged preferences of CYP2C9 (acids) and CYP2D6 (basic amines) (Kerns and Di, 2008) to be distinguishing physicochemical features in our data set (data not shown).
Fig. 2.

Property distributions including (A) molecular weight, (B) TPSA, and (C) Slog P for the entire data set compared with substrates (t1/2 < 30 minutes) of CYP3A4, CYP2C9, and CYP2D6 in the supersome clearance assay.
Cytochrome P450 Inhibitors and Activators Identified in qHTS Assays.
The highest percentage of P450-Glo hits were obtained in the CYP3A4 screen (29%), followed by CYP2C9 (23%) and CYP2D6 (19%) (Table 7). In contrast to the clearance assay, only 5% overlap was found among the three enzymes. It should be noted that both inhibitors and substrates decrease the luminescent signal in this assay by occluding the probe from the substrate pocket, and therefore a P450-Glo hit may not be a cytochrome P450 inhibitor in the true sense.
TABLE 7.
Percentage of P450-Glo hits across three enzymes
| CYP3A4 | CYP2C9 | CYP2D6 | |
|---|---|---|---|
| % | |||
| CYP3A4 | 35 | 19 | 13 |
| CYP2C9 | 29 | 10 | |
| CYP2D6 | 25 | ||
The P450-Glo hits used in this study for the development of predictive QSAR models exclude compounds that increase cytochrome P450 metabolism of the probe substrate, observed through an elevated luminescence readout. As current knowledge would lead us to expect, CYP3A4 exhibited the greatest number of molecules (118) that increased luciferin production. Notably, from the 37 compounds that stimulated CYP2C9 metabolism, two molecules were indiscriminate against CYP3A4, i.e., proscillaridin and hematopoietic prostaglandin D synthase-inhibitor-1 (although at varying half-maximal activity concentration values). Although the half-maximal concentration range of CYP2C9, between 0.025 and ∼45 µM, was comparable to that of CYP3A4, spanning from 0.001 to ∼39 µM, the sole two molecules that increased CYP2D6 activity both had a potency of ∼30 µM. Furthermore, only CYP2D6 and CYP3A4 had a single discrete compound with increased probe metabolism that also exhibited high clearance: tenatoprazole (a putative proton pump inhibitor) and SCHEMBL17791590 (an aldehyde dehydrogenase inhibitor), respectively. However, predictive QSAR models for activators were not feasible, given that a larger data set of activator compounds would be needed to power the model; the complete list of activating compounds has been provided in the Supplemental Material.
Parsing of Substrates and Inhibitors by Process of Elimination.
Although the P450-Glo assay alone cannot distinguish inhibitors and substrates, when stratified with the clearance assay, the parsing of probable inhibitors and substrates is feasible. The number of putative inhibitors identified in the P450-Glo assay decreased significantly (77%, 32%, and 66% for CYP3A4, 2C9, and 2D6, respectively). The Venn diagrams in Fig. 3, A and B show the overlap of substrates and inhibitors, highlighting the broad substrate/inhibitor recognition capabilities of the three enzymes. The predisposition of CYP3A4 to metabolize xenobiotics is apparent, with the number of substrates being 3 times greater than that of inhibitors. Although CYP2D6 also exhibited a higher tendency to metabolize compounds, at a substrate:inhibitor ratio of ∼2, CYP2C9 is clearly more susceptible to inhibition, with a corresponding ratio of ∼0.5. Nonetheless, the annotation of these enzymes as xenobiotic metabolizers is validated through the observation of notably higher counts and overlap among substrates as compared with the parsed inhibitors.
Fig. 3.

Substrate (A) and inhibitor (B) overlap among three enzymes. (C) Chemical space plot example for CYP2C9.
Figure 3C displays an example of a chemical space plot for all CYP2C9 data based on visual clustering (Optibrium; www.optibrium.com/stardrop). We find that the compounds are widely scattered, pointing to the diversity of our data set.
It is important to note this approach overlooks compounds that have divergent enzymatic mechanisms based on the presence of additional ligands, by which binding alone leads to the oxidation of the molecule but can lead to a purely inhibitory enzyme-ligand complex in the presence of the probe (Fig. 1, schemes Bi, Bii middle, and Bii right, can be true for the same compound). The compounds in reference cannot be distinguished from those in category 1 per the rationale used (Table 5) and, for the purposes of this, study will remain in that category given that they fall within the high clearance threshold. Ketoconazole is a prime example of this case, as it is a well established substrate while also considered a potent inhibitor of cytochrome P450 catalytic activity (Boulenc et al., 2016; https://www.accessdata.fda.gov/drugsatfda_docs/label/2014/018533s041lbl.pdf, 2020). We reviewed the literature for several of the category 1 compounds and found that some historical compounds can be further annotated with this data set, such as the assignment of tripelennamine as a substrate for CYP2D6. Although it is not surprising that this first-generation antihistamine is cleared by the enzyme, the reported metabolism of this archaic compound is limited (Chaudhuri et al., 1976; Yeh, 1991) and is solely categorized as an inhibitor of CYP2D6 in the Drug Interactions Flockhart Table (https://drug-interactions.medicine.iu.edu/MainTable.aspx), illustrating the presence of missing substrate/inhibitor annotations for familiar older compounds.
Predictive Models: SB and MT-DNN.
Once data analysis and curation were complete, we focused our attention on building classification models that can effectively distinguish actives from inactives using a machine-learning approach. For this, a panel of classifiers were trained on all data sets using different combinations of descriptors. To avoid bias that might occur as a result of the splitting schemes employed, all models were evaluated in a 5-fold external crossvalidation scheme. Considering the average prediction performance across 5-folds, models for all six data sets (three substrate + three inhibitor) showed BACC values close to or above 70%. For the CYP3A4 substrate data set, a DNN-based model with dragon descriptors performed the best (BACC = 76%; MCC = 0.51), closely followed by SB with a combination of Morgan fingerprints and five physicochemical properties (BACC = 75%; MCC = 0.49), which was found to be the best-performing method for the five remaining data sets. Taking a consensus between different descriptor combinations and/or machine-learning approaches did not improve the model performance (data not shown). Considering that the two approaches did not yield significantly different BACC and MCC values (Fig. 4; Table 8), SB with Morgan fingerprints and five physicochemical properties was chosen as the default model for all data sets because of its accessibility (i.e., open source). Supplemental Table 1 reports prediction performance measures for all data sets used in this study.
TABLE 8.
Summary of crossvalidation results
| BACC | |||||||
|---|---|---|---|---|---|---|---|
| Substrates | Inhibitors | ||||||
| Classifiers | Descriptors | CYP2C9 | CYP2D6 | CYP3A4 | CYP2C9 | CYP2D6 | CYP3A4 |
| SB | Morgan + PhysChem | 0.69 | 0.71 | 0.75 | 0.74 | 0.64 | 0.68 |
| SB | Avalon + PhysChem | 0.67 | 0.69 | 0.72 | 0.73 | 0.63 | 0.64 |
| SB | Dragon_Normalized | 0.68 | 0.69 | 0.76 | 0.72 | 0.62 | 0.67 |
| DNN | Morgan + PhysChem | 0.62 | 0.67 | 0.72 | 0.63 | 0.58 | 0.58 |
| DNN | Avalon + PhysChem | 0.64 | 0.66 | 0.68 | 0.63 | 0.59 | 0.58 |
| DNN | Dragon_Normalized | 0.66 | 0.68 | 0.72 | 0.63 | 0.57 | 0.59 |
Fig. 4.

The 5-fold crossvalidation results from SB with Morgan fingerprints: (A) AUC and (B) BACC.
Applicability Domain Assessment.
The applicability domain (AD) of a QSAR model defines the limitation in its structural domain and response space. In other words, this principle for model validation restricts the applicability of a model to reliably predict test compounds that are structurally similar to training compounds used while building the model. Historically, several approaches have been proposed to calculate the applicability of a QSAR model (Sushko et al., 2010; Sahigara et al., 2013; Yun et al., 2017; Patel et al., 2018). In this study, for estimation of the model’s AD, the Tanimoto similarity was assessed between test set compounds and its nearest neighbor in the training set using Morgan fingerprints. The calculations were performed separately for all six data sets. For each fold within each data set, we filtered out compounds that were below a certain similarity threshold and further calculated the BACC and the coverage of predictions as the percentage of compounds that fall within the model’s AD. The distribution of BACC, and corresponding coverage values, for test sets (5-fold average) versus AD cutoffs are presented in Fig. 5 using the CYP2C9 substrate data set as an example. The data reveal a positive trend between AD and prediction accuracy, wherein the AD threshold value increases with the prediction accuracy of the model. The coverage of prediction correlates inversely with AD, as shown by the dramatic decrease in coverage as AD values rise.
Fig. 5.

Distribution of prediction results for test set over AD cutoffs and coverage values using the CYP2C9 substrate data set as an example.
The best prediction results for CYP2C9 substrates were achieved with an AD equal to 0.8, resulting in a BACC of 0.79, although with a very low coverage value of ∼1%. The coverage achieved with an AD cutoff of 0.7 was not significantly better. The optimal ratio of both the accuracy of prediction and coverage was achieved with an AD cutoff value of 0.6. Similar results were obtained for all other data sets (Supplemental Table 2). Given the clear trend between the accuracy of prediction and AD values, this approach can be used to establish the confidence level of predictions.
Analysis of Uncertainty of Prediction/Class Probability.
In addition to the category, the classification approach provides an output for class probability, a numerical value between 0 and 1, which corresponds to the probability of a compound being active. Class probability is an estimation of the reliability of predictions and is referred to as uncertainty of prediction. Values close to 1 indicate active compounds, whereas values close to 0 indicate inactive compounds. Analysis of class probability showed most of the misclassification was in the class probability range of 0.5 to 0.6. In the case of the CYP2C9 substrate data set, the models predicted more than 80% of the compounds correctly for the class probability range between 0–0.4 and 0.7–1 (Fig. 6). The same trend was observed for all six prediction models (Supplemental Table 3), reinforcing increased confidence in model predictions when the 0.5–0.6 class probability range is excluded.
Fig. 6.

Distribution of substrates and inhibitors over class probability prediction values.
Comparison with the Reference Tools/Models.
After completion of the models, an external validation test set was sought to ascertain their utility, a challenging endeavor considering that clearance and inhibition data for individual enzymes is limited and scattered in literature. In addition, we pursued to compare our model performance with other open-source models that exist in literature. Although a few open-source websites offer cytochrome P450–specific substrate and inhibitor models, most were developed using compounds from literature, i.e., essentially a subset of the NPC. Given that our models were developed on the entire NPC data set, the effort to compare model performance metrics was abandoned.
The focus was then shifted to comparing against commercial models. ADMET Predictor from Simulations Plus is one of the leading software packages for ADMET predictions and is regularly used at NCATS. The software includes substrate and inhibitor classification models for nine cytochrome P450 enzymes using data obtained from Biovia metabolite database, DrugBank, and other literature sources. The total number of compounds used to build the CYP2C9, CYP2D6, and CYP3A4 substrate models ranged from 1400 to 1600, whereas the inhibition models were developed using ∼700 compounds. To compare against ADMET Predictor, our models developed herein were retrained using only the NPC library. Since SB with Morgan fingerprints and five physicochemical properties showed superior performance in comparison with other techniques, we used this combination to develop prediction models on the NPC library. These models were then used to predict the NCATS annotated library, and model performances were compared against predictions from ADMET Predictor. Model statistics on the training set (NPC) can be found in the Supplemental Table 4. As shown in Fig. 7, the models developed from this work outperformed those from ADMET Predictor in terms of both BACC and MCC.
Fig. 7.

Comparison of model performance on NCATS annotated library.
To ascertain the robustness of our model, we identified singletons in the NCATS annotated library and compared prediction results/model statistics for those compounds. We found 615 singletons in the NCATS annotated library, and once again, our models outperformed ADMET Predictor (Supplemental Table 4). Although our models exhibited superior performance on this test set as compared with ADMET Predictor, it must be noted that the data used by the models in ADMET Predictor may not have been generated in assays that are the same or similar to those employed in this study. Therefore, these results are provided only for a comparative assessment and must be cautiously inferred.
Discussion
HLMs are the gold standard for studying phase I/cytochrome P450–mediated metabolism. An abundance of HLM data exist in literature, and several groups have used this data to publish QSAR models (Lee et al., 2007; Sakiyama et al., 2008; Hu et al., 2010; Zakharov et al., 2012; Liu et al., 2015). The majority of established, respected models, and (importantly) source data sets, are proprietary, which limits their public accessibility. Although several HLM clearance models exist, the QSAR knowledge for individual cytochromes P450 remains limited. A joint effort by NCATS and members from the IQ Consortium commenced the mission to publish a database of clearance values, which will not only help advance drug design efforts but also provide a better understanding of the structure-activity relationship for major cytochrome P450 enzymes. The benefits gained by the scientific community from this effort include 1) enhancing lead optimization by guiding structure modification, 2) improving hit selection by high-throughput and computational screening, and 3) enabling advanced computational human metabolic models for individual metabolic enzymes.
At ∼4000 molecules, it is the largest library of compounds screened for individual cytochrome P450 enzymes. Notably, the database includes the majority of investigational and regulatory agency–approved drugs, making it the most publicly available, comprehensive list of CYP2C9, 2D6, and 3A4 substrates and inhibitors for clinically used small molecules, which has been founded on single-source empirical data. The complex kinetics of cytochrome P450 enzymes creates a multitude of enzyme-ligand scenarios, which could make designations of substrate or inhibitor ambiguous. Stemming from observations of nonlinear kinetics with cytochrome P450–mediated reactions, Korzekwa et al. (1998) provided some of the first evidence that enzymes bind multiple ligands simultaneously. The issue of cytochrome P450 cooperativity and allosteric interactions have since been reviewed extensively (Davydov and Halpert, 2008; Denisov et al., 2009). However, it is still necessary to evaluate the cytochrome P450–ligand binding empirically, as predictive models do not exist for all the possibilities. Importantly, the data sets and models generated from this effort cannot be extended beyond the simple categorical assignment of substrates and inhibitors, considering that an extensive amount of additional investigation is necessary to fully characterize binding modes (Guengerich et al., 2019), which is more appropriately evaluated spectroscopically using the shift in the quintessential P450 absorbance band. Further, cytochrome P450 inhibition models are complexed by multiligand interactions in which different probe substrates may lead to alternate structure-activity relationships. Nonetheless, we consider the data sets and models reported herein widely applicable, as they were developed using P450-Glo system, which is a commonly used assay in the drug discovery screening paradigm.
The predictive machine-learning models developed from these studies are fortified by the use of reliable data that are unhindered by assay and laboratory-to-laboratory variability, an inherent affliction of most commercial and open-source models that is introduced by sourcing data from compiled literature. We employed SB and multitask deep learning models to classify compounds as substrates or inhibitors for three predominant xenobiotic metabolizing enzymes (CYP3A4, CYP2C9, and CYP2D6). Despite the imbalance of the data sets in our study, especially the inhibitor data sets, we were able to achieve classification accuracies (BACC) around 70% (Fig. 4; Supplemental Table 1). Comparison with the widely used commercial software, i.e., ADMET Predictor, demonstrates the value of our model and the quality of our data. Since 2012, chemists at NCATS have synthesized >20,000 compounds for more than 250 drug discovery projects that cover a wide range of disease areas, pharmacological targets, and cellular pathways. A high degree of similarity in physicochemical properties (molecular weight, Slog P, TPSA, H-Bond Acceptor, and H-Bond Donor) was observed between this data set (Siramshetty et al., 2020) and our NCATS-ADME 5K library. Thus, our models can be used during the compound design phase as well as after synthesis as a filtering mechanism to rank order compounds for phenotyping and cytochrome P450 inhibition assays in drug discovery.
Clustering is a powerful approach that allows the grouping of “similar” compounds to distinguish chemical series within a data set of diverse compounds, analyze the SAR, and identify “regions” of chemistry that may yield good properties. Manual inspection of our data sets revealed great structural diversity. With the aim to quantify as well as qualitatively describe this structural diversity, we performed 1) clustering analysis based on Morgan fingerprints (KNIME) and 2) clustering based on maximum common substructure (StarDrop). From 3584 compounds that encompassed the substrate data across the three enzymes, 1829 different clusters were identified using Morgan fingerprints. From those, 1067 were singletons, and only 24 clusters contained ≥10 compounds. The most populated cluster contained 30 compounds. Clustering based on maximum common substructure algorithm, as implemented in StarDrop (similarity threshold = 0.70), also revealed high structural diversity in the data set, yielding 2059 singletons and a maximum cluster size of 33 compounds. Additionally, the Murcko (Bemis and Murcko, 1996) scaffold algorithm used identified over 2617 different Murcko scaffolds within the aforementioned 3584 compound data set. The Murcko analysis produced an average scaffold-to-compound ratio of 0.73, once again signifying the large structural diversity of our data set. Analysis showing the most frequent scaffolds is presented in Fig. 8. Benzene scaffolds were found with very low frequency (∼6% of the data set), and no other scaffolds reached prevalence values above 0.5%.
Fig. 8.

Plot presenting the most frequent Bemis-Murcko scaffolds in our data set and relative percentages of frequencies within.
Considering the central role CYP450 enzymes play in the clearance of small-molecule therapeutics, evaluation of the specific cytochrome P450 enzymes that catalyze the metabolism of new chemical entities is essential at the preclinical phase of the drug discovery and development process. For more than 20 years, health authorities have provided guidance toward the characterization of in vitro drug interactions involving cytochromes P450, as well as other enzymes involved in the human disposition of xenobiotics (Huang et al., 2008; Prueksaritanont et al., 2013). The focus is on reducing the risk that a novel molecule will act as a DDI “perpetrator” in the clinic through the interactions, i.e., substrate and inhibitor, with cytochrome P450 enzymes. However, DDI assessments are typically conducted long after the concept has been synthesized and now poised as a developmental molecule. Although the models provided in this report are not adequate to replace the studies necessary to predict clinical DDI, the prediction of cytochrome P450 substrate and inhibitors can be fully exploited early in the discovery phase for ranking or selecting compounds for experimental validation.
One of the first examples of cytochrome P450 polymorphism was reported by Eichelbaum et al. (1975), who showed that N-oxidation of sparteine was subject to a high degree of interindividual variability. Since then, it has been well established that almost all drug-metabolizing cytochrome P450 enzymes are polymorphic. Johansson and Ingelman-Sundberg (2011) have summarized the most important cytochrome P450 alleles related to drug toxicity and the classes of drugs most commonly affected by these polymorphisms. The polymorphic variability in pharmacokinetics, which drives pharmacodynamics, can lead to toxicity or inefficacy, with both results being detrimental to patients. The use of our models in early drug discovery could enable the flagging of compounds/series that rely heavily on one of these three enzymes for clearance, as well as those with inhibition potential against these enzymes, prompting medicinal chemists to produce compounds that avoid potential future development problems. The application of this level of detailed information early in the drug discovery process will be invaluable.
In summary, we report the first systemic attempt to profile and generate a substrate and inhibitor database of this scope and size for major CYP450 enzymes. This collaborative effort between NCATS and IQ Consortium yielded several useful tools, including 1) a high-throughput automated incubation method for metabolic stability screening; 2) two different automated data acquisition methods with two different mass spectrometry systems; 3) an automated method of assigning t1/2 via the Validator software [code publicly available (Shah et al., 2016)]; 4) large, publicly available data sets (>4000 compounds) for three major cytochrome P450 enzymes; and 5) robust predictive models for cytochrome P450 substrates and inhibitors (https://opendata.ncats.nih.gov/adme). We look forward to the prospect that the knowledge gained, and the tools developed, from this venture will accelerate drug translational research in academia, small biotech, and pharmaceutical companies.
Acknowledgments
The authors would like to acknowledge compound management, especially Paul Shinn and Misha Itkin, for their support. The authors would also like to thank Jorge Neyra for his help with implementing the QSAR models. The authors would also like to acknowledge all working group members from the IQ Consortium, especially Dr. Fabio Broccatelli, Dr. Susanne Winiwarter, Dr. Prashant Desai, and Dr. Matthew Cerny, for their valuable insights.
Abbreviations
- AD
applicability domain
- ADMET
absorption, distribution, metabolism, elimination, and toxicity
- BACC
balanced accuracy
- BLQ
below limit of quantitation
- BSA
bovine serum albumin
- DNN
deep neural networks
- FRD
Flying Reagent Dispenser
- HLM
human liver microsome
- INC
inconclusive
- IS
internal standard
- MCC
Matthews correlation coefficient
- NCATS
National Center for Advancing Translational Sciences
- N/F
not found
- NPC
NCATS Pharmaceutical Collection
- qHTS
quantitative High-Throughput Screening
- QSAR
quantitative structure-activity relationship
- RT
room temperature
- SB
stratified bagging
- t 1/2
half-life
- TPSA
topological polar surface area
Authorship Contributions
Participated in research design: Gonzalez, Shah, Torimoto-Katori, Zakharov, Nguyễn, Obach, Hop, Xu.
Conducted experiments: Gonzalez, Shah, Torimoto-Katori, Sakamuru.
Contributed new reagents or analytic tools: Xia, Xu.
Performed data analysis: Gonzalez, Jain, Shah, Zakharov, Huang.
Wrote or contributed to the writing of the manuscript: Gonzalez, Jain, Shah, Torimoto-Katori, Zakharov, Nguyễn, Sakamuru, Huang, Xia, Obach, Hop, Simeonov, Xu.
Footnotes
This research was supported by the Intramural Research Program of the National Institutes of Health [National Center for Advancing Translational Sciences].
The authors declare no conflict of interest.
References
- Bajusz D, Rácz A, Héberger K (2015) Why is Tanimoto index an appropriate choice for fingerprint-based similarity calculations? J Cheminform 7:20 10.1186/s13321-015-0069-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bemis GW, Murcko MA (1996). The properties of known drugs. 1. Molecular frameworks. J Med Chem 39:2887–2893. [DOI] [PubMed] [Google Scholar]
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kotter T, Meinl T, Ohl P, Sieb C, Thiel K, Wiswedel B (2008) KNIME: the Konstanz information miner, in Data Analysis, Machine Learning and Applications (Burkhardt H, Schmidt-Thieme L, Decker R, eds) pp 319–326, Springer Berlin Heidelberg. [Google Scholar]
- Boulenc X, Nicolas O, Hermabessiere S, Zobouyan I, Martin V, Donazzolo Y, Ollier C (2016) CYP3A4-based drug-drug interaction: CYP3A4 substrates' pharmacokinetic properties and ketoconazole dose regimen effect. Eur J Drug Metab Pharmacokinet 41:45–54. [DOI] [PubMed] [Google Scholar]
- Breiman L (2001) Random Forests. Mach Learn 45:5–32 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Chaudhuri, N. K., Servando, O. A., Manniello, M. J., Luders, R. C., Chao, D. K., Bartlett, M. F. (1976). Metabolism of tripelennamine in man. Drug Metab Dispos 4:372–378. [PubMed] [Google Scholar]
- Davydov DR, Halpert JR (2008) Allosteric P450 mechanisms: multiple binding sites, multiple conformers or both? Expert Opin Drug Metab Toxicol 4:1523–1535 10.1517/17425250802500028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denisov IG, Frank DJ, Sligar SG (2009). Cooperative properties of cytochromes P450. Pharmacol Ther 124:151–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di L (2017). Reaction phenotyping to assess victim drug-drug interaction risks. Expert Opin Drug Discov 12:1105–1115. [DOI] [PubMed] [Google Scholar]
- Eichelbaum M, Spannbrucker N, Dengler HJ (1975). Proceedings: N-oxidation of sparteine in man and its interindividual differences. Naunyn Schmiedebergs Arch Pharmacol 287. [PubMed] [Google Scholar]
- Guengerich FP (2015). Human cytochrome P450 enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano PR, ed) pp 523–785. Springer International Publishing, Switzerland. [Google Scholar]
- Guengerich FP (2018) Mechanisms of Cytochrome P450-Catalyzed Oxidations. ACS Catal 8:10964–10976 10.1021/acscatal.8b03401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guengerich FP, Wilkey CJ, Phan TTN (2019) Human cytochrome P450 enzymes bind drugs and other substrates mainly through conformational-selection modes. J Biol Chem 294:10928–10941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He H, Garcia EA (2009) Learning from Imbalanced Data. IEEE Trans Knowl Data Eng 21:1263–1284 10.1109/TKDE.2008.239. [DOI] [Google Scholar]
- Hu Y, Unwalla R, Denny RA, Bikker J, Di L, Humblet C (2010) Development of QSAR models for microsomal stability: identification of good and bad structural features for rat, human and mouse microsomal stability. J Comput Aided Mol Des 24:23–35 10.1007/s10822-009-9309-9. [DOI] [PubMed] [Google Scholar]
- Huang R, Southall N, Wang Y, Yasgar A, Shinn P, Jadhav A, Nguyen DT, Austin CP (2011) The NCGC pharmaceutical collection: a comprehensive resource of clinically approved drugs enabling repurposing and chemical genomics. Sci Transl Med 3:80ps16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S-M, Strong JM, Zhang L, Reynolds KS, Nallani S, Temple R, Abraham S, Habet SA, Baweja RK, Burckart GJ, et al. (2008) New era in drug interaction evaluation: US Food and Drug Administration update on CYP enzymes, transporters, and the guidance process. J Clin Pharmacol 48:662–670 10.1177/0091270007312153. [DOI] [PubMed] [Google Scholar]
- Ingelman-Sundberg M (2005) Genetic polymorphisms of cytochrome P450 2D6 (CYP2D6): clinical consequences, evolutionary aspects and functional diversity. Pharmacogenomics J 5:6–13. [DOI] [PubMed] [Google Scholar]
- Jain S, Kotsampasakou E, Ecker GF (2018) Comparing the performance of meta-classifiers-a case study on selected imbalanced data sets relevant for prediction of liver toxicity. J Comput Aided Mol Des 32:583–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansson I, Ingelman-Sundberg M (2011) Genetic polymorphism and toxicology–with emphasis on cytochrome p450. Toxicol Sci 120:1–13. [DOI] [PubMed] [Google Scholar]
- Kaminsky LS, Zhang ZY (1997) Human P450 metabolism of warfarin. Pharmacol Ther 73:67–74 10.1016/s0163-7258(96)00140-4. [DOI] [PubMed] [Google Scholar]
- Kerns EH, Di L (2008) Drug-like properties: concepts, structure design and methods from ADME to toxicity optimization. Academic Press: Amsterdam ; Boston, 2008; p xix, 526 pages. 1–528. [Google Scholar]
- Korotcov A, Tkachenko V, Russo DP, Ekins S (2017) Comparison of Deep Learning With Multiple Machine Learning Methods and Metrics Using Diverse Drug Discovery Data Sets. Mol Pharm 14:4462–4475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korzekwa KR, Krishnamachary N, Shou M, Ogai A, Parise RA, Rettie AE, Gonzalez FJ, Tracy TS (1998) Evaluation of atypical cytochrome P450 kinetics with two-substrate models: evidence that multiple substrates can simultaneously bind to cytochrome P450 active sites. Biochemistry 37:4137–4147. [DOI] [PubMed] [Google Scholar]
- Kotsiantis SB (2008) Handling imbalanced data sets with a modification of Decorate algorithm. Int J Comput Appl Technol 33:91–98 10.1504/Ijcat.2008.021931. [DOI] [Google Scholar]
- Layton D, Key C, Shakir SAW (2003) Prolongation of the QT interval and cardiac arrhythmias associated with cisapride: limitations of the pharmacoepidemiological studies conducted and proposals for the future. Pharmacoepidemiol Drug Saf 12:31–40. [DOI] [PubMed] [Google Scholar]
- Lee PH, Cucurull-Sanchez L, Lu J, Du YJ (2007) Development of in silico models for human liver microsomal stability. J Comput Aided Mol Des 21:665–673 10.1007/s10822-007-9124-0. [DOI] [PubMed] [Google Scholar]
- Liu R, Schyman P, Wallqvist A (2015) Critically Assessing the Predictive Power of QSAR Models for Human Liver Microsomal Stability. J Chem Inf Model 55:1566–1575 10.1021/acs.jcim.5b00255. [DOI] [PubMed] [Google Scholar]
- Oshiro TM, Perez PS, Baranauskas JA (2012) How Many Trees in a Random Forest? In Perner P. (ed) Machine Learning and Data Mining in Pattern Recognition. MLDM; 2012; Vol 7376, Lecture Notes in Computer Science, Springer, Berlin, Heidelberg. [Google Scholar]
- Patel M, Chilton ML, Sartini A, Gibson L, Barber C, Covey-Crump L, Przybylak KR, Cronin MTD, Madden JC (2018) Assessment and Reproducibility of Quantitative Structure-Activity Relationship Models by the Nonexpert. J Chem Inf Model 58:673–682 10.1021/acs.jcim.7b00523. [DOI] [PubMed] [Google Scholar]
- Prueksaritanont T, Chu X, Gibson C, Cui D, Yee KL, Ballard J, Cabalu T, Hochman J (2013) Drug-drug interaction studies: regulatory guidance and an industry perspective. AAPS J 15:629–645 10.1208/s12248-013-9470-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roden DM, GeorgeAL, Jr (2002) The genetic basis of variability in drug responses. Nat Rev Drug Discov 1:37–44. [DOI] [PubMed] [Google Scholar]
- Sahigara F, Ballabio D, Todeschini R, Consonni V (2013) Defining a novel k-nearest neighbours approach to assess the applicability domain of a QSAR model for reliable predictions. J Cheminform 5:27 10.1186/1758-2946-5-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakiyama Y, Yuki H, Moriya T, Hattori K, Suzuki M, Shimada K, Honma T (2008) Predicting human liver microsomal stability with machine learning techniques. J Mol Graph Model 26:907–915. [DOI] [PubMed] [Google Scholar]
- Sansone-Parsons A, Krishna G, Simon J, Soni P, Kantesaria B, Herron J, Stoltz R (2007) Effects of age, gender, and race/ethnicity on the pharmacokinetics of posaconazole in healthy volunteers. Antimicrob Agents Chemother 51:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah P, Kerns E, Nguyen D-T, Obach RS, Wang AQ, Zakharov A, McKew J, Simeonov A, Hop CECA, Xu X (2016) An automated high-throughput metabolic stability assay using an integrated high-resolution accurate mass method and automated data analysis software. Drug Metab Dispos 44:1653–1661 10.1124/dmd.116.072017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siramshetty VBShah PKerns ENguyen KYu KRKabir MWilliams JNeyra JSouthall NNguyễn Ð-T, et al. (2020) Retrospective assessment of rat liver microsomal stability at NCATS: data and QSAR models. Sci Rep 10:20713 10.1038/s41598-020-77327-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sushko INovotarskyi SKörner RPandey AKCherkasov ALi JGramatica PHansen KSchroeter TMüller KR, et al. (2010) Applicability domains for classification problems: benchmarking of distance to models for Ames mutagenicity set. J Chem Inf Model 50:2094–2111 10.1021/ci100253r. [DOI] [PubMed] [Google Scholar]
- Tetko IV, Novotarskyi S, Sushko I, Ivanov V, Petrenko AE, Dieden R, Lebon F, Mathieu B (2013) Development of dimethyl sulfoxide solubility models using 163,000 molecules: using a domain applicability metric to select more reliable predictions. J Chem Inf Model 53:1990–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tropsha A (2010) Best Practices for QSAR Model Development, Validation, and Exploitation. Mol Inform 29:476–488. [DOI] [PubMed] [Google Scholar]
- Veith HSouthall NHuang RJames TFayne DArtemenko NShen MInglese JAustin CPLloyd DG, et al. (2009) Comprehensive characterization of cytochrome P450 isozyme selectivity across chemical libraries. Nat Biotechnol 27:1050–1055 10.1038/nbt.1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J, Matter H, Schmidt F (2019) Predictive Multitask Deep Neural Network Models for ADME-Tox Properties: Learning from Large Data Sets. J Chem Inf Model 59:1253–1268 10.1021/acs.jcim.8b00785. [DOI] [PubMed] [Google Scholar]
- Wildman SA, Crippen G (1999) Prediction of Physicochemical Parameters by Atomic Contributions. J Chem Inf Comput Sci 39:868–873. [Google Scholar]
- Yeh SY (1991) Metabolic profile of tripelennamine in humans. J Pharm Sci 80:815–819. [DOI] [PubMed] [Google Scholar]
- Yun YH, Wu DM, Li GY, Zhang QY, Yang X, Li QF, Cao DS, Xu QS (2017) A strategy on the definition of applicability domain of model based on population analysis. Chemom Intell Lab Syst 170:77–83 10.1016/j.chemolab.2017.09.007. [DOI] [Google Scholar]
- Zakharov A, Gonzalez E, Shah P, Nguyen DT, Southall N, Torimoto-Katori N, Sakamuru S, Xia MH, Zhao TG, Obach RS, Hop C, Simeonov A, Xu X (2019a) AI-driven QSAR modeling of P450-mediated drug metabolism. Abstracts of Papers of the American Chemical Society, 257. Retrieved from <Go to ISI>://WOS:000478860504830 [Google Scholar]
- Zakharov AV, Peach ML, Sitzmann M, Filippov IV, McCartney HJ, Smith LH, Pugliese A, Nicklaus MC (2012) Computational tools and resources for metabolism-related property predictions. 2. Application to prediction of half-life time in human liver microsomes. Future Med Chem 4:1933–1944 10.4155/fmc.12.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zakharov AV, Zhao T, Nguyen D-T, Peryea T, Sheils T, Yasgar A, Huang R, Southall N, Simeonov A (2019b) Novel consensus architecture to improve performance of large-scale multitask deep learning QSAR models. J Chem Inf Model 59:4613–4624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zientek MA, Youdim K (2015) Reaction phenotyping: advances in the experimental strategies used to characterize the contribution of drug-metabolizing enzymes. Drug Metab Dispos 43:163–181. [DOI] [PubMed] [Google Scholar]