

Abstract
Background:
Platinum-based neoadjuvant chemotherapy (NAC) is standard for patients with muscle-invasive bladder cancer (MIBC). Pathologic response (complete: ypT0N0 and partial: <ypT2N0) to NAC is associated with improved survival with ypT0N0 achieved in 3040% of cases. Strategies to increase response to NAC are needed. Incorporation of immune checkpoint inhibitors (ICIs) has demonstrated promise, and better spatial understanding of the tumor microenvironment may help predict NAC/ICI response.
Objective:
Using the NanoString GeoMx platform, we performed proteomic digital spatial profiling (DSP) on transurethral resections of bladder tumors from 18 responders (<ypT2) and 18 nonresponders (ypT2) from two studies of NAC (gemcitabine and cisplatin) plus ICI (LCCC1520 [pembrolizumab] and BLASST-1 [nivolumab]).
Design, setting, and participants:
Pretreatment tumor samples were stained by hematoxylin and eosin and immunofluorescence (panCK and CD45) to select four regions of interest (ROIs): tumor enriched (TE), immune enriched (IE), tumor/immune interface (tumor interface = TX and immune interface = IX).
Outcome measurements and statistical analysis:
DSP was performed with 52 protein markers from immune cell profiling, immunotherapy drug target, immune activation status, immune cell typing, and pan-tumor panels.
Results and limitations:
Protein marker expression patterns were analyzed to determine their association with pathologic response, incorporating or agnostic of their ROI designation (TE/IE/TX/IX). Overall, DSP-based marker expression showed high intratumoral heterogeneity; however, response was associated with markers including PD-L1 (ROI agnostic), Ki-67 (ROI agnostic, TE, IE, and TX), HLA-DR (TX), and HER2 (TE). An elastic net model of response with ROI-inclusive markers demonstrated better validation set performance (area under the curve [AUC] = 0.827) than an ROI-agnostic model (AUC = 0.432). A model including DSP, tumor mutational burden, and clinical data performed no better (AUC = 0.821) than the DSP-only model.
Conclusions:
Despite high intratumoral heterogeneity of DSP-based marker expression, we observed associations between pathologic response and specific DSP-based markers in a spatially dependent context. Further exploration of tumor regionspecific biomarkers may help predict response to neoadjuvant chemoimmunotherapy in MIBC.
Patient summary:
In this study, we used the GeoMx platform to perform proteomic digital spatial profiling on transurethral resections of bladder tumors from 18 responders and 18 nonresponders from two studies of neoadjuvant chemotherapy (gemcitabine and cisplatin) plus immune checkpoint inhibitor therapy (LCCC1520 [pembrolizumab] and BLASST-1 [nivolumab]). We found that assessing protein marker expression in the context of tumor architecture improved response prediction.
Keywords: Bladder cancer, Neoadjuvant, chemoimmunotherapy, Digital spatial profiling, Elastic net regression, LCCC1520, BLASST-1, GeoMx
1. Introduction
Standard-of-care platinum-based neoadjuvant chemotherapy (NAC) followed by radical cystectomy is associated with improved overall survival (OS) in muscle-invasive bladder cancer (MIBC), with pathologic response (complete: ypT0N0 and partial: <ypT2N0) associated with improved outcomes [1,3]. Despite NAC, MIBC still has poor 5-yr survival rates (5060%) [2,3]. Immune checkpoint inhibitors (ICIs) have improved MIBC outcomes for patients with high-risk localized disease receiving adjuvant treatment, for locally advanced/metastatic disease, and for bacillus Calmette-Gu Õrinunresponsive non-MIBC [4,7]. Promising results with ICIs have spurred several neoadjuvant trials with ICIs and combined with chemotherapy [8,13]. Two recent neoadjuvant trials demonstrated that patients who received gemcitabine plus cisplatin with pembrolizumab (LCCC1520) or nivolumab (BLASST-1) achieved pathologic response rates of 3666% (ypT0: 36% and 37%, and <ypT2: 56% and 66%, respectively) and manageable toxicity profiles, with improved recurrence-free survival in responders (LCCC1520, <ypT2N0) [11,12].
Two neoadjuvant trials of single-agent ICIsABACUS (ate-zolizumab) and PURE-01 (pembrolizumab)have reported baseline biomarkers associated with ICI response and resistance [8,9]; however, the ICI-associated biomarkers are inconsistent, possibly due to intratumoral heterogeneity within and between trials. Owing to current widespread use of bulk RNAseq for biomarker identification, studies have been unable to leverage immune-tumor interactions as predictive model features.
The tumor microenvironment (TME) and its architecture have shown promise to help elucidate dynamics of tumor initiation, progression, and treatment response [14]. This was highlighted by analyses from the IMvigor210 trial that evaluated clinical activity of atezolizumab in metastatic urothelial carcinoma (UC), wherein decreased pan-fibroblast TGF- response gene signature (F-TBRS) was associated with response to atezolizumab, but only in immune excluded tumors (not desert or inflamed) [15].
Here, we integrate spatial-oriented tumor architecture data with other clinicogenomic features to train and test a predictive model of response to neoadjuvant chemoimmunotherapy (NACI). Using samples and data from LCCC1520 and BLASST-1, we demonstrate that the use of spatially anchored TME features improves NACI response prediction.
2. Patients and methods
2.1. Sample selection and institutional review board approvals
Pretreatment samples were selected from patients who consented to LCCC1520 (NCT02690558) or BLASST-1 (NCT03294304), and use of biospecimens for a correlative analysis. Nine responders (complete/partial response) and nonresponders (stable/progressive disease) were randomly selected per trial. Selected samples were confirmed as UC by a genitourinary pathologist (S.W.). LCCC1520 was conducted at UNC and Duke. BLASST-1 was conducted at University of Minnesota Masonic Cancer Center, Dana-Farber Cancer Institute, University of Utah Huntsman Cancer Institute, and Beth Israel Deaconess Medical Center. Both trials were approved by the respective institutional review boards.
2.2. Digital spatial profiling processing
Digital spatial profiling (DSP) was performed using the NanoString GeoMx platform, enabling protein quantification within user-defined areas of interest via UV cleavage and simultaneous collection of DNA barcodes previously conjugated to antibodies (Supplementary material) [34]. Regions of interest (ROIs) were selected by a genitourinary pathologist (S.W.) from hematoxylin and eosinstained sections using GeoMx DSP overlay function, including four samples of four ROIs per tumor: tumor enriched (TE), immune enriched (IE), and mixed tumor-immune (interface; Fig. 1A and 1B). The pathologist was blinded to patient outcome. ROIs were aligned with immunofluorescence (IF) staining (panCK and CD45). GeoMx masking was used to select tumor-predominant (TX) and immune-predominant (IX) components of interface. ROIs were interrogated for 52 DSP protein markers associated with immune cell profiling, immunotherapy drug target, immune activation status, immune cell typing, and pan-tumor (Supplementary Table 1). Background normalization was performed using GeoMx software to six housekeeping/negative control markers and masked area. For an ROI-agnostic analysis, mean marker expression was calculated for each tumor across ROIs. For ROI-inclusive analysis, the median expression value of each DSP-ROI combination was calculated.
Fig. 1 –
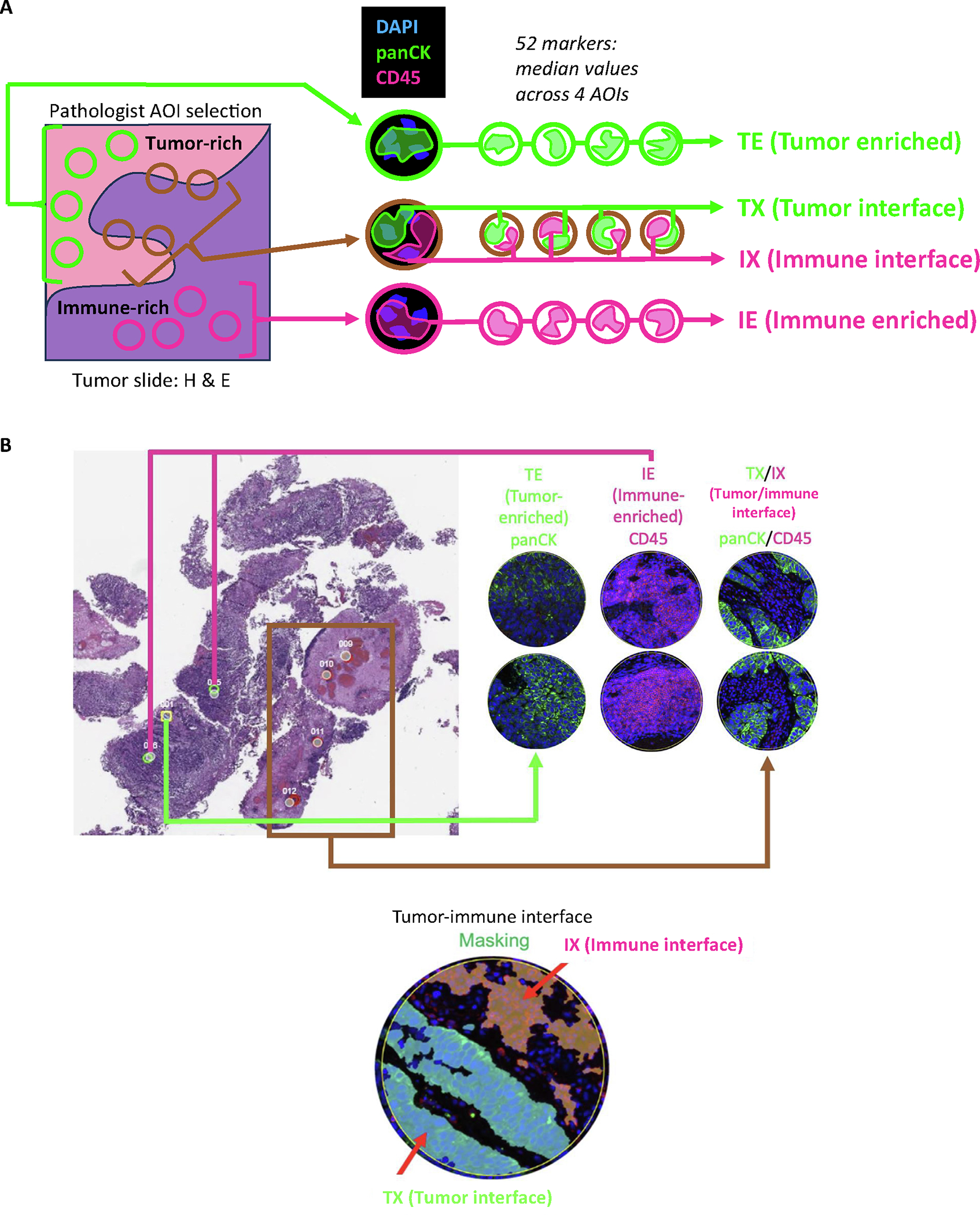
DSP processing. (A) ROIs were selected from hematoxylin and eosin (H&E)-stained sections to include 12 independent, randomly sampled ROIs (four each of tumor enriched [TE], immune enriched [IE], and mixed tumor-immune [interface]) for analysis. Following pathologist selection of ROIs, the H&E selections were aligned with immunofluorescence staining (panCK and CD45). Masking was performed in the GeoMx software to select the tumor-predominant component of interface regions (TX) and immune-predominant component (IX). Individual ROIs were then interrogated for 52 DSP-based markers. (B) An H&E slide with selected ROIs is shown along with the panCK and CD45 immunofluorescence masking images. AOI = area of interest; DSP = digital spatial profiling; ROI = region of interest.
2.3. DNA sequencing
Whole-exome sequencing was performed on formalin-fixed paraffin-embedded (FFPE) tumor tissue collected before NACI, with PBMC-matched normal. Sequencing libraries were prepared from 100 ng of DNA using Illumina (San Diego, California, USA) DNA Prep with Enrichment kit and Illumina Exome Panel. During library preparation, DNA was amplified with IDT for Illumina DNA Unique Dual Indexes, resulting in a pool of 12 dual-indexed paired-end libraries sequenced on Illuminas NovaSeq 6000 (2 æ 100) version 1.5.
2.4. Tumor mutational burden calculation
For LCCC1520, BAM files from Abra were passed through Strelka [16], Mutect2 [17,18], and Cadabra [19]. High confidence variants were kept. Tumor mutational burden (TMB) was calculated from small indels, and single base pair substitutions were identified by whole-exome sequencing. Samtools [20] provided coverage depth estimates at each exome location, and bases adequately covered by reads were counted. For BLASST-1, TMB was calculated with the R maftools package [21]. High TMB was defined as more than ten mutations per megabase [22]. We assessed TMB as a continuous variable and dichotomized into high/low because some studies suggest that TMB has a dichotomous effect on response [22].
2.5. Elastic net
Independently within BLASST-1 and LCCC1520, continuous variables were standardized (mean = 0, standard deviation = 1) and samples with nonavailable values were omitted. Generalized linear regression with elastic net (EN) regularization and ten-fold cross-validation was used to build optimal logistic models for response using the R package caret [35] (tuneLength = 15). Predictor -coefficient means and 95% confidence intervals were calculated using ten-fold cross-validation with optimal models tuning parameters.
2.6. Statistical analysis
Fishers exact (categorical variables) and Wilcoxon (continuous) tests were used for comparisons, with Benjamini-Hochberg false discovery rate (FDR) correction. Model performance to predict NACI response was compared using DeLong’s tests of area under the receiver operating characteristic the curve. Differences between patient groups in terms of survival with NACI were compared by log-rank tests of Kaplan-Meier curves of OS (time from trial therapy initiation) censored at the last follow-up.
2.7. Data availability
The Supplementary material contains data and code for figures.
3. Results
3.1. DSP-based markers including PD-L1, CD34, FAP-alpha, and CD127 are differentially expressed by NACI response
We utilized DSP to quantify expression of 52 protein markers in specific ROIs from FFPE slides. An ROI-agnostic assessment, without FDR correction, revealed numerous markers differentially expressed by response (n = 15; Fig. 2A). Responders exhibited higher PD-L1 (significant after FDR correction; fold change of responders over nonresponders = 1.3) as well as CTLA4 (1.2), HLA-DR (1.3), and beta-2-microglobulin (1.2; not significant after FDR correction), suggesting upregulation of immune checkpoints and enhanced antigen presentation. Nonresponders had higher (significant after FDR correction) CD34 (fold change, responders over nonresponders = 0.63), FAP-alpha (0.76), and CD127 (0.73), along with fibronectin (0.70) and CD25 (0.83; not significant after FDR correction), suggesting increased vascularity, more T regulatory (Treg) cells, and immunosuppressive stroma.
Fig. 2 –
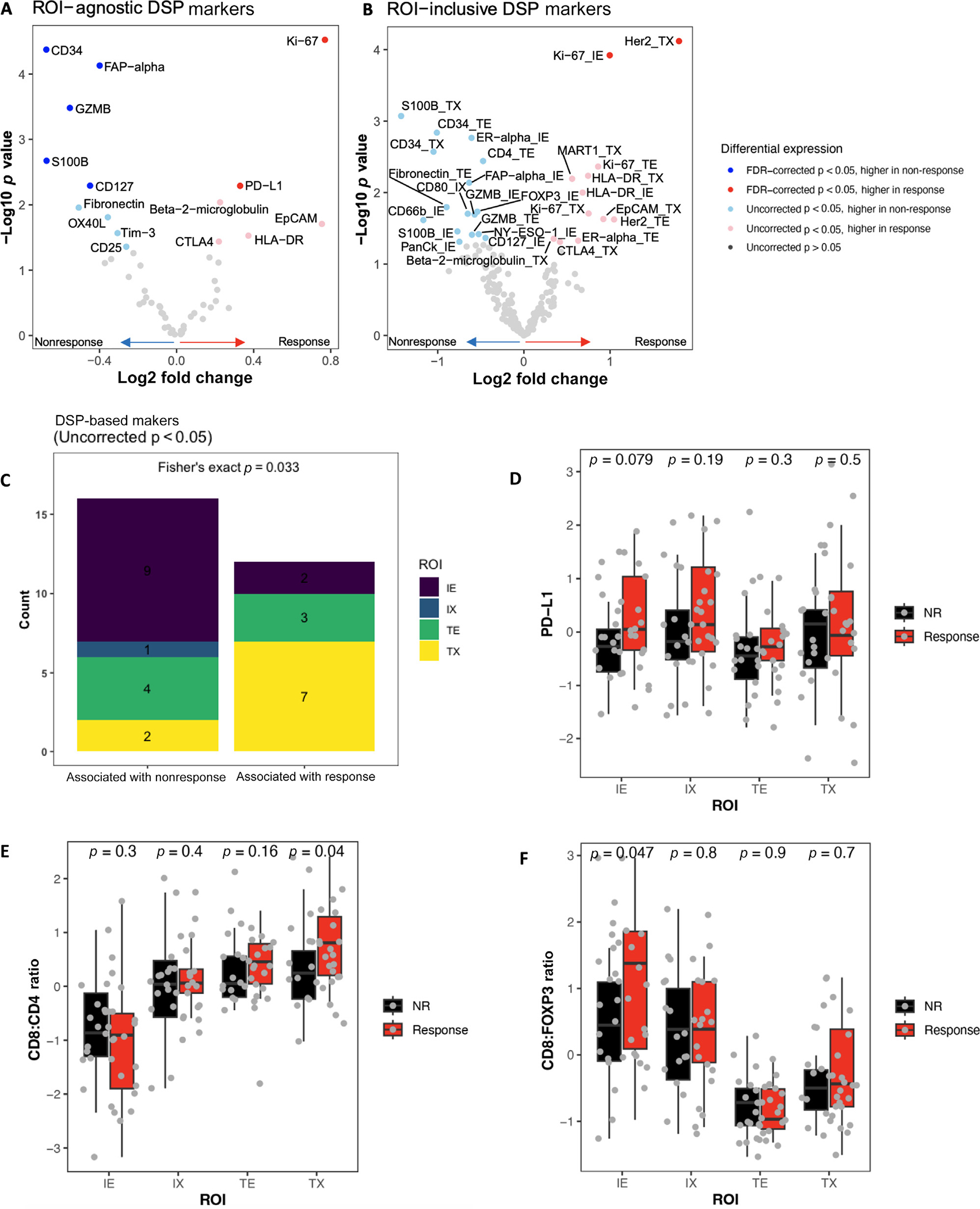
DSP-based markers are associated with response. The differential expression in responders versus nonresponders of DSP-based markers from BLASST-1 and LCCC1520 was compared in an (A) ROI-agnostic or (B) ROI-inclusive manner. (C) DSP-based markers with DE by response were compared by ROI type. (D) The levels of PD-L1, and the ratios of (E) CD8 to CD4 and (F) CD8 to FOXP3 were compared by ROI in responders versus nonresponders. DE = differentially expressed; DSP = digital spatial profiling; FDR = false discovery rate; IE = immune enriched; IX = immune interface; NR = nonresponse; ROI = region of interest; TE = tumor enriched; TX = tumor interface.
3.2. DSP-based markers including Ki-67 and Her2 have spatial-specific context
To evaluate associations of DSP-based markers with response within specific ROIs, markers were binned by ROI type (TE/IE/TX/IX). Following FDR correction, only Ki-67_IE (fold change of responders over nonresponders = 2.0) and Her2_TX (3.5) were associated with response. Prior to correction, 28 marker-ROI combinations were significant (Fig. 2B and Supplementary Table 2). Analyzing these associations, we found that most markers associated with nonresponse were in IE, while most markers associated with response were in TX (Fig. 2C).
We considered the possibility that the lower number of significant associations in the ROI-inclusive analysis was due to the stringent correction because of the larger number of comparisons. To address this, we directly investigated the known candidate biomarkers of NACI response. Immune cell PD-L1 expression, a putative predictive biomarker for atezolizumab response in metastatic UC [23], was significantly associated with response in an ROI-agnostic context. When PD-L1 was analyzed across ROIs, no significant differences in abundance between individual ROIs were observed (Fig. 2D), suggesting that PD-L1 may be a biomarker for ICI response independent of its spatial distribution. Additionally, we analyzed immune effector-to-suppressor ratios and found that TX CD8:CD4 and IE CD8:FOXP3 were higher in responders (Fig. 2E and 2F), suggesting that the effector-to-suppressor ratio is associated with response in specific ROIs. In conclusion, both ROI-inclusive and ROI-agnostic protein expression may be valuable putative predictive biomarkers for NACI response.
3.3. EN modeling using ROI-inclusive DSP defines features associated with response
In prior work, we found that multivariable EN models--a statistical methodology that selects features that, together, best predict a response variable--predicted ICI response better than single features alone (eg, TMB) in metastatic UC [24]. Most features associated with response were RNA signatures related to tumor immune milieu. We hypothesized that the inclusion of ROI could improve the predictive ability of our model. We found no significant differences in LCCC1520 and BLASST-1 patient characteristics (Table 1 and Supplementary Fig. 1), so we used both cohorts to perform EN generalized linear modeling of ROI-inclusive DSP-based markers to predict NACI response, training on BLASST-1, and testing validation performance on LCCC1520. Predictors with the largest coefficients associated with response included Ki-67_TE, Ki-67_TX, Her2_TE, and HLA-DR_IE, many of which are in line with our findings above (Fig. 3A and Supplementary Fig. 2). In contrast, CD14_TE expression was associated with nonresponse, perhaps implicating a suppressive role for a subset of tumor-associated macrophages. The BLASST-1 trained ROI-inclusive model performance was validated on LCCC1520 (Fig. 3B) and predicted response well (area under the curve [AUC] = 0.827, p = 0.009 vs null [AUC = 0.5]).
Table 1 -.
Clinical characteristics
| BLASST1 |
LCCC1520 |
|||
|---|---|---|---|---|
| Nonresponder (N = 9) | Responder (N = 9) | Nonresponder (N = 9) | Responder (N = 9) | |
|
| ||||
| Age | ||||
| Median (Q1, Q3) | 67 (59, 69) | 63 (57, 71) | 71 (63, 73) | 64 (61, 66) |
| Female gender, n (%) | 5 (56) | 3 (33) | 2 (22) | 0 |
| Stage, n (%) | ||||
| T2 | 8 (89) | 8 (89) | 6 (67) | 7 (77) |
| T3 | 1 (11) | 0 | 3 (33) | 1 (11) |
| T4 | 0 | 1 (11) | 0 | 1 (11) |
| Positive N stage, n (%) | 1 (11) | 1 (11) | 0 | 0 |
| ECOG restricted (1), n (%) | 1 (11) | 5 (56) | 3 (33) | 4 (44) |
| TMB | ||||
| Median (Q1, Q3) | 4.47 (2.43, 6.13) | 8.77 (4.20, 10.6) | 8.56 (5.02, 10.6) | 6.86 (6.00, 9.91) |
| Missing, n (%) | 0 | 0 | 2 (22) | 1 (11) |
ECOG = Eastern Cooperative Oncology Group; TMB = tumor mutational burden.
Fig. 3 –
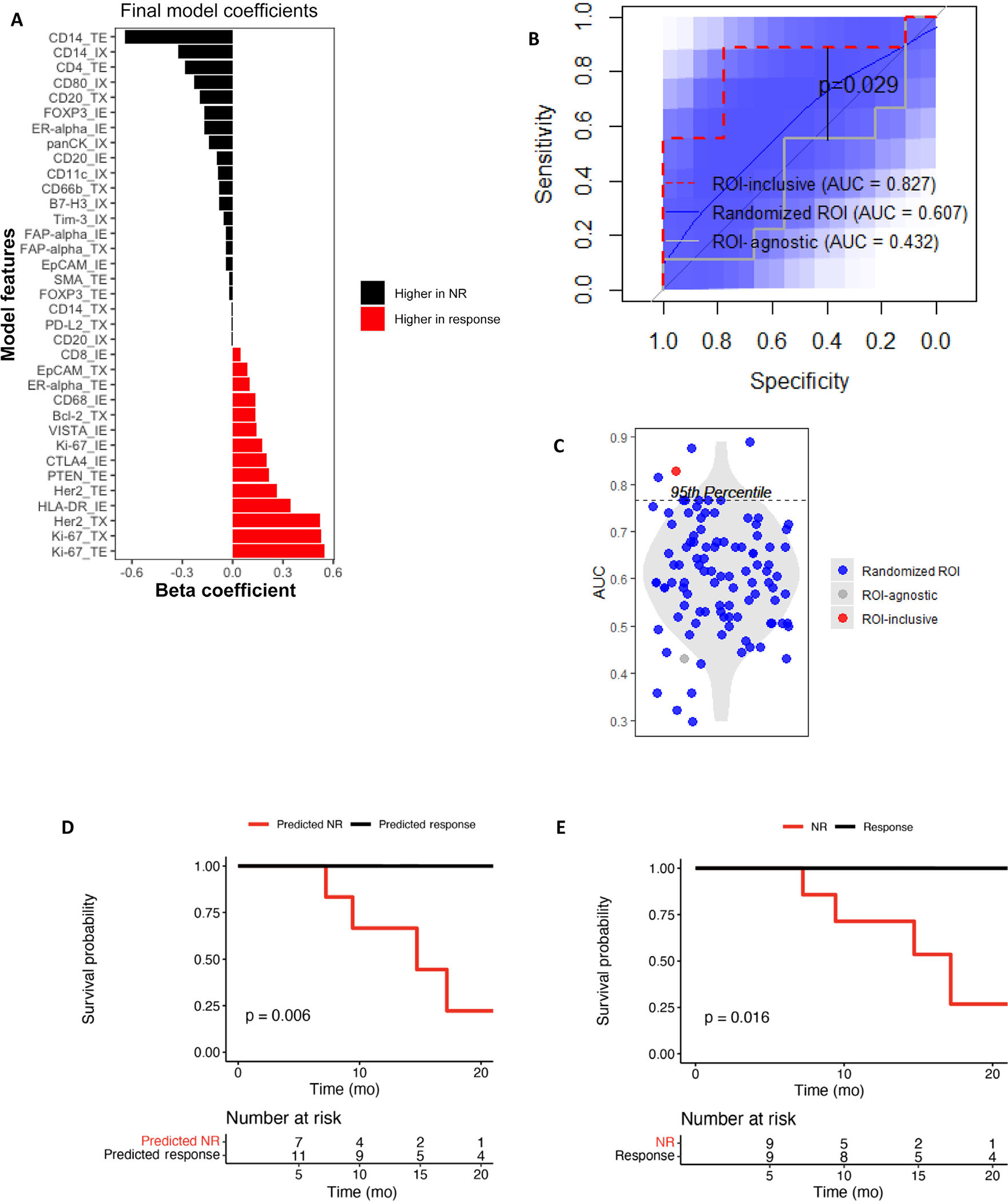
Including ROI improves outcome prediction. The predictor coefficients are shown for an elastic net model trained on BLASST-1 to predict response from the (A) DSP-based markers by ROI. (B) Performance in predicting response in LCCC1520 is compared for three elastic net models: (1) incorporating the actual ROI associated with each DSP-based marker, (2) incorporating randomized ROIs for each DSP-based marker in LCCC1520 with 100 repeat randomizations, and (3) averaging the DSP-based marker levels across ROIs in BLASST-1 and LCCC1520. 95% confidence intervals of the randomized ROI ROC curves are shown in blue. (C) The AUCs of ROC curves from Figure 3A are compared. The (D) survival curves of the response predictions from the EN model and (E) actual patient responses are shown with log-rank p-values. AUC = area under the curve; DSP = digital spatial profiling; EN = elastic net; NR = nonresponse; ROI = region of interest.
To determine whether the ROIs of DSP-based markers improved model accuracy, we trained and tested an ROI-agnostic EN model, which performed poorly (AUC = 0.432; Fig. 3B). Given that the ROI-inclusive model had four times more features, we controlled for feature numbers by randomly reassigning ROIs to DSP-based markers. These control models also performed poorly (mean AUC = 0.607; Fig. 3B) and significantly worse than the ROI-inclusive model (Fig. 3C), suggesting that ROI improves performance of EN models predicting NACI response from DSP-based markers.
3.4. EN model of response is prognostic
It is well established that NAC pathologic response is associated with OS. To determine whether our NACI response EN model held prognostic value, we examined OS based on the model predictions and found that OS of predicted responders was longer than that of predicted nonresponders (p = 0.006; Fig. 3D). Interestingly, the difference in OS by EN modelpredicted response was comparable with the difference in OS by actual pathologic response (p = 0.016; Fig. 3E), suggesting that pretreatment DSP also predicts OS well.
3.5. Intratumoral heterogeneity of DSP-based marker expression influences the number of assessed ROIs required for accurate assessment
ROI selection requires time-intensive pathologist selection of TE, IE, and interface ROIs. We sought to determine the minimum of each ROI type (TE, IE, TX, and IX) necessary for accurate prediction. If DSP-based marker variation between ROIs is low, sampling fewer ROIs might maintain accuracy while reducing labor. We found high within-sample variation in DSP-based markers between ROIs correlated with intertumoral variation (Fig. 4A), suggesting that reducing sampled ROIs per type could compromise model accuracy. Additionally, within-ROI variation is less than between-ROI variation for each DSP-based marker (Fig. 4B), suggesting that inclusion of ROIs can improve model predictive accuracy. In testing EN models using fewer samples per ROI in the validation set (Supplementary Fig. 3AC), we observed an improvement in accuracy as the number of samples per ROI increased, with an AUC of 0.827 when all four samples per ROI were used (Fig. 4C). These findings highlight the importance of increasing training cohort size to account for inherent heterogeneity and develop a model that accurately predicts NACI response across diverse MIBC tumors seen in clinical practice.
Fig. 4 –
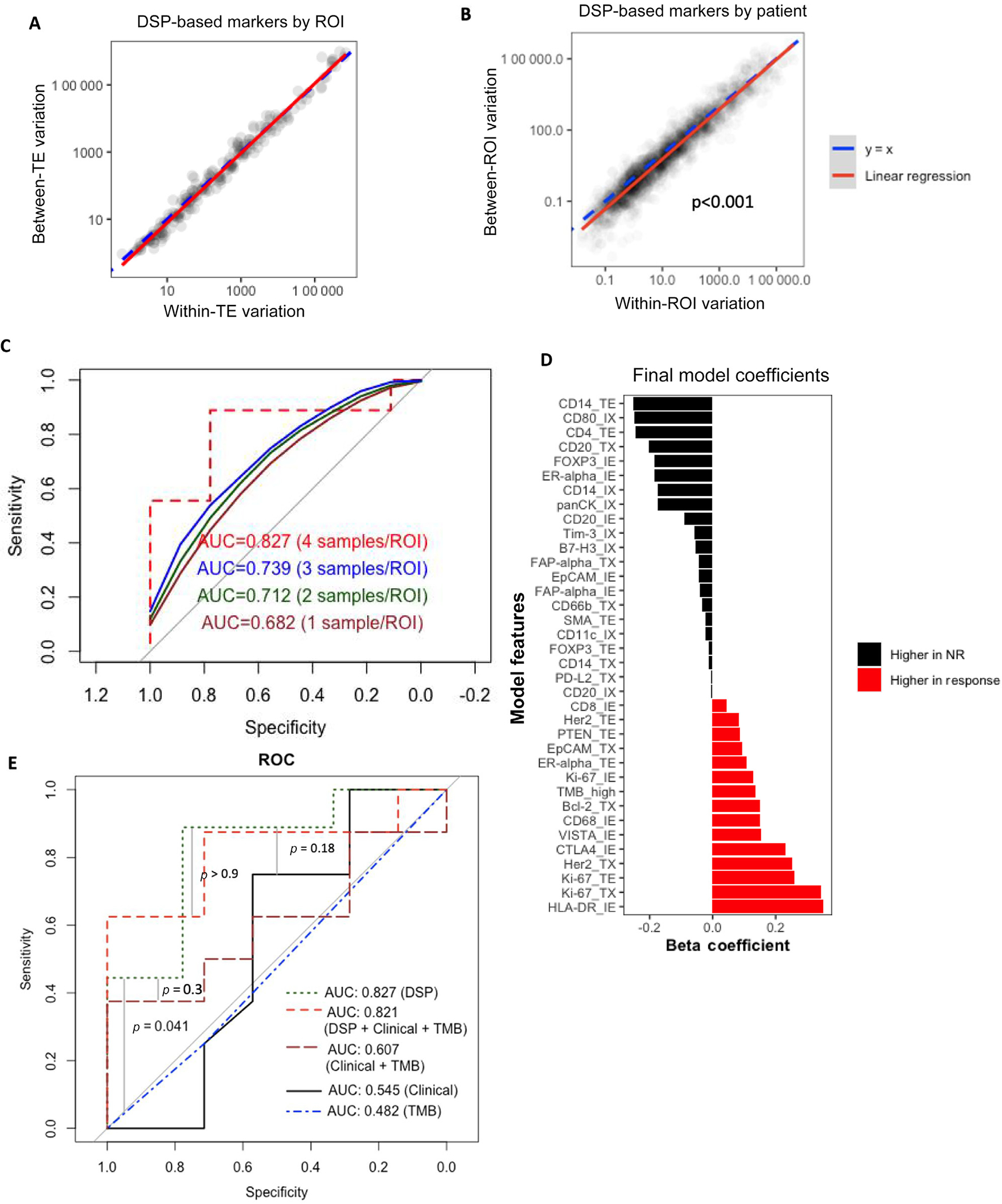
Accurate response prediction requires DSP with multiple samples per ROI but does not require clinical data or TMB. (A) The variations between tumors and within tumors were compared for each DSP-based markerROI pair. (B) The variations between ROIs and within ROIs were compared for each DSP-based marker-patient pair. (C) Samples were randomly selected 100 times for each ROI in each patient in LCCC1520. The performance of the elastic net model generated in BLASST-1 is shown for the LCCC1520 data with only one, two, or three samples per ROI per patient, with the average ROC curves across the 100 selections shown. (D) The predictor coefficients are shown for an elastic net model trained on BLASST-1 to predict response from the DSP-based markers by ROI plus clinical variables. (E) The performance in predicting response in LCCC1520 is shown for five elastic net models: the full model incorporating DSP-based markers by ROI plus the clinical variables and TMB, the model incorporating only DSP-based markers by ROI, a model with only TMB, a model with only the clinical variables, and a model with clinical variables plus TMB. AUC = area under the curve; DSP = digital spatial profiling; NR = nonresponse; ROC = receiver operating characteristics; ROI = region of interest; TE = tumor enriched; TMB = tumor mutational burden.
3.6. DSP-based marker exclusive predictor is more accurate than TMB alone or a clinicoproteomic model
Current nomograms mainly rely on clinical variables for risk/survival stratification; we asked whether the inclusion of available clinical features (age, Eastern Cooperative Oncology Group performance status, T stage, N stage, and sex) and TMB (continuous and dichotomized TMB high/low) in a clinicoproteomic EN response model would improve prediction accuracy. Training the model on BLASST-1, the only non-DSP feature among the cross-validated coefficients (Supplementary Fig. 4) and final model coefficients (Fig. 4D) was dichotomized TMB (high/low). The integrated clinicoproteomic EN model outperformed random in LCCC1520 (AUC = 0.821, p = 0.014). TMB alone has been suggested as a biomarker in other trials, but a TMB-only model performed worse than the integrated clinicoproteomic EN model (p = 0.041). Overall, the ROI-inclusive DSP-only model was no worseif not betterat predicting response than models with combinations of DSP, clinical, and TMB features (Fig. 4E), suggesting that DSP has potential to predict NACI response accurately.
3.7. A multiplex immunohistochemistry or IF panel could predict NACI response
Using lasso regularization instead of EN, we identified a smaller panel of nine DSP-based markers to predict NACI response (Fig. 5A). The lasso model (AUC = 0.753) predicted response comparably with the EN model (AUC = 0.827, lasso vs EN, p = 0.4; Fig. 5B). The lasso model also predicted survival (log-rank p = 0.001; Fig. 5C). The lasso model could be utilized with multiplex immunohistochemistry or IF by (1) staining one or multiple slides with DAPI, panCK, and CD45; (2) identifying panCK- and CD45-rich tumor regions; (3) calculating expression of the nine lasso markers in panCK-high (TE), CD45-high (IE), and interface regions (TX and IX); and then (4) multiplying marker expression by the lasso model coefficients (Fig. 5D).
Fig. 5 –
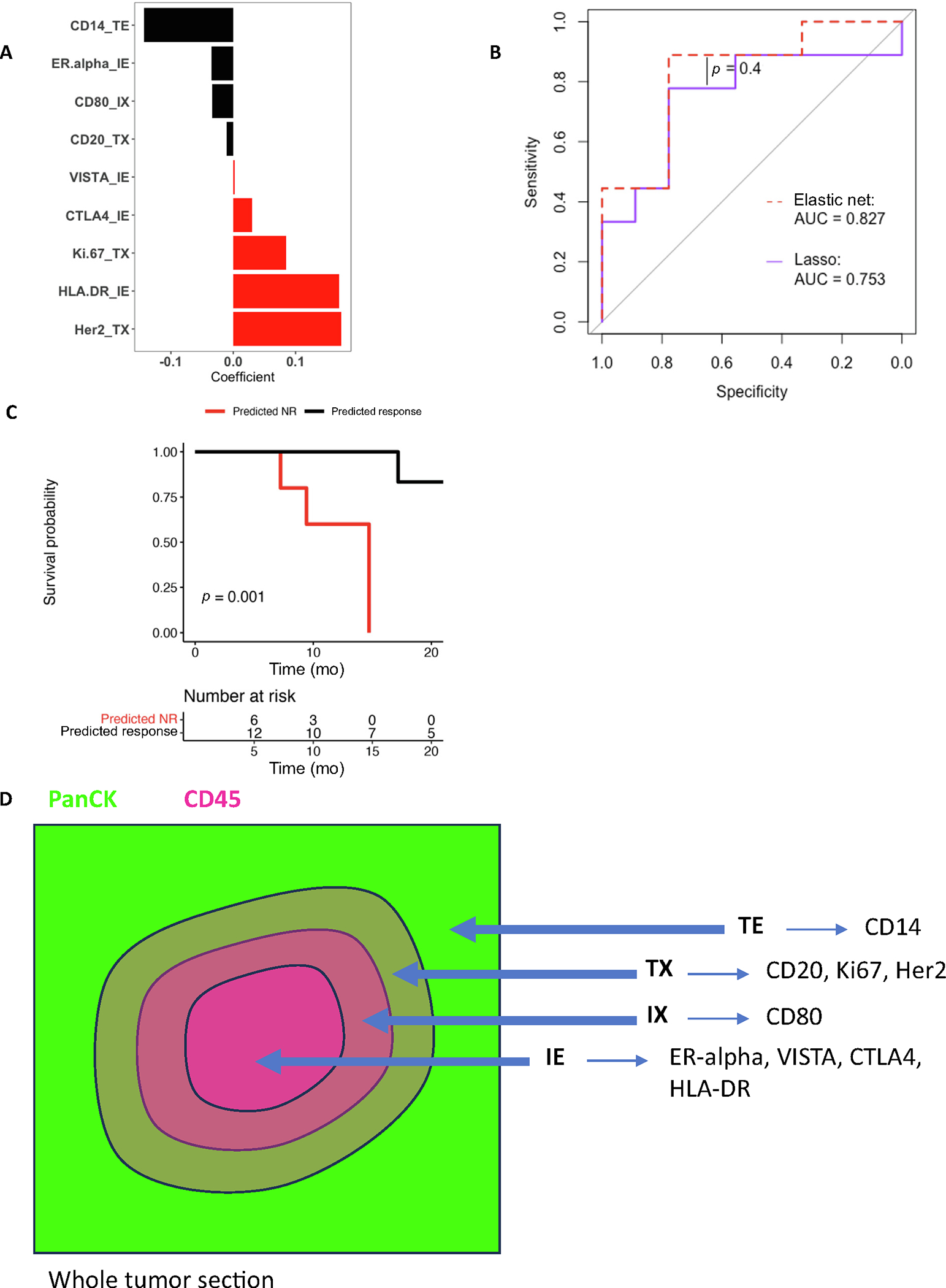
Lasso regression identifies features for multiplex IHC/IF. (A) The predictor coefficients are shown for a lasso model trained on BLASST-1 to predict response from the DSP-based markers by ROI. (B) Performance in predicting response in LCCC1520 is compared between the DSP-only EN model and the lasso model. (C) Survival is compared between lasso modelpredicted responders and nonresponders in LCCC1520. (D) A schematic for ROI approximation and lasso model prediction using multiplex IF/IHC is shown. AUC = area under the curve; DSP = digital spatial profiling; EN = elastic net; IE = immune enriched; IF = immunofluorescence; IHC = immunohistochemistry; IX = immune interface; NR = nonresponse; ROI = region of interest; TE = tumor enriched; TX = tumor interface.
4. Discussion
Multiple studies have examined NACI effectiveness in MIBC, consistently demonstrating pathologic downstaging (<ypT2) rates of 5570% [10,11,25], but toxicity can occur and many patients do not benefit. Recent technology enables protein expression assessment in specific ROIs, permitting multiple measurements of different tumor ROIs. We performed DSP to analyze 58 protein tumor and immunobiology markers in a clinical-proteomic NACI response model. Intratumoral heterogeneity was high, but Ki67 in tumor and immune enriched ROIs and MHC class II (HLA-DR) in immune enriched ROIs were associated with response. Potentially, myeloid cells and Tregs play a role in suppressing response. An integrated clinicoproteomic EN model predicted response with high accuracy (AUC = 0.827), and DSP alone outperformed clinical variables and TMB.
While intratumoral heterogeneity at histologic and genomic levels in UC is documented, immunobiology heterogeneity is less explored [26–28]. Tumor stromal interactions appear important in immunotherapy response, with TGF-beta and EMT/stromal gene signatures correlating with decreased response to single-agent PD-1 or PD-L1 blockade [15,29]. However, presence of increased stroma was inconsistently associated with resistance in neoadjuvant immunotherapy trials [8,30]. In our EN model of NACI response, FAP-alpha in immune and tumor ROIs and SMA expression in the interface-panCK ROI were associated with decreased response. FAP-alpha and SMA are associated with cancer-associated fibroblasts [31], tumor development [32], and ICI resistance [32]. Our work extends previous studies and demonstrates the importance of spatial profiling tumor stroma in mediating NACI response. A recent study involving the PURE-01 trial of neoadjuvant pembrolizumab for MIBC identified five genetic/transcriptomic signatures associated with response [33]. TMEs were evaluated by proteomic DSP in pretreatment tumors (five complete responders and three nonresponders). TMEs of complete responders were relatively concordant, while TMEs from nonresponders exhibited significant inter-TME heterogeneity among subtypes.
DSP predicted response with high accuracy (AUC = 0.827) without clinical variables or TMB. None of these variables, or in combination, predicted response as well as DSP alone. Additionally, the only non-DSP variable included in the clinicoproteomic EN model was dichotomous TMB, possibly because EN modeling may remove features such as clinical T stage or age that have a correlation with DSP-based markers (Supplementary Fig. 5). Alternatively, DSP-based markers may have stronger associations with NACI response than several clinical variables previously linked with ICI response. Future efforts should explore whether a multiplex immunofluorescent staining approach using features from our final EN model can match DSPs predictive accuracy. Additionally, promising putative response biomarkers such as gut and bladder microbiome features should be considered.
Our study suggests that patient NACI response prediction from protein markers depends on tumor architecture as identified through DSP. Increasing the samples of ROIs per patient improved our response model. Sampling more ROIs per tumor, although more costly, could improve response prediction, and whole-slide high-resolution DSP platforms could maximize response prediction.
Our study has several limitations. Our hypothesis-generating small studys sample size (and event number) limits statistical power. However, we collected samples from two independent similarly designed trials, and our EN response model was trained and validated independently. Another limitation is the limited number of DSP-based markers. We delineated broad markers for CD4, CD8, fibroblast, myeloid, and tumor cells, but were limited in subsetting these cells into more specific populations (eg, MDSC). Possible biases in sampling and patient selection may affect our results. Ultimately, the EN model of predictive DSP biomarkers should undergo further refinement, prospective clinical validation, and assessment of clinical utility before potential implementation in clinical trials or practice. While our model performed well on an independent validation dataset (LCCC1520), additional, larger retrospective and prospective datasets would be desirable.
5. Conclusions
In summary, we present a DSP analysis of pretreatment tumors from two MIBC NACI trials, shedding light on intratumoral heterogeneity of DSP-based marker expression and the importance of marker location within tumor architecture for predicting NACI response. Our DSP EN response model represents a first step in developing predictive biomarkers that could become integral components of future clinical trials.
Supplementary Material
Acknowledgments:
We thank the members of the Kim Laboratory for useful discussions. We thank the UNC Pathology Services Core (PSC) for the technical assistance.
Funding/Support and role of the sponsor:
This work was supported by The Leo & Anne Albert Institute for Bladder Cancer Care and Research, the University Cancer Research Fund (UCRF; William Y. Kim), the National Cancer Institute R01-CA241810 (William Y. Kim and Benjamin G. Vincent), the UNC Integrated Translational Oncology Program (iTOP) T32-CA244125 (Wolfgang Beckabir), and funding from the Thomas M. Mohr Fund for Bladder Cancer Research and the Waterproof Foundation. Additional funding was provided through the Cancer Center Core Support Grant (P30- CA016086; William Y. Kim and Matthew I. Milowsky).
Financial disclosures:
Wolfgang Beckabir certifies that all conflicts of interest, including specific financial interests and relationships and affiliations relevant to the subject matter or materials discussed in the manuscript (eg, employment/affiliation, grants or funding, consultancies, honoraria, stock ownership or options, expert testimony, royalties, or patents filed, received, or pending), are the following: Petros Grivas: consulting for 4D Pharma, Aadi Bioscience, Astellas, Asieris Pharmaceuticals, AstraZeneca, BostonGene, Bristol Myers Squibb, CG Oncology, Dyania Health, Exelixis, Fresenius Kabi, Genentech, Gilead Sciences, Guardant Health, ImmunityBio, Infinity Pharmaceuticals, Janssen, Lucence, Merck KGaA, Mirati Therapeutics, MSD, Pfizer, PureTech, QED Therapeutics, Regeneron, Roche, Seattle Genetics, Silverback Therapeutics, Strata Oncology, and UroGen Pharma; and institutional research funding from Bavarian Nordic, Bristol Myers Squibb, Clovis Oncology, Debiopharm Group, G1 Therapeutics, Gilead Sciences, GlaxoSmithKline, Merck KGaA, Mirati Therapeutics, MSD, Pfizer, and QED Therapeutics. Marc Bjurlin: clinical Investigator for Urogen, ImmunityBio, Janssen Research & Development, LLC, and Anchiano Therapeutics; paid consultant for Urogen; and paid proctor for Intuitive. Matthew Milowsky: stock and other ownership interests at Pfizer, Merck, and Gilead Sciences; consulting or advisory role at Loxo/Lilly; research funding from Merck, Roche/Genentech, Bristol-Myers Squibb, Mirati Therapeutics, Incyte, Seagen, G1 Therapeutics, Alliance Foundation Trials, Alliance for Clinical Trials in Oncology, Clovis Oncology, Arvinas, ALX Oncology, Loxo, and Hoosier Cancer Research Network; and other relationship with Elsevier, Medscape, and Research to Practice. William Kim: consulting for Focal Medical, Oncorev, Janssen. Stock ownership: Abbvie, Amgen, BeiGene, Bristol Myers Squibb, Moderna, Natera, Revolution Medicines, Tango Therapeutics, Zentalis, Focal Medical. Research Funding: Merck.
Footnotes
This study was presented in part at the 8th Annual Leo & Anne Albert Institute Bladder Cancer Symposium.
Peer Review Summary
Peer Review Summary and Supplementary data to this article can be found online at https://doi.org/10.1016/j.eururo.2023.11.008.
References
- [1].International collaboration of Trialists, Medical Research Council Advanced Bladder Cancer Working Party (now the National Cancer Research Institute Bladder Cancer Clinical Studies Group), European Organisation for Research and Treatment of Cancer Genito-Urinary Tract Cancer Group, et al. International phase III trial assessing neoadjuvant cisplatin, methotrexate, and vinblastine chemotherapy for muscle-invasive bladder cancer: long-term results of the BA06 30894 trial. J Clin Oncol 2011;29:2171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Grossman HB, Natale RB, Tangen CM, et al. Neoadjuvant chemotherapy plus cystectomy compared with cystectomy alone for locally advanced bladder cancer. N Engl J Med 2003;349:859–66. [DOI] [PubMed] [Google Scholar]
- [3].Iyer G, Tully CM, Zabor EC, et al. Neoadjuvant gemcitabine-cisplatin plus radical cystectomy-pelvic lymph node dissection for muscle-invasive bladder cancer: a 12-year experience. Clin Genitourin Cancer 2020;18:387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Balar AV, Kamat AM, Kulkarni GS, et al. Pembrolizumab monotherapy for the treatment of high-risk non-muscle-invasive bladder cancer unresponsive to BCG (KEYNOTE-057): an open-label, single-arm, multicentre, phase 2 study. Lancet Oncol 2021;22:919–30. [DOI] [PubMed] [Google Scholar]
- [5].Bajorin DF, Witjes JA, Gschwend JE, et al. Adjuvant nivolumab versus placebo in muscle-invasive urothelial carcinoma. N Engl J Med 2021;384:2102–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Powles T, Park SH, Voog E, et al. Avelumab maintenance therapy for advanced or metastatic urothelial carcinoma. N Engl J Med 2020;383:1218–30. [DOI] [PubMed] [Google Scholar]
- [7].Bellmunt J, de Wit R, Vaughn DJ, et al. Pembrolizumab as second-line therapy for advanced urothelial carcinoma. N Engl J Med 2017;376:1015–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Powles T, Kockx M, Rodriguez-Vida A, et al. Clinical efficacy and biomarker analysis of neoadjuvant atezolizumab in operable urothelial carcinoma in the ABACUS trial. Nat Med 2019;25:1706–14. [DOI] [PubMed] [Google Scholar]
- [9].Necchi A, Anichini A, Raggi D, et al. Pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive urothelial bladder carcinoma (PURE-01): an open-label, single-arm, phase II study. J Clin Oncol 2018;36:3353–60. [DOI] [PubMed] [Google Scholar]
- [10].Funt SA, Lattanzi M, Whiting K, et al. Neoadjuvant atezolizumab with gemcitabine and cisplatin in patients with muscle-invasive bladder cancer: a multicenter, single-arm, phase II trial. J Clin Oncol 2022;40:1312–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Rose TL, Harrison MR, Deal AM, et al. Phase II study of gemcitabine and split-dose cisplatin plus pembrolizumab as neoadjuvant therapy before radical cystectomy in patients with muscle-invasive bladder cancer. J Clin Oncol 2021;39:3140–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Gupta S Results from BLASST-1 (Bladder Cancer Signal Seeking Trial) of nivolumab, gemcitabine, and cisplatin in muscle invasive bladder cancer (MIBC) undergoing cystectomy. Genitourinary Cancers Symposium, 2020. [Google Scholar]
- [13].Cathomas R. Safety and efficacy of perioperative cisplatin/gemcitabine and durvalumab for operable muscle-invasive urothelial carcinoma: SAKK 06/17. American Society of Clinical Oncology Genitourinary Cancers Symposium, 2021. [Google Scholar]
- [14].Almagro J, Messal HA, Elosegui-Artola A, van Rheenen J, Behrens A. Tissue architecture in tumor initiation and progression. Trends Cancer 2022;8:494–505. [DOI] [PubMed] [Google Scholar]
- [15].Mariathasan S, Turley SJ, Nickles D, et al. TGF attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Saunders CT, Wong WSW, Swamy S, Becq J, Murray LJ, Cheetham RK. Strelka: accurate somatic small-variant calling from sequenced tumornormal sample pairs. Bioinformatics 2012;28:1811–7. [DOI] [PubMed] [Google Scholar]
- [17].Benjamin D, Sato T, Cibulskis K, Getz G, Stewart C, Lichtenstein L. Calling somatic SNVs and indels with Mutect2. Biorxiv 2019;861054. [Google Scholar]
- [18].Cibulskis K, Lawrence MS, Carter SL, et al. Sensitive detection of somatic point mutations in impure and heterogeneous cancer samples. Nat Biotechnol 2013;31:213–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Mose LE, Perou CM, Parker JS. Improved indel detection in DNA and RNA via realignment with ABRA2. Bioinformatics 2019;35:2966–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009;25:1754–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Mayakonda A, Lin DC, Assenov Y, Plass C, Koeffler HP. Maftools: efficient and comprehensive analysis of somatic variants in cancer. Genome Res 2018;28:1747–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Sha D, Jin Z, Budczies J, Kluck K, Stenzinger A, Sinicrope FA. Tumor mutational burden as a predictive biomarker in solid tumors. Cancer Discov 2020;10:1808–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Powles T, Eder JP, Fine GD, et al. MPDL3280A (anti-PD-L1) treatment leads to clinical activity in metastatic bladder cancer. Nature 2014;515:558–62. [DOI] [PubMed] [Google Scholar]
- [24].Damrauer JS, Beckabir W, Klomp J, et al. Collaborative study from the Bladder Cancer Advocacy Network for the genomic analysis of metastatic urothelial cancer. Nat Commun 2022;13:6658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Peyrottes A, Ouzaid I, Califano G, Hermieu JF, Xylinas E. Neoadjuvant immunotherapy for muscle-invasive bladder cancer. Medicina 2021;57:769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Faltas BM, Prandi D, Tagawa ST, et al. Clonal evolution of chemotherapy-resistant urothelial carcinoma. Nat Genet 2016;48:1490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Christensen E, Nordentoft I, Vang S, et al. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci Rep 2018;8:1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Warrick JI, Hu W, Yamashita H, et al. FOXA1 repression drives lineage plasticity and immune heterogeneity in bladder cancers with squamous differentiation. Nat Commun 2022;13:6575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Wang L, Saci A, Szabo PM, et al. EMT- and stroma-related gene expression and resistance to PD-1 blockade in urothelial cancer. Nat Commun 2018;9:3503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Necchi A, Raggi D, Gallina A, et al. Impact of molecular subtyping and immune infiltration on pathological response and outcome following neoadjuvant pembrolizumab in muscle-invasive bladder cancer. Eur Urol 2020;77:701–10. [DOI] [PubMed] [Google Scholar]
- [31].Eskandari-Malayeri F, Rezaei M. Immune checkpoint inhibitors as mediators for immunosuppression by cancer-associated fibroblasts: a comprehensive review. Front Immunol 2022;13:996145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Xin L, Gao J, Zheng Z, et al. Fibroblast activation protein- as a target in the bench-to-bedside diagnosis and treatment of tumors: a narrative review. Frontiers Oncol 2021;11:648187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Robertson AG, Meghani K, Cooley LF, et al. Expression-based subtypes define pathologic response to neoadjuvant immune-checkpoint inhibitors in muscle-invasive bladder cancer. Nat Commun 2023;14:2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Gupta S, Zugazagoitia J, Martinez-Morilla S, Fuhrman K, Rimm DL. Digital quantitative assessment of PD-L1 using digital spatial profiling. Lab Invest 2020;100:1311–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kuhn M Building predictive models in R using the caret package. J Stat Softw 2008;28:1–26.27774042 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The Supplementary material contains data and code for figures.