

Abstract
Background
Bronchopulmonary dysplasia (BPD) is a common complication in preterm infants. BPD is associated with poor long‐term respiratory and neurodevelopmental outcome and increased mortality. The prophylactic use of agents that modulate inflammation such as pentoxifylline, a synthetic methylxanthine and phosphodiesterase inhibitor, may reduce the incidence of BPD.
Objectives
The primary objective of this review was to determine the effect of pentoxifylline on the incidence of BPD, death prior to 36 weeks postmenstrual age (PMA), and BPD or death prior to 36 weeks PMA in preterm neonates.
Search methods
We searched the Cochrane Neonatal Review Group Specialized Register, CENTRAL (The Cochrane Library Issue 9, 2012), EMBASE (January 1974 to September 2012), PubMed (January 1966 to September 2012), and CINAHL (January 1982 to September 2012) in September 2012. We checked references and cross‐references from identified studies. We handsearched abstracts from the proceedings of the Pediatric Academic Societies Meetings (from January 1990 to September 2012). We placed no restrictions on language.
Selection criteria
Randomised or quasi‐randomised clinical trials of systemic or nebulised pentoxifylline in preterm neonates less than 32 weeks gestational age or less than 1500 g birth weight, reporting on at least one outcome of interest, were eligible for inclusion in the review.
Data collection and analysis
We used the standard methods of the Cochrane Neonatal Review Group and The Cochrane Collaboration. Two review authors (SMS and SK) independently searched the literature as described above and selected studies. Any disagreements were resolved by discussion involving all review authors.
Main results
We identified one randomised clinical trial eligible for inclusion in this review. This study compared the use of nebulised pentoxifylline versus placebo for prevention of BPD in 100 preterm infants and was at high risk of bias due to lack of blinding of intervention and outcome assessors, and incomplete outcome data. There was no statistically significant effect of nebulised pentoxifylline versus placebo on individual outcomes of BPD at 36 weeks PMA or on death prior to 36 weeks PMA. There was no significant effect of nebulised pentoxifylline on intraventricular haemorrhage, periventricular leukomalacia, sepsis, or patent ductus arteriosus (PDA) requiring ligation. The study did not report any of the other secondary outcomes considered for this review. Reporting of adverse events was very limited and did not allow for reliable judgement on the incidence of such events. No long‐term outcomes were reported.
Authors' conclusions
There is insufficient evidence to determine the safety and efficacy of pentoxifylline for prevention of BPD in preterm neonates. We encourage researchers to conduct clinical trials to confirm or refute the role of pentoxifylline for prevention of BPD in preterm neonates. These trials should report on clinically important outcomes and, ideally, on long‐term neurodevelopmental outcome.
Keywords: Humans; Infant, Newborn; Bronchopulmonary Dysplasia; Bronchopulmonary Dysplasia/mortality; Bronchopulmonary Dysplasia/prevention & control; Gestational Age; Infant, Premature; Infant, Premature, Diseases; Infant, Premature, Diseases/mortality; Infant, Premature, Diseases/prevention & control; Pentoxifylline; Pentoxifylline/therapeutic use; Phosphodiesterase Inhibitors; Phosphodiesterase Inhibitors/therapeutic use; Randomized Controlled Trials as Topic
Plain language summary
Pentoxifylline for the prevention of bronchopulmonary dysplasia in preterm infants
Babies who are born early (preterm) often suffer from long‐lasting breathing problems known as bronchopulmonary dysplasia, which can lead to poor health in childhood and adulthood. Drugs that act on the body's self‐defense system may help to lower the risk of long‐lasting breathing problems. Pentoxifylline is one such drug. The main aim of this review was to find out whether pentoxifylline compared with placebo (an inactive drug) or no drug offers important advantages to babies born early. Only one study of moderate size and quality was identified in this review. This study did not show strong evidence that pentoxifylline offers important advantages to babies born early. We have therefore been unable to determine the effects of pentoxifylline in preventing long‐lasting breathing problems in babies born early. Future high‐quality studies are needed to answer this question.
Background
Description of the condition
Bronchopulmonary dysplasia (BPD) is a common complication in preterm infants, especially in those born below 28 weeks gestational age (GA) who require ventilatory assistance for respiratory distress syndrome (RDS). Different definitions of BPD have been used in the past. The simple definition of this chronic respiratory disorder as oxygen dependency at 36 weeks postmenstrual age (PMA) was validated as a modifiable outcome in large randomised controlled trials (RCT) and as an important predictor of death and long‐term outcome after preterm birth (summarised in Schmidt 2008). Owing to advances in perinatal care, the absolute number of extremely preterm survivors has increased, but these advances have not reduced the incidence of BPD, which continues to remain between 20% and 40% in this population (Allen 2003). BPD is associated with significant morbidity, including prolonged hospital stay, need for long‐term oxygen administration, impaired lung function, and poor neurodevelopmental outcome (Jobe 2001; Ehrenkranz 2005; Schmidt 2008). Major risk factors for BPD include low GA (Sinkin 1990), need for supplemental oxygen and positive pressure ventilation (Coalson 1995; Coalson 1999), PDA (Oh 2005), and sepsis (Hentschel 2005). Pathophysiology of BPD is complex and incompletely understood (Jobe 2001). Inflammation plays an important role in the pathogenesis of BPD (summarised in Speer 2001). In many infants with BPD, an inflammatory reaction showing high amounts of neutrophils in tracheal aspirate is evident shortly after birth, suggesting that the process may have been triggered in the perinatal period by chorioamnionitis (Watterberg 1996). A pulmonary infiltration of inflammatory cells (including activated neutrophils) is seen, associated with increased pro‐inflammatory cytokines such as tumour necrosis factor‐alpha (TNF‐α), interleukin‐8 (IL‐8), and intercellular adhesion molecule‐1 (ICAM‐1) (Speer 2003). Activation of neutrophils mediates endothelial cytotoxicity and free‐radical production (Jobe 2001), while activated complement (C5a) and IL‐8 contribute to increased levels of platelet‐activating factor (PAF), a potent lipid mediator with known injurious effects on the lungs (Speer 2001).
Description of the intervention
Pharmacological interventions aimed at reducing or modulating the inflammatory response may decrease the incidence and severity of BPD. Prophylaxis of BPD using corticosteroids is attractive due to their strong anti‐inflammatory properties. Systematic reviews on the use of systemic postnatal corticosteroids have demonstrated a reduction in BPD (Bhuta 1998; Halliday 2009; Halliday 2010). However, there are serious concerns about the short‐ and long‐term adverse effects of systemic postnatal corticosteroid therapy in preterm infants. These include hyperglycaemia, hypertension, hypertrophic obstructive cardiomyopathy, gastrointestinal bleeding and perforation, growth failure, and, most alarmingly, neurodevelopmental impairment (Halliday 2009; Halliday 2010). A systematic review of seven randomised trials on nebulised corticosteroids for prevention of BPD in preterm infants found no significant benefits or harms associated with the use of nebulised corticosteroids (Shah 2007). Given the adverse effects of systemic corticosteroids and the lack of evidence to support the use of nebulised corticosteroids, alternative treatment options are desirable.
Two other drugs that are efficacious in reducing the rate of BPD are caffeine and vitamin A. One large (n = 2006) multicentre RCT in infants of 500 g to 1250 g birth weight showed that caffeine therapy for apnoea of prematurity introduced within the first 10 days of life reduced the risk of BPD without significant adverse events (Schmidt 2006). Of 963 infants who were assigned to caffeine and who remained alive at a PMA of 36 weeks, 350 (36%) received supplemental oxygen, as did 447 of the 954 infants (47%) assigned to placebo (adjusted odds ratio (OR) 0.63; 95% confidence interval (CI) 0.52 to 0.76; P < 0.001). Positive airway pressure was discontinued one week earlier in the infants assigned to caffeine (median PMA 31.0 weeks; interquartile range 29.4 to 33.0 weeks) than in the infants in the placebo group (median PMA 32.0 weeks; interquartile range 30.3 to 34.0 weeks; P < 0.001). Furthermore, caffeine significantly improved the rate of survival without neurodevelopmental disability at a corrected age of 18 to 21 months (adjusted OR 0.77; 95% CI 0.64 to 0.93; P = 0.008) (Schmidt 2007), although statistical significance of this finding could not be demonstrated at five years of age (adjusted OR 0.82; 95% CI 0.65 to 1.03; P = 0.09) (Schmidt 2012). Extremely low birth weight infants often have low plasma concentrations of vitamin A during the first few weeks of life, and these are associated with the development of BPD (Ambalavanan 2004). In one large multicentre RCT (n = 807), intramuscular injections of vitamin A 5000 IU versus sham treatment three times a week for four weeks reduced the rate of death or BPD at 36 weeks PMA in extremely low birth weight infants (55% with vitamin A versus 62% with sham; risk ratio (RR) 0.89; 95% CI 0.80 to 0.99) (Tyson 1999). There was no difference in neurodevelopmental outcomes at 18 to 22 months (Ambalavanan 2005).
Pentoxifylline is a drug that may be an alternative for prophylaxis of BPD. Pentoxifylline is a synthetic methylxanthine derivative and non‐selective phosphodiesterase inhibitor with anti‐inflammatory and immunomodulatory properties. Pentoxifylline does not show significant bronchodilating, central nervous stimulatory, or cardiac effects at therapeutic doses compared to other methylxanthines such as caffeine or theophylline (Harris 2010). Several clinical trials indicated that intravenous pentoxifylline as an adjunct to antibiotics reduces mortality in preterm neonates with blood culture‐positive sepsis (Lauterbach 1996; Lauterbach 1999a). One systematic review of RCTs suggested safety and efficacy of intravenous pentoxifylline on mortality in preterm neonates with sepsis or necrotizing enterocolitis (NEC) (Haque 2011). The two RCTs enrolled a total of 140 preterm (less than 36 weeks PMA) neonates with suspected late‐onset (greater than seven days) sepsis to evaluate the effect of pentoxifylline on neonatal outcomes. However, the two studies reported outcomes of only the 107 randomised patients with confirmed sepsis. Some evidence suggests that the use of pentoxifylline as an adjunct to antibiotics in neonatal sepsis reduces mortality without any adverse effects, but the number of neonates studied was small and considerable methodological weaknesses existed in the included trials. Hence these results should be interpreted with caution. Reduced oxygen requirements related to the use of nebulised pentoxifylline for treatment of BPD in preterm infants were reported in one small observational study (Lauterbach 1999).
Pentoxifylline has a multitude of effects at cellular, endothelial, and vascular levels (Haque 2011). Although the use of pentoxifylline in neonates with sepsis was not associated with increased risk of bleeding (Haque 2011), its stimulating effects on fibrinolysis (Schröer 1985), in vitro inhibition of platelet aggregation (Magnusson 2008), and two case reports relating pentoxifylline to thrombocytopenia (Acharya 1997; Vedes 2011), suggest consideration of bleeding as a potential adverse event. In one meta‐analysis of trials assessing the efficacy of pentoxifylline for treatment of venous leg ulcers in adults, the use of pentoxifylline was associated with gastrointestinal disturbance in 72% of the patients (Jull 2007). No other significant adverse effects have been reported in animal or human studies (Haque 2011).
How the intervention might work
Pentoxifylline down‐regulates the production of powerful inflammatory cytokines including TNF‐α, ICAM‐1, interleukin 6 (IL‐6), and interferon‐γ (Ward 1987; Krakauer 2000). One randomised study suggests that intravenous pentoxifylline compared to placebo decreases the pulmonary inflammatory response (bronchoalveolar levels of PAF, lung tissue myeloperoxidase, and lung wet‐to‐dry weight ratio) in ventilated newborn piglets (Smalling 2004). Given its anti‐inflammatory effects, pentoxifylline may be a non‐steroidal treatment option for conditions involving an inflammatory response, including BPD, sepsis, and NEC (Harris 2010). Using nebulised rather than systemic pentoxifylline for preventing BPD may limit potential systemic adverse events and allow for BPD prophylaxis in infants who do not have intravenous access. However, drug delivery via aerosol may not be effective owing to low levels of lung deposition or lack of beneficial systemic effects, or both, of pentoxifylline. Additionally, aerosol delivery of pentoxifylline may interfere with assisted ventilation and may cause difficulty in terms of adequate triggering of the ventilator or airway obstruction.
The use of caffeine to treat apnoea of prematurity has become standard practice in many jurisdictions. Caffeine is partially metabolised to theophylline. Given that a small study of nine healthy volunteers found a 30% mean increase in theophylline levels when simultaneously administering systemic pentoxifylline (Ellison 1990), there may be interactions or additive effects, or both, of caffeine and pentoxifylline both with regards to their effectiveness and their potential side effects. We planned to perform a sensitivity analysis to explore whether co‐treatment with caffeine affects outcomes of this review.
Why it is important to do this review
Safe and effective pharmacological interventions aimed at prevention of BPD in preterm infants are limited. Significant adverse effects related to the use of corticosteroids warrant study of alternative treatment modalities such as pentoxifylline. Despite the efficacy and widespread use of caffeine, BPD is still a significant complication of preterm birth and further treatment options are desirable. The use of pentoxifylline for prevention of BPD needs to be formally assessed in order to provide caregivers with clinically relevant data on its efficacy and safety. The aim of this systematic review was to summarise current evidence on benefits and harms of pentoxifylline for prevention of BPD in preterm infants.
Objectives
The primary objective of this review was to determine the effect of pentoxifylline on the cumulative incidence of BPD (supplemental oxygen at 36 weeks PMA), death prior to 36 weeks PMA, and the composite outcome of BPD or death prior to 36 weeks PMA in preterm neonates less than 32 weeks GA or less than 1500 g birth weight.
Secondary objectives included potential benefits of pentoxifylline in terms of mortality, respiratory morbidity prior to hospital discharge, and complications of preterm birth. Furthermore, secondary outcomes included potential adverse effects of pentoxifylline.
Subgroup analyses were planned considering whether GA at birth (extremely preterm neonates less than 28 weeks GA, very preterm neonates 29 to 31 weeks GA) or co‐administration of pentoxifylline with caffeine influence outcomes of the intervention.
Methods
Criteria for considering studies for this review
Types of studies
Randomised or quasi‐randomised clinical trials of pentoxifylline in preterm neonates were eligible for inclusion in the review. Parallel group but not cross‐over trials were eligible for inclusion.
Types of participants
Preterm neonates (GA less than 32 weeks or birth weight less than 1500 g, postnatal age less than 14 days) requiring respiratory support including endotracheal ventilation, continuous positive airway pressure (CPAP), and nasal cannula, or supplemental oxygen.
Types of interventions
Systemic pentoxifylline at any dose and any duration versus placebo or no intervention.
Nebulised pentoxifylline at any dose and any duration versus placebo or no intervention.
Types of outcome measures
Primary outcomes
Death is a competing outcome with BPD, thus, primary outcomes were defined as follows:
BPD (supplemental oxygen at 36 weeks PMA);
Death prior to 36 weeks PMA;
BPD or death prior to 36 weeks PMA.
Secondary outcomes
Mortality during initial hospital stay.
Oxygen requirement at 28 days of life.
Mild, moderate, or severe BPD at 36 weeks PMA or discharge (whichever comes first) based on the classification suggested by Jobe and Bancalari (Jobe 2001). Table 1 summarises definitions of severity of BPD for infants born at less than 32 weeks GA:
Table 1 BPD classification
| Severity | Entry criterion | Oxygen requirement at 36 weeks PMA or discharge |
| Mild BPD | Supplemental oxygen for at least 28 days (a day of O2 treatment is defined as O2 > 12 hours/day) |
Room air |
| Moderate BPD | 22‐29% O2 | |
| Severe BPD | ≥ 30% O2 and/or positive pressure support |
Oxygen requirements are calibrated by an oxygen‐reduction test as described previously (Walsh 2003). Briefly, infants in supplemental oxygen of less than 30% undergo a stepwise reduction of supplemental oxygen to room air. Those who fail this reduction are diagnosed with mild BPD. Failure is defined as oxygen saturation 80% to 87% for five minutes, or less than 80% for one minute. No BPD is defined as treatment with room air with an oxygen saturation greater than or equal to 88%, or passing the oxygen‐reduction test. There are two methods for passing: a rapid pass and pass after full monitoring. Rapid‐pass criteria are met when an infant is successfully weaned to room air with all saturation values greater than or equal to 96% over a 15‐minute period. If instead oxygen saturations are between 88% and 95%, the infant is monitored for a full 60 minutes in room air and defined as a pass when all saturations exceed 88% in that 60‐minute period.
Duration of endotracheal ventilation (days).
Duration of total respiratory support (sum of endotracheal ventilation, CPAP, and heated humidified high‐flow cannula therapy) (days).
Duration of supplemental oxygen administration (days).
Supplemental home oxygen requirement.
Exposure to postnatal systemic corticosteroids.
Duration of hospital stay during initial hospitalisation (days).
-
Complications of preterm birth including:
intraventricular haemorrhage (IVH) grade 1 to 4 (Papile classification) (Papile 1978);
severe IVH grade 3 to 4 (Papile classification) (Papile 1978);
periventricular leukomalacia (PVL) (Valcamonico 2007; Mercier 2010);
neurodevelopmental impairment (assessed at ≥ 12 months corrected age by a validated scale, e.g. Griffiths or Bayley Scales of Infant Development) (Griffiths 1954; Bayley 1993; Mercier 2010);
NEC ≥ stage 2 (modified Bell's criteria) (Bell 1978);
retinopathy of prematurity (ROP) stage 1 to 5 (international classification of ROP) (International Committee Retinopathy 2005);
severe ROP stage 3 to 5 (international classification of ROP) (International Committee Retinopathy 2005);
sepsis (positive culture in otherwise sterile body fluid);
patent ductus arteriosus (PDA) (echocardiographically diagnosed).
-
Potential adverse events directly attributable to pentoxifylline (occurring within 24 hours following study drug administration) including:
thrombocytopenia (platelet count < 100,000 x 109/L);
increased gastric residuals (gastric aspirate > 30% of oral feed);
vomiting (forceful expulsion of gastric content associated with apparent displeasure);
cholestatic jaundice requiring therapy;
major bleeding requiring therapy;
-
adverse events directly attributable to, and occurring during, nebulised administration of study drug:
hypoxaemia (PaO2 < 50 mmHg);
hypercarbia (PaCO2 > 60 mmHg);
bradycardia (< 80 beats/minute);
failed triggering or auto‐triggering of ventilator;
airway obstruction requiring suction or termination of nebulisation.
Search methods for identification of studies
Electronic searches
We searched the Cochrane Neonatal Review Group Specialized Register, the Cochrane Central Register of Controlled Trials (CENTRAL, Issue 9, 2012), EMBASE (January 1974 to September 2012), PubMed (January 1966 to September 2012), and CINAHL (January 1982 to September 2012).
Appendix 1 lists the search terms used.
Searching other resources
We checked references and cross‐references from identified studies. Abstracts from the proceedings of the Pediatric Academic Societies Meetings (from January 1990 to September 2012) were handsearched. We placed no restrictions on language. We searched clinical trials registries for ongoing or recently completed trials (clinicaltrials.gov; controlled‐trials.com; and who.int/ictrp).
Data collection and analysis
We used the standard methods of the Cochrane Neonatal Review Group and The Cochrane Collaboration.
Selection of studies
Two review authors (SMS and SK) independently searched the literature as described above. We considered only RCTs and quasi‐RCTs fulfilling the above criteria for inclusion in the review. We planned to not include studies published only in abstract form unless the final results of the trial were reported and all necessary information could be ascertained from the abstract or authors, or both. Selection of studies was done separately by two review authors (SMS and SK). Any disagreements were resolved by discussion involving all review authors.
Data extraction and management
Two review authors (SMS and SK) independently extracted, assessed, and coded data using standardised data extraction forms. One review author (SMS) entered data into Review Manager software (RevMan 2011). All review authors checked data for accuracy. We contacted authors where any queries arose or where additional data were required.
Assessment of risk of bias in included studies
We assessed risk of bias according to selection bias (quality of randomisation, allocation concealment/blinding of randomisation), performance bias (blinding of intervention), attrition bias (completeness of follow‐up), and detection bias (blinding of outcome measurement). Assessments were specified as 'Yes', 'No', or 'Unclear'. Any disagreements were resolved by discussion involving all review authors. We included this information in the 'Characteristics of included studies' table. We evaluated the following issues and entered the information into the 'Risk of bias' table (Higgins 2011).
-
Sequence generation (checking for possible selection bias). Was the allocation sequence adequately generated? For each included study, we categorised the method used to generate the allocation sequence as:
adequate (any truly random process, e.g. random number table, computer random number generator);
inadequate (any non‐random process, e.g. odd or even date of birth, hospital, or clinic record number);
unclear.
-
Allocation concealment (checking for possible selection bias). Was allocation adequately concealed? For each included study, we categorised the method used to conceal the allocation sequence as:
adequate (e.g. telephone or central randomisation, consecutively numbered sealed, opaque envelopes);
inadequate (open random allocation, unsealed or non‐opaque envelopes, alternation, date of birth);
unclear.
-
Blinding (checking for possible performance bias). Was knowledge of the allocated intervention adequately prevented during the study? At study entry? At the time of outcome assessment? For each included study, we categorised the methods used to blind study participants and personnel from knowledge of which intervention a participant received. Blinding was assessed separately for different outcomes or classes of outcomes. In some situations there may be partial blinding, for example where outcomes are self‐reported by unblinded participants, but they are recorded by blinded personnel without knowledge of group assignment. Where needed, 'partial' was added to the list of options for assessing quality of blinding. We categorised the methods as:
adequate, inadequate, or unclear for participants;
adequate, inadequate, or unclear for personnel;
adequate, inadequate, or unclear for outcome assessors.
-
Incomplete outcome data (checking for possible attrition bias through withdrawals, dropouts, protocol deviations). Were incomplete outcome data adequately addressed? For each included study and for each outcome, we described the completeness of data including attrition and exclusions from the analysis. We noted whether attrition and exclusions were reported, the numbers included in the analysis at each stage (compared with the total randomised participants), reasons for attrition or exclusion where reported, and whether missing data were balanced across groups or were related to outcomes. Where sufficient information was reported or supplied by the trial authors, we re‐included missing data in the analyses. We categorised the methods as:
adequate (< 20% missing data);
inadequate (≥ 20% missing data);
unclear.
-
Selective reporting bias. Are reports of the study free of suggestion of selective outcome reporting? For each included study, we described how we investigated the possibility of selective outcome reporting bias and what we found. We assessed the methods as:
adequate (where it is clear that all of the study's pre‐specified outcomes and all expected outcomes of interest to the review have been reported);
inadequate (where not all the study's pre‐specified outcomes have been reported; one or more reported primary outcomes were not pre‐specified; outcomes of interest were reported incompletely and so cannot be used; study failed to include results of a key outcome that would have been expected to have been reported);
unclear.
-
Other sources of bias. Was the study apparently free of other problems that could put it at a high risk of bias? For each included study, we described any important concerns we had about other possible sources of bias (for example, whether there was a potential source of bias related to the specific study design or whether the trial was stopped early owing to some data‐dependent process). We assessed whether each study was free of other problems that could put it at risk of bias as:
yes;
no;
unclear.
We planned to explore the impact of the level of bias through undertaking sensitivity analyses if needed.
Measures of treatment effect
We performed statistical analyses using the standard methods of the Cochrane Neonatal Review Group. We used relative risk (RR), risk difference (RD), number needed to treat for an additional beneficial outcome (NNTB) or number needed to treat for an additional harmful outcome (NNTH) for categorical variables and weighted mean difference (WMD) for continuous variables. Any within‐group standard error of the mean (SEM) reported in a trial was to be replaced by its corresponding standard deviation (SD) using the formula SD = SEM x √N. We reported 95% CI for each statistic.
Unit of analysis issues
We included all RCTs and quasi‐RCTs in which the unit of allocation was the individual infant.
Assessment of heterogeneity
We planned to assess the magnitude of heterogeneity of treatment effects using the I2 statistic. An I2 greater than 60% is considered an indication of high heterogeneity. Additionally, we planned to inspect each forest plot carefully for heterogeneity, as indicated by lack of overlapping CIs of individual trials.
Assessment of reporting biases
We examined the possibility of within‐study selective outcome reporting for each study included in the review. We searched for trial protocols of included trials on PubMed, ClinicalTrials.gov, controlled‐trials.com, and the Clinical Trials Search Portal of the World Health Organization (www.who.int/ictrp) in order to assess whether outcome reporting seems to be sufficiently complete and transparent.
Data synthesis
We planned to use a fixed‐effect model to pool data for meta‐analyses.
Subgroup analysis and investigation of heterogeneity
We aimed to perform subgroup analyses of infants less than 28 weeks GA and those born from 28 weeks and 0 days to 31 weeks and 6 days GA given that the baseline risk of BPD is closely related to GA at birth. We further planned subgroup analyses on presence or absence of caffeine therapy to find out whether co‐administration of pentoxifylline with caffeine results in changes in adverse events of caffeine (irritability, tachycardia) and whether such co‐administration influences beneficial effects and adverse events of pentoxifylline.
Sensitivity analysis
Differences in study design of included trials might affect the results of the systematic review. We planned to do a sensitivity analysis to compare the effects of pentoxifylline in truly randomised trials as opposed to quasi‐randomised trials.
Results
Description of studies
See: Included studies; Excluded studies.
Results of the search
The search identified 17 reports. We detected six potentially eligible studies. Of these, five were excluded. Details of the excluded studies along with the reasons for exclusion are listed in the 'Characteristics of excluded studies' table. One study was included in this review (Lauterbach 2006). One ongoing trial was identified.
Included studies
Details on included studies are given in the 'Characteristics of included studies' table.
Types of participants
Lauterbach 2006 was a randomised clinical single‐centre trial involving 150 preterm neonates with a mean (range) gestational age of 28 (24 to 33) weeks. Eligibility criteria included birth weight less than 1500 g and need for supplemental oxygen on day four of life, independent of presence and type of further respiratory support. Exclusion criteria included major congenital malformation or intraventricular haemorrhage grade 3 or 4 detected within the first three days of life.
Types of interventions
Lauterbach 2006 allocated infants to one of the following three interventions: a) nebulised pentoxifylline every six hours for three days; b) nebulised sterile water (placebo) at a volume equal to that of pentoxifylline every six hours for three days; c) intravenous dexamethasone every 12 hours for three days. If participants required positive pressure support to maintain oxygen saturation targets (oxygen saturation range, 86% to 95%) one week after starting study drug, interventions were repeated weekly until 36 weeks PMA.
Types of outcome measures
Primary outcome in Lauterbach 2006 was the incidence of BPD (supplemental oxygen at 36 weeks PMA). Secondary outcomes included mortality, pneumothorax, IVH, PVL, PDA requiring ligation, and plasma pentoxifylline concentration.
Excluded studies
We excluded five identified studies from this review because they enrolled predominantly term and late preterm infants, did not report on respiratory outcomes, or were not randomised/quasi‐randomised trials (Lauterbach 1996, Lauterbach 1999, Lauterbach 1999a, Ali 2006, Adel 2010). Details of the excluded studies along with the reasons for exclusion are listed in the 'Characteristics of excluded studies' table.
Risk of bias in included studies
The single included study (Lauterbach 2006) was at high risk of bias. Random sequence generation and allocation concealment for pentoxifylline and placebo groups were adequate. The study lacked blinding of intervention and outcome assessment. A total of 19 of 150 infants were excluded post‐randomisation. Data were primarily reported per protocol. Intention‐to‐treat analysis was reported only for the primary outcomes of this review. Ratings of methodological quality are given in the 'Characteristics of included studies' table.
Allocation
Lauterbach 2006 allocated infants using a computer‐generated random sequence. The investigators had pre‐planned to allocate 50 patients to each of the three study arms: nebulised pentoxifylline (n = 50); nebulised sterile water (n = 50); intravenous dexamethasone (n = 50). There was no intravenous placebo group. Allocation was concealed by using sealed, opaque, numbered containers.
Blinding
Lauterbach 2006 did not attempt to blind study personnel, outcome assessors, or participants from knowledge of which intervention a participant received.
Incomplete outcome data
Lauterbach 2006 randomised 150 infants. Thirty‐four infants died prior to 36 weeks PMA (22.7%) and 97 of 116 survivors (83.6%) were analysed at 36 weeks PMA. Among the 19 infants who dropped out, Lauterbach 2006 excluded 16 infants post‐allocation due to receiving open‐label pentoxifylline (n = 10) or dexamethasone (n = 2) or development of surgical NEC (n = 4). A further three patients were excluded in the placebo group due to withdrawal of parental consent. Numbers of dropouts in each study arm due to open‐label treatment or surgical NEC were as follows: pentoxifylline group, 3 of 50 (6%); placebo group, 5 of 50 (10%); dexamethasone group, 8 of 50 (16%). Specific reasons for dropouts within each study arm were not given. Outcomes were primarily reported per protocol. Lauterbach 2006 provided additional data on BPD at 36 weeks PMA and death prior to 36 weeks PMA, allowing for an intention‐to‐treat analysis of these outcomes.
Selective reporting
Reporting of outcomes of infants who died prior to 36 weeks PMA or were excluded due to withdrawal of parental consent was incomplete.
Other potential sources of bias
None detected.
Effects of interventions
Systemic pentoxifylline at any dose and any duration versus placebo or no intervention (Comparison 1)
No trials assessing this comparison qualified for inclusion in this review.
Nebulised pentoxifylline at any dose and any duration versus placebo or no intervention (Comparison 2)
Lauterbach 2006 tested this comparison. Data on BPD at 36 weeks PMA and death prior to 36 weeks PMA were from an intention‐to‐treat analysis and were provided by the contact author. All secondary outcomes were based on per‐protocol analyses.
Primary outcomes
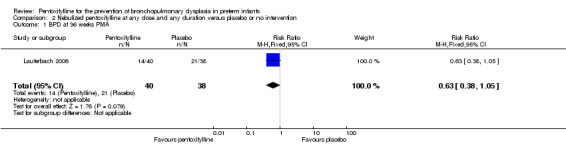
BPD at 36 weeks PMA (Outcome 2.1)
Lauterbach 2006 reported on the incidence of BPD among all infants randomised to pentoxifylline or placebo (n = 100). There was no statistically significant difference in the incidence of BPD at 36 weeks PMA in the nebulised pentoxifylline group versus the placebo group (risk ratio (RR) 0.63, 95% CI 0.38 to 1.05; risk difference (RD) ‐0.20, 95% CI ‐0.42 to 0.01).
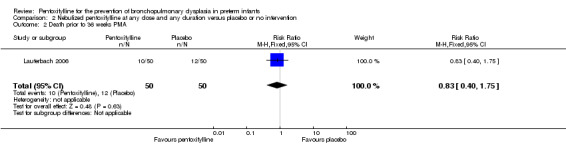
Death prior to 36 weeks PMA (Outcome 2.2)
Lauterbach 2006 reported on death prior to 36 weeks PMA among 99 of 100 infants randomised to pentoxifylline or placebo. There was no statistically significant difference in death prior to 36 weeks PMA in the nebulised pentoxifylline group versus the placebo group (RR 0.83, 95% CI 0.40 to 1.75; RD ‐0.04, 95% CI ‐0.20 to 0.12).
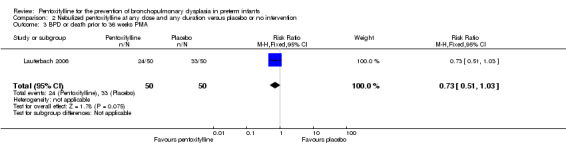
BPD or death prior to 36 weeks PMA (Outcome 2.3)
Lauterbach 2006 reported on BPD or death prior to 36 weeks PMA among 99 of 100 infants randomised to pentoxifylline or placebo. There was no statistically significant difference in BPD or death prior to 36 weeks PMA in the nebulised pentoxifylline group versus the placebo group (RR 0.73, 95% CI 0.51 to 1.03; RD ‐0.18, 95% CI ‐0.37 to 0.01).
Secondary outcomes
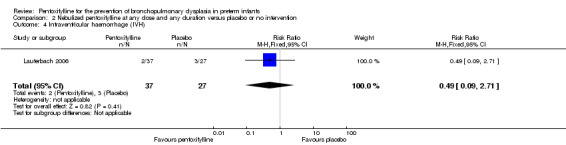
Intraventricular haemorrhage (IVH) (Outcome 2.4)
Lauterbach 2006 reported on IVH (no grading) among 64 of 100 infants randomised to pentoxifylline or placebo. There was no statistically significant difference in IVH in the nebulised pentoxifylline group versus the placebo group (RR 0.49, 95% CI 0.09 to 2.71; RD ‐0.06, 95% CI ‐0.20 to 0.08).
Periventricular leukomalacia (PVL) (Outcome 2.5)
Lauterbach 2006 reported on PVL among 64 of 100 infants randomised to pentoxifylline or placebo. There was no statistically significant difference in PVL in the nebulised pentoxifylline group versus the placebo group (RR 0.49, 95% CI 0.09 to 2.71; RD ‐0.06, 95% CI ‐0.20 to 0.08).
Sepsis (Outcome 2.6)
Lauterbach 2006 reported on sepsis among 64 of 100 infants randomised to pentoxifylline or placebo. There was no statistically significant difference in sepsis in the nebulised pentoxifylline group versus the placebo group (RR 1.22, 95% CI 0.63 to 2.36; RD 0.07, 95% CI ‐0.17 to 0.31).
Patent ductus arteriosus (PDA) requiring ligation (Outcome 2.7)
Lauterbach 2006 reported on PDA requiring ligation among 64 of 100 infants randomised to pentoxifylline or placebo. There was no statistically significant difference in PDA requiring ligation in the nebulised pentoxifylline group versus the placebo group (RR 1.22, 95% CI 0.32 to 4.66; RD 0.02, 95% CI ‐0.14 to 0.19).
Adverse events
Lauterbach 2006 reported that administration of nebulised pentoxifylline resulted in occasional feeding intolerance. No further details on adverse events were given. Lauterbach 2006 did not report any of the other secondary outcomes considered for this review.
Discussion
Summary of main results
Only one RCT qualified for inclusion in this review (Lauterbach 2006). This study was at high risk of bias and compared the use of nebulised pentoxifylline versus placebo for prevention of BPD in preterm infants. No trials were found that studied the use of systemic pentoxifylline for prevention of BPD. On intention‐to‐treat analysis of data from Lauterbach 2006, there was no statistically significant effect of nebulised pentoxifylline versus placebo on BPD at 36 weeks PMA, death prior to 36 weeks PMA, or the composite outcome of BPD or death prior to 36 weeks PMA. All secondary outcomes of this review reported in Lauterbach 2006 were reported per protocol and did not suggest a significant effect of nebulised pentoxifylline on IVH, PVL, sepsis, or PDA requiring ligation. Reporting of adverse events in Lauterbach 2006 was very limited and did not allow for reliable judgement on the incidence of such events. No long‐term outcomes were reported.
Overall completeness and applicability of evidence
The objective of this review was to determine the effect of pentoxifylline on the incidence of BPD or death prior to 36 weeks PMA in preterm neonates. The single‐centre study from Poland included in this review provided insufficient evidence to address the objectives of this review. This study (Lauterbach 2006) did not show significant effects associated with the use of nebulised pentoxifylline versus placebo but a trend towards a reduced incidence of BPD or death prior to 36 weeks PMA. No long‐term outcomes were reported. Given the paucity of data available, the high risk of bias in Lauterbach 2006, and a 67% baseline risk of BPD or death prior to 36 weeks PMA in the placebo group of infants with a mean gestational age of 29 weeks, applicability and generalisability of the results of this review is very limited.
Quality of the evidence
Only one study qualified for inclusion in this review (Lauterbach 2006). This study randomised 100 participants to nebulised pentoxifylline or placebo and was at high risk of bias. Method of allocation and allocation concealment were probably adequate, but investigators did not attempt to mask caregivers, assessors of outcomes, or families to study group, and outcome reporting was incomplete. Thus, internal validity of the results of this review is very limited.
Potential biases in the review process
Although we did an extensive literature search, we cannot exclude the possibility that we missed relevant studies. We applied no language restrictions to reduce publication bias and contacted authors of potentially eligible studies to clarify study methodology. We further contacted authors of the single included study to understand methodological details of the trial and to obtain relevant outcome data in order to reduce attrition bias.
Agreements and disagreements with other studies or reviews
We did not find other systematic reviews addressing the use of pentoxifylline for prevention of BPD in preterm neonates.
Authors' conclusions
Implications for practice.
There is insufficient evidence to determine the safety and efficacy of pentoxifylline for prevention of BPD in preterm neonates.
Implications for research.
We encourage researchers to undertake well‐designed RCTs to confirm or refute the role of pentoxifylline for prevention of BPD in preterm neonates. These trials should report on clinically important outcomes (BPD or death prior to 36 weeks PMA, complications of prematurity, adverse events, among others) and, ideally, on long‐term neurodevelopmental outcome. In case of nebulised administration of pentoxifylline, a standardised method of nebulisation with particular attention to the nominal dose delivered to the lung should be used in order to optimise drug delivery.
Acknowledgements
We thank Professor Lauterbach for clarification of study methodology and provision of additional outcome data.
We thank the Cochrane Neonatal Group for providing valuable comments and advice at the protocol and review stages.
Appendices
Appendix 1. Search strategy
("Infant, newborn" [MeSH] OR "Infant, premature" [MeSH] OR infant* OR neonate* OR newborn*) AND ("Pentoxifylline" [MeSH] OR pentoxif* OR Trental OR Torental OR PTF OR PTX OR PTX F OR agapurin OR BL‐191 OR oxpentif*) AND ("controlled clinical trial" [Publication Type] OR "randomized controlled trial" [Publication Type]).
Data and analyses
Comparison 2. Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 BPD at 36 weeks PMA | 1 | 78 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.63 [0.38, 1.05] |
| 2 Death prior to 36 weeks PMA | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.83 [0.40, 1.75] |
| 3 BPD or death prior to 36 weeks PMA | 1 | 100 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.73 [0.51, 1.03] |
| 4 Intraventricular haemorrhage (IVH) | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.09, 2.71] |
| 5 Periventricular leukomalacia (PVL) | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.49 [0.09, 2.71] |
| 6 Sepsis | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.63, 2.36] |
| 7 PDA requiring ligation | 1 | 64 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.22 [0.32, 4.66] |
2.1. Analysis.
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 1 BPD at 36 weeks PMA.
2.2. Analysis.
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 2 Death prior to 36 weeks PMA.
2.3. Analysis.
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 3 BPD or death prior to 36 weeks PMA.
2.4. Analysis.
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 4 Intraventricular haemorrhage (IVH).
2.5. Analysis.
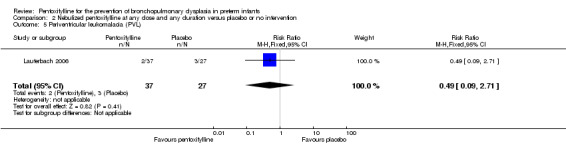
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 5 Periventricular leukomalacia (PVL).
2.6. Analysis.
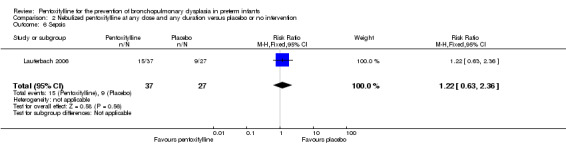
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 6 Sepsis.
2.7. Analysis.
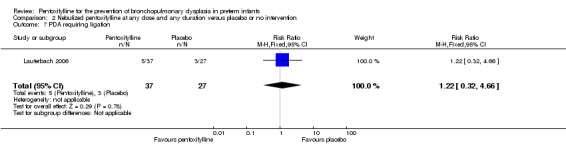
Comparison 2 Nebulized pentoxifylline at any dose and any duration versus placebo or no intervention, Outcome 7 PDA requiring ligation.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Lauterbach 2006.
| Methods | Single‐centre study performed in Kraków, Poland Allocation concealment: Yes Blinding of caregivers to intervention: No Blinding of outcome ascertainment: No Complete follow‐up: No |
|
| Participants | Preterm neonates (n = 150) with birth weight < 1500 g who required supplemental oxygen on day four of life with or without positive pressure support were eligible. Mean (range) gestational age was 28 (24 to 33) weeks, mean (range) birth weight was 1090 (580 to 1500) g. | |
| Interventions | Patients were randomly assigned to receive one of the following three interventions: Nebulised pentoxifylline (Polfilin, Polfa, Poland; n = 50). Application: 10 mg/kg if intubated; 20 mg/kg if on continuous positive airway pressure or without positive pressure support; administered with a nebuliser connected between y‐piece and endotracheal tube (Porta‐Neb, Medic‐Aid Ltd, West Sussex, UK), delivered over 10 min and diluted with sterile water every six hours for three days. Nebulised sterile water (placebo) at a volume equal to that of pentoxifylline (n = 50). Application: 1.5 mL/kg if intubated; 3.0 mL/kg if on continuous positive airway pressure or without positive pressure support; administered with a nebuliser connected between y‐piece and endotracheal tube (Porta‐Neb, Medic‐Aid Ltd, West Sussex, UK), delivered over 10 min every six hours for three days. Intravenous dexamethasone 0.25 mg/kg/dose every 12 hours for three days (Dexaven, Polfa, Poland; n = 50). If participants required positive pressure support to maintain oxygen saturation targets (range, 86% to 95%) one week after starting study drug, interventions were repeated weekly until 36 weeks PMA. |
|
| Outcomes | Primary outcome: BPD (supplemental oxygen at 36 weeks PMA) Secondary outcomes: Mortality, pneumothorax, IVH, PVL, PDA requiring ligation, sepsis Plasma pentoxifylline concentration |
|
| Notes | Prof Lauterbach (contact author) provided additional information on study methodology and outcomes at 36 weeks PMA. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random sequence |
| Allocation concealment (selection bias) | Low risk | This study compared three interventions. Study drug for nebulisation (i.e. pentoxifylline and sterile water placebo) was delivered in closed, numbered, opaque containers. Dexamethasone was given intravenously. Given that this review focuses on comparison of pentoxifylline with placebo, allocation concealment is considered adequate. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Blinding of the intervention was not attempted. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Blinding of outcome assessors was not attempted. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 150 infants were randomised, 34 infants died prior to 36 weeks PMA (22.7%), 97/116 survivors (83.6%) survived to 36 weeks PMA and were analysed per protocol. 16 infants were excluded post‐ allocation due to receiving open‐label pentoxifylline (n = 10) or dexamethasone (n = 2) or development of surgical NEC (n = 4) (pentoxifylline group, 3/50 (6%); placebo group, 5/50 (10%); dexamethasone group, 8/50 (16%)). A further 3/50 (6%) infants were excluded post‐ allocation in the placebo group due to withdrawal of parental consent. Outcomes were reported on a per‐protocol basis, i.e. outcomes of infants who dropped out due to open‐ label treatment with pentoxifylline or dexamethasone, development of surgical NEC, or withdrawal of parental consent were not reported. Only data on the rate of BPD at 36 weeks PMA and death prior to 36 weeks PMA in all randomised infants were reported, allowing for an intention‐to‐treat analysis of the primary outcomes. |
| Selective reporting (reporting bias) | High risk | Reporting of outcomes of infants who died prior to 36 weeks PMA or were excluded due to withdrawal of parental consent was incomplete. |
| Other bias | Low risk | None detected |
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Adel 2010 | Quasi‐randomised clinical trial (n = 37) assessing effects of IV pentoxifylline versus placebo on coagulation in neonates with sepsis. Excluded because a) study participants were predominantly term and late preterm neonates and b) respiratory outcomes were not reported. Outcomes of this trial are reported in another systematic review (Haque 2011). |
| Ali 2006 | Randomised clinical trial (n = 50) assessing effects of IV pentoxifylline versus placebo in neonates with sepsis. Excluded because gestational age range of participants was 32 to 37 weeks. Outcomes of this trial are reported in another systematic review (Haque 2011). |
| Lauterbach 1996 | Randomised clinical trial (n = 40) assessing effects of IV pentoxifylline versus placebo in preterm neonates < 36 weeks gestational age with sepsis. Excluded because a) gestational age of a significant proportion of participants was > 32 weeks and b) respiratory outcomes were not reported. Outcomes of this trial are reported in another systematic review (Haque 2011). |
| Lauterbach 1999 | Case series describing the use of nebulised pentoxifylline in five preterm neonates with established BPD. Excluded because the study was not a randomised or quasi‐randomised trial. |
| Lauterbach 1999a | Randomised clinical trial (n = 100) assessing effects of IV pentoxifylline versus placebo in preterm neonates < 36 weeks gestational age with late‐onset sepsis. Excluded because a) gestational age of a significant proportion of participants was > 32 weeks and b) respiratory outcomes were not reported. Outcomes of this trial are reported in another systematic review (Haque 2011). |
Characteristics of ongoing studies [ordered by study ID]
Schulzke 2011.
| Trial name or title | Nebulised pentoxifylline for prevention of chronic lung disease in extremely preterm infants |
| Methods | Randomised, masked, placebo‐controlled single‐centre trial |
| Participants | Preterm neonates < 28 weeks gestational age, mechanically ventilated or requiring ≥ 30% of supplemental oxygen between 72 and 168 hours of age |
| Interventions | Nebulised pentoxifylline 1 mL/kg (20 mg/kg) or an equal volume of placebo (sterile water) every six hours for 10 consecutive days |
| Outcomes | Primary outcomes: duration of supplemental oxygen; death; chronic lung disease; death or chronic lung disease |
| Starting date | 2011 |
| Contact information | Sven Schulzke, sven.schulzke@unibas.ch; Sanjay Patole, Sanjay.Patole@health.wa.gov.au |
| Notes | Australian New Zealand Clinical Trials Registry No: ACTRN12611000145909 |
Differences between protocol and review
Co‐administration of caffeine moved from sensitivity analysis to subgroup analysis in the final review.
Contributions of authors
SMS: designed the review, searched for studies, assessed studies, wrote the report. SK: contributed to design of protocol, searched for studies, assessed studies, contributed to writing of report. SKP: contributed to overall design, assessed studies, critically reviewed report.
Sources of support
Internal sources
-
University Children's Hospital Basel, Switzerland.
Protected research time
External sources
-
Eunice Kennedy Shriver National Institute of Child Health and Human Development National Institutes of Health, Department of Health and Human Services, USA.
Editorial support of the Cochrane Neonatal Review Group has been funded with federal funds from the Eunice Kennedy Shriver National Institute of Child Health and Human Development National Institutes of Health, Department of Health and Human Services, USA, under Contract No. HHSN275201100016C.
Declarations of interest
Drs. S. Schulzke and S. Patole are the principal investigators of an ongoing randomised trial entitled 'Nebulised pentoxifylline for the prevention of chronic lung disease in extremely preterm infants'. This trial is registered in the Australian New Zealand Clinical Trials Registry (No ACTRN12611000145909).
New
References
References to studies included in this review
Lauterbach 2006 {published and unpublished data}
- Lauterbach R, Szymura‐Oleksiak J, Pawlik D, Warchol J, Lisowska‐Miszczyk I, Rytlewski K. Nebulized pentoxifylline for prevention of bronchopulmonary dysplasia in very low birth weight infants: a pilot clinical study. The Journal of Maternal‐Fetal and Neonatal Medicine 2006;19(7):433‐8. [PUBMED: 16923699] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Adel 2010 {published data only}
- Adel M, Awad HA, Abdel‐Naim AB, Al‐Azizi MM. Effects of pentoxifylline on coagulation profile and disseminated intravascular coagulation incidence in Egyptian septic neonates. Journal of Clinical Pharmacy and Therapeutics 2010;35(3):257‐65. [PUBMED: 20831528] [DOI] [PubMed] [Google Scholar]
Ali 2006 {published data only}
- Ali W, Ahmed P, Bhat MA, Mushtaq AB, Mushtaq S. Pentoxifylline in the treatment of sepsis in premature infants. JK Practitioner 2006;13:204‐7. [Google Scholar]
Lauterbach 1996 {published data only}
- Lauterbach R, Zembala M. Pentoxifylline reduces plasma tumour necrosis factor‐alpha concentration in premature infants with sepsis. European Journal of Pediatrics 1996;155(5):404‐9. [PUBMED: 8741040] [DOI] [PubMed] [Google Scholar]
Lauterbach 1999 {published data only}
- Lauterbach R, Szymura‐Oleksiak J. Nebulized pentoxifylline in successful treatment of five premature neonates with bronchopulmonary dysplasia. European Journal of Pediatrics 1999;158(7):607. [PUBMED: 10412826] [DOI] [PubMed] [Google Scholar]
Lauterbach 1999a {published data only}
- Lauterbach R, Pawlik D, Kowalczyk D, Ksycinski W, Helwich E, Zembala M. Effect of the immunomodulating agent, pentoxifylline, in the treatment of sepsis in prematurely delivered infants: a placebo‐controlled, double‐blind trial. Critical Care Medicine 1999;27(4):807‐14. [PUBMED: 10321674] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
Schulzke 2011 {unpublished data only}
- Nebulised pentoxifylline for prevention of chronic lung disease in extremely preterm infants. Ongoing study 2011.
Additional references
Acharya 1997
- Acharya S, Nair BC. Pentoxifylline‐induced thrombocytopenia. International Journal of Dermatology 1997;36(8):635‐6. [DOI] [PubMed] [Google Scholar]
Allen 2003
- Allen J, Zwerdling R, Ehrenkranz R, Gaultier C, Geggel R, Greenough A, et al. Statement on the care of the child with chronic lung disease of infancy and childhood. American Journal of Respiratory and Critical Care Medicine 2003;168(3):356‐96. [DOI] [PubMed] [Google Scholar]
Ambalavanan 2004
- Ambalavanan N, Kennedy K, Tyson J, Carlo WA. Survey of vitamin A supplementation for extremely‐low‐birth‐weight infants: is clinical practice consistent with the evidence?. The Journal of Pediatrics 2004;145(3):304‐7. [DOI] [PubMed] [Google Scholar]
Ambalavanan 2005
- Ambalavanan N, Tyson JE, Kennedy KA, Hansen NI, Vohr BR, Wright L, et al. National Institute of Child Health and Human Development Neonatal Research Network. Vitamin A supplementation for extremely low birth weight infants: outcome at 18 to 22 months. Pediatrics 2005;115(3):e249‐54. [DOI] [PubMed] [Google Scholar]
Bayley 1993
- Bayley N. Bayley Scales of Infant Development. 2nd Edition. San Antonio, TX: Psychological Corporation, 1993. [Google Scholar]
Bell 1978
- Bell MJ, Ternberg JL, Feigin RD, Keating JP, Mashall R, Barton L, et al. Neonatal necrotizing enterocolitis. Therapeutic decisions based upon clinical staging. Annals of Surgery 1978;187(1):1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bhuta 1998
- Bhuta T, Ohlsson A. Systematic review and meta‐analysis of early postnatal dexamethasone for prevention of chronic lung disease. Archives of Disease in Childhood. Fetal and Neonatal Edition 1998;79(1):F26‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
Coalson 1995
- Coalson JJ, Winter V, deLemos RA. Decreased alveolarization in baboon survivors with bronchopulmonary dysplasia. American Journal of Respiratory and Critical Care Medicine 1995;152(2):640‐6. [DOI] [PubMed] [Google Scholar]
Coalson 1999
- Coalson JJ, Winter VT, Siler‐Khodr T, Yoder BA. Neonatal chronic lung disease in extremely immature baboons. American Journal of Respiratory and Critical Care Medicine 1999;160(4):1333‐46. [DOI] [PubMed] [Google Scholar]
Ehrenkranz 2005
- Ehrenkranz RA, Walsh MC, Vohr BR, Jobe AH, Wright LL, Fanaroff AA, et al. Validation of the National Institutes of Health consensus definition of bronchopulmonary dysplasia. Pediatrics 2005;116(6):1353‐60. [DOI] [PubMed] [Google Scholar]
Ellison 1990
- Ellison MJ, Horner RD, Willis SE, Cummings DM. Influence of pentoxifylline on steady‐state theophylline serum concentrations from sustained‐release formulations. Pharmacotherapy 1990;10(6):383‐6. [PubMed] [Google Scholar]
Griffiths 1954
- Griffiths R. The Abilities of Babies: a Study in Mental Measurement. New York, US: McGraw‐Hill Book Company, Inc., 1954. [Google Scholar]
Halliday 2009
- Halliday HL, Ehrenkranz RA, Doyle LW. Late (>7 days) postnatal corticosteroids for chronic lung disease in preterm infants. Cochrane Database of Systematic Reviews 2009, Issue 1. [DOI: 10.1002/14651858.CD001145.pub2] [DOI] [PubMed] [Google Scholar]
Halliday 2010
- Halliday HL, Ehrenkranz RA, Doyle LW. Early (< 8 days) postnatal corticosteroids for preventing chronic lung disease in preterm infants. Cochrane Database of Systematic Reviews 2010, Issue 1. [DOI: 10.1002/14651858.CD001146.pub3] [DOI] [PubMed] [Google Scholar]
Haque 2011
- Haque KN, Pammi M. Pentoxifylline for treatment of sepsis and necrotizing enterocolitis in neonates. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD004205.pub2] [DOI] [PubMed] [Google Scholar]
Harris 2010
- Harris E, Schulzke SM, Patole SK. Pentoxifylline in preterm neonates: a systematic review. Paediatric Drugs 2010;12(5):301‐11. [DOI] [PubMed] [Google Scholar]
Hentschel 2005
- Hentschel J, Berger TM, Tschopp A, Muller M, Adams M, Bucher HU. Population‐based study of bronchopulmonary dysplasia in very low birth weight infants in Switzerland. European Journal of Pediatrics 2005;164(5):292‐7. [DOI] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 [updated March 2011]. The Cochrane Collaboration, 2011. www.cochrane‐handbook.org.
International Committee Retinopathy 2005
- International Committee for the Classification of Retinopathy of Prematurity. The International Classification of Retinopathy of Prematurity revisited. Archives of Ophthalmology 2005;123(7):991‐9. [DOI] [PubMed] [Google Scholar]
Jobe 2001
- Jobe AH, Bancalari E. Bronchopulmonary dysplasia. American Journal of Respiratory and Critical Care Medicine 2001;163(7):1723‐9. [DOI] [PubMed] [Google Scholar]
Jull 2007
- Jull A, Arroll B, Parag V, Waters J. Pentoxifylline for treating venous leg ulcers. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD001733.pub2] [DOI] [PubMed] [Google Scholar]
Krakauer 2000
- Krakauer T. Pentoxifylline inhibits ICAM‐1 expression and chemokine production induced by proinflammatory cytokines in human pulmonary epithelial cells. Immunopharmacology 2000;46(3):253‐61. [DOI] [PubMed] [Google Scholar]
Magnusson 2008
- Magnusson M, Gunnarsson M, Berntorp E, Björkman S, Höglund P. Effects of pentoxifylline and its metabolites on platelet aggregation in whole blood from healthy humans. European Journal of Pharmacology 2008;581(3):290‐5. [DOI] [PubMed] [Google Scholar]
Mercier 2010
- Mercier CE, Dunn MS, Ferrelli KR, Howard DB, Soll RF, Vermont Oxford Network ELBW Infant Follow‐Up Study Group. Neurodevelopmental outcome of extremely low birth weight infants from the Vermont Oxford Network: 1998‐2003. Neonatology 2010;97(4):329‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Oh 2005
- Oh W, Poindexter BB, Perritt R, Lemons JA, Bauer CR, Ehrenkranz RA, et al. Association between fluid intake and weight loss during the first ten days of life and risk of bronchopulmonary dysplasia in extremely low birth weight infants. The Journal of Pediatrics 2005;147(6):786‐90. [DOI] [PubMed] [Google Scholar]
Papile 1978
- Papile LA, Burstein J, Burstein R, Koffler H. Incidence and evolution of subependymal and intraventricular hemorrhage: a study of infants with birth weights less than 1,500 gm. The Journal of Pediatrics 1978;92(4):529‐34. [DOI] [PubMed] [Google Scholar]
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Schmidt 2006
- Schmidt B, Roberts RS, Davis P, Doyle LW, Barrington KJ, Ohlsson A, et al. Caffeine therapy for apnea of prematurity. The New England Journal of Medicine 2006;354(20):2112‐21. [DOI] [PubMed] [Google Scholar]
Schmidt 2007
- Schmidt B, Roberts RS, Davis P, Doyle LW, Barrington KJ, Ohlsson A, et al. Long‐term effects of caffeine therapy for apnea of prematurity. The New England Journal of Medicine 2007;357(19):1893‐902. [DOI] [PubMed] [Google Scholar]
Schmidt 2008
- Schmidt B, Roberts R, Millar D, Kirpalani H. Evidence‐based neonatal drug therapy for prevention of bronchopulmonary dysplasia in very‐low‐birth‐weight infants. Neonatology 2008;93(4):284‐7. [DOI] [PubMed] [Google Scholar]
Schmidt 2012
- Schmidt B, Anderson PJ, Doyle LW, Dewey D, Grunau RE, Asztalos EV, et al. Survival without disability to age 5 years after neonatal caffeine therapy for apnea of prematurity. Journal of the American Medical Association 2012;307(3):275‐82. [DOI] [PubMed] [Google Scholar]
Schröer 1985
- Schröer RH. Antithrombotic potential of pentoxifylline. A hemorheologically active drug. Angiology 1985;36(6):387‐98. [DOI] [PubMed] [Google Scholar]
Shah 2007
- Shah V, Ohlsson A, Halliday HL, Dunn MS. Early administration of inhaled corticosteroids for preventing chronic lung disease in ventilated very low birth weight preterm neonates. Cochrane Database of Systematic Reviews 2007, Issue 4. [DOI: 10.1002/14651858.CD001969.pub2] [DOI] [PubMed] [Google Scholar]
Sinkin 1990
- Sinkin RA, Cox C, Phelps DL. Predicting risk for bronchopulmonary dysplasia: selection criteria for clinical trials. Pediatrics 1990;86(5):728‐36. [PubMed] [Google Scholar]
Smalling 2004
- Smalling WE Jr, Suguihara C, Huang J, Rodriguez MM, Bancalari E. Protective effect of pentoxifylline on volume‐induced lung injury in newborn piglets. Biology of the Neonate 2004;86(1):15‐21. [DOI] [PubMed] [Google Scholar]
Speer 2001
- Speer CP. New insights into the pathogenesis of pulmonary inflammation in preterm infants. Biology of the Neonate 2001;79(3‐4):205‐9. [DOI] [PubMed] [Google Scholar]
Speer 2003
- Speer CP. Inflammation and bronchopulmonary dysplasia. Seminars in Neonatology 2003;8(1):29‐38. [DOI] [PubMed] [Google Scholar]
Tyson 1999
- Tyson JE, Wright LL, Oh W, Kennedy KA, Mele L, Ehrenkranz RA, et al. National Institute of Child Health and Human Development Neonatal Research Network. Vitamin A supplementation for extremely‐low‐birth‐weight infants. The New England Journal of Medicine 1999;340(25):1962‐8. [DOI] [PubMed] [Google Scholar]
Valcamonico 2007
- Valcamonico A, Accorsi P, Sanzeni C, Martelli P, Boria P, Cavazza A, et al. Mid‐ and long‐term outcome of extremely low birth weight (ELBW) infants: an analysis of prognostic factors. The Journal of Maternal‐Fetal and Neonatal Medicine 2007;20(6):465‐71. [DOI] [PubMed] [Google Scholar]
Vedes 2011
- Vedes EC, Marques LD, Toscano Rico MC. Severe isolated thrombocytopenia after clopidogrel and pentoxifylline therapy: a case report. Journal of Medical Case Reports 2011;5:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
Walsh 2003
- Walsh MC, Wilson‐Costello D, Zadell A, Newman N, Fanaroff A. Safety, reliability, and validity of a physiologic definition of bronchopulmonary dysplasia. Journal of Perinatology 2003;23(6):451‐6. [DOI] [PubMed] [Google Scholar]
Ward 1987
- Ward A, Clissold SP. Pentoxifylline. A review of its pharmacodynamic and pharmacokinetic properties, and its therapeutic efficacy. Drugs 1987;34(1):50‐97. [DOI] [PubMed] [Google Scholar]
Watterberg 1996
- Watterberg KL, Demers LM, Scott SM, Murphy S. Chorioamnionitis and early lung inflammation in infants in whom bronchopulmonary dysplasia develops. Pediatrics 1996;97(2):210‐5. [PubMed] [Google Scholar]