

Summary
Mutations in proteasome β-subunits or their chaperone and regulatory proteins are associated with proteasome-associated autoinflammatory disorders (PRAAS). We studied six unrelated infants with three de novo heterozygous missense variants in PSMB10, encoding the proteasome β2i-subunit. Individuals presented with T-B-NK± severe combined immunodeficiency (SCID) and clinical features suggestive of Omenn syndrome, including diarrhea, alopecia, and desquamating erythematous rash. Remaining T cells had limited T cell receptor repertoires, a skewed memory phenotype, and an elevated CD4/CD8 ratio. Bone marrow examination indicated severely impaired B cell maturation with limited V(D)J recombination. All infants received an allogeneic stem cell transplant and exhibited a variety of severe inflammatory complications thereafter, with 2 peri-transplant and 2 delayed deaths. The single long-term transplant survivor showed evidence for genetic rescue through revertant mosaicism overlapping the affected PSMB10 locus. The identified variants (c.166G>C [p.Asp56His] and c.601G>A/c.601G>C [p.Gly201Arg]) were predicted in silico to profoundly disrupt 20S immunoproteasome structure through impaired β-ring/β-ring interaction. Our identification of PSMB10 mutations as a cause of SCID-Omenn syndrome reinforces the connection between PRAAS-related diseases and SCID.
Keywords: severe combined immune deficiency, Omenn syndrome, PSMB10, immunoproteasome, revertant somatic mosaicism, uniparental disomy
Graphical abstract
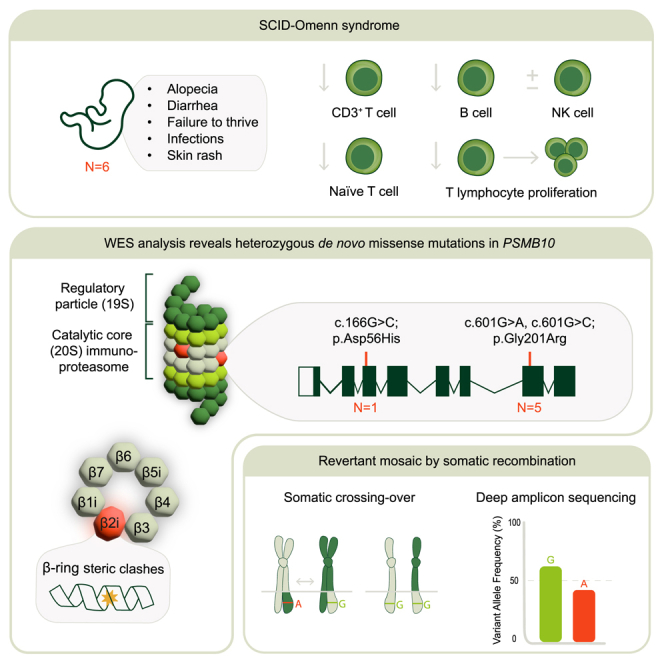
This study highlights the identification of recurrent heterozygous de novo mutations in immunoproteasome subunit β2i (PSMB10) as a cause for SCID-Omenn syndrome. These variants are predicted to profoundly disrupt immunoproteasome structure and function, emphasizing its importance for lymphocyte development. Pathogenic variants in PSMB10 should be sought in SCID newborn screening.
Main text
The human proteasome facilitates the controlled degradation of intracellular proteins that are targeted for breakdown by ubiquitination. The standard proteasome (SP) is composed of an enclosed cylinder-shaped central 20S core complex harboring the acidic, basic, and hydrophobic cleaving β-subunits β1, β2 ,and β5, which together with two 19S regulatory units or the PA28 regulator form the 26S proteasome.1 The proteolytic β-subunits can be exchanged for their inducible counterparts β1i (LMP2/PSMB9), β2i (MECL1/PSMB10), and β5i (LMP7/PSMB8) to form the immunoproteasome (IP).2 The IP is constitutively expressed in hematopoietic cells and can be induced in non-immune cells upon exposure to proinflammatory cytokines.3 Compared to the SP, IP assembly is faster with more efficient antigen processing, explaining a fundamental role in the generation of pathogen-derived peptides and major histocompatibility complex (MHC) class I antigen presentation.3,4,5 Moreover, thymic cortical epithelial cells express thymoproteasomes that are essential in establishing central tolerance, containing the β1i and β2i subunits together with the thymus-specific β5t subunit (PSMB11) instead of β5i.6 Dysfunction of several proteasome β-subunits or their respective chaperone and regulatory proteins leads to proteasome-associated autoinflammatory syndrome (PRAAS), characterized by nodular dermatitis, lipodystrophy, recurrent fever, and type-I interferon-induced immune dysregulation.
The majority of individuals with PRAAS have underlying autosomal-recessive or digenic heterozygous mutations impairing the 20S core particle (please refer to Table 1 for an overview of PRAAS-related diseases).7,8,9,10,11 Autosomal-dominant mutations in proteasome subunits or accessory proteins have been associated with phenotypes distinct from classical PRAAS. Heterozygous (de novo) mutations affecting 19S subunits have been described in persons with primary neurodevelopmental syndromes with a type-I interferon (IFN) signature. Moreover, individuals with de novo nonsense-mediated decay (NMD)-escaping mutations affecting proteasome assembly protein POMP exhibited signs of autoinflammation, immune dysregulation, and combined immunodeficiency (PRAID).12 Recently, a recurrent de novo mutation in PSMB9 was described in two infants presenting with combined immunodeficiency.13
Table 1.
Current genetic and clinical spectrum of monogenic proteasome-associated inflammatory diseases
| Gene | PSMB87,10,14,15,16 | POMP12,17 | PSMB48,18 | PSMG29 | PSMB1011,19 | PSMB120 | PSMD1221,22,23,24 | PSMC3,25 | PSMC326, | PSMB913 |
|---|---|---|---|---|---|---|---|---|---|---|
| Disease | PRAAS1 (MIM: 256040 | PRAAS2 (MIM: 618048) | PRAAS3 (MIM: 617591) | PRAAS4 (MIM: 619183) | PRAAS5 (MIM: 619175) | NDD (MIM: 620038) | Stankiewicz–Isidor syndrome (MIM: 617516) | NDD | DCIDP (MIM: 619354) | PRAAS-ID |
| Mutational mechanism | AR LoF | AD LoF DN | AR LoF | AR LoF | AR LoF | AR LoF | AD LoF HI | AD LoF | AR LoF | De novo LoF DN |
| Encoded protein | Subunit β5i | proteasome maturation protein | subunit β7 | assembly chaperone 2 | subunit β2i | subunit β6 | 19S/26S subunit, non-ATPase 12 | 19S/26S subunit, ATPase 3 | 19S/26S subunit, ATPase 3 | subunit β1i |
| Proteasome defect | 20S, 26S IP defect | 20S, 26S IP + SP defect | 20S, 26S IP + SP defect | 20S, 26S IP + SP defect | 20S, 26S IP defect | 20S, 26S SP defect | 20S IP + SP defect | 20S, 26S SP + IP defect | decreased ubiquitylation, proteotoxic stress | 20S IP defect |
| Clinical findings | ||||||||||
| Periodic fever | + | + | + | + | + | – | – | – | – | + |
| Skin rash | + | + | + | + | + | – | + | – | – | + |
| Myositis/muscle dystrophy | + | – | + | + | – | N/A | N/A | – | – | + |
| Arthritis | + | + | + | – | – | – | – | – | – | – |
| Liver dysfunction | + | N/A | + | N/A | N/A | N/A | N/A | N/A | N/A | + |
| Infections | +/− | + | + | – | N/A | – | – | – | – | + |
| Pneumonia | + | + | + | – | – | – | – | – | – | + |
| Splenomegaly | + | – | + | + | + | – | – | – | – | +/− |
| Lipodystrophy | + | + | + | + | N/A | – | – | – | – | – |
| Basal ganglia calcification | +/− | – | – | + | N/A | N/A | – | N/A | N/A | + |
| IFN-I signature | + | ++ | + | + | + | N/A | + | + | N/A | =/+ |
| Viremia | N/A | N/A | – | – | N/A | N/A | – | – | – | + |
| Congenital malformations (incl. facial dysmorphism) | + | + | N/A | N/A | + | ++ (short stature, deafness) | +++ (deafness) | +++ (deafness) | +++ (cataract, deafness) | N/A |
| Neurological abnormalities | + | + | N/A | + | N/A | +++ (IDD) | +++ (IDD, autism) | +++ (IDD) | +++ (IDD, PNP) | – |
| Laboratory evaluation | ||||||||||
| Elevated inflammatory markers | + | + | + | + | + | N/A | + | N/A | N/A | + |
| Microcytic anemia | + | N/A | +/− | – | + | N/A | N/A | N/A | N/A | N/A |
| Thrombocytopenia | =/↑ | + | + | + | =/↑ | N/A | N/A | N/A | N/A | + |
| T cell | N/A | CD4 ↑, CD8 ↓, CD4/CD8 ratio ↑, naive T cell phenotype | low CD8, CD4/CD8 ratio ↑ | N/A | N/A | N/A | variable | N/A | N/A | variable |
| B cell | N/A | ↓ | variable | N/A | N/A | N/A | =/↑ | N/A | N/A | =/↓ |
| Serum Ig | ↑ | Dysgamma-globulinemia | =/↑ | N/A | normal | N/A | N/A | N/A | N/A | IgG ↓ |
| Auto-antibodies | variable | + | + | + | N/A | N/A | N/A | N/A | N/A | – |
AD, autosomal-dominant; AR, autosomal-recessive; DCIDP, deafness, cataract, impaired intellectual development, and polyneuropathy; DN, dominant-negative; HI, haploinsufficiency; IDD, intellectual and developmental disability; IFN-I, type-I interferon; LoF, loss-of-function; NDD, neurodevelopmental disorders; PNP, polyneuropathy.
Individuals with typical severe combined immunodeficiency (SCID) lack T cells altogether and present during early infancy with recurrent opportunistic infections and failure to thrive (FTT). In atypical forms of SCID, immunodeficiency is similar in severity, but the block in T cell development is incomplete, leading to low naive T cells (<20%) usually within a reduced total T cell number. Omenn syndrome (OS [MIM 603554]) is a specific form of atypical SCID, in which T cells expand oligoclonally in the periphery, infiltrating end-organs such as the skin. The clinical diagnosis of OS requires a generalized erythrodermic rash, exclusion of maternofetal engraftment, and 2 or more of hepatosplenomegaly and lymphadenopathy, eosinophilia or high IgE levels.27 These disorders can be detected based on clinical presentation or newborn screening (NBS) programs for SCID. In this study, we describe six unrelated infants with predominant signs of SCID and clinically diagnosed Omenn(-like) syndrome who carried heterozygous de novo missense variants in PSMB10, with an overlapping revertant mosaicism (RM) in one infant. The latter individual was previously reported as part of a trio sequencing study in subjects with inborn errors of immunity (IEI).28 Molecular modeling suggests that the encoded PSMB10 missense variants cause profound disruption of the 20S proteasome structure through impaired β-ring/β-ring interaction, similar to previously reported human mutations in PSMB9. These results identify de novo variants in PSMB10 as a monogenic cause of autosomal-dominant SCID-OS within the spectrum of PRAAS-related diseases. All six individuals presented with early-onset erythroderma, FTT, diarrhea, alopecia, and opportunistic infections (detailed clinical history and laboratory values are available in Table 2; Figure 1A; Tables S1–S3 and Supplemental note: Case reports).
Table 2.
Genetic and clinical characteristics of individuals with monoallelic PSMB10 variants
| Individual 1 | Individual 2 | Individual 3 | Individual 4 | Individual 5 | Individual 6 | |
|---|---|---|---|---|---|---|
| Genetics | ||||||
| Ancestry | European | Jewish-Sepharadi | European | European | European | European |
| Variant | c.601G>A (p.Gly201Arg) | c.601G>A (p.Gly201Arg) | c.601G>C (p.Gly201Arg) | c.601G>A (p.Gly201Arg) | c.166G>C (p.Asp56His) | c.601G>A (p.Gly201Arg) |
| Allele frequencya | 0 | 0 | 0 | 0 | 0 | 0 |
| CADD-Phred score | 35 | 35 | 34 | 35 | 28 | 35 |
| Clinical presentation | ||||||
| Age at investigation (weeks) | 8 | 2 | 6 | 13 | 4 | 0c |
| Sex | M | M | M | F | M | M |
| Failure to thrive | + | + | + | + | + | – |
| Diarrhea | + | +++ | + | + | +++ | + |
| Skin rash | +++ | + | +++ | +++ | ++ | + |
| Age at onset rash (weeks) | <1 | 2 | 3 | 1 | 8 | <1 |
| Recurrent infections | + | + | + | + | + | – |
| Systemic inflammation | – | + | – | – | – | – |
| Hepatomegaly | – | – | + | + | – | – |
| Lymphadenopathy | + | – | – | – | + | – |
| Alopecia | + | + | + | + | N/A | – |
| Dysmorphology | + | + | – | – | – | – |
| Laboratory investigationb | ||||||
| Eosinophils (/μL) (40–800) | 896 | 2,930 | 1,700 | 1,700 | 720 | 1,000 |
| IgG (g/L) (3.7–12.6) | 1.35 | 0.974 | 2.6 | 2.76 | 5.1 | 2.4 |
| IgA (g/L) (0.02–0.15) | <0.07 | <0.01 | <0.07 | 0.23 | 0.41 | <0.04 |
| IgM (g/L) (0.05–0.29) | <0.07 | <0.02 | 0.09 | 0.12 | 0.98 | <0.04 |
| CD3 (/μL) (1,700–3,600) | 1,300 | 1,552 | 595 | 1,239 | 188 | 309 |
| CD4 (/μL) (1,700–2,800) | 1,100 | 730 | 551 | 1,143 | 137 | 272 |
| CD8 (/μL) (800–1,200) | 40 | 820 | 72 | 83 | 26 | 60 |
| CD4:CD8 ratio | 27.5 | 0.89 | 7.7 | 13.8 | 5.3 | 4.5 |
| CD19 (/μL) (500–1500) (%) | 40 | 430 (cells/mm3) | 0 | <1 | 19 | 34 |
| CD3/CD56 (/μL) (300–700) | 70 | 1,826 | 46 | 414 | 22 | 368 |
| CD45RA (%CD3) | 8.6 | N/Ac | 1 | 0 | 8 | 0 |
| CD45RO (%CD3) | 95 | N/Ac | 91 | N/A | N/A | 4 |
| TCR αβ (%CD3) | 98 | N/A | 97 | 99 | 74 | 96 |
| TCR γδ (%CD3) | 2 | N/A | 3 | 1 | 26 | 4 |
| Mitogen response (PHA) | decreased | decreased | decreased | decreased | decreased | decreased |
| Therapy–Hematopoietic stem cell transplantation (HSCT) | ||||||
| Age at transplant (weeks) | 12 | 130 | 11 | 16 | 12 | 9 |
| Donor information | HLA identical sibling | URD | URD cord | maternal haplo | 9/10 mM cord blood | paternal haplo |
| Serotherapy | ATG | ATG | alemtuzumab | ATG | none | ATG + Rituximab |
| Chemotherapy | Cyclo | Cyclo + MMF | Flu + Mel | Bu + Cyclo | Treo + Flu | Treo + Flu |
| Outcome and follow up | alive, age 18 years | died, age 2 years | died, age 16 years | died, age 11 weeks | died, age 4 years | alive, age 0–1 year |
| 100% donor skin/gut GVHD marked infection- and drug-induced hyperresponsivity of the skin |
100% donor fatal transplant-associated TMA |
100% donor skin GVHD severe VOD long-term enteropathy liver cirrhosis ESRD (hemodialysis) |
pneumonitis with capillary leak peri-engraftment GVHD skin and gut recurrence of VZV with fatal encephalopathy |
100% donor skin GVHD (late) marked mucositis and skin toxicity adenoviraemia long-term enteropathy with norovirus infections |
100% donor no GVHD episode of acute encephalopathy currently <3 months post-HSCT |
|
ATG, antithymocyte globulin; Bu, busulfan; CADD, combined annotation dependent depletion; Cyclo, cyclophosphamide; ESRD, end-stage renal disease; Flu, fludarabine; GVHD, graft-versus-host disease; Mel, melphalan; MMF, mycophenolate mofetil; TREC, T cell receptor excision circles; Treo, treosulfan; URD, unrelated donor; VOD, veno-occlusive disease; VZV, varicella zoster virus.
Allele frequency in GnomAD, dbSNP or ExAC databases.
Parameters are presented with units and normal reference ranges if applicable.
For this individual, TREC copies were available with significantly reduced levels.
Figure 1.

Clinical features and identification of PSMB10 de novo missense variants
(A) PSMB10 variants in six infants with SCID.
(B) Erythematosquamous rash in individual 1 at 12 weeks after birth.
(C) Histology of the initial skin biopsy of individual 1 showed a graft-versus-host-disease-like pattern with vacuolar interface inflammation, multiple scattered apoptotic keratinocytes, and involvement of the adnexal structures, in the presence of a limited lymphocytic infiltrate (hematoxylin and eosin staining; original magnification ×41).
(D) Histopathological evaluation of an inguinal lymph node extracted from individual 5 showed a paucicellular, stroma-rich lymph node.
(E) A jejunal biopsy from individual 3 was hallmarked by partial villous atrophy and crypt hyperplasia with relatively few lymphocytes.
(F) Colonic mucosa biopsied from individual 5 showed preserved crypt architecture with an empty lamina propria with few lymphocytes, in keeping with an immunodeficiency-related enteropathy.
(G) Immunohistochemistry showed the absence of CD3+ and CD20+ positive cells in the colon samples from individual 5, although significant numbers of CD4+ cells were observed that may be of a macrophage/monocyte lineage.
(H) Visualization of the three identified PSMB10 variants at the cDNA and protein level. The conservation across species is shown and scaled by color. The asterisk indicates the position of a previously studied Psmb10 variant in TUB6 mice.
(I) Immunoblot for PSMB10 in dermal fibroblasts of 3 controls, individual 3 (F3.II.1; p.Gly201Arg), individual 4 (F4.II.1; p.Gly201Arg), and individual 5 (F5.II.1; p.Asp56His), with or without prior IFN-gamma induction. Upper band (white arrowhead) represents immature and lower band (black arrowhead), mature, PSMB10; subject samples also show an additional, intermediate band (black arrow). Representative of 4 independent experiments.
The skin rash manifested within two months after birth and was characterized as a generalized erythroderma with desquamation (individual 1 [Figure 1B], 3, and 4), a raised generalized maculopapular rash (individuals 5 and 6), or only a mild facial erythroderma diagnosed as acne neonatorum (individual 2). The infections consisted of oral candidiasis (individuals 1, 3, and 4), disseminated and chronic viral infections (varicella zoster virus [VZV]), adenovirus, cytomegalovirus (CMV) (individuals 2, 4, and 5), Pneumocystis pneumonia (individual 4), or secondary skin infections (individuals 1 and 4). Individuals 2 and 5 displayed severe, intractable diarrhea that required total parenteral nutrition (TPN). A sixth infant (individual 6) was identified via NBS as having low T cell receptor excision circles (TRECs) and thrived despite developing blood and mucus in his stools and generalized dry skin by 2 months of age. On laboratory testing, T lymphocytes were low in number with an elevated CD4:CD8 ratio (except individual 2) and skewing toward a memory phenotype with low CD45RA and/or elevated CD45RO expression and reduced proliferative capacity (Table 2). T cell receptor (TCR) repertoires were limited (tested in individuals 1, 2, and 3). Circulating B lymphocytes were reduced or absent, associated with marked hypogammaglobulinemia. Natural killer (NK) cell numbers were low-normal. Individuals 1–4 had hypereosinophilia, which is typical for SCID-OS. Bone marrow (BM) examination showed a near complete block of B lymphocyte development in individuals 1 and 5, although slightly less severe when compared with other individuals with OS caused by RAG1, RAG2, or Artemis deficiency (Figure S1). In individual 6, a non-accredited interferon-stimulated gene transcriptional signature was assessed pre-transplant and was not markedly raised (5 of 6 transcripts showed normal abundance while IFI27 transcripts were 3× above the upper limit of normal).
Histological examination of the skin indicated a flattened epidermis with vacuolization of the basal epidermal layer and hyperparakeratosis in individuals 1 (Figure 1C) and 3 and vacuolar dermatitis with eosinophils and pigment laden macrophages in individual 2. Lymph node biopsy in individual 5 was stroma-rich with a paucity of (CD4+) lymphocytes, abortive primary follicle formation, and absence of germinal centers (Figures 1D and S2A–S2C). Small bowel biopsies in individuals 3 and 5 showed partial villous atrophy and crypt hyperplasia with apoptotic bodies, while colon biopsies showed preserved crypt architecture (Figures 1E, 1F, S2D, and S2E). Immunohistochemistry demonstrated an empty lamina propria with lack of plasma cells, low T cell numbers, and absence of B lymphocytes, consistent with an immunodeficiency-related enteropathy (Figures 1G and S2F–S2H).
All affected infants received allogeneic hematopoietic stem cell transplantation (HSCT) with different donor types and pre-conditioning regimens (Table 2). Post-transplant outcomes were characterized by severe inflammatory complications, including graft-versus-host disease (GVHD) in individuals 1, 3, 4, and 5 and fatal thrombotic micro-angiopathy (TMA) in individual 2. Shortly after HSCT, individual 4 died following encephalopathy caused by treatment-refractory reactivation of VZV and associated encephalitis confirmed at autopsy. Individual 6 developed an acute neurological deterioration one month after transplant with evidence of widespread white matter changes on magnetic resonance imaging and the main differential diagnosis of chemotherapy-related neurotoxicity or immune-mediated encephalitis. He improved with high-dose corticosteroids and supportive care, but the long-term neurological prognosis remains guarded. Individuals 3 and 5 had signs of severe chronic enteropathy during long-term follow up. Immunosuppressive treatment yielded a partial response in individual 5 that was complicated by chronic norovirus infection, and he died due to sepsis at age 4. Individual 3, who had learning difficulties, acquired other chronic comorbidities, including liver cirrhosis and hemodialysis-dependent end-stage renal disease and died at the age of 16. Individual 1 suffered from severe cyclosporin-related toxicity and still experiences notable infection- and drug-induced hypersensitivity, resulting in toxic skin reactions. He is currently 18 years after HSCT and has normal cognitive and intellectual development.
Clinical whole-exome sequencing (WES) was performed in all individuals, but in silico analysis did not reveal any actionable variants within known genes associated with IEI.29 Written informed consent and publication consent were obtained from individuals 1–2 and/or their parents and were approved by the local ethics committees. Parents of individuals 3–6 provided generic consent for future research through ethically approved procedures (REC ref. 10/H0906/22). Subsequently, exome-wide research-based analysis identified three heterozygous missense variants in PSMB10: two variants affected the same nucleotide in exon 7 (c.601G>A [GenBank: NM_002801] [p.Gly201Arg] and c.601G>C [GenBank: NM_002801] [p.Gly201Arg]) and one was located in exon 3 (c.166G>C [GenBank: NM_002801] [p.Asp56His]) (Figure 1A). All variants impacted highly conserved nucleotides and amino acid residues in the protein structure. The variants were predicted by in silico tools to be deleterious and were absent from population and our in-house WES databases (Table 1; Figure 1H). Trio-based exome sequencing in individuals 1 and 2 and segregation analysis in individuals 3, 4, 5, and 6 determined all variants to be de novo, while no other disease-causing candidate variants were identified (Table S4). Using a commercial antibody, we performed immunoblotting for PSMB10 protein on dermal fibroblasts that were available from individuals 3, 4, and 5 and controls, with or without prior IFN-gamma treatment (Figure 1I). There was no difference in overall PSMB10 protein accumulation nor the ratio of immature to mature forms, but we noted an additional band of intermediate size present only in samples from individuals bearing a PSMB10 variant. This implies that mutated protein is expressed but shows altered physico-chemical characteristics, consistent with (although not pathognomonic of) dominant-negative behavior.
Moreover, a genome-wide single-nucleotide polymorphism (SNP) microarray in individual 1, performed on blood-derived DNA before transplant at the age of 2 months, identified a partial somatic uniparental disomy (UPD) of chromosome 16 (UPD16) overlapping the PSMB10 locus, indicating an aberrant B-allele frequency (BAF) profile on the q arm of chromosome 16 spanning ∼24 Mb from 16q21 to the terminal end of 16q (16qter) (Figure 2A). Further validation in buccal tissue at the age of 8 and 13 years demonstrated a different UPD, roughly ∼10 Mb more proximal (16q12.1) (Figure 2A). The two independent UPDs presumably originated during mitotic recombination where in a single progenitor cell, the germline mutation was restored with a wild-type copy of the unaffected parental chromosome. Ultra-deep amplicon sequencing demonstrated a slightly higher rate of RM in blood, with respective variant allele frequencies (VAFs) of 39.98% for blood-derived DNA obtained before transplant and 42.90% for buccal-swab DNA collected at age 13, suggesting that 79.96% and 85.80% of cells are heterozygous for the PSMB10 mutation (Figure 2B). We cannot exclude the fact that this slight difference might be due to contamination of buccal swab DNA with post-HSCT wild-type blood-derived DNA. Subsequently, we have adapted our haplarithmisis method for trio WES data to confirm the somatic UPD and to determine its parental origin. The level of mosaicism was estimated by calculating the distortion of segmented haplarithm values from the expected 1:1 allelic ratio, i.e., 0.5:0.5 vertical distance in each segmented parental haplarithm. This approach confirmed that the p arm of chromosome 16 shows both parental haplotypes present in 0.5:0.5 vertical distance (green vertical arrows), but also suggests a somatic UPD by RM in blood-derived DNA, resulting in a paternal allele bias of 0.59 (9% distortion from BAF, upper orange vertical bar) and maternal allele bias of 0.44 (6% distortion from BAF, lower orange vertical bar), resulting in a 15% mosaic maternal UPD on chromosome 16q21–qter (Figure 2C). The proportion of cells that were estimated to be restored to wild-type following the 15% maternal UPD event correlated with the 20% of cells that were homozygous wild-type based on amplicon sequencing data. There was no indication of the presence of RM in the other individuals.
Figure 2.

Acquired segmental UPD overlapping the PSMB10 locus shows evidence of RM
(A) Aberrant BAF profile of the genome-wide SNP-array analysis in individual 1 reveals two independent UPD events at the q arm of chromosome 16 in blood (pre-HSCT at 2 months of age; 16q12.1) and in the buccal mucosa (16q12.1) spanning to the terminal end of 16q (16qter). Arrows indicate the respective breakpoints of UPD and are color-coded for each tissue; the red bar in the ideogram represents the distinct location of PSMB10.
(B) Exome inclusion of the unaffected parents of individual 1 resulted in the identification of a unique heterozygous de novo missense mutation in PSMB10 (c.601G>A [GenBank: NM_002801.3] [p.Gly201Arg]). De novo status was verified by Sanger sequencing, while deep amplicon sequencing using the Ion Torrent accurately determined the respective mosaic levels in both tissues.
(C) Results from haplarithmisis on blood-derived DNA from individual 1 (pre-HSCT at 2 months of age) and parents. From top to bottom we depict BAF, paternal haplarithm, maternal haplarithm, and logR (relative copy number) values of the child, followed by BAF and logR-values of the parents. BAF of a single-nucleotide variant (SNV) is the number of allele B over the number of alleles A and B for that SNV, and logR is the base 2 logarithm of the summed normalized number of both alleles in a window of 100 kb over the expected signal intensity values.
Next, we modeled the structural impact of both missense variants using experimentally determined 3D structures of PSMB10 and the 20S proteasome (Figure 3A). Both the aspartic acid at position 56 and the glycine at position 201 hold highly conserved positions in β2i/PSMB10 and are positioned at the β-ring interface in close proximity to residues in regulating domains that facilitate incorporation into the proteasome 20S complex through interaction with surrounding subunits (Figure 3B). Substitutions of the acidic aspartic acid to the larger, basic histidine and the highly flexible wild-type glycine for the substantially larger mutant arginine are expected to cause significant disruption to the local structural environment due to the inability to fit within the same spatial constraints (Figure 3B). In particular, the β-ring/β-ring interaction between β2i and β7 (PSMB4) is predicted to be affected by steric clashes, leading to misfolding of the β2i subunit. This is supported by energy calculations of protein stability, indicating that the variants induce a high change in Gibbs free energy (ΔΔG) and are thus highly destabilizing for the 20S IP structure (Figures 3C–3F).
Figure 3.

Predicted structural consequences of the PSMB10 variants
(A) Crystal structure of human IP 20S particle (PDB: 6E5B) is shown with its alpha subunits in shades of yellow/orange and beta subunits in shades of blue/green.
(B) Positions of interest, Gly201 and Asp56, in the β2i subunits are highlighted as red spheres. The local structural environment of Gly201 (sticks) is depicted in (C) including distances from its interacting residues in the α-helix, which is located close to the β-ring interface.
(D) Local structural clashes (in red discs) potentially brought about by p.Gly201Arg are shown. Together with the drastic changes in free energy of the complex, p.Gly201Arg appears to be structurally damaging. Similar structural representations are illustrated for the variant p.Asp56His in (E) and (F), which also include the position Gly201 for visual reference.
Subsequently, we compared the effect of the p.Gly201Arg variant to a published TUB6 mouse model harboring the ENU (N-ethyl-N-nitrosourea) mutagenesis-generated c.625G>T (p.Gly209Trp) variant in Psmb10 (reported as p.Gly170Trp based on sequence alignment to Thermoplasma acidophilum), causing a phenotype reminiscent of human OS and sterile autoinflammation.30,31,32,33 When superposing the structures of the human and mouse PSMB10 and 20S IP, we observed a high degree of structural similarity (Figure 4A). Structural alignment showed that both glycine variants were in close proximity (Figure 4B). We noted that p.Gly209Trp conferred similar destabilizing effects as p.Gly201Arg, owing to the bulky nature of tryptophan amino acid at position 209 that introduces steric clashes with the neighboring residues (Figure 4C). The variant was predicted to have a more destabilizing impact on 20S IP structure as compared to the human variants (Figure 4D). Moreover, the p.Asp56His and p.Gly201Arg variants were predicted to be slightly less destabilizing for the full 26S proteasome as compared to the 20S IP (Figure 4D).
Figure 4.

Comparison of predicted structural impact of variants on human and mouse PSMB10
(A) Superposition of human (PDB: 6E5B) and mouse (PDB: 3UNH, purple) 20S IP crystal structures is shown (root-mean-square deviation [RMSD] = 0.59 Å), with the positions of interest highlighted in red spheres. The local structural environment of Gly201 and Gly209 is depicted in (B), which highlights the clear overlap of residues between human (cyan) and mouse (purple) PSMB10.
(C) The steric clashes potentially brought about by Gly209 in human 20S IP are illustrated as red discs, suggesting a structurally damaging outcome of the p.Gly209Trp variant. The table in (D) lists the predicted differences in free energies of the proteasome complexes for each variant type for each protein system.
The clinical and immunological phenotypes of the individuals we studied contrast with classical PRAAS (Table 1) and instead fulfills diagnostic criteria for SCID-OS syndrome.27 Infants presented early with FTT, erythroderma, diarrhea, oral candidiasis, opportunistic infections, and inflammatory phenomena involving the skin and GI tract. Consistent with OS, they had a reduced number and function of T and B lymphocytes, hypereosinophilia, variability in NK lymphocyte numbers, T cell clonal expansion, and impaired T cell proliferation. Histological features in skin were compatible with OS without PRAAS-characteristic signs of neonatal-onset neutrophilic dermatosis or lipoatrophy.11 B cell maturation in the BM was severely reduced in a pattern similar to that seen in classical OS caused by defects of V(D)J recombination (Figure S1). Compared to OS, an important difference is the poor HSCT outcome in our studied individuals, with higher incidence of GVHD and mortality.34,35 In particular, the chronic enteropathy and skin hypersensitivity remaining post-GVHD in these tissues were remarkable. Previously, persons with PRAAS due to POMP deficiency and recessive mutations in PSMB4 have successfully undergone HSCT without severe post-transplant complications.17,18,36,37 Although potential benefit of treatment with JAK inhibitors has been demonstrated in individuals with type I interferonopathies, including PRAAS, the role of dysregulated type I IFN signaling in individuals with (severe) combined immunodeficiency due to proteasome-associated mutations remains to be elucidated.13,38
Autosomal-recessive mutations in PSMB10 have been described in individuals with PRAAS without immunodeficiency.11,19 All four reported individuals carried compound heterozygous or homozygous mutations affecting the phenylalanine residue at position 14, located in the N-terminal pro-peptide sequence. These mutations interfered with cleavage of PSMB10, thereby impairing β2i maturation, proteasome assembly, and trypsin-like catalytic function.11,19 In contrast, the clinical features of our studied individuals, with in part recurrent de novo mutations, recall the recent description of two immunodeficient children with de novo missense mutation in PSMB9, which the authors termed PRAAS with immunodeficiency (PRAAS-ID) to distinguish it from the classic PRAAS phenotype.13 Shared features with our PSMB10-mutated infants included infections and chronic viremia, liver dysfunction, skin rash, absence of lipoatrophy and a combined T and B cell defect with an increased CD4/CD8 ratio, skewing toward a CD8 T cell memory phenotype, and hypogammaglobulinemia. The authors demonstrated that the c.494G>A (p.Gly156Asp) substitution impaired maturation of the PSMB9/β1i, β2i, and β5i subunits and abrogated activity of the 20S but not the 26S IP.13,39 The clinical and functional phenotype was recapitulated in mice that had a knockin of the identified p.Gly156Asp variant, except for the autoinflammatory symptoms including fever and myositis.13 In silico structure modeling indicated that the p.Gly156Asp variant disturbed the β-ring/β-ring interaction without affecting the active site conformation. Similar variant effects were predicted for our PSMB10 mutations. Given the co-dependent incorporation of the β1i and β2i subunits that together facilitate β5i incorporation, overall misfolding of all three βi-subunits was predicted, rather than a selective impact on β2i′s enzymatic (trypsin-like) activity.4 Since the IP incorporates two of each subunit into the catalytic core, we hypothesize that the p.Asp56His and p.Gly201Arg PSMB10 variants would exert similar dominant-negative effects.
The observed p.Gly201Arg substitution is located in close proximity to the Psmb10 p.Gly209Trp variant in TUB6 mice.30 Homozygous mice presented with T-B-NK-SCID, sterile autoinflammation, hyperkeratosis of the skin, alopecia, neutrophilic infiltration, lipoatrophy, and a reduced life span, while heterozygous mice exhibited an isolated T cell defect with decreased T cell number, elevated CD4/CD8 ratio, and susceptibility to Listeria infection.30 The overlap of these features in mice compared with our studied individuals is striking, although the heterozygous mice exhibited less severe features, likely due to a more robust compensation of heterozygous mutations generally observed in mice. Although mice did show neutrophilic infiltration and lipoatrophy at 8 weeks, the apparent lack of autoinflammatory symptoms in affected children could be due to the early intervention with HSCT. Interestingly, individual 2 displayed hemophagocytic lymphohistiocytosis (HLH)-like inflammation and was transplanted last at the age of 2.5 years. In addition, the severe post-HSCT inflammatory complications might be related to the underlying IP defect, perhaps resulting from an impaired function in extra-hematopoietic tissues such as in the epithelial cells of the skin and gut. Furthermore, the p.Gly209Trp Psmb10 variant was predicted to disrupt β-ring/β-ring interaction similar to p.Gly201Arg and was expected to dramatically impact 20S IP complex formation and, to a lesser extent, the 26S proteasome. Since β2i is incorporated into the thymoproteasome along with β1i and β5t, and TUB6 mice lacked cortical thymic epithelial cells that are important for T cell development, thymic dysfunction might contribute to the observed T cell phenotype in our individuals.40 In addition, dominant-negative missense mutations are hypothesized to prevent substitution of the mutated subunits by their constitutive counterparts and affect the stability of core 20S proteasome that interferes with homeostatic IP functions beyond CD8 T cell function and MHC-I antigen presentation as observed in single, double, or triple PSMB8/β5i, PSMB9/β1i and PSMB10/β2i knockout mouse models.3,41,42,43 These include removal of unfolded and oxidized proteins and T cell proliferation, differentiation and survival independent of MHC-I antigen presentation capabilities, especially in the context of inflammation.44,45,46 Therefore, the observed defects in T and B cell development and maturation in our individuals might suggest a mutation-specific defect in these homeostatic functions cumulating in proteotoxic stress.47 In addition, PSMB10 might have a direct role in V(D)J recombination.
This study also demonstrates that the evaluation of RM in trio-based exome data using the haplarithmisis method offers a unique, sensitive mapping strategy that, depending on the quality of UPD mapping and the detection sensitivity, has the potential to trace previously undetected mutations. RM delineates the rare phenomenon in which the occurrence and/or accumulation of spontaneous somatic mutations coincides with the direct or indirect rescue of a pathogenic germline mutation,48 in essence suggesting natural evidence for the repair of a functional defect caused by the de novo mutation in PSMB10. RM has been observed to have a beneficial clinical impact for a range of IEI, including OS.49,50,51 The co-occurrence of a de novo PSMB10 mutation with RM is unlikely to be a chance finding, demonstrates an elegant but possibly underestimated disease gene mapping strategy, and is suggestive for a strong cellular effect of the respective PSMB10 mutation and an associated evolutionary pressure of mutated cell lineages.48 To our knowledge, exome data have not been used to detect and visualize haplotype revertant mosaics, and we propose that this approach should be applied more often to individuals with IEI as a mapping strategy for disease gene identification. It is intriguing that the only person to achieve full T cell reconstitution post-transplant also demonstrated partial somatic reversion of the PSMB10 mutation.
In summary, our study reports de novo autosomal-dominant, suspected dominant-negative, PSMB10 missense mutations as a cause of SCID-OS (PSMB10-OS) that should be sought in NBS. Furthermore, these findings expand the spectrum of proteasome-associated monogenic diseases.
Data and code availability
The article includes all datasets generated or analyzed during this study. The exome-sequencing data of the five families supporting the current study have not been deposited in a public repository because of privacy issues but are available from the corresponding author on request.
Acknowledgments
We would like to thank the individuals and their families for participation in this study. We acknowledge colleagues from our diagnostic division (Genome Diagnostics Nijmegen), the Radboud Genomics Technology Center, and all members of the Radboud University Medical Center multidisciplinary immune-disease board and colleagues at Great North Children’s Hospital, Great Ormond St Hospital for Children, and sister hospitals. C.I.v.d.M., S.K., and A.H. were supported by the Radboud Institute for Molecular Life Sciences. G.R. was supported by the Europees Fonds voor Regionale Ontwikkeling (EFRO) (R0005582). T.M. was supported by the Sigrid Jusélius Foundation. M.G.N. was supported by the Nederlandse Organisatie voor Wetenschappelijk Onderzoek (Spinoza grant) and the European Research Council (grant agreement no. 310372). F.L.v.d.V. was supported by a ZonMW Vidi grant. R.S. is supported by the Israel Science Foundation (ISF) under the Israel Precision Medicine Program (IPMP), grant agreement no. 3115/19. S.H. was supported by the Wellcome Trust (grant number 207556_Z_17_Z). A.H. was supported by the Solve-RD project, which has received funding from the European Union’s Horizon 2020 research and innovation programme under grant agreement no. 779257.
Author contributions
Conceptualization: C.I.v.d.M., S.K., O.C., R.S., S.H., S.S.V.H., and A.H.; data curation: C.I.v.d.M., S.K., O.C., K.R.E., G.R., H.G., and I.S.v.d.L.; formal analysis: C.I.v.d.M., S.K., G.R., H.V., R.E., and M.Z.E.; funding acquisition: M.G.N., F.L.v.d.V., R.S., S.H., A.H.; investigation: C.I.v.d.M., S.K., O.C., K.R.E., G.R., H.G., I.S.v.d.L., H.V., R.S., M.Z.E., M.v.d.B., and S.H.; resources: A.R.R., M.S., J.H.M.S., T.M., M.Z.E., A.L.A., P.S.L., A.S., R.P., A.W., M.M.S., M.A.S., T.F., A.R.G., A.J.S., A.L., S.F., O.B., M.v.d.B., R.S., S.H., and A.H.; supervision: F.L.v.d.V., M.G.N., R.S., S.H., S.S.V.H., and A.H.; visualization: C.I.v.d.M., S.K., G.R., R.E., and M.Z.E.; writing – original draft: C.I.v.d.M. and S.K.; writing – review and editing: all authors. All authors read and approve the final manuscript.
Declaration of interests
The authors declare no competing interests.
Published: March 18, 2024
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ajhg.2024.02.013.
Supplemental information
References
- 1.Murata S., Takahama Y., Kasahara M., Tanaka K. The immunoproteasome and thymoproteasome: functions, evolution and human disease. Nat. Immunol. 2018;19:923–931. doi: 10.1038/s41590-018-0186-z. [DOI] [PubMed] [Google Scholar]
- 2.McCarthy M.K., Weinberg J.B. The immunoproteasome and viral infection: a complex regulator of inflammation. Front. Microbiol. 2015;6 doi: 10.3389/fmicb.2015.00021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kincaid E.Z., Che J.W., York I., Escobar H., Reyes-Vargas E., Delgado J.C., Welsh R.M., Karow M.L., Murphy A.J., Valenzuela D.M., et al. Mice completely lacking immunoproteasomes show major changes in antigen presentation. Nat. Immunol. 2011;13:129–135. doi: 10.1038/ni.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Van den Eynde B.J., Morel S. Differential processing of class-I-restricted epitopes by the standard proteasome and the immunoproteasome. Curr. Opin. Immunol. 2001;13:147–153. doi: 10.1016/S0952-7915(00)00197-7. [DOI] [PubMed] [Google Scholar]
- 5.Heink S., Ludwig D., Kloetzel P.-M., Krüger E. Vol. 102. 2005. IFN-γ-induced immune adaptation of the proteasome system is an accelerated and transient response; pp. 9241–9246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nitta T., Murata S., Sasaki K., Fujii H., Ripen A.M., Ishimaru N., Koyasu S., Tanaka K., Takahama Y. Thymoproteasome Shapes Immunocompetent Repertoire of CD8+ T Cells. Immunity. 2010;32:29–40. doi: 10.1016/j.immuni.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 7.Arima K., Kinoshita A., Mishima H., Kanazawa N., Kaneko T., Mizushima T., Ichinose K., Nakamura H., Tsujino A., Kawakami A., et al. Vol. 108. 2011. Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome; pp. 14914–14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brehm A., Liu Y., Sheikh A., Marrero B., Omoyinmi E., Zhou Q., Montealegre G., Biancotto A., Reinhardt A., Almeida De Jesus A., et al. Additive loss-of-function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J. Clin. Invest. 2015;125:4196–4211. doi: 10.1172/JCI81260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Jesus A.A., Brehm A., VanTries R., Pillet P., Parentelli A.-S., Montealegre Sanchez G.A., Deng Z., Paut I.K., Goldbach-Mansky R., Krüger E. Novel proteasome assembly chaperone mutations in PSMG2/PAC2 cause the autoinflammatory interferonopathy CANDLE/PRAAS4. J. Allergy Clin. Immunol. 2019;143:1939–1943.e8. doi: 10.1016/j.jaci.2018.12.1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kitamura A., Maekawa Y., Uehara H., Izumi K., Kawachi I., Nishizawa M., Toyoshima Y., Takahashi H., Standley D.M., Tanaka K., et al. A mutation in the immunoproteasome subunit PSMB8 causes autoinflammation and lipodystrophy in humans. J. Clin. Invest. 2011;121:4150–4160. doi: 10.1172/JCI58414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Papendorf J.J., Ebstein F., Alehashemi S., Piotto D.G.P., Kozlova A., Terreri M.T., Shcherbina A., Rastegar A., Rodrigues M., Pereira R., et al. Identification of eight novel proteasome variants in five unrelated cases of proteasome-associated autoinflammatory syndromes (PRAAS) Front. Immunol. 2023;14 doi: 10.3389/fimmu.2023.1190104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Poli M.C., Ebstein F., Nicholas S.K., de Guzman M.M., Forbes L.R., Chinn I.K., Mace E.M., Vogel T.P., Carisey A.F., Benavides F., et al. Heterozygous Truncating Variants in POMP Escape Nonsense-Mediated Decay and Cause a Unique Immune Dysregulatory Syndrome. Am. J. Hum. Genet. 2018;102:1126–1142. doi: 10.1016/j.ajhg.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kanazawa N., Hemmi H., Kinjo N., Ohnishi H., Hamazaki J., Mishima H., Kinoshita A., Mizushima T., Hamada S., Hamada K., et al. Heterozygous missense variant of the proteasome subunit β-type 9 causes neonatal-onset autoinflammation and immunodeficiency. Nat. Commun. 2021;12:6819. doi: 10.1038/s41467-021-27085-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Agarwal A.K., Xing C., DeMartino G.N., Mizrachi D., Hernandez M.D., Sousa A.B., Martínez de Villarreal L., Dos Santos H.G., Garg A. PSMB8 Encoding the β5i Proteasome Subunit Is Mutated in Joint Contractures, Muscle Atrophy, Microcytic Anemia, and Panniculitis-Induced Lipodystrophy Syndrome. Am. J. Hum. Genet. 2010;87:866–872. doi: 10.1016/j.ajhg.2010.10.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Garg A., Hernandez M.D., Sousa A.B., Subramanyam L., Martínez de Villarreal L., dos Santos H.G., Barboza O. An Autosomal Recessive Syndrome of Joint Contractures, Muscular Atrophy, Microcytic Anemia, and Panniculitis-Associated Lipodystrophy. J. Clin. Endocrinol. Metab. 2010;95:E58–E63. doi: 10.1210/jc.2010-0488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu Y., Ramot Y., Torrelo A., Paller A.S., Si N., Babay S., Kim P.W., Sheikh A., Lee C.C.R., Chen Y., et al. Mutations in proteasome subunit β type 8 cause chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature with evidence of genetic and phenotypic heterogeneity. Arthritis Rheum. 2012;64:895–907. doi: 10.1002/art.33368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gatz S.A., Salles D., Jacobsen E.-M., Dörk T., Rausch T., Aydin S., Surowy H., Volcic M., Vogel W., Debatin K.-M., et al. MCM3AP and POMP Mutations Cause a DNA-Repair and DNA-Damage-Signaling Defect in an Immunodeficient Child. Hum. Mutat. 2016;37:257–268. doi: 10.1002/humu.22939. [DOI] [PubMed] [Google Scholar]
- 18.Verhoeven D., Schonenberg-Meinema D., Ebstein F., Papendorf J.J., Baars P.A., van Leeuwen E.M.M., Jansen M.H., Lankester A.C., van der Burg M., Florquin S., et al. Hematopoietic stem cell transplantation in a patient with proteasome-associated autoinflammatory syndrome (PRAAS) J. Allergy Clin. Immunol. 2022;149:1120–1127.e8. doi: 10.1016/j.jaci.2021.07.039. [DOI] [PubMed] [Google Scholar]
- 19.Sarrabay G., Méchin D., Salhi A., Boursier G., Rittore C., Crow Y., Rice G., Tran T.-A., Cezar R., Duffy D., et al. PSMB10, the last immunoproteasome gene missing for PRAAS. J. Allergy Clin. Immunol. 2020;145:1015–1017.e6. doi: 10.1016/j.jaci.2019.11.024. [DOI] [PubMed] [Google Scholar]
- 20.Ansar M., Ebstein F., Özkoç H., Paracha S.A., Iwaszkiewicz J., Gesemann M., Zoete V., Ranza E., Santoni F.A., Sarwar M.T., et al. Biallelic variants in PSMB1 encoding the proteasome subunit β6 cause impairment of proteasome function, microcephaly, intellectual disability, developmental delay and short stature. Hum. Mol. Genet. 2020;29:1132–1143. doi: 10.1093/hmg/ddaa032. [DOI] [PubMed] [Google Scholar]
- 21.Küry S., Besnard T., Ebstein F., Khan T.N., Gambin T., Douglas J., Bacino C.A., Craigen W.J., Sanders S.J., Lehmann A., et al. De Novo Disruption of the Proteasome Regulatory Subunit PSMD12 Causes a Syndromic Neurodevelopmental Disorder. Am. J. Hum. Genet. 2017;100:352–363. doi: 10.1016/j.ajhg.2017.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yan K., Zhang J., Lee P.Y., Tao P., Wang J., Wang S., Zhou Q., Dong M. Haploinsufficiency of PSMD12 Causes Proteasome Dysfunction and Subclinical Autoinflammation. Arthritis Rheumatol. 2022;74:1083–1090. doi: 10.1002/art.42070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mégarbané A., Sanders A., Chouery E., Delague V., Medlej-Hashim M., Torbey P.-H. An unknown autoinflammatory syndrome associated with short stature and dysmorphic features in a young boy. J. Rheumatol. 2002;29:1084–1087. [PubMed] [Google Scholar]
- 24.Khalil R., Kenny C., Hill R.S., Mochida G.H., Nasir R., Partlow J.N., Barry B.J., Al-Saffar M., Egan C., Stevens C.R., et al. PSMD12 haploinsufficiency in a neurodevelopmental disorder with autistic features. American J of Med Genetics Pt B. 2018;177:736–745. doi: 10.1002/ajmg.b.32688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ebstein F., Küry S., Rosenfelt C., Scott-Boyer M., van Woerden M., Besnard T., Papendorf J.J., Studencka-Turski M., Wang T., Hsieh T., et al. PSMC3 proteasome subunit variants are associated with neurodevelopmental delay and type I interferon production. Sci.Transl.Med. 2023;15:eabo3189. doi: 10.1126/scitranslmed.abo3189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kröll-Hermi A., Ebstein F., Stoetzel C., Geoffroy V., Schaefer E., Scheidecker S., Bär S., Takamiya M., Kawakami K., Zieba B.A., et al. Proteasome subunit PSMC3 variants cause neurosensory syndrome combining deafness and cataract due to proteotoxic stress. EMBO Mol. Med. 2020;12 doi: 10.15252/emmm.201911861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dvorak C.C., Haddad E., Heimall J., Dunn E., Cowan M.J., Pai S.-Y., Kapoor N., Satter L.F., Buckley R.H., O’Reilly R.J., et al. The diagnosis of severe combined immunodeficiency: Implementation of the PIDTC 2022 Definitions. J. Allergy Clin. Immunol. 2023;151:547–555.e5. doi: 10.1016/j.jaci.2022.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hebert A., Simons A., Schuurs-Hoeijmakers J.H.M., Koenen H.J.P.M., Zonneveld-Huijssoon E., Henriet S.S.V., Schatorjé E.J.H., Hoppenreijs E.P.A.H., Leenders E.K.S.M., Janssen E.J.M., et al. Trio-based whole exome sequencing in patients with suspected sporadic inborn errors of immunity: a retrospective cohort study. Elife. 2022;11 doi: 10.7554/eLife.78469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tangye S.G., Al-Herz W., Bousfiha A., Cunningham-Rundles C., Franco J.L., Holland S.M., Klein C., Morio T., Oksenhendler E., Picard C., et al. Human Inborn Errors of Immunity: 2022 Update on the Classification from the International Union of Immunological Societies Expert Committee. J. Clin. Immunol. 2022;42:1473–1507. doi: 10.1007/s10875-022-01289-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Treise I., Huber E.M., Klein-Rodewald T., Heinemeyer W., Grassmann S.A., Basler M., Adler T., Rathkolb B., Helming L., Andres C., et al. Defective immuno- and thymoproteasome assembly causes severe immunodeficiency. Sci. Rep. 2018;8:5975. doi: 10.1038/s41598-018-24199-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zamani Esteki M., Viltrop T., Tšuiko O., Tiirats A., Koel M., Nõukas M., Žilina O., Teearu K., Marjonen H., Kahila H., et al. In vitro fertilization does not increase the incidence of de novo copy number alterations in fetal and placental lineages. Nat. Med. 2019;25:1699–1705. doi: 10.1038/s41591-019-0620-2. [DOI] [PubMed] [Google Scholar]
- 32.Zamani Esteki M., Dimitriadou E., Mateiu L., Melotte C., Van der Aa N., Kumar P., Das R., Theunis K., Cheng J., Legius E., et al. Concurrent Whole-Genome Haplotyping and Copy-Number Profiling of Single Cells. Am. J. Hum. Genet. 2015;96:894–912. doi: 10.1016/j.ajhg.2015.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Essers R., Lebedev I.N., Kurg A., Fonova E.A., Stevens S.J.C., Koeck R.M., Von Rango U., Brandts L., Deligiannis S.P., Nikitina T.V., et al. Prevalence of chromosomal alterations in first-trimester spontaneous pregnancy loss. Nat. Med. 2023;29:3233–3242. doi: 10.1038/s41591-023-02645-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mazzolari E., Moshous D., Forino C., De Martiis D., Offer C., Lanfranchi A., Giliani S., Imberti L., Pasic S., Ugazio A.G., et al. Hematopoietic stem cell transplantation in Omenn syndrome: a single-center experience. Bone Marrow Transplant. 2005;36:107–114. doi: 10.1038/sj.bmt.1705017. [DOI] [PubMed] [Google Scholar]
- 35.Schuetz C., Gerke J., Ege M., Walter J., Kusters M., Worth A., Kanakry J.A., Dimitrova D., Wolska-Kuśnierz B., Chen K., et al. Hypomorphic RAG deficiency: impact of disease burden on survival and thymic recovery argues for early diagnosis and HSCT. Blood. 2023;141:713–724. doi: 10.1182/blood.2022017667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Martinez C., Ebstein F., Nicholas S.K., De Guzman M., Forbes L.R., Delmonte O.M., Bosticardo M., Castagnoli R., Krance R., Notarangelo L.D., et al. HSCT corrects primary immunodeficiency and immune dysregulation in patients with POMP-related autoinflammatory disease. Blood. 2021;138:1896–1901. doi: 10.1182/blood.2021011005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Meinhardt A., Ramos P.C., Dohmen R.J., Lucas N., Lee-Kirsch M.A., Becker B., De Laffolie J., Cunha T., Niehues T., Salzer U., et al. Curative Treatment of POMP-Related Autoinflammation and Immune Dysregulation (PRAID) by Hematopoietic Stem Cell Transplantation. J. Clin. Immunol. 2021;41:1664–1667. doi: 10.1007/s10875-021-01067-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sanchez G.A.M., Reinhardt A., Ramsey S., Wittkowski H., Hashkes P.J., Berkun Y., Schalm S., Murias S., Dare J.A., Brown D., et al. JAK1/2 inhibition with baricitinib in the treatment of autoinflammatory interferonopathies. J. Clin. Invest. 2018;128:3041–3052. doi: 10.1172/JCI98814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kataoka S., Kawashima N., Okuno Y., Muramatsu H., Miwata S., Narita K., Hamada M., Murakami N., Taniguchi R., Ichikawa D., et al. Successful treatment of a novel type I interferonopathy due to a de novo PSMB9 gene mutation with a Janus kinase inhibitor. J. Allergy Clin. Immunol. 2021;148:639–644. doi: 10.1016/j.jaci.2021.03.010. [DOI] [PubMed] [Google Scholar]
- 40.Murata S., Sasaki K., Kishimoto T., Niwa S.I., Hayashi H., Takahama Y., Tanaka K. Regulation of CD8 + T Cell Development by Thymus-Specific Proteasomes. Science. 2007;316:1349–1353. doi: 10.1126/science.1141915. [DOI] [PubMed] [Google Scholar]
- 41.Fehling H.J., Swat W., Laplace C., Kühn R., Rajewsky K., Müller U., Von Boehmer H. MHC Class I Expression in Mice Lacking the Proteasome Subunit LMP-7. Science. 1994;265:1234–1237. doi: 10.1126/science.8066463. [DOI] [PubMed] [Google Scholar]
- 42.Van Kaer L., Ashton-Rickardt P.G., Eichelberger M., Gaczynska M., Nagashima K., Rock K.L., Goldberg A.L., Doherty P.C., Tonegawa S. Altered peptidase and viral-specific T cell response in LMP2 mutant mice. Immunity. 1994;1:533–541. doi: 10.1016/1074-7613(94)90043-4. [DOI] [PubMed] [Google Scholar]
- 43.Basler M., Beck U., Kirk C.J., Groettrup M. The Antiviral Immune Response in Mice Devoid of Immunoproteasome Activity. J. Immunol. 2011;187:5548–5557. doi: 10.4049/jimmunol.1101064. [DOI] [PubMed] [Google Scholar]
- 44.Moebius J., Van Den Broek M., Groettrup M., Basler M. Immunoproteasomes are essential for survival and expansion of T cells in virus-infected mice. Eur. J. Immunol. 2010;40:3439–3449. doi: 10.1002/eji.201040620. [DOI] [PubMed] [Google Scholar]
- 45.Muchamuel T., Basler M., Aujay M.A., Suzuki E., Kalim K.W., Lauer C., Sylvain C., Ring E.R., Shields J., Jiang J., et al. A selective inhibitor of the immunoproteasome subunit LMP7 blocks cytokine production and attenuates progression of experimental arthritis. Nat. Med. 2009;15:781–787. doi: 10.1038/nm.1978. [DOI] [PubMed] [Google Scholar]
- 46.Seifert U., Bialy L.P., Ebstein F., Bech-Otschir D., Voigt A., Schröter F., Prozorovski T., Lange N., Steffen J., Rieger M., et al. Immunoproteasomes Preserve Protein Homeostasis upon Interferon-Induced Oxidative Stress. Cell. 2010;142:613–624. doi: 10.1016/j.cell.2010.07.036. [DOI] [PubMed] [Google Scholar]
- 47.Kim M., Serwa R.A., Samluk L., Suppanz I., Kodroń A., Stępkowski T.M., Elancheliyan P., Tsegaye B., Oeljeklaus S., Wasilewski M., et al. Immunoproteasome-specific subunit PSMB9 induction is required to regulate cellular proteostasis upon mitochondrial dysfunction. Nat. Commun. 2023;14:4092. doi: 10.1038/s41467-023-39642-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Revy P., Kannengiesser C., Fischer A. Somatic genetic rescue in Mendelian haematopoietic diseases. Nat. Rev. Genet. 2019;20:582–598. doi: 10.1038/s41576-019-0139-x. [DOI] [PubMed] [Google Scholar]
- 49.Buonocore F., Kühnen P., Suntharalingham J.P., Del Valle I., Digweed M., Stachelscheid H., Khajavi N., Didi M., Brady A.F., Blankenstein O., et al. Somatic mutations and progressive monosomy modify SAMD9-related phenotypes in humans. J. Clin. Invest. 2017;127:1700–1713. doi: 10.1172/JCI91913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Crestani E., Choo S., Frugoni F., Lee Y.N., Richards S., Smart J., Notarangelo L.D. RAG1 Reversion Mosaicism in a Patient with Omenn Syndrome. J. Clin. Immunol. 2014;34:551–554. doi: 10.1007/s10875-014-0051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hirschhorn R., Yang D.R., Puck J.M., Huie M.L., Jiang C.-K., Kurlandsky L.E. Spontaneous in vivo reversion to normal of an inherited mutation in a patient with adenosine deaminase deficiency. Nat. Genet. 1996;13:290–295. doi: 10.1038/ng0796-290. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The article includes all datasets generated or analyzed during this study. The exome-sequencing data of the five families supporting the current study have not been deposited in a public repository because of privacy issues but are available from the corresponding author on request.