

Abstract
The recent symposium on “Target-Site” Drug Metabolism and Transport that was sponsored by the American Society for Pharmacology and Experimental Therapeutics at the 2014 Experimental Biology meeting in San Diego is summarized in this report. Emerging evidence has demonstrated that drug-metabolizing enzyme and transporter activity at the site of therapeutic action can affect the efficacy, safety, and metabolic properties of a given drug, with potential outcomes including altered dosing regimens, stricter exclusion criteria, or even the failure of a new chemical entity in clinical trials. Drug metabolism within the brain, for example, can contribute to metabolic activation of therapeutic drugs such as codeine as well as the elimination of potential neurotoxins in the brain. Similarly, the activity of oxidative and conjugative drug-metabolizing enzymes in the lung can have an effect on the efficacy of compounds such as resveratrol. In addition to metabolism, the active transport of compounds into or away from the site of action can also influence the outcome of a given therapeutic regimen or disease progression. For example, organic anion transporter 3 is involved in the initiation of pancreatic β-cell dysfunction and may have a role in how uremic toxins enter pancreatic β-cells and ultimately contribute to the pathogenesis of gestational diabetes. Finally, it is likely that a combination of target-specific metabolism and cellular internalization may have a significant role in determining the pharmacokinetics and efficacy of antibody-drug conjugates, a finding which has resulted in the development of a host of new analytical methods that are now used for characterizing the metabolism and disposition of antibody-drug conjugates. Taken together, the research summarized herein can provide for an increased understanding of potential barriers to drug efficacy and allow for a more rational approach for developing safe and effective therapeutics.
Introduction
Although drug metabolism is most often thought of as a mechanism to eliminate toxic substances and therapeutic entities from systemic circulation, emerging evidence has highlighted the importance of drug-metabolizing enzymes and transporters at the site of therapeutic action. These enzymes and transporters are widely expressed throughout the body and often serve as detoxification mechanisms at a cellular level. In an attempt to maintain a homeostatic environment, however, the enzymes and transporters may also serve to decrease the amount of drug available to reach a given target, ultimately attenuating the desired pharmacodynamic response of the drug (Urquhart et al., 2007; Chen et al., 2012). In many cases, their expression is sensitive to physiologic and pathologic changes, with altered regulation of drug-metabolizing enzymes and transporters observed in both oncology and inflammatory diseases (Renton, 2004, 2005; Morgan et al., 2008).
In general, the liver, intestine, and kidney are believed to be the major organs involved in drug elimination, with the first two often having a significant role in the first-pass metabolism of orally administered xenobiotics (Pond and Tozer, 1984). However, the expression of drug-metabolizing enzymes such as cytochrome P450s and UDP-glucuronosyltransferases is not limited to these organs, but rather occurs in organs throughout the body (Guengerich, 2005; Remmel and Zhou, 2009; Foti and Fisher, 2012). Significant expression levels of drug-metabolizing enzymes have been found in the brain, lung, nasal mucosa, trachea, esophagus, stomach, and colon (Ding and Kaminsky, 2003). Although these additional sites of expression may not always represent significant clearance pathways, they can play a role in affecting local tissue concentrations of a given drug and, in turn, the efficacy of the drug against a target located in that tissue. For example, many drugs against neurologic targets are also metabolized by cytochrome P450s that are located in the brain (Ferguson and Tyndale, 2011; Miksys and Tyndale, 2013; Ravindranath and Strobel, 2013). Similarly, the activity of inhaled drugs such as ciclesonide or 17-beclomethasone dipropionate is dependent upon metabolic activation by drug-metabolizing enzymes located within the lungs (Nave et al., 2007, 2010). Conversely, intestinal and hepatic metabolism and transport play an important role in limiting the systemic exposure of drugs such as loperamide, budesonide, or sulfasalazine which bind to therapeutic targets located in the intestine (Peppercorn and Goldman, 1972; Schinkel et al., 1996; Kim et al., 2004; Acharya et al., 2006; O'Donnell and O'Morain, 2010).
The role of drug transporters in affecting the efficacy of clinical therapeutics has also received a great deal of interest. The effect of transporter-mediated multidrug resistance on the outcome of therapeutic treatments is perhaps the quintessential example of absorption, distribution, metabolism, excretion and toxicity factors that influence the efficacy of a drug at the site of action (Gottesman et al., 2002). Efflux transporters such as P-glycoprotein (P-gp), the breast cancer resistance protein (BCRP), and the multidrug resistance–associated proteins (MRPs) have been implicated in the therapeutic limitations of drugs such as doxorubicin, vinblastine, and topotecan (Awasthi et al., 1994; Kruijtzer et al., 2002). Similarly, activity of uptake transporters, including members of the solute carrier 22 (SLC22) family, influence the therapeutic activity of antibiotics (penicillin G), diuretics (furosemide), and antineoplastic agents (cisplatin) (Sweet, 2010). As a testament to the importance of overcoming the ramifications of transporter-mediated drug resistance, multiple clinical evaluations aimed at identifying inhibitors of these transporters have been undertaken (Morrissey et al., 2013; Videira et al., 2014). While representing a key research focus, drug resistance or altered efficacy due to transporters at the site of therapeutic action is not limited to oncology indications. For example, the expression of P-gp on proinflammatory Th17 cells is a determining factor in the resistance of these cells to glucocorticoid treatment in autoimmune diseases (Ramesh et al., 2014). In vitro results suggest that MRP1 and BCRP expression in synovial tissue of rheumatic patients may impact the clinical efficacy of disease-modifying antirheumatic drugs such as methotrexate and leflunomide (van der Heijden et al., 2009). Finally, some patients taking pravastatin and gemfibrozil in combination experienced increased pravastatin plasma levels and decreased renal clearance (altered pharmacokinetics) resulting in increased pravastatin-associated adverse events, and preliminary evidence suggests this drug-drug interaction may involve inhibition of human organic anion transporter 3 (hOAT3)–mediated pravastatin transport by gemfibrozil (Kyrklund et al., 2003; Nakagomi-Hagihara et al., 2007).
The effects of metabolism and transport at the site of therapeutic action are not limited to small molecules but also play a role in determining the pharmacokinetics and efficacy of protein therapeutics. In the case of antibody-drug conjugates, for example, the mechanism of action begins with binding and internalization of the conjugate through receptor-mediated endocytosis (Ghose and Blair, 1987; Chari, 2008). Lysosomal metabolism and release of the cytotoxic agent result in the observed pharmacodynamic effects of the antibody-drug conjugate (Erickson et al., 2006). As such, optimization efforts for antibody-drug conjugates are often focused on factors which influence the internalization and subsequent degradation at the site of action (Alley et al., 2010; Perez et al., 2014).
The 2014 Experimental Biology symposium entitled “Target-Site” Drug Metabolism and Transport was sponsored by the American Society for Pharmacology and Experimental Therapeutics and brought together experts whose research focused on the localized effects of drug-metabolizing enzymes and transporters. This report serves to summarize the research and overarching themes which were presented and discussed during the symposium.
Drug Metabolism within the Brain Changes Drug Response In Vivo (K.L.P.G. and R.F.T.)
Drug metabolism by cytochrome P450 enzymes (P450s) occurs mainly in the liver, and it is this hepatic expression that is a major contributor to pharmacokinetics and resulting systemic drug and metabolite levels. However, drug-metabolizing P450s are also expressed extrahepatically and may play a role in local metabolism within these specific organs. These P450s are expressed in the brain (Miksys et al., 2000; Ferguson and Tyndale, 2011), where protein levels in certain cell types can be as high as in hepatocytes (Miksys et al., 2000). They are functionally active ex vivo, but more importantly in vivo, indicating that sufficient levels of cofactors and coenzymes are colocalized within the brain (Miksys and Tyndale, 2009). This suggests that metabolism of drugs that act on the central nervous system (CNS) could occur in the brain independent of liver metabolism. This is particularly relevant since the clinical response to CNS-acting drugs does not always correlate with plasma drug levels (Michels and Marzuk, 1993). As examples, we will describe two well known drug-metabolizing enzymes which are expressed in the brain, CYP2B and CYP2D, both of which can metabolize several CNS-acting drugs and neurotoxins. By selectively manipulating these enzymes in the brain through induction and inhibition, the CNS activity and subsequent responses to CYP2B and CYP2D substrates can be altered. Thus, the expression of P450s in the brain may play a role in drug response, and as they are highly variable, may also contribute to interindividual variation in drug response to CNS-acting drugs.
CYP2B can metabolize many CNS-acting drugs, such as bupropion (Hesse et al., 2000), methadone (Gerber et al., 2004), and nicotine (Yamazaki et al., 1999; Yamanaka et al., 2005), the primary psychoactive ingredient in cigarettes. Large variations in CYP2B isoform levels have been found in the brain (Miksys et al., 2003), in part due to induction by common drugs such as nicotine. Brain CYP2B expression can be increased in primates and rodents: higher CYP2B6 and CYP2B1 levels are found after chronic nicotine treatment in monkeys (Lee et al., 2006) and rats (Miksys et al., 2000), respectively, consistent with higher levels in brains of smokers (Miksys et al., 2003). The human CYP2B6 gene is highly polymorphic (Lang et al., 2001); one common decreased expression variant, CYP2B6*6, was associated with reduced protein levels in the human brain (Miksys et al., 2003). Thus, variation in CYP2B expressed in the brain may influence the levels of its substrates and their resulting drug and toxic responses.
CYP2B metabolically inactivates the anesthetic propofol. In humans, brain propofol levels were a better predictor of the behavioral response (anesthesia or sleep time) when compared with plasma propofol levels (Liu et al., 2009), providing a rationale for examining the role of brain CYP2B in metabolizing propofol and the behavioral response to this drug. To do this, mechanism-based inhibitors (MBIs), substrates that are metabolized to a reactive intermediate that subsequently inactivates the enzyme, were used to inhibit CYP2B activity specifically in the brain (Fig. 1). C8-xanthate and 8-methoxypsoralen are two structurally distinct MBIs that can selectively inhibit CYP2B (Koenigs and Trager, 1998; Yanev et al., 1999). Delivery of either of these CYP2B MBIs into rat brain via i.c.v. or regional injection was used to demonstrate that P450s within the brain were functional in vivo (Miksys and Tyndale, 2009). Intracerebroventricular injections of CYP2B MBIs increased rat brain propofol levels (Fig. 1B) and prolonged sleep time (Fig. 2A); this was found with both MBIs, supporting the selectivity of CYP2B inhibition (Khokhar and Tyndale, 2011). The effect on brain propofol levels and response increased with increasing doses of one of the MBIs. Plasma propofol levels in vivo and hepatic CYP2B activity ex vivo were not influenced by MBI i.c.v. treatment, supporting the contention that CYP2B inhibition via i.c.v. administration was selectively achieved within the brain and had little/no impact on hepatic metabolism. CYP2B induction within the brain and not in the liver increased brain CYP2B enzyme levels and propofol inactivation. CYP2B can be induced in the brain, but not the liver, by a week of nicotine treatment (Miksys et al., 2000; Khokhar et al., 2010); the treatment resulted in lower propofol levels and reduced sleep times, which could be reversed by an i.c.v. injection of a MBI (Fig. 2B). Thus, having increased CYP2B enzyme in the brain resulted in lower brain propofol levels and shorter sleep times, whereas having inhibited enzyme in the brain resulted in increased brain propofol levels and longer sleep times. Together this suggests that variable CYP2B activity within the brain can alter the rate of inactivation of propofol and influence drug response. Recently, it has also been shown that inhibiting CYP2B in rat brain can also alter nicotine levels in the brain, measured by microdialysis, and resulting nicotine self-administration (Garcia et al., 2015).
Fig. 1.

Inhibition of CYP2 activity in the brain. Inhibiting activity of CYP2 enzymes specifically in the brain via MBI pretreatment can alter substrate and metabolite brain levels. (A) MBIs are substrates for their target enzymes, which are metabolized into intermediate compounds that bind irreversibly to the enzyme, thus inactivating it. MBIs are injected into the brain via i.c.v. injections. (B) Brain P450 inhibition increases the parent drug, illustrated for the active CYP2B substrate propofol: brain CYP2B inhibition by the i.c.v. MBI C8-xanthate (C8-X) increased propofol brain levels relative to vehicle [artificial cerebrospinal fluid (ACSF)] pretreatment; there was no difference in plasma propofol levels. (C) Brain P450 inhibition reduces metabolite levels, illustrated for the metabolically activated metabolite morphine from CYP2D substrate codeine: brain CYP2D inhibition by MBI propranolol (PL) pretreatment decreased morphine levels in the brain; there was no difference in plasma morphine levels (*P < 0.05). Data compiled from previously published reports (Khokhar and Tyndale, 2011; Zhou et al., 2013).
Fig. 2.

Effect of CYP2 inhibition on the efficacy of CNS-acting substrates. Inhibiting the specific activity of CYP2 enzymes in the brain can influence the behavioral response to CNS-acting substrates. (A) CYP2B inhibition by the CYP2B MBI C8-xanthate (C8-X) in the brain increased propofol-induced sleep times in rats. (B) Inducing CYP2B in the brain by 7 days of nicotine treatment reduced propofol-induced sleep times, and this effect was reversed when rats were given C8-X. (C) CYP2D inhibition by the CYP2D MBI propranolol (PL) in the brain reduced analgesia at early time points after administration of its substrate codeine, but had no effect at later time points when morphine made peripherally enters the brain. (D) CYP2D inhibition by propranolol in the brain after a morphine injection did not have an effect on analgesia at any time; morphine is not a substrate for CYP2D. Data compiled from previously published reports (Khokhar and Tyndale, 2011; Zhou et al., 2013). *P < 0.05. ACSF, artificial cerebrospinal fluid; %MPE, percent maximal possible effect; SC, subcutaneous.
P450s also metabolize substrates from inactive parent compounds into active metabolites. For example, CYP2B metabolically activates the pesticide chlorpyrifos to chlorpyrifos-oxon (Tang et al., 2001), a toxic metabolite that inhibits acetylcholinesterase activity (Sultatos, 1994). Excess acetylcholine from reduced acetylcholinesterase activity leads to cholinergic system overstimulation, resulting in neurotoxicity and cognitive deficits, indicating the brain is one target of this toxicity (Steenland et al., 2000). There is negligible oxon detectable in the plasma after chlorpyrifos exposure due to its rapid metabolism to 3,5,6-trichloro-2-pyridinol in the blood by esterases (Costa et al., 1990; Drevenkar et al., 1993); therefore, the oxon formed from the liver might not reach the brain. In vitro brain homogenates can metabolize chlorpyrifos to the oxon (Chambers and Chambers, 1989), suggesting that brain-specific formation of this metabolite may be important in producing chlorpyrifos-induced neurotoxicity. This hypothesis was tested by administration of CYP2B MBI in the brain: rats were treated i.c.v. with MBI or vehicle and given a peripheral injection of chlorpyrifos (Khokhar and Tyndale, 2012). With MBI CNS treatment, brain chlorpyrifos levels increased while chlorpyrifos-oxon levels decreased, indicating that the MBI blocked CYP2B activation of the oxon metabolite. Remaining brain acetylcholine esterase activity was higher in MBI-treated rats, demonstrating that reducing CYP2B enzyme activity in the brain lowered the metabolite-mediated inhibition of the esterase. In rats, chlorpyrifos-oxon toxicity causes acute reductions in body temperature (hypothermia) and lasting behavioral impairments (e.g., reduced gait and aerial righting reflexes). MBI i.c.v. pretreatment reduced both the hypothermia and the behavioral impairments in animals given chlorpyrifos while having no impact in the absence of chlorpyrifos and no effect on systemic chlorpyrifos levels. Initially, MBI i.c.v. treatment had been given prior to chlorpyrifos to determine whether inhibiting brain CYP2B can prevent or reduce neurotoxicity. Subsequently, it was shown that, even following chlorpyrifos exposure, MBIs given i.c.v. could ameliorate toxicity days after chlorpyrifos treatment due to the long duration of chlorpyrifos in the body (Khokhar and Tyndale, 2014). This work demonstrated that MBI delivered directly to the brain can reduce the behavioral impairments, suggesting that in humans, inhibition of CYP2B6 might be a useful adjunct for treating chlorpyrifos toxicity. These results, together with the findings for propofol, demonstrate that brain-specific metabolism by CYP2B can significantly influence the response to CNS-acting substrates.
CYP2D can metabolize a variety of CNS-acting compounds including opiates, amphetamines, and antidepressants, as well as endogenous substances including numerous neurotransmitters (Zanger et al., 2004). Similar to CYP2B6, the gene that encodes human CYP2D6 is highly polymorphic, and genetic variants have been associated with lower or higher metabolism of CYP2D6 substrates (Gaedigk et al., 2008). CYP2D6 has been identified in the brain; individuals with variants which disrupt the gene have lower CYP2D6 protein levels in the brain as well as in the liver (Miksys et al., 2002). Behavioral traits have been associated with CYP2D6 genetic variation where genetically slower metabolizers score higher on anxiety and socialization indexes, suggesting a possible role for the enzyme within the brain (Bertilsson et al., 1989; Llerena et al., 1989, 1993; Gan et al., 2004; Roberts et al., 2004). CYP2D6 genetic variation is also associated with different levels of human resting cerebral brain perfusion, suggesting that fast versus slow metabolizers have an altered response to environmental stimuli (Kirchheiner et al., 2011). Thus, in addition to a potential role in response to drugs and toxins within the brain, CYP2D activity may influence endogenous brain processes.
CYP2D metabolizes codeine to morphine, which is the active compound that elicits analgesia (Adler et al., 1955). CYP2D6 genetic variation can alter codeine-induced analgesia: genetically poor metabolizers do not exhibit an analgesic response to codeine due to their inability to convert codeine to morphine (Sindrup et al., 1990, 1992; Chen et al., 1991). This metabolic activation is mediated mainly by CYP2D6 in the liver, and morphine subsequently crosses the blood-brain barrier and elicits analgesia. However, codeine is more lipophilic than morphine and crosses the blood-brain barrier faster than morphine, and morphine is also actively transported out of the brain (Oldendorf et al., 1972; Bouw et al., 2000); therefore, the early time points of analgesia following codeine administration could be due to codeine entering the brain and being converted to morphine within the brain by CYP2D. This was tested by measuring analgesia after i.c.v. injection of competitive (propafenone) or mechanism-based (propranolol) inhibitors of CYP2D directly into the brain, as described earlier for CYP2B (Zhou et al., 2013). Intracerebroventricular injection of either inhibitor resulted in a reduction of codeine-mediated analgesia in rats. This reduction occurred for the first 30 minutes after codeine administration, whereas at later time points there was no effect of inhibition, suggesting that morphine from the periphery had sufficient time to enter the brain (Fig. 2C). Intracerebroventricular inhibitor pretreatment reduced brain morphine levels, whereas plasma morphine levels were unchanged (Fig. 1C), providing evidence that the inhibitor did not influence hepatic CYP2D activity. The inhibitor had no effect on either morphine’s ability to enter the brain or morphine-induced analgesia, which supports the selectivity of the inhibitor’s effect on codeine metabolism and resulting analgesia (Fig. 2D). Smokers have higher CYP2D6 protein in the basal ganglia and cerebellar regions, and chronic nicotine treatment can increase CYP2D6 expression in monkeys (Mann et al., 2008) and CYP2D1 expression in rats (Yue et al., 2008). Thus, it is likely that induced levels of CYP2D within the brain will increase the formation rate of morphine and subsequently increase analgesia—which was recently confirmed (McMillan and Tyndale, 2015).
CYP2D can metabolically inactivate the neurotoxin 1-methyl-4-phenylpyridinium (MPP+), the active metabolite from the precursor 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine. MPP+ interferes with oxidative phosphorylation in mitochondria, resulting in cell death (Ramsay et al., 1991), and exposure to this toxin can elicit neurodegenerative effects similar to the symptoms observed in Parkinson’s disease (Deng et al., 2004). Consistent with this metabolic inactivation, CYP2D6 genetic poor metabolizers have an increased risk of developing Parkinson’s disease (McCann et al., 1997). Thus, it is possible that CYP2D6 inactivation of toxins in the brain could be important in protecting against neurotoxicity and neurodegeneration. The influence of CYP2D6 in MPP+ toxicity in neuronal tissue has been evinced in vitro: applying MPP+ to human brain cell lines dose dependently increased cell death, whereas overexpression of CYP2D6 in these cell lines protected them from MPP+ toxicity (Matoh et al., 2003). Conversely, inhibiting CYP2D6 in the presence of MPP+ increased this cytotoxic effect likely by reducing its inactivation, analogous to the increased risk for Parkinson’s disease seen in individuals without this enzyme (Mann and Tyndale, 2010). Consistent with this, brain CYP2D6 protein levels are lower in Parkinson’s disease patients compared with age-matched healthy controls (Mann et al., 2012). The ex vivo and postmortem brain results suggest that having lower CYP2D6 activity in the brain may increase vulnerability to neurochemical insults by reducing the ability to inactivate certain classes of neurotoxins. Thus, CYP2D6 expression in the brain may play a role in protecting the CNS from toxins as well as mediating drug response.
Altering brain activity of either CYP2B or CYP2D can influence the CNS levels of drugs and toxins and make a significant impact on their subsequent behavioral effects. These proof-of-concept findings suggest that CNS expression of P450s contributes to drug and toxin metabolism within the brain and the resulting response. This not only demonstrates the potential importance of extrahepatic expression of enzymes in drug response but also provides some understanding of the sources of variation in drug response, including genetic and environmental alterations of brain P450 activity. Thus, although early in our understanding of the role of P450 enzymes within the brain, these studies provide evidence that under some circumstances this may produce important variation in drug and toxin response.
The SLC22 Transporter Family: Impact on Drug Efficacy, Drug-Drug Interactions, and Pathophysiology (D.H.S.)
In the kidney, the proximal tubule region of the mature nephron is tasked with performing much of the secretion and reabsorption of charged organic solutes. The limited passive diffusibility of such compounds necessitates utilization of dozens of membrane-bound transporters for efficient renal transcellular organic solute flux. Two major transporter superfamilies overseeing this transcellular flux are the ATP binding cassette (ABC) transporters (directly bind and hydrolyze ATP as an energy source) and the SLCs (driven by membrane potential and/or energy stored in concentration gradients) (Sweet, 2005, 2010). The human SLC superfamily is composed of ∼55 unique gene families with ∼400 confirmed or putative transporters (He et al., 2009). The SLC22 family (organic cation/anion/zwitterion transporter family) encompasses ∼30 identified and putative members and includes the organic cation transporters (OCTs and organic cation transporters-novel) and the organic anion transporters (OATs) (Sweet, 2005, 2010; VanWert et al., 2010). Results from a variety of comparative models, e.g., intestinal sacs, renal and hepatic tissue slices and membrane vesicles, intact isolated renal tubules, liver, choroid plexus, and brain capillaries, demonstrated that the “physiological footprint” of mediated organic cation and anion flux across each of these tissues was similar (Pritchard and Miller, 1993), and it has been established that “renal” OCTs and OATs are expressed and function in barrier tissues throughout the body (Pritchard et al., 1999; Sweet et al., 2001; Sweet, 2010; VanWert et al., 2010; Farthing and Sweet, 2014). SLC22 transporters interact with a plethora of structurally diverse endogenous and foreign organic solutes such as metabolic byproducts, hormones, mycotoxins, pesticides, and a number of the “top 200” prescribed drugs, including diuretics, antibiotics, antivirals, and antidiabetic and chemotherapeutic agents; thus, SLC22 transporter activity is a major determinant of the pharmacokinetic, pharmacodynamic, and toxicity profiles of these compounds. Clearly, a fundamental understanding of SLC22 transporters is required to properly evaluate their impact on drug efficacy and exposure, drug-drug interactions (DDIs), and the pathophysiology of some disease states.
Review of the clinical literature revealed a number of apparent DDIs between probenecid (OAT inhibitor) or cimetidine (OCT inhibitor) and numerous therapeutics such as ciprofloxacin, cefaclor, famotidine, acyclovir, cidofovir, furosemide, and metformin, suggesting SLC22 transporter function impacts their clinical profiles (Morrissey et al., 2013). Indeed, a 4- to 5-fold decrease in in vivo efficacy of furosemide was observed in organic anion transporter 1 knockout mice (Eraly et al., 2006). Thus, if renal proximal tubule cells and/or the urinary space is the intended therapeutic target, loss of OAT1 function (via DDI or genetics) might be associated with decreased efficacy. When probenecid was coadministered with mesna, an agent that reduces the renal toxicity of some chemotherapeutic agents, mesna excretion was decreased ∼70% and total renal clearance was decreased ∼55% (Cutler et al., 2012). Follow-up in vitro studies with cloned hOAT1, hOAT3, and hOAT4 demonstrated probenecid-sensitive transport of mesna with inhibitory constants (Ki) below the clinical concentration, strongly implicating OATs as having a role in this DDI (Cutler et al., 2012). Similarly, the pharmacological action and associated toxicity of platinum antineoplastic agents (e.g., cisplatin and oxaliplatin) involves human OCT (hOCT) transport activity, and cotreatment with hOCT inhibitors (e.g., cimetidine) blunted accumulation and toxicity of cisplatin in renal proximal tubule cells and stably transfected transporter-expressing cells (Nelson et al., 1984; Pan et al., 1999; Ciarimboli et al., 2005; Yokoo et al., 2007, 2008). Recently, regulatory and scientific experts proposed guidelines for assessing the potential clinical relevance of such DDIs for about a dozen transporters, including the SLC22 family members OCT2, OAT1, and OAT3. As part of these recommendations, a DDI index was defined as the maximal unbound plasma concentration (Cmax or Css) of a compound after administering the highest clinical dose divided by the compound’s in vitro inhibition potency (Ki or IC50) on the transporter of interest (Giacomini et al., 2010). A resultant DDI index value ≥0.1 (∼9% inhibition) was conservatively selected as indicating the need for follow-up clinical DDI investigations.
Such analysis was used to assess the DDI potential of the antituberculosis drug ethambutol dihydrochloride (EMB) on hOCTs (Pan et al., 2013). The World Health Organization recommends EMB, along with pyrazinamide, isoniazid, and rifampin, as a front-line antituberculosis drug. Treatment of multidrug-resistant tuberculosis (TB; estimated to be 3.7% of new cases and 20% of previously treated cases) includes EMB to reduce the emergence of isoniazid- or rifampin-resistant mycobacteria (http://apps.who.int/iris/bitstream/10665/75938/1/9789241564502_eng.pdf). In vitro characterization demonstrated that EMB inhibits hOCT1-, hOCT2-, and hOCT3-mediated transport (Pan et al., 2013). Using an estimated EMB concentration of 67 µM in the portal system after oral dosing, DDI index analysis indicated a rank order of DDI potencies on hOCT1 as enterocyte (0.6) = hepatocyte (0.6) > proximal tubule cell (0.2), all above the 0.1 threshold value (Pan et al., 2013). These values are indicative of high potential for EMB-mediated DDIs on hOCT1 affecting disposition and clearance. The largest DDI index (8.3, ∼90% inhibition) was predicted for hOCT3 in enterocytes, suggesting orally dosed EMB could negatively impact cationic drug and/or nutrient absorption. DDIs involving EMB on hOCT2 expressed in the renal proximal tubule were predicted to be of low likelihood (DDI index = 0.1).
Further, many antidiabetic (metformin) and anti–human immunodeficiency virus (HIV; lamivudine, raltegravir, tenofovir, and zalcitabine) therapeutics are known substrates and/or inhibitors of SLC22s (Koepsell et al., 2007; Sweet, 2010; VanWert et al., 2010; Wang and Sweet, 2013). Thus, TB therapy is further complicated by the rise in clinical association of TB with diabetes or HIV, with TB now listed as a leading cause of mortality among HIV-infected patients (in 2011: 430,000 out of the 1.4 million TB deaths were HIV-associated) (Faurholt-Jepsen et al., 2012; Pawlowski et al., 2012). Overall, TB, TB-diabetes, and TB-HIV patients are subjected to polypharmacy to minimize development of drug resistance or treat these comorbidities, greatly increasing the risk of experiencing SLC22 transporter–mediated DDIs. For example, a patient population expressing a mutant form of hOCT1 with reduced transport function exhibited significantly altered metformin pharmacokinetics and experienced poorer glucose-lowering effects, i.e., loss of efficacy (Shu et al., 2008). The DDI index of 0.6 for hOCT1 in enterocytes and hepatocytes predicts as much as a 38% loss of hOCT1 transport activity in these cell types during routine EMB therapy (Pan et al., 2013). Together, these observations indicate a strong potential for EMB-metformin interactions on hOCT1 that result in altered drug efficacy in TB-diabetes patients.
In addition to altered xenobiotic disposition, renal toxicity and/or insufficiency is often clinically associated with a systemic increase in endogenous molecules, including the so-called uremic retention solutes or uremic toxins. More than 150 compounds that are elevated in the biofluids of uremic patients have been identified, and their urinary excretion has been demonstrated to be largely a consequence of active tubular secretion as opposed to glomerular filtration (Masereeuw et al., 2014). Therefore, the pathophysiological progression of chronic kidney disease often includes the simultaneous increase in the systemic concentration of a variety of uremic toxins that interact with the organic cation and anion (i.e., SLC22-mediated) transport pathways, potentially leading to competitive interactions for OCT- and OAT-mediated transporter function.
In human kidneys, OCT2 appears to be the major OCT expressed; however, its role in uremic toxin accumulation/clearance is largely unknown. A number of cationic uremic toxins are known to effectively inhibit OCT2-mediated transport in vitro, and transporter expression was reduced in in vivo rodent models of acute and chronic kidney failure, supporting a link between transporter function and disease progression (Masereeuw et al., 2014). Similarly, it was observed decades ago that exposing rat kidney slices in vitro to the serum of uremic patients blocked accumulation of p-aminohippurate (prototypical organic anion substrate), and now dozens of uremic toxins are known substrates and/or inhibitors of OAT1, OAT2, OAT3, OAT4, and urate transporter 1 (White, 1966; VanWert et al., 2010; Wang and Sweet, 2013). Moreover, loss of renal OAT1 transport function was linked to elevated serum levels of several uremic toxins, i.e., decreased uremic retention solute elimination, via metabolomic profiling of organic anion transporter 1 knockout mice (Wikoff et al., 2011).
In humans, the uremic toxin hippuric acid is predicted to have high DDI potential on hOAT1 (DDI index = 12.3, ∼92% inhibition) and on hOAT3 (DDI index = 7.5, ∼88% inhibition) (Masereeuw et al., 2014). Indoxyl sulfate, strongly suspected as a causal factor in chronic renal failure, has a 10-fold stronger affinity for hOAT1 (∼18 µM) than for hOAT3 (VanWert et al., 2010; Wang and Sweet, 2013). Given the 15 µM unbound plasma concentration in uremic patients, indoxyl sulfate could produce ∼50% inhibition of hOAT1, correlating with the predicted DDI index of 0.8 or ∼44% inhibition (Masereeuw et al., 2014). Finally, affinity of 3-carboxy-4-methyl-5-propyl-2-furanopropanoic acid (CMPF) for hOAT1 (Km = ∼140 µM) appears too low to warrant any DDI concerns (DDI index = 0); however, the DDI index on hOAT3 (Km = ∼27 µM) is 0.9, suggesting potential for marked inhibition of hOAT3 (47%) during chronic kidney disease (VanWert et al., 2010; Wang and Sweet, 2013; Masereeuw et al., 2014). As indicated, dozens of uremic toxins increase in concentration simultaneously, most likely leading to additive or synergistic effects that are greater than those produced by any single compound and indicating significant potential for SLC22-mediated drug–uremic toxin interactions in renal insufficiency patients.
Interestingly, elevated CMPF levels were recently observed in plasma from gestational diabetes mellitus, type 2 diabetes, and prediabetic patients versus individuals exhibiting normal glucose tolerance (Prentice et al., 2014). This suggested that CMPF might be linked to both gestational diabetes mellitus and the progression to type 2 diabetes. Supporting this connection, artificially elevating plasma CMPF in mice in vivo to human diabetic levels (150–300 µM) induced glucose intolerance, impaired glucose-stimulated insulin secretion, and decreased glucose utilization (Prentice et al., 2014). Additionally, in vitro treatment of isolated murine or human islet cells with 200 µM CMPF for 24 hours significantly decreased insulin secretion. Taken together with the fact that hOAT3′s affinity for CMPF is 7- to 10-fold higher than human diabetic CMPF plasma levels, it was hypothesized that OAT3 expressed in islet cells might mediate the accumulation of CMPF and, thus, directly impact its β-cell toxicity and the development of gestational diabetes, as well as subsequent progression to type 2 diabetes (Prentice et al., 2014).
In support of this contention, β-cell–specific expression of hOAT1, hOAT3, and hOAT4 was detected by microarray, quantitative polymerase chain reaction, immunoblot, and immunohistochemistry (Prentice et al., 2014). Moreover, treatment with probenecid (pan OAT inhibitor) or penicillin G (OAT3 substrate), but not p-aminohippurate (OAT1 substrate), prevented CMPF-induced impairment of insulin secretion (200 µM CMPF for 24 hours) in isolated murine islets. Importantly, islets isolated from organic anion transporter 3 knockout mice were insensitive to CMPF-induced β-cell dysfunction, retaining glucose-stimulated insulin secretion and normal total insulin content despite 24-hour exposure to 200 µM CMPF (Prentice et al., 2014). Thus, OAT3 mediates β-cell entry of CMPF, and inhibition of this function prevents the deleterious effects of CMPF, suggesting CMPF and hOAT3 provide a link between β-cell dysfunction and gestational diabetes mellitus/type 2 diabetes that could be targeted therapeutically.
In conclusion, it is well established that members of the SLC22 transporter family represent major transport pathways for the absorption, tissue distribution, elimination, and reabsorption of a variety of xenobiotic and endogenous compounds. The same SLC22 transporters often have functional roles in multiple barrier tissues. OCTs and OATs are emerging as sites for many clinically relevant DDIs, and as such, activity of these transporters markedly influences the pharmacokinetic profiles of many therapeutics, significantly impacting dosing regimen, pharmacodynamics, efficacy, and toxicity. Loss of specific SLC22 transporter function leads to alterations in drug pharmacokinetics and is associated with accumulation of uremic retention solutes. Subsequent SLC22 interactions with uremic toxins may provide a link between renal insufficiency/failure and progression of uremic syndrome, development of associated uremic encephalopathies, etc. In particular, recent evidence strongly implicates a link between the uremic toxin, CMPF, hOAT3 function in β-cells, and manifestation of gestational diabetes mellitus and progression to type 2 diabetes. Clearly, the SLC22 transporter family markedly impacts drug efficacy, drug-drug interactions, and pathophysiology.
Pulmonary Metabolism of Resveratrol: In Vitro and In Vivo Evidence (S.S. and S.N.)
Prediction of drug and xenobiotic absorption, distribution, metabolism and excretion requires knowledge of disposition in major organs of absorption, distribution, and metabolism. Although the liver, intestine, kidney, and brain are commonly evaluated for xenobiotic distribution and metabolism, additional organs may play a major role in transport as well as metabolism of compounds. Organ-specific disposition is especially important for specific organs that are the target site for the compound. The human lung is one such organ with respect to disposition of drugs (e.g., lung cancer chemotherapy), xenobiotics (e.g., tobacco-specific carcinogens), and endobiotics (e.g., steroids) (Hecht, 2008; Gundert-Remy et al., 2014).
Expression and activity of xenobiotic-metabolizing enzymes (XMEs) in the lung have been evaluated extensively. The expression of various P450 isozymes has been reported in the lung (Fig. 3). P450 expression in various pulmonary cell lines representing the complex heterogenous cell layers of the lung has been reported, and variable P450 isozyme levels are thought to be expressed across the pulmonary cell layers (Gundert-Remy et al., 2014). Thus, Somers et al. (2007) reported modest human lung parenchymal levels of CYP1A1, 2J2, and 2A5, but lower expression levels compared with the liver. Numerous studies have shown modest expression of CYP1A1 in the bronchus and lung, with highly induced levels of the enzyme in smokers (Anttila et al., 2011). P450 expression in the lung is highly relevant in the context of endogenous steroid homeostasis as well as activation of tobacco-specific lung carcinogens (Hecht, 2008; Gundert-Remy et al., 2014).
Fig. 3.

XME and transporter expression in human lung. The key P450, UGT, SULT, and drug transporters known to be expressed in the human lung are depicted (PEPT, peptide transporter; SLCO, solute carrier organic anion transporter; OATP, organic anion–transporting polypeptide). Data compiled from previously published reports (Gallagher et al., 2007; Somers et al., 2007; Bosquillon, 2010; Anttila et al., 2011; Sharan and Nagar, 2013; Jones and Lazarus, 2014).
The human lung additionally expresses “phase II” enzymes, among which the XMEs uridine diphosphoglucuronosyltransferases (UGTs) and sulfotransferases (SULTs) are important enzyme superfamilies. These enzymes play important roles in steroid homeostasis as well as in the detoxification and elimination of procarcinogens and carcinogens (Dalhoff et al., 2005; Nagar and Remmel, 2006). Nakamura et al. (2008) detected UGT2B4 and 2B7 in normal human lung samples, whereas UGT1A1 and 1A3 were additionally reported in human lung adenocarcinoma A549 cell lines (Zhang et al., 2013). In another study, relatively high UGT1A6 and 2A1 were reported in human lung parenchyma, comparable to human liver expression (Somers et al., 2007). A recent study evaluated UGT2B expression in the upper respiratory tract, and reported low expression of various UGT2B isozymes (2B4, 2B7, 2B10, 2B11, 2B15, and 2B17) in the human lung (Jones and Lazarus, 2014). SULT pulmonary expression has similarly been reported, with low expression overall; SULT1E1 levels in the lung were the highest, followed by SULT1A1, 1A3, 1B1, and 2A1 (Riches et al., 2009). Somers et al. (2007) reported robust expression of several SULT isozymes in human lung parenchyma relative to human liver. Pharmacogenetic variability in XMEs can impact their role in the overall disposition of drugs and xenobiotics, and may predict the clinical outcome. An example is the UGT2B17 gene deletion, which was found to correlate with increased incidence of lung adenocarcinoma in women, possibly due to decreased glucuronidation (detoxification) of the tobacco-specific carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol (Gallagher et al., 2007).
Trans-resveratrol (RES) is a polyphenolic compound present in various dietary sources, and has been shown to have numerous health effects via different mechanisms (Baur and Sinclair, 2006). RES is extensively metabolized into its sulfated and glucuronidated metabolites, with trans-resveratrol-3-sulfate (R3S) and trans-resveratrol-3-glucuronide (R3G) as major metabolites (Juan et al., 2010; Iwuchukwu et al., 2012; Sharan et al., 2012). RES is sulfated mainly by SULT1A1 with minor contribution from SULT1E1 (Ung and Nagar, 2007). RES is glucuronidated at its 3-OH position via UGT1A1, UGT1A6, UGT1A7, UGT1A8, UGT1A9, UGT1A10, and UGT2B7 (Brill et al., 2006). Metabolism of RES is dependent on the site of metabolism. Human intestinal microsomes have a higher capacity for glucuronidation of RES (Iwuchukwu and Nagar, 2008) as compared with human liver microsomes. Systemic clearance of RES in mouse is much higher than the hepatic blood flow, indicating extrahepatic conjugation in the mouse (Sharan et al., 2012). Liver and gut as sites of RES metabolism have been well documented (Brill et al., 2006; Iwuchukwu and Nagar, 2008; van de Wetering et al., 2009), but the role of lungs in the metabolism of RES was not previously known. For a compound to be absorbed when given orally, it has to cross gut, liver, and lungs before it is available to the entire body. Metabolism by lungs in addition to liver and gut can significantly influence drug disposition, as the lung receives the entire cardiac output. Phenols are known to have higher phase II metabolism in lungs as compared with liver (Cassidy and Houston, 1980). In comparison with human cryopreserved hepatocytes, human lung parenchymal cells have higher sulfation activity (Somers et al., 2007). SULT1A1, SULT1A3, and SULT1E1 are the major SULT enzymes present in human lungs (80% of all SULTs expressed in human lungs) (Riches et al., 2009), which are also involved in the sulfation of RES (Miksits et al., 2005). There are conflicting reports about UGT expression in human lungs, showing low to no UGT activity in normal lung tissue (Zheng et al., 2002; Somers et al., 2007; Nakamura et al., 2008). Mouse lungs have been shown to express Ugt1a6 (Buckley and Klaassen, 2007), which is also involved in glucuronidation of RES. The aforementioned information about expression of metabolizing enzymes and their involvement in RES metabolism suggests the possibility of pulmonary contribution in the metabolism of RES.
Pulmonary metabolism of RES in vivo can be identified using multiple sites of administration and a single site of sampling approach (Cassidy and Houston, 1980). To evaluate the pulmonary contribution of RES, the right carotid artery and jugular vein of male C57BL/6 mice were cannulated under anesthesia with 1.5% isoflurane and 2 l/min oxygen. RES solubilized in 20% 2-hydroxylpropyl-β-cyclodextrin in saline was administered by i.v. or i.a. route at a dose of 15 mg/kg (Sharan et al., 2012; Sharan and Nagar, 2013). A carotid artery cannula was used for blood sampling. Blood samples were centrifuged at 14,000 rpm for 2 minutes, and plasma was stored at −80°C until analysis. Plasma samples were analyzed using electrospray ionization liquid chromatography–tandem mass spectrometry set in negative ion scan mode as per a published protocol (Iwuchukwu et al., 2012). Phoenix WinNonlin (version 6.1; Pharsight Corporation, Palo Alto, CA) was used for noncompartmental pharmacokinetic analysis of RES, R3G, and R3S. RES area under the curve exposure (AUC) and its half-life (t1/2) were significantly lower (AUC: P = 0.01, t1/2: P = 0.04) when administered by venous route (294.98 ± 137.87 minutes*µM, 101.30 ± 43.41 minutes) as compared with arterial route (591.08 ± 167.29 minutes*µM, 190.58 ± 69.65 minutes). The bioavailability of RES after i.v. administration, defined as a ratio of mean AUC0-inf after i.v. and i.a. route of administration, was 0.49. Exposure of R3G (2268.35 ± 517.00 minutes*µM) after RES i.v. administration was significantly higher (P = 0.004) as compared with R3G exposure (921.23 ± 457.07 minutes*µM) after RES i.a. administration. In vivo results were further confirmed by in vitro experiments in both mouse and human lung fractions. Mouse lungs showed both sulfation and glucuronidation with a partial substrate inhibition profile. Glucuronidation (Vmax: 324.40 ± 13.05 pmol/min/mg, Km: 7.34 ± 1.60 µM, Ki: 6632 ± 1198 µM) was more pronounced in mouse than sulfation (Vmax: 7.05 ± 0.28 pmol/min/mg, Km: 2.69 ± 0.45 µM, Ki: 2021 ± 717.6 µM). Interestingly, in experiments with human lung fractions, sulfation (Vmax: 16.15 ± 0.48 pmol/min/mg, Km: 4.45 ± 0.79 µM, Ki: 23,238 ± 7305 µM) was observed to be the major metabolic process. No R3G was observed above the limit of quantification (10 ng/ml) at the end of 60-minute incubations, indicating minimal contribution of pulmonary glucuronidation of RES in humans. The differences in pulmonary metabolism of RES in mice and humans can be a possible explanation for the observed differences in the disposition of RES in rodents and humans. R3G has been reported as the major metabolite in rodents (Juan et al., 2010; Iwuchukwu et al., 2012; Sharan et al., 2012) in terms of exposure as compared with R3S in humans (Boocock et al., 2007; Brown et al., 2010). This also reaffirms the differences in the way drugs are handled among species and emphasizes the need for proper understanding of interspecies differences to effectively use preclinical data for extrapolation to humans.
The importance of drug transporters in the disposition of compounds and their metabolites is increasingly recognized (Hillgren et al., 2013; Zamek-Gliszczynski et al., 2014). RES has been reported to be a substrate for BCRP, whereas its conjugated metabolites are effluxed by BCRP, MRP2, and MRP3 (van de Wetering et al., 2009; Planas et al., 2012). Interestingly, these and many other transporters have been shown to be expressed in the lung (Fig. 3) (Bosquillon, 2010; Gumbleton et al., 2011; Doring and Petzinger, 2014). Thus, various family members of the ABC transporter family as well as SLC and solute carrier organic anion transporters are expressed in the lung (Bosquillon, 2010). The solute carrier organic anion transporter OATP1B3 is expressed in normal lung as well as in solid lung tumors, and was shown to correlate with disposition and toxicity of platinum cancer chemotherapy drugs (Lancaster et al., 2013).
The interplay between XMEs and transporters is of great interest, especially in the context of metabolism with subsequent efflux of metabolites. This interplay has been evaluated at the level of common regulatory pathways for XMEs and transporters, and in regard to the pharmacokinetics of compounds that are substrates for both XMEs and transporters (Nies et al., 2008; Pang et al., 2009; Zhang et al., 2009; Fan et al., 2010). XMEs as well as drug transporters share common regulatory pathways; for example, pregnane X receptor, constitutive androstane receptor, AhR/ARNT, and Nrf2-Keap mediated regulation of protein expression. Overlapping roles of metabolism and transport impact the overall disposition of RES and its conjugates (Maier-Salamon et al., 2013). It therefore becomes important to integrate metabolism and transport of RES across all eliminating organs to understand the target tissue concentrations of the active compound. Figure 4 depicts proposed pathways for the disposition of RES and its major metabolites in the mouse lung, liver, and gut.
Fig. 4.
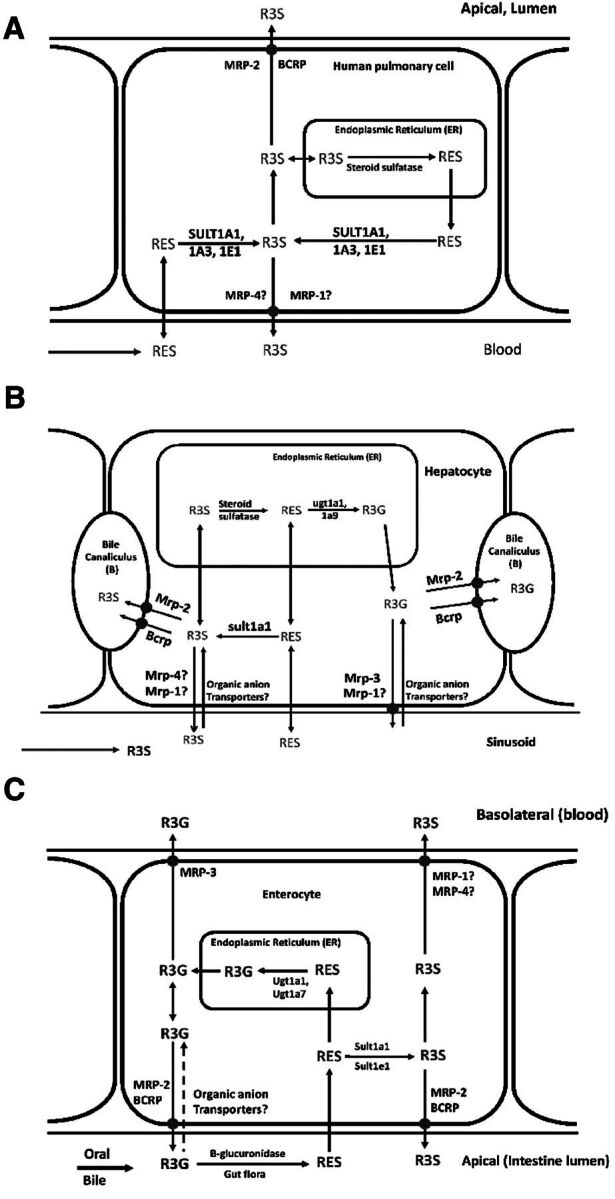
Proposed scheme of transporter and metabolizing enzyme interaction mediating the disposition of RES and its metabolites in mouse lung cells (A), hepatocytes (B), and gut (C). Solid arrows represent pathways based on published reports for metabolism or transport of RES, R3S, and R3G. Dashed line represents pathways based on reports of similar substrates and transporters. Transporters followed by “?” indicate that the role of these transporters has not been established for transport of R3S and R3G but is hypothesized based on literature for other glucuronidated and sulfated metabolites.
Several XMEs and drug transporters are expressed in the lung. Their relative expression and activity across various pulmonary cells and experimental pulmonary cell lines are not completely understood. Pulmonary XME expression is inducible, with common regulatory pathways between XMEs and transporters. The present case study used RES as a model substrate, which was shown to be conjugated in the mouse as well as human lung in vitro, with clear species differences in the kinetics of RES conjugation. The reversible conjugation of RES along with its transport is tissue-dependent. It is clear that, in addition to hepatic metabolism and transport, the extrahepatic disposition of compounds and their relevant metabolites must be evaluated in an integrated manner before target-site drug concentrations and pharmacodynamics can be better predicted.
Tumor Metabolism of Antibody-Drug Conjugates: Effect on Pharmacokinetics and Efficacy (D.R.)
Therapeutics based upon antibody-drug conjugates (ADCs) are being pursued with a renewed vigor based largely upon advances in the next generation of linkers and payloads that have increased selectivity and potency, respectively, in this class of molecules. This is evidenced with the recent approval of Adcetris (brentuximab vedotin) to treat Hodgkin lymphoma and Kadcyla (ado-trastuzumab emtansine) to treat patients with human epidermal growth factor receptor 2-positive, late-stage breast cancer (http://www.fda.gov/NewsEvents/Newsroom/PressAnnouncements/ucm268781.htm; http://www.fda.gov/newsevents/newsroom/pressannouncements/ucm340704.htm). Of particular interest is the prediction of released payload concentrations at the site of therapeutic action, which may be significantly higher than the concentration observed in plasma (Alley et al., 2009). As direct clinical measurement of released payload concentrations through the use of tumor distribution studies is often logistically challenging, a significant effort has recently been placed on the development of in vitro or in silico techniques to predict the effect of therapeutic site-specific catabolism on the observed target-site concentrations of ADC payloads (Shah et al., 2014). The final aspect of this symposium focused on the use of in vitro cellular assays to determine the effect of ADC catabolism at the site of therapeutic action on the observed pharmacodynamics of the ADC.
The stability of the cytotoxin conjugated to the antibody is paramount to achieving the therapeutic window required for the success of novel ADCs. Due to the long half-life of antibodies, stability testing with ADCs requires evaluation over the course of days as opposed to hours as is typical for small molecules. This represents a significant challenge in testing ADC stability in preclinical systems. For example, plasma may not be a suitable matrix for assessing stability in vitro beyond 48 hours due to changes in pH, altered reduction potential, and protein aggregation (Rossi et al., 2002). This poses a particular challenge for noncleavable ADCs where the linker is nonreducibly attached to the antibody with a covalent bond that is designed to be highly stable. Moreover, the size of ADCs (>150 kDa) precludes catabolism by drug-metabolizing enzymes found in hepatic liver microsomes.
Cell-based assays may be more suitable for stability and catabolism assessment needs for ADCs given the ability to culture cells for days. However, even in vitro uptake of antibodies in non–target-expressing cells via endocytosis or phagocytosis is slow and can limit the detection of antibody or ADC catabolism. Therefore, cells expressing the antigen, such as tumor cells or engineered cells transfected with tumor antigen, could be considered to interrogate ADC stability (Erickson et al., 2006). Furthermore, the cell-based assay format can mimic the in vivo conditions in which the ADC is expected to bind and undergo receptor-mediated endocytosis en route to lysosomal processing (Fig. 5).
Fig. 5.

Schematic of ADC catabolism. The catabolic pathway includes ADC (A) binding to tumor antigen (B); internalization of the ADC-tumor antigen complex into the endosome (noncleavable linker is stable) (C); a portion of the endosome is sorted to the lysosome where ADC is catabolized (D); and the catabolite exits the lysosome (E) and binds to microtubulin (F).
Prior to conducting cell-based experiments with ADCs, the integrity of the physical properties of the ADC should be assessed. That is, the conjugated antibody should retain similar physical and biosphysical properties (binding to the antigen and to the neonatal Fc receptor) as compared with the unconjugated antibody. ADC conjugation to the antibody most commonly occurs through the ε-amine of the lysine side chain or via cysteine residues, either from reduction of the interchain disulfides or engineered cysteine residues. The resultant conjugated antibody material is composed of a heterogeneous population of species, each with varying conjugation stoichiometry. Each species could behave uniquely relative to the unconjugated antibody (Hamblett et al., 2004). Although any biophysical measurement of a heterogeneous population cannot read distinct properties of binding interactions within a mixture, the composite binding isotherm may indicate the presence of a strong outlier within the distribution (typically >2-fold change from unconjugated antibody). These results provide further confidence that subsequent cellular experiments are not selecting a subset of the ADC population for cellular catabolism, which may limit the accurate interpretation of the stability results. This should also be factored into the cellular experimental design wherein saturating the antigen-expressing cells with excess ADC to antigen creates a preference for the higher affinity species (unconjugated antibody) to limit binding of the lower affinity (high drug to antibody-derived material), thus preventing unbiased ruling on the in vitro stability. This can be overcome by the high antigen-to-antibody ratios possible in the test system and under conditions which consume >50% of dosed ADC to help ensure greater sampling of the mixture.
Cell-based stability experiments with antigen-positive cells can be used with ADCs to facilitate the uptake of the ADC into the cell, where it is exposed to endosome lysosome sorting pathways similar to those which will be encountered in vivo. There is a time component to the experiment findings given the need for antibody binding, internalization, and proteolytic processing. Incubations require a balance in concentrations used due to cellular toxicity upon ADC degradation. Some noncleavable ADCs are more stable to proteolysis depending upon the site of conjugation (Shen et al., 2012). Furthermore, the compartment of degradation can be tuned for protease-cleavable linkers. Okeley et al. (2014) designed a series of linkers with distinct processing within the cell leading to increased cellular cytotoxicity. This is another advantage to the cell-based stability evaluation in that it allows for catabolism to be investigated in different cellular compartments. However, most noncleavable linker systems require lysosomal processing. To demonstrate the time dependence and importance of target expression in the design of in vitro stability assays, varying concentrations of a mertansine (DM1)-conjugated monoclonal antibody were incubated for 48 or 96 hours in antigen-positive target cells (T-ADC) and compared with incubations with cells devoid of the target antigen (NT-control). Cells were plated (96-well) and allowed to adhere overnight, after which new culture medium containing the DM1 conjugate was added to the cells. Cellular survival was determined using a luciferase-based ATP quantification assay and visualized as luminescence output versus DM1 equivalents. The time course for the degradation process is shown in Fig. 6 by tracking cell viability for this antigen-positive cell. The nontargeting control provides corresponding control for the lack of activity with antibody without cell surface antigen conjugated with the cytotoxin through the same linker.
Fig. 6.
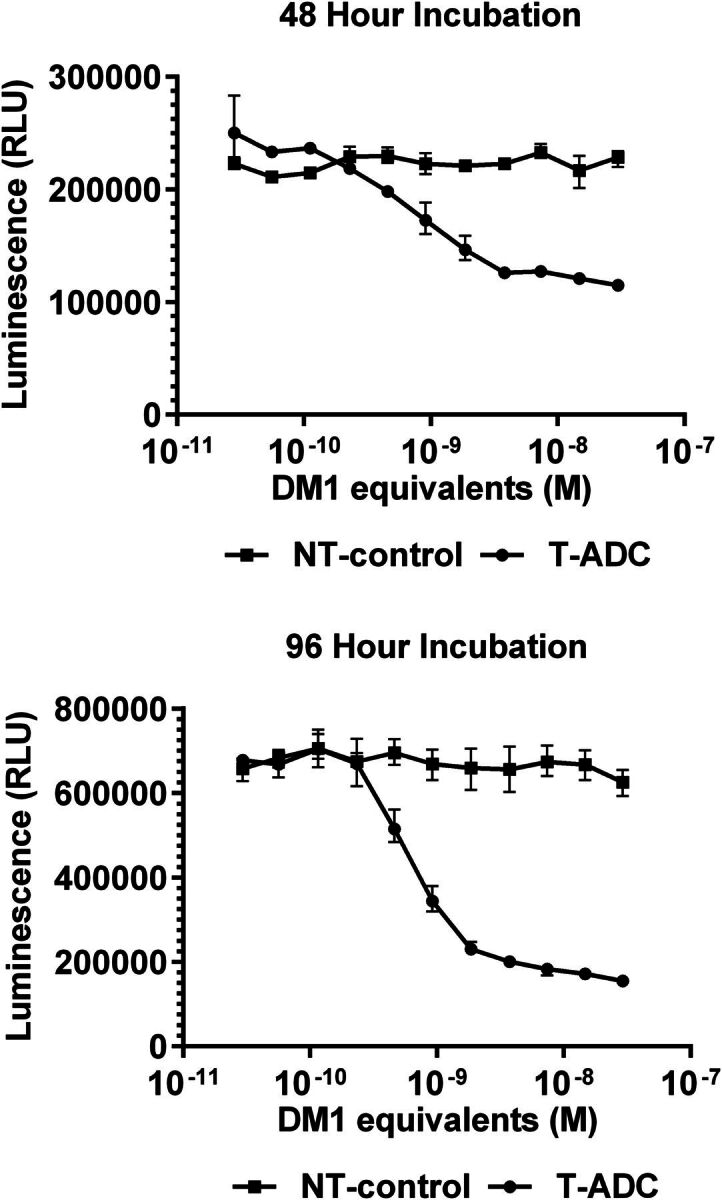
Effect of tumor antigen on the cytotoxicity profile of ADCs. Example ADC incubated in cells transfected with the tumor antigen (T), whereas the same ADC incubated with control cells without tumor antigen (NT). Time-dependent cellular cytotoxicity at 48 hours (top) and 96 hours (bottom) illustrating the selectivity of toxicity toward tumor antigen–expressing cells.
The cellular supernatant and cell lysis can be evaluated for ADC catabolites. As most cell types share overlapping proteases in their lysosome, it is assumed the catabolism would represent that detected from a variety of cell types upon in vivo processing. To test this hypothesis, an antibody linked via succinimidyl 4-[N-maleimidomethyl]cyclohexane-1-carboxylate to DM1 was incubated for 24 or 48 hours in isolated lysosomal fractions from HepG2 cells, cellular lysate, nonadherent HepG2 cells, and control cell media. Lysosomes were isolated from HepG2 cells with the use of a lysosome isolation kit (Thermo Scientific, Waltham, MA) and resuspended in 100 mM sodium acetate buffer (pH 5.5) containing sodium chloride (100 mM), diethyldithiocarbamate (2 mM), EDTA (1 mM), and Triton X-100 (0.05%). Formation of lysine-[N-maleimidomethyl]-cyclohexane-1-carboxylate-DM1 (lysine-MCC-DM1) and other potential metabolites was assessed using a liquid chromatography–tandem mass spectrometry approach as previously described (Davis et al., 2012). Incubations resulted in the identification of a sole detectable catabolite of lysine-MCC-DM1. More detailed processing of the cells to isolate lysosome was conducted to isolate the subcellular compartment thought to be responsible for catabolite formation. As shown in Fig. 7, upon lysosome isolation, there is clear detection of lysine-MCC-DM1 in this compartment, consistent with this organelle as the origin of the catabolite. Not only is it present, but it is found in excess when compared with the cell supernatant and half of that detected in the lysate of the cells.
Fig. 7.

Effect of tumor antigen on the distribution of ADCs in vitro. Cell lysate from cells transfected with tumor antigen contains the majority of catabolite, with lysosome harboring approximately half of all cell-generated catabolite. No catabolite was detected in cell media.
The absence of lysine-MCC-DM1 in the supernatant of cells may be due to the low passive permeability of lysine-MCC-DM1 which was determined in LLC-PK1 cells. Furthermore, as this is expected to be the primary ADC catabolite from succinimidyl 4-[N-maleimidomethyl]-cyclohexane-1-carboxylate linked DM1 to antibodies the efflux due to P-gp was also assessed in LLC-PK1 cells transfected with the human MDR1 gene (P-gp, ABCB1). In brief, cell culture medium containing l-glutamine (2 mM), penicillin (50 units/ml), streptomycin (50 µg/ml), and fetal bovine serum (10%, v/v) was used to culture and plate cells in 96-well plates (90,000 cells per 150 µl) as previously described (Schinkel et al., 1995; Booth-Genthe et al., 2006). After 5 days of plating, cell medium was replaced with 150 µl of Hanks’ buffered saline solution containing 10 mM Hepes and various concentrations of 14C-lysine-MCC-DM1. 14C-mannitol was used as a marker of monolayer integrity, 14C-cyclosporin A as a positive control for P-gp–mediated efflux, and elacridar as an inhibitor of P-gp activity in vitro. Plates were incubated for 2 hours, after which sample radioactivity was measured from the apical and basolateral compartments using liquid scintillation counting. Calculation of efflux ratios [ratio of apparent permeability coefficient (Papp) of basolateral to apical compartment divided by the Papp of the apical to basolateral compartment] suggests lysine-MCC-DM1 exhibits low passive permeability and a low potential for the catabolite to serve as a substrate for P-gp (Fig. 8).
Fig. 8.

Lysine-MCC-DM1 catabolite permeability at 0.1, 1.0, 5.0, and 10 µM. Data reveal low passive permeability of lysine-MCC-DM1. There is no evidence implicating lysine-MCC-DM1 as a P-gp substrate. CsA, cyclosporin A; GG, elacridar; LMD, lysine-MCC-DM1; Mann, mannitol.
The data presented support the use of cellular systems to evaluate ADC catabolism and aid in the characterization of ADC absorption, distribution, metabolism and excretion in preclinical testing.
Summary
The research which was presented at the “Target-Site” Drug Metabolism and Transport symposium at the 2014 Experimental Biology meeting and which is summarized in this report highlights the importance of drug-metabolizing enzymes and transporters at the therapeutic site of action for a given drug. An increased understanding of such phenomena should allow for a more rational approach to drug optimization while increasing our knowledge of the potential barriers to drug efficacy. Ultimately, such research should serve to increase our ability to provide more effective clinical therapies in a safe and timely manner.
Abbreviations
- ABC
ATP binding cassette
- ADC
antibody-drug conjugate
- AUC
area under the curve
- BCRP
breast cancer resistance protein
- CMPF
3-carboxy-4-methyl-5-propyl-2-furanopropanoic acid
- CNS
central nervous system
- DDI
drug-drug interaction
- DM1
mertansine
- EMB
ethambutol dihydrochloride
- HIV
human immunodeficiency virus
- hOAT
human organic anion transporter
- hOCT
human organic cation transporter
- ICV
intracerebroventricular
- MBI
mechanism-based inhibitor
- MPP+
1-methyl-4-phenylpyridinium
- MRP
multidrug resistance–associated protein
- OAT
organic anion transporter
- OCT
organic cation transporter
- P450
cytochrome P450
- P-gp
P-glycoprotein
- RES
trans-resveratrol
- R3G
trans-resveratrol-3-glucuronide
- R3S
trans-resveratrol-3-sulfate
- SLC
solute carrier
- SULT
sulfotransferase
- TB
tuberculosis
- UGT
UDP-glucuronosyltransferase
- XME
xenobiotic-metabolizing enzyme
Authorship Contributions
Wrote or contributed to the writing of the manuscript: Foti, Tyndale, Garcia, Sweet, Nagar, Sharan, Rock.
Footnotes
References
- Acharya P, Tran TT, Polli JW, Ayrton A, Ellens H, Bentz J. (2006) P-Glycoprotein (P-gp) expressed in a confluent monolayer of hMDR1-MDCKII cells has more than one efflux pathway with cooperative binding sites. Biochemistry 45:15505–15519. [DOI] [PubMed] [Google Scholar]
- Adler TK, Fujimoto JM, Way EL, Baker EM. (1955) The metabolic fate of codeine in man. J Pharmacol Exp Ther 114:251–262. [PubMed] [Google Scholar]
- Alley SC, Okeley NM, Senter PD. (2010) Antibody-drug conjugates: targeted drug delivery for cancer. Curr Opin Chem Biol 14:529–537. [DOI] [PubMed] [Google Scholar]
- Alley SC, Zhang X, Okeley NM, Anderson M, Law CL, Senter PD, Benjamin DR. (2009) The pharmacologic basis for antibody-auristatin conjugate activity. J Pharmacol Exp Ther 330:932–938. [DOI] [PubMed] [Google Scholar]
- Anttila S, Raunio H, Hakkola J. (2011) Cytochrome P450-mediated pulmonary metabolism of carcinogens: regulation and cross-talk in lung carcinogenesis. Am J Respir Cell Mol Biol 44:583–590. [DOI] [PubMed] [Google Scholar]
- Awasthi S, Singhal SS, Srivastava SK, Zimniak P, Bajpai KK, Saxena M, Sharma R, Ziller SA, 3rd, Frenkel EP, Singh SV, et al. (1994) Adenosine triphosphate-dependent transport of doxorubicin, daunomycin, and vinblastine in human tissues by a mechanism distinct from the P-glycoprotein. J Clin Invest 93:958–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baur JA, Sinclair DA. (2006) Therapeutic potential of resveratrol: the in vivo evidence. Nat Rev Drug Discov 5:493–506. [DOI] [PubMed] [Google Scholar]
- Bertilsson L, Alm C, De Las Carreras C, Widen J, Edman G, Schalling D. (1989) Debrisoquine hydroxylation polymorphism and personality. Lancet 1:555. [DOI] [PubMed] [Google Scholar]
- Boocock DJ, Faust GE, Patel KR, Schinas AM, Brown VA, Ducharme MP, Booth TD, Crowell JA, Perloff M, Gescher AJ, et al. (2007) Phase I dose escalation pharmacokinetic study in healthy volunteers of resveratrol, a potential cancer chemopreventive agent. Cancer Epidemiol Biomarkers Prev 16:1246–1252. [DOI] [PubMed] [Google Scholar]
- Booth-Genthe CL, Louie SW, Carlini EJ, Li B, Leake BF, Eisenhandler R, Hochman JH, Mei Q, Kim RB, Rushmore TH, et al. (2006) Development and characterization of LLC-PK1 cells containing Sprague-Dawley rat Abcb1a (Mdr1a): comparison of rat P-glycoprotein transport to human and mouse. J Pharmacol Toxicol Methods 54:78–89. [DOI] [PubMed] [Google Scholar]
- Bosquillon C. (2010) Drug transporters in the lung—do they play a role in the biopharmaceutics of inhaled drugs? J Pharm Sci 99:2240–2255. [DOI] [PubMed] [Google Scholar]
- Bouw MR, Gårdmark M, Hammarlund-Udenaes M. (2000) Pharmacokinetic-pharmacodynamic modelling of morphine transport across the blood-brain barrier as a cause of the antinociceptive effect delay in rats—a microdialysis study. Pharm Res 17:1220–1227. [DOI] [PubMed] [Google Scholar]
- Brill SS, Furimsky AM, Ho MN, Furniss MJ, Li Y, Green AG, Bradford WW, Green CE, Kapetanovic IM, Iyer LV. (2006) Glucuronidation of trans-resveratrol by human liver and intestinal microsomes and UGT isoforms. J Pharm Pharmacol 58:469–479. [DOI] [PubMed] [Google Scholar]
- Brown VA, Patel KR, Viskaduraki M, Crowell JA, Perloff M, Booth TD, Vasilinin G, Sen A, Schinas AM, Piccirilli G, et al. (2010) Repeat dose study of the cancer chemopreventive agent resveratrol in healthy volunteers: safety, pharmacokinetics, and effect on the insulin-like growth factor axis. Cancer Res 70:9003–9011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckley DB, Klaassen CD. (2007) Tissue- and gender-specific mRNA expression of UDP-glucuronosyltransferases (UGTs) in mice. Drug Metab Dispos 35:121–127. [DOI] [PubMed] [Google Scholar]
- Cassidy MK, Houston JB. (1980) Phenol conjugation by lung in vivo. Biochem Pharmacol 29:471–474. [DOI] [PubMed] [Google Scholar]
- Chambers JE, Chambers HW. (1989) Oxidative desulfuration of chlorpyrifos, chlorpyrifos-methyl, and leptophos by rat brain and liver. J Biochem Toxicol 4:201–203. [DOI] [PubMed] [Google Scholar]
- Chari RV. (2008) Targeted cancer therapy: conferring specificity to cytotoxic drugs. Acc Chem Res 41:98–107. [DOI] [PubMed] [Google Scholar]
- Chen Y, Tang Y, Guo C, Wang J, Boral D, Nie D. (2012) Nuclear receptors in the multidrug resistance through the regulation of drug-metabolizing enzymes and drug transporters. Biochem Pharmacol 83:1112–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ZR, Somogyi AA, Reynolds G, Bochner F. (1991) Disposition and metabolism of codeine after single and chronic doses in one poor and seven extensive metabolisers. Br J Clin Pharmacol 31:381–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciarimboli G, Ludwig T, Lang D, Pavenstädt H, Koepsell H, Piechota HJ, Haier J, Jaehde U, Zisowsky J, Schlatter E. (2005) Cisplatin nephrotoxicity is critically mediated via the human organic cation transporter 2. Am J Pathol 167:1477–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa LG, McDonald BE, Murphy SD, Omenn GS, Richter RJ, Motulsky AG, Furlong CE. (1990) Serum paraoxonase and its influence on paraoxon and chlorpyrifos-oxon toxicity in rats. Toxicol Appl Pharmacol 103:66–76. [DOI] [PubMed] [Google Scholar]
- Cutler MJ, Urquhart BL, Velenosi TJ, Meyer Zu Schwabedissen HE, Dresser GK, Leake BF, Tirona RG, Kim RB, Freeman DJ. (2012) In vitro and in vivo assessment of renal drug transporters in the disposition of mesna and dimesna. J Clin Pharmacol 52:530–542. [DOI] [PubMed] [Google Scholar]
- Dalhoff K, Buus Jensen K, Enghusen Poulsen H. (2005) Cancer and molecular biomarkers of phase 2. Methods Enzymol 400:618–627. [DOI] [PubMed] [Google Scholar]
- Davis JA, Rock DA, Wienkers LC, Pearson JT. (2012) In vitro characterization of the drug-drug interaction potential of catabolites of antibody-maytansinoid conjugates. Drug Metab Dispos 40:1927–1934. [DOI] [PubMed] [Google Scholar]
- Deng Y, Newman B, Dunne MP, Silburn PA, Mellick GD. (2004) Further evidence that interactions between CYP2D6 and pesticide exposure increase risk for Parkinson’s disease. Ann Neurol 55:897. [DOI] [PubMed] [Google Scholar]
- Ding X, Kaminsky LS. (2003) Human extrahepatic cytochromes P450: function in xenobiotic metabolism and tissue-selective chemical toxicity in the respiratory and gastrointestinal tracts. Annu Rev Pharmacol Toxicol 43:149–173. [DOI] [PubMed] [Google Scholar]
- Döring B, Petzinger E. (2014) Phase 0 and phase III transport in various organs: combined concept of phases in xenobiotic transport and metabolism. Drug Metab Rev 46:261–282. [DOI] [PubMed] [Google Scholar]
- Drevenkar V, Vasilić Z, Stengl B, Fröbe Z, Rumenjak V. (1993) Chlorpyrifos metabolites in serum and urine of poisoned persons. Chem Biol Interact 87:315–322. [DOI] [PubMed] [Google Scholar]
- Eraly SA, Vallon V, Vaughn DA, Gangoiti JA, Richter K, Nagle M, Monte JC, Rieg T, Truong DM, Long JM, et al. (2006) Decreased renal organic anion secretion and plasma accumulation of endogenous organic anions in OAT1 knock-out mice. J Biol Chem 281:5072–5083. [DOI] [PubMed] [Google Scholar]
- Erickson HK, Park PU, Widdison WC, Kovtun YV, Garrett LM, Hoffman K, Lutz RJ, Goldmacher VS, Blättler WA. (2006) Antibody-maytansinoid conjugates are activated in targeted cancer cells by lysosomal degradation and linker-dependent intracellular processing. Cancer Res 66:4426–4433. [DOI] [PubMed] [Google Scholar]
- Fan J, Chen S, Chow EC, Pang KS. (2010) PBPK modeling of intestinal and liver enzymes and transporters in drug absorption and sequential metabolism. Curr Drug Metab 11:743–761. [DOI] [PubMed] [Google Scholar]
- Farthing CA, Sweet DH. (2014) Expression and function of organic cation and anion transporters (SLC22 family) in the CNS. Curr Pharm Des 20:1472–1486. [DOI] [PubMed] [Google Scholar]
- Faurholt-Jepsen D, Range N, PrayGod G, Jeremiah K, Faurholt-Jepsen M, Aabye MG, Changalucha J, Christensen DL, Krarup H, Witte DR, et al. (2012) The role of diabetes on the clinical manifestations of pulmonary tuberculosis. Trop Med Int Health 17:877–883. [DOI] [PubMed] [Google Scholar]
- Ferguson CS, Tyndale RF. (2011) Cytochrome P450 enzymes in the brain: emerging evidence of biological significance. Trends Pharmacol Sci 32:708–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foti RS, Fisher MB. (2012) UDP-Glucuronosyltransferases: Pharmacogenetics, Functional Characterization, and Clinical Relevance, in Encyclopedia of Drug Metabolism and Interactions (Lyubimov AV. ed) John Wiley & Sons, Inc. Malden, MA [Google Scholar]
- Gaedigk A, Simon SD, Pearce RE, Bradford LD, Kennedy MJ, Leeder JS. (2008) The CYP2D6 activity score: translating genotype information into a qualitative measure of phenotype. Clin Pharmacol Ther 83:234–242. [DOI] [PubMed] [Google Scholar]
- Gallagher CJ, Muscat JE, Hicks AN, Zheng Y, Dyer AM, Chase GA, Richie J, Lazarus P. (2007) The UDP-glucuronosyltransferase 2B17 gene deletion polymorphism: sex-specific association with urinary 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanol glucuronidation phenotype and risk for lung cancer. Cancer Epidemiol Biomarkers Prev 16:823–828. [DOI] [PubMed] [Google Scholar]
- Gan SH, Ismail R, Wan Adnan WA, Zulmi W, Kumaraswamy N, Larmie ET. (2004) Relationship between Type A and B personality and debrisoquine hydroxylation capacity. Br J Clin Pharmacol 57:785–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garcia KL, Coen K, Miksys S, Dzung Le A and Tyndale RF (2015) Effect of brain CYP2B inhibition on brain nicotine levels and nicotine self-administration. Neuropsychopharmacology DOI: 10.1038/npp.2015.40 [published ahead of print]. [DOI] [PMC free article] [PubMed]
- Gerber JG, Rhodes RJ, Gal J. (2004) Stereoselective metabolism of methadone N-demethylation by cytochrome P4502B6 and 2C19. Chirality 16:36–44. [DOI] [PubMed] [Google Scholar]
- Ghose T, Blair AH. (1987) The design of cytotoxic-agent-antibody conjugates. Crit Rev Ther Drug Carrier Syst 3:263–359. [PubMed] [Google Scholar]
- Giacomini KM, Huang SM, Tweedie DJ, Benet LZ, Brouwer KL, Chu X, Dahlin A, Evers R, Fischer V, Hillgren KM, et al. International Transporter Consortium (2010) Membrane transporters in drug development. Nat Rev Drug Discov 9:215–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottesman MM, Fojo T, Bates SE. (2002) Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2:48–58. [DOI] [PubMed] [Google Scholar]
- Guengerich FP.(2005) Human cytochrome P450 enzymes, in Cytochrome P450: Structure, Mechanism, and Biochemistry (Ortiz de Montellano PR. ed), Kluwer Academic/Plenum, New York. [Google Scholar]
- Gumbleton M, Al-Jayyoussi G, Crandon-Lewis A, Francombe D, Kreitmeyr K, Morris CJ, Smith MW. (2011) Spatial expression and functionality of drug transporters in the intact lung: objectives for further research. Adv Drug Deliv Rev 63:110–118. [DOI] [PubMed] [Google Scholar]
- Gundert-Remy U, Bernauer U, Blömeke B, Döring B, Fabian E, Goebel C, Hessel S, Jäckh C, Lampen A, Oesch F, et al. (2014) Extrahepatic metabolism at the body’s internal-external interfaces. Drug Metab Rev 46:291–324. [DOI] [PubMed] [Google Scholar]
- Hamblett KJ, Senter PD, Chace DF, Sun MM, Lenox J, Cerveny CG, Kissler KM, Bernhardt SX, Kopcha AK, Zabinski RF, et al. (2004) Effects of drug loading on the antitumor activity of a monoclonal antibody drug conjugate. Clin Cancer Res 10:7063–7070. [DOI] [PubMed] [Google Scholar]
- He L, Vasiliou K, Nebert DW. (2009) Analysis and update of the human solute carrier (SLC) gene superfamily. Hum Genomics 3:195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecht SS. (2008) Progress and challenges in selected areas of tobacco carcinogenesis. Chem Res Toxicol 21:160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hesse LM, Venkatakrishnan K, Court MH, von Moltke LL, Duan SX, Shader RI, Greenblatt DJ. (2000) CYP2B6 mediates the in vitro hydroxylation of bupropion: potential drug interactions with other antidepressants. Drug Metab Dispos 28:1176–1183. [PubMed] [Google Scholar]
- Hillgren KM, Keppler D, Zur AA, Giacomini KM, Stieger B, Cass CE, Zhang L, International Transporter Consortium (2013) Emerging transporters of clinical importance: an update from the International Transporter Consortium. Clin Pharmacol Ther 94:52–63. [DOI] [PubMed] [Google Scholar]
- Iwuchukwu OF, Nagar S. (2008) Resveratrol (trans-resveratrol, 3,5,4′-trihydroxy-trans-stilbene) glucuronidation exhibits atypical enzyme kinetics in various protein sources. Drug Metab Dispos 36:322–330. [DOI] [PubMed] [Google Scholar]
- Iwuchukwu OF, Sharan S, Canney DJ, Nagar S. (2012) Analytical method development for synthesized conjugated metabolites of trans-resveratrol, and application to pharmacokinetic studies. J Pharm Biomed Anal 63:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones NR, Lazarus P. (2014) UGT2B gene expression analysis in multiple tobacco carcinogen-targeted tissues. Drug Metab Dispos 42:529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juan ME, Maijó M, Planas JM. (2010) Quantification of trans-resveratrol and its metabolites in rat plasma and tissues by HPLC. J Pharm Biomed Anal 51:391–398. [DOI] [PubMed] [Google Scholar]
- Khokhar JY, Miksys SL, Tyndale RF. (2010) Rat brain CYP2B induction by nicotine is persistent and does not involve nicotinic acetylcholine receptors. Brain Res 1348:1–9. [DOI] [PubMed] [Google Scholar]
- Khokhar JY, Tyndale RF. (2011) Drug metabolism within the brain changes drug response: selective manipulation of brain CYP2B alters propofol effects. Neuropsychopharmacology 36:692–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khokhar JY, Tyndale RF. (2012) Rat brain CYP2B-enzymatic activation of chlorpyrifos to the oxon mediates cholinergic neurotoxicity. Toxicol Sci 126:325–335. [DOI] [PubMed] [Google Scholar]
- Khokhar JY, Tyndale RF. (2014) Intracerebroventricularly and systemically delivered inhibitor of brain CYP2B (C8-Xanthate), even following chlorpyrifos exposure, reduces chlorpyrifos activation and toxicity in male rats. Toxicol Sci 140:49–60. [DOI] [PubMed] [Google Scholar]
- Kim KA, Chung J, Jung DH, Park JY. (2004) Identification of cytochrome P450 isoforms involved in the metabolism of loperamide in human liver microsomes. Eur J Clin Pharmacol 60:575–581. [DOI] [PubMed] [Google Scholar]
- Kirchheiner J, Seeringer A, Godoy AL, Ohmle B, Maier C, Beschoner P, Sim EJ, Viviani R. (2011) CYP2D6 in the brain: genotype effects on resting brain perfusion. Mol Psychiatry 16:237–, 333–341.. [DOI] [PubMed] [Google Scholar]
- Koenigs LL, Trager WF. (1998) Mechanism-based inactivation of cytochrome P450 2B1 by 8-methoxypsoralen and several other furanocoumarins. Biochemistry 37:13184–13193. [DOI] [PubMed] [Google Scholar]
- Koepsell H, Lips K, Volk C. (2007) Polyspecific organic cation transporters: structure, function, physiological roles, and biopharmaceutical implications. Pharm Res 24:1227–1251. [DOI] [PubMed] [Google Scholar]
- Kruijtzer CM, Beijnen JH, Rosing H, ten Bokkel Huinink WW, Schot M, Jewell RC, Paul EM, Schellens JH. (2002) Increased oral bioavailability of topotecan in combination with the breast cancer resistance protein and P-glycoprotein inhibitor GF120918. J Clin Oncol 20:2943–2950. [DOI] [PubMed] [Google Scholar]
- Kyrklund C, Backman JT, Neuvonen M, Neuvonen PJ. (2003) Gemfibrozil increases plasma pravastatin concentrations and reduces pravastatin renal clearance. Clin Pharmacol Ther 73:538–544. [DOI] [PubMed] [Google Scholar]
- Lancaster CS, Sprowl JA, Walker AL, Hu S, Gibson AA, Sparreboom A. (2013) Modulation of OATP1B-type transporter function alters cellular uptake and disposition of platinum chemotherapeutics. Mol Cancer Ther 12:1537–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang T, Klein K, Fischer J, Nüssler AK, Neuhaus P, Hofmann U, Eichelbaum M, Schwab M, Zanger UM. (2001) Extensive genetic polymorphism in the human CYP2B6 gene with impact on expression and function in human liver. Pharmacogenetics 11:399–415. [DOI] [PubMed] [Google Scholar]
- Lee AM, Miksys S, Palmour R, Tyndale RF. (2006) CYP2B6 is expressed in African Green monkey brain and is induced by chronic nicotine treatment. Neuropharmacology 50:441–450. [DOI] [PubMed] [Google Scholar]
- Liu SH, Wei W, Ding GN, Ke JD, Hong FX, Tian M. (2009) Relationship between depth of anesthesia and effect-site concentration of propofol during induction with the target-controlled infusion technique in elderly patients. Chin Med J (Engl) 122:935–940. [PubMed] [Google Scholar]
- Llerena A, Cobaleda J, Benítez J. (1989) Debrisoquine hydroxylation phenotypes in healthy volunteers. Lancet 1:1398. [DOI] [PubMed] [Google Scholar]
- LLerena A, Herraíz AG, Cobaleda J, Johansson I, Dahl ML. (1993) Debrisoquin and mephenytoin hydroxylation phenotypes and CYP2D6 genotype in patients treated with neuroleptic and antidepressant agents. Clin Pharmacol Ther 54:606–611. [DOI] [PubMed] [Google Scholar]
- Maier-Salamon A, Böhmdorfer M, Riha J, Thalhammer T, Szekeres T, Jaeger W. (2013) Interplay between metabolism and transport of resveratrol. Ann N Y Acad Sci 1290:98–106. [DOI] [PubMed] [Google Scholar]
- Mann A, Miksys S, Lee A, Mash DC, Tyndale RF. (2008) Induction of the drug metabolizing enzyme CYP2D in monkey brain by chronic nicotine treatment. Neuropharmacology 55:1147–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann A, Miksys SL, Gaedigk A, Kish SJ, Mash DC, Tyndale RF. (2012) The neuroprotective enzyme CYP2D6 increases in the brain with age and is lower in Parkinson’s disease patients. Neurobiol Aging 33:2160–2171. [DOI] [PubMed] [Google Scholar]
- Mann A, Tyndale RF. (2010) Cytochrome P450 2D6 enzyme neuroprotects against 1-methyl-4-phenylpyridinium toxicity in SH-SY5Y neuronal cells. Eur J Neurosci 31:1185–1193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masereeuw R, Mutsaers HA, Toyohara T, Abe T, Jhawar S, Sweet DH, Lowenstein J. (2014) The kidney and uremic toxin removal: glomerulus or tubule? Semin Nephrol 34:191–208. [DOI] [PubMed] [Google Scholar]
- Matoh N, Tanaka S, Takehashi M, Banasik M, Stedeford T, Masliah E, Suzuki S, Nishimura Y, Ueda K. (2003) Overexpression of CYP2D6 attenuates the toxicity of MPP+ in actively dividing and differentiated PC12 cells. Gene Expr 11:117–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCann SJ, Pond SM, James KM, Le Couteur DG. (1997) The association between polymorphisms in the cytochrome P-450 2D6 gene and Parkinson’s disease: a case-control study and meta-analysis. J Neurol Sci 153:50–53. [DOI] [PubMed] [Google Scholar]
- McMillan DM, Tyndale RF. (2015) Nicotine Increases Codeine Analgesia Through the Induction of Brain CYP2D and Central Activation of Codeine to Morphine. Neuropsychopharmacology 40:1804–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michels R, Marzuk PM. (1993) Progress in psychiatry (1). N Engl J Med 329:552–560. [DOI] [PubMed] [Google Scholar]
- Miksits M, Maier-Salamon A, Aust S, Thalhammer T, Reznicek G, Kunert O, Haslinger E, Szekeres T, Jaeger W. (2005) Sulfation of resveratrol in human liver: evidence of a major role for the sulfotransferases SULT1A1 and SULT1E1. Xenobiotica 35:1101–1119. [DOI] [PubMed] [Google Scholar]
- Miksys S, Hoffmann E, Tyndale RF. (2000) Regional and cellular induction of nicotine-metabolizing CYP2B1 in rat brain by chronic nicotine treatment. Biochem Pharmacol 59:1501–1511. [DOI] [PubMed] [Google Scholar]
- Miksys S, Lerman C, Shields PG, Mash DC, Tyndale RF. (2003) Smoking, alcoholism and genetic polymorphisms alter CYP2B6 levels in human brain. Neuropharmacology 45:122–132. [DOI] [PubMed] [Google Scholar]
- Miksys S, Rao Y, Hoffmann E, Mash DC, Tyndale RF. (2002) Regional and cellular expression of CYP2D6 in human brain: higher levels in alcoholics. J Neurochem 82:1376–1387. [DOI] [PubMed] [Google Scholar]
- Miksys S, Tyndale RF. (2009) Brain drug-metabolizing cytochrome P450 enzymes are active in vivo, demonstrated by mechanism-based enzyme inhibition. Neuropsychopharmacology 34:634–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miksys S, Tyndale RF. (2013) Cytochrome P450-mediated drug metabolism in the brain. J Psychiatry Neurosci 38:152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan ET, Goralski KB, Piquette-Miller M, Renton KW, Robertson GR, Chaluvadi MR, Charles KA, Clarke SJ, Kacevska M, Liddle C, et al. (2008) Regulation of drug-metabolizing enzymes and transporters in infection, inflammation, and cancer. Drug Metab Dispos 36:205–216. [DOI] [PubMed] [Google Scholar]
- Morrissey KM, Stocker SL, Wittwer MB, Xu L, Giacomini KM. (2013) Renal transporters in drug development. Annu Rev Pharmacol Toxicol 53:503–529. [DOI] [PubMed] [Google Scholar]
- Nagar S, Remmel RP. (2006) Uridine diphosphoglucuronosyltransferase pharmacogenetics and cancer. Oncogene 25:1659–1672. [DOI] [PubMed] [Google Scholar]
- Nakagomi-Hagihara R, Nakai D, Tokui T. (2007) Inhibition of human organic anion transporter 3 mediated pravastatin transport by gemfibrozil and the metabolites in humans. Xenobiotica 37:416–426. [DOI] [PubMed] [Google Scholar]
- Nakamura A, Nakajima M, Yamanaka H, Fujiwara R, Yokoi T. (2008) Expression of UGT1A and UGT2B mRNA in human normal tissues and various cell lines. Drug Metab Dispos 36:1461–1464. [DOI] [PubMed] [Google Scholar]
- Nave R, Fisher R, McCracken N. (2007) In vitro metabolism of beclomethasone dipropionate, budesonide, ciclesonide, and fluticasone propionate in human lung precision-cut tissue slices. Respir Res 8:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave R, Watz H, Hoffmann H, Boss H, Magnussen H. (2010) Deposition and metabolism of inhaled ciclesonide in the human lung. Eur Respir J 36:1113–1119. [DOI] [PubMed] [Google Scholar]
- Nelson JA, Santos G, Herbert BH. (1984) Mechanisms for the renal secretion of cisplatin. Cancer Treat Rep 68:849–853. [PubMed] [Google Scholar]
- Nies AT, Schwab M, Keppler D. (2008) Interplay of conjugating enzymes with OATP uptake transporters and ABCC/MRP efflux pumps in the elimination of drugs. Expert Opin Drug Metab Toxicol 4:545–568. [DOI] [PubMed] [Google Scholar]
- O'Donnell S, O'Morain CA. (2010) Therapeutic benefits of budesonide in gastroenterology. Ther Adv Chronic Dis 1:177–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okeley NM, VanEpps HA, Zhang X, Setter JR, Burke PJ, Hamilton JZ, Lyon RP. (2014) Abstract 4465: Differential MMAE delivery from ADCs utilizing the valine-citrulline-PAB and β-glucuronide cleavable linker systems. Cancer Res 74:4465. [Google Scholar]
- Oldendorf WH, Hyman S, Braun L, Oldendorf SZ. (1972) Blood-brain barrier: penetration of morphine, codeine, heroin, and methadone after carotid injection. Science 178:984–986. [DOI] [PubMed] [Google Scholar]
- Pan BF, Sweet DH, Pritchard JB, Chen R, Nelson JA. (1999) A transfected cell model for the renal toxin transporter, rOCT2. Toxicol Sci 47:181–186. [DOI] [PubMed] [Google Scholar]
- Pan X, Wang L, Gründemann D, Sweet DH. (2013) Interaction of Ethambutol with human organic cation transporters of the SLC22 family indicates potential for drug-drug interactions during antituberculosis therapy. Antimicrob Agents Chemother 57:5053–5059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pang KS, Maeng HJ, Fan J. (2009) Interplay of transporters and enzymes in drug and metabolite processing. Mol Pharm 6:1734–1755. [DOI] [PubMed] [Google Scholar]
- Pawlowski A, Jansson M, Sköld M, Rottenberg ME, Källenius G. (2012) Tuberculosis and HIV co-infection. PLoS Pathog 8:e1002464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peppercorn MA, Goldman P. (1972) The role of intestinal bacteria in the metabolism of salicylazosulfapyridine. J Pharmacol Exp Ther 181:555–562. [PubMed] [Google Scholar]
- Perez HL, Cardarelli PM, Deshpande S, Gangwar S, Schroeder GM, Vite GD, Borzilleri RM. (2014) Antibody-drug conjugates: current status and future directions. Drug Discov Today 19:869–881. [DOI] [PubMed] [Google Scholar]
- Planas JM, Alfaras I, Colom H, Juan ME. (2012) The bioavailability and distribution of trans-resveratrol are constrained by ABC transporters. Arch Biochem Biophys 527:67–73. [DOI] [PubMed] [Google Scholar]
- Pond SM, Tozer TN. (1984) First-pass elimination. Basic concepts and clinical consequences. Clin Pharmacokinet 9:1–25. [DOI] [PubMed] [Google Scholar]
- Prentice KJ, Luu L, Allister EM, Liu Y, Jun LS, Sloop KW, Hardy AB, Wei L, Jia W, Fantus IG, et al. (2014) The furan fatty acid metabolite CMPF is elevated in diabetes and induces β cell dysfunction. Cell Metab 19:653–666. [DOI] [PubMed] [Google Scholar]
- Pritchard JB, Miller DS. (1993) Mechanisms mediating renal secretion of organic anions and cations. Physiol Rev 73:765–796. [DOI] [PubMed] [Google Scholar]
- Pritchard JB, Sweet DH, Miller DS, Walden R. (1999) Mechanism of organic anion transport across the apical membrane of choroid plexus. J Biol Chem 274:33382–33387. [DOI] [PubMed] [Google Scholar]
- Ramesh R, Kozhaya L, McKevitt K, Djuretic IM, Carlson TJ, Quintero MA, McCauley JL, Abreu MT, Unutmaz D, Sundrud MS. (2014) Pro-inflammatory human Th17 cells selectively express P-glycoprotein and are refractory to glucocorticoids. J Exp Med 211:89–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsay RR, Krueger MJ, Youngster SK, Gluck MR, Casida JE, Singer TP. (1991) Interaction of 1-methyl-4-phenylpyridinium ion (MPP+) and its analogs with the rotenone/piericidin binding site of NADH dehydrogenase. J Neurochem 56:1184–1190. [DOI] [PubMed] [Google Scholar]
- Ravindranath V, Strobel HW. (2013) Cytochrome P450-mediated metabolism in brain: functional roles and their implications. Expert Opin Drug Metab Toxicol 9:551–558. [DOI] [PubMed] [Google Scholar]
- Remmel RP and Zhou J(2009) UDP-glucuronosyltransferases, in Handbook of Drug Metabolism (Pearson PG and Wienkers LC eds) pp 137–178, Informa Healthcare, New York. [Google Scholar]
- Renton KW. (2004) Cytochrome P450 regulation and drug biotransformation during inflammation and infection. Curr Drug Metab 5:235–243. [DOI] [PubMed] [Google Scholar]
- Renton KW. (2005) Regulation of drug metabolism and disposition during inflammation and infection. Expert Opin Drug Metab Toxicol 1:629–640. [DOI] [PubMed] [Google Scholar]
- Riches Z, Stanley EL, Bloomer JC, Coughtrie MW. (2009) Quantitative evaluation of the expression and activity of five major sulfotransferases (SULTs) in human tissues: the SULT “pie”. Drug Metab Dispos 37:2255–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RL, Luty SE, Mulder RT, Joyce PR, Kennedy MA. (2004) Association between cytochrome P450 2D6 genotype and harm avoidance. Am J Med Genet B Neuropsychiatr Genet 127B:90–93. [DOI] [PubMed] [Google Scholar]
- Rossi R, Milzani A, Dalle-Donne I, Giustarini D, Lusini L, Colombo R, Di Simplicio P. (2002) Blood glutathione disulfide: in vivo factor or in vitro artifact? Clin Chem 48:742–753. [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, Mol CA, van Deemter L. (1996) P-glycoprotein in the blood-brain barrier of mice influences the brain penetration and pharmacological activity of many drugs. J Clin Invest 97:2517–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schinkel AH, Wagenaar E, van Deemter L, Mol CA, Borst P. (1995) Absence of the mdr1a P-Glycoprotein in mice affects tissue distribution and pharmacokinetics of dexamethasone, digoxin, and cyclosporin A. J Clin Invest 96:1698–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah DK, King LE, Han X, Wentland JA, Zhang Y, Lucas J, Haddish-Berhane N, Betts A, Leal M. (2014) A priori prediction of tumor payload concentrations: preclinical case study with an auristatin-based anti-5T4 antibody-drug conjugate. AAPS J 16:452–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan S, Iwuchukwu OF, Canney DJ, Zimmerman CL, Nagar S. (2012) In vivo-formed versus preformed metabolite kinetics of trans-resveratrol-3-sulfate and trans-resveratrol-3-glucuronide. Drug Metab Dispos 40:1993–2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharan S, Nagar S. (2013) Pulmonary metabolism of resveratrol: in vitro and in vivo evidence. Drug Metab Dispos 41:1163–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen BQ, Xu K, Liu L, Raab H, Bhakta S, Kenrick M, Parsons-Reponte KL, Tien J, Yu SF, Mai E, et al. (2012) Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat Biotechnol 30:184–189. [DOI] [PubMed] [Google Scholar]
- Shu Y, Brown C, Castro RA, Shi RJ, Lin ET, Owen RP, Sheardown SA, Yue L, Burchard EG, Brett CM, et al. (2008) Effect of genetic variation in the organic cation transporter 1, OCT1, on metformin pharmacokinetics. Clin Pharmacol Ther 83:273–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sindrup SH, Arendt-Nielsen L, Brøsen K, Bjerring P, Angelo HR, Eriksen B, Gram LF. (1992) The effect of quinidine on the analgesic effect of codeine. Eur J Clin Pharmacol 42:587–591. [DOI] [PubMed] [Google Scholar]
- Sindrup SH, Brøsen K, Bjerring P, Arendt-Nielsen L, Larsen U, Angelo HR, Gram LF. (1990) Codeine increases pain thresholds to copper vapor laser stimuli in extensive but not poor metabolizers of sparteine. Clin Pharmacol Ther 48:686–693. [DOI] [PubMed] [Google Scholar]
- Somers GI, Lindsay N, Lowdon BM, Jones AE, Freathy C, Ho S, Woodrooffe AJ, Bayliss MK, Manchee GR. (2007) A comparison of the expression and metabolizing activities of phase I and II enzymes in freshly isolated human lung parenchymal cells and cryopreserved human hepatocytes. Drug Metab Dispos 35:1797–1805. [DOI] [PubMed] [Google Scholar]
- Steenland K, Dick RB, Howell RJ, Chrislip DW, Hines CJ, Reid TM, Lehman E, Laber P, Krieg EFJ, Jr, Knott C. (2000) Neurologic function among termiticide applicators exposed to chlorpyrifos. Environ Health Perspect 108:293–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultatos LG. (1994) Mammalian toxicology of organophosphorus pesticides. J Toxicol Environ Health 43:271–289. [DOI] [PubMed] [Google Scholar]
- Sweet DH. (2005) Organic anion transporter (Slc22a) family members as mediators of toxicity. Toxicol Appl Pharmacol 204:198–215. [DOI] [PubMed] [Google Scholar]
- Sweet DH. (2010) Renal organic cation and anion transport: From physiology to genes, in Comprehensive Toxicology (McQueen CA. ed) pp 23–53, Elsevier, Ltd., Oxford. [Google Scholar]
- Sweet DH, Miller DS, Pritchard JB. (2001) Ventricular choline transport: a role for organic cation transporter 2 expressed in choroid plexus. J Biol Chem 276:41611–41619. [DOI] [PubMed] [Google Scholar]
- Tang J, Cao Y, Rose RL, Brimfield AA, Dai D, Goldstein JA, Hodgson E. (2001) Metabolism of chlorpyrifos by human cytochrome P450 isoforms and human, mouse, and rat liver microsomes. Drug Metab Dispos 29:1201–1204. [PubMed] [Google Scholar]
- Ung D, Nagar S. (2007) Variable sulfation of dietary polyphenols by recombinant human sulfotransferase (SULT) 1A1 genetic variants and SULT1E1. Drug Metab Dispos 35:740–746. [DOI] [PubMed] [Google Scholar]
- Urquhart BL, Tirona RG, Kim RB. (2007) Nuclear receptors and the regulation of drug-metabolizing enzymes and drug transporters: implications for interindividual variability in response to drugs. J Clin Pharmacol 47:566–578. [DOI] [PubMed] [Google Scholar]
- van der Heijden JW, Oerlemans R, Tak PP, Assaraf YG, Kraan MC, Scheffer GL, van der Laken CJ, Lems WF, Scheper RJ, Dijkmans BA, et al. (2009) Involvement of breast cancer resistance protein expression on rheumatoid arthritis synovial tissue macrophages in resistance to methotrexate and leflunomide. Arthritis Rheum 60:669–677. [DOI] [PubMed] [Google Scholar]
- van de Wetering K, Burkon A, Feddema W, Bot A, de Jonge H, Somoza V, Borst P. (2009) Intestinal breast cancer resistance protein (BCRP)/Bcrp1 and multidrug resistance protein 3 (MRP3)/Mrp3 are involved in the pharmacokinetics of resveratrol. Mol Pharmacol 75:876–885. [DOI] [PubMed] [Google Scholar]
- VanWert AL, Gionfriddo MR, Sweet DH. (2010) Organic anion transporters: discovery, pharmacology, regulation and roles in pathophysiology. Biopharm Drug Dispos 31:1–71. [DOI] [PubMed] [Google Scholar]
- Videira M, Reis RL, Brito MA. (2014) Deconstructing breast cancer cell biology and the mechanisms of multidrug resistance. Biochim Biophys Acta 1846:312–325. [DOI] [PubMed] [Google Scholar]
- Wang L, Sweet DH. (2013) Renal organic anion transporters (SLC22 family): expression, regulation, roles in toxicity, and impact on injury and disease. AAPS J 15:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White AG. (1966) Uremic serum inhibition of renal paraaminohippurate transport. Proc Soc Exp Biol Med 123:309–310. [DOI] [PubMed] [Google Scholar]
- Wikoff WR, Nagle MA, Kouznetsova VL, Tsigelny IF, Nigam SK. (2011) Untargeted metabolomics identifies enterobiome metabolites and putative uremic toxins as substrates of organic anion transporter 1 (Oat1). J Proteome Res 10:2842–2851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka H, Nakajima M, Fukami T, Sakai H, Nakamura A, Katoh M, Takamiya M, Aoki Y, Yokoi T. (2005) CYP2A6 AND CYP2B6 are involved in nornicotine formation from nicotine in humans: interindividual differences in these contributions. Drug Metab Dispos 33:1811–1818. [DOI] [PubMed] [Google Scholar]
- Yamazaki H, Inoue K, Hashimoto M, Shimada T. (1999) Roles of CYP2A6 and CYP2B6 in nicotine C-oxidation by human liver microsomes. Arch Toxicol 73:65–70. [DOI] [PubMed] [Google Scholar]
- Yanev S, Kent UM, Pandova B, Hollenberg PF. (1999) Selective mechanism-based inactivation of cytochromes P-450 2B1 and P-450 2B6 by a series of xanthates. Drug Metab Dispos 27:600–604. [PubMed] [Google Scholar]
- Yokoo S, Masuda S, Yonezawa A, Terada T, Katsura T, Inui K. (2008) Significance of organic cation transporter 3 (SLC22A3) expression for the cytotoxic effect of oxaliplatin in colorectal cancer. Drug Metab Dispos 36:2299–2306. [DOI] [PubMed] [Google Scholar]
- Yokoo S, Yonezawa A, Masuda S, Fukatsu A, Katsura T, Inui K. (2007) Differential contribution of organic cation transporters, OCT2 and MATE1, in platinum agent-induced nephrotoxicity. Biochem Pharmacol 74:477–487. [DOI] [PubMed] [Google Scholar]
- Yue J, Miksys S, Hoffmann E, Tyndale RF. (2008) Chronic nicotine treatment induces rat CYP2D in the brain but not in the liver: an investigation of induction and time course. J Psychiatry Neurosci 33:54–63. [PMC free article] [PubMed] [Google Scholar]
- Zamek-Gliszczynski MJ, Chu X, Polli JW, Paine MF, Galetin A. (2014) Understanding the transport properties of metabolites: case studies and considerations for drug development. Drug Metab Dispos 42:650–664. [DOI] [PubMed] [Google Scholar]
- Zanger UM, Raimundo S, Eichelbaum M. (2004) Cytochrome P450 2D6: overview and update on pharmacology, genetics, biochemistry. Naunyn Schmiedebergs Arch Pharmacol 369:23–37. [DOI] [PubMed] [Google Scholar]
- Zhang L, Huang M, Blair IA, Penning TM. (2013) Interception of benzo[a]pyrene-7,8-dione by UDP glucuronosyltransferases (UGTs) in human lung cells. Chem Res Toxicol 26:1570–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Zhang Y, Huang SM. (2009) Scientific and regulatory perspectives on metabolizing enzyme-transporter interplay and its role in drug interactions: challenges in predicting drug interactions. Mol Pharm 6:1766–1774. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Fang JL, Lazarus P. (2002) Glucuronidation: an important mechanism for detoxification of benzo[a]pyrene metabolites in aerodigestive tract tissues. Drug Metab Dispos 30:397–403. [DOI] [PubMed] [Google Scholar]
- Zhou K, Khokhar JY, Zhao B, Tyndale RF. (2013) First demonstration that brain CYP2D-mediated opiate metabolic activation alters analgesia in vivo. Biochem Pharmacol 85:1848–1855. [DOI] [PMC free article] [PubMed] [Google Scholar]